

Supplemental Digital Content is available in the text.
Keywords: aging, arteries, endoplasmic reticulum, vascular calcification, warfarin
Objective:
Vascular calcification is common among aging populations and mediated by vascular smooth muscle cells (VSMCs). The endoplasmic reticulum (ER) is involved in protein folding and ER stress has been implicated in bone mineralization. The role of ER stress in VSMC-mediated calcification is less clear.
Approach and Results:
mRNA expression of the ER stress markers PERK (PKR (protein kinase RNA)-like ER kinase), ATF (activating transcription factor) 4, ATF6, and Grp78 (glucose-regulated protein, 78 kDa) was detectable in human vessels with levels of PERK decreased in calcified plaques compared to healthy vessels. Protein deposition of Grp78/Grp94 was increased in the matrix of calcified arteries. Induction of ER stress accelerated human primary VSMC-mediated calcification, elevated expression of some osteogenic markers (Runx2 [RUNX family transcription factor 2], OSX [Osterix], ALP [alkaline phosphatse], BSP [bone sialoprotein], and OPG [osteoprotegerin]), and decreased expression of SMC markers. ER stress potentiated extracellular vesicle (EV) release via SMPD3 (sphingomyelin phosphodiesterase 3). EVs from ER stress-treated VSMCs showed increased Grp78 levels and calcification. Electron microscopy confirmed the presence of Grp78/Grp94 in EVs. siRNA (short interfering RNA) knock-down of Grp78 decreased calcification. Warfarin-induced Grp78 and ATF4 expression in rat aortas and VSMCs and increased calcification in an ER stress-dependent manner via increased EV release.
Conclusions:
ER stress induces vascular calcification by increasing release of Grp78-loaded EVs. Our results reveal a novel mechanism of action of warfarin, involving increased EV release via the PERK-ATF4 pathway, contributing to calcification. This study is the first to show that warfarin induces ER stress and to link ER stress to cargo loading of EVs.
Highlights.
The chaperone Grp78 (glucose-regulated protein, 78 kDa) is deposited in calcified human arteries in a pattern consistent with extracellular vesicles.
Under endoplasmic reticulum stress conditions, there is increased release of Grp78 loaded extracellular vesicles.
Grp78 mediates endoplasmic reticulum stress–induced calcification of human primary vascular smooth muscle cells by enhancing the calcific properties of extracellular vesicles.
The widely used anticoagulant warfarin induces endoplasmic reticulum stress specifically via the PERK (PKR [protein kinase RNA]-like ER kinase)-ATF (activating transcription factor) 4 pathway, both in rats in vivo and human primary vascular smooth muscle cells.
Mechanisms previously credited with mediating endoplasmic reticulum stress-driven calcification (osteogenic differentiation, oxidative stress, and apoptosis) are not relevant in all contexts of endoplasmic reticulum stress activation.
Vascular calcification (VC) is the process of deposition of calcium phosphate crystals in the ECM (extracellular matrix) of the blood vessel wall.1 The presence of VC poses an increased risk of cardiovascular and all-cause mortality and is an independent risk factor predictive of cardiac events.2 Nearly all patients with cardiovascular disease have some degree of calcification, and in asymptomatic adults, prevalence of coronary calcification corresponds with age; among 60-year-olds, ≈60% have VC.3,4
VC can be categorized into 2 types: medial and intimal, depending on its localization in the vessel wall.5 Medial calcification is associated primarily with aging, diabetes, and chronic kidney disease. Intimal calcification predominantly colocalizes with lipid-rich/necrotic regions of atherosclerotic plaques.6
Vascular smooth muscle cells (VSMCs) play a crucial role in regulating atherosclerotic plaque formation and VC.7–9 In response to cellular stress, VSMCs undergo calcification via several mechanisms including apoptosis,10 release of extracellular vesicles (EVs),11,12 loss of calcification inhibitors,13,14 aging-related DNA damage,15 oxidative stress,16 and osteogenic differentiation.17 Many of these processes have parallels with physiological bone formation, where chondrocyte apoptosis, downregulation of calcification inhibitors, release of EVs, and expression of bone-specific genes by osteoblasts lead to bone mineralization.18
The endoplasmic reticulum (ER) is the first organelle of the secretory pathway, where most secreted and transmembrane proteins are folded and mature. ER stress occurs when the influx of unfolded proteins to the ER exceeds its capacity to fold them, resulting in activation of a signaling pathway called the unfolded protein response (UPR).19,20 Three ER stress transducers are responsible for sensing an increased load of unfolded proteins and define 3 branches of the UPR: IRE (inositol-requiring protein) 1, ATF (activating transcription factor) 6, and PERK (PKR [protein kinase RNA]-like ER kinase).19 Activation of the UPR results in a decrease in protein influx to the ER and expression of various genes encoding chaperones (eg, Grp78 [glucose-regulated protein, 78 kDa] and Grp94) that mitigate the effects of increased load of unfolded proteins.21,22 However, if ER stress is overwhelming and cannot be resolved it may lead to cell death.23
Importantly, ER stress and the UPR have been shown to be crucial for bone development. All 3 branches of the UPR are activated during stages of bone formation to regulate expression of osteogenic genes.24–28 Because bone mineralization and VSMC calcification share mechanistic pathways, we hypothesized ER stress might also be involved in regulating pathological VC. Indeed, studies to date suggest that ER stress can accompany VC in mice and rats and increase VSMC osteogenic gene expression and calcification in vitro.29–37 However, other key molecular mechanisms involved in VSMC calcification have not been studied. Therefore, we investigated how ER stress influences VC and define a role for this signaling pathway in regulating EV release and cargo loading.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Cell Culture and Treatments
Human aortic VSMCs were isolated and cultured, as described previously.38 At passage 3, expression of SM22α (smooth muscle protein 22α), CNN1 (calponin 1), and p-MLC (phosphorylated myosin light chain) was confirmed by immunocytochemistry. Cells were used for experiments at passage 4 to 12. VSMCs were collected from deceased adult organ donors with local ethics committee approval (Cambridge Local Research Ethics Committee LREC 97/084), characterized, and archived in the laboratory. All human materials were handled in compliance with the Human Tissue Act (2004, United Kingdom). Additionally, human aortic samples were obtained from patients undergoing open aortic surgery at Maastricht University Medical Centre. Collection, storage, and use of tissue and patient data were performed in agreement with the Dutch Code for Proper Secondary Use of Human Tissue (http://www.fmwv.nl). This study complies with the Declaration of Helsinki.
VSMCs were treated with the following compounds at stated final concentrations: tunicamycin (T7765, 0.04–0.4 µg/mL; Sigma), thapsigargin (T9033, 0.01–0.2 µg/mL; Sigma), warfarin (P5104, 10–200 µmol/L; Sigma), 4-phenylbutyric acid (PBA, P2,100-5, 0.5–1 mmol/L; Sigma), CaCl2 (C7902, 2.7 mmol/L; Sigma), NaK2PO4 (S-5011, 2.5 mmol/L; Sigma), H2O2 (H1009, 1–20 µmol/L; Sigma), IRE1 inhibitor CB5305630 (AOB4004, 2–25 µM39; Aobius), PERK inhibitor GSK2656157 (B2175, 0.1–10 µmol/L40; ApexBio), SMPD3 (sphingomyelin phosphodiesterase 3) inhibitor spiroexpoxide (0.5 µmol/L, SC-20272; Santa Cruz). For 24 hour assays, M199 with 0.5% FBS was used, for longer treatments with 2.5% or 5% FBS.
Transient Gene Knock-Down With siRNA
Cells were incubated in the complete medium for 24 hours and then transfection medium was applied for 48 hours. For one well of a 48 well plate, the transfection medium contained 250 μL M199 with 20% FBS and PSG, 25 μL OptiMEM (Gibco), 3 μL HiPerfect (Qiagen), and 3 pmol siRNA (short interfering RNA) oligonucleotide smartpool (Grp78 siRNA L-008198-00-0005, GE Dharmacon; Nontargetting siRNA 1022076, Qiagen).
Immunohistochemistry
Eight human carotid arteries and 14 human aortic samples were analyzed. The samples were collected from adult organ transplant donors, characterized, and archived in the laboratory. All human materials were handled in compliance with the Human Tissue Act (2004, United Kingdom) with approval of Cambridge Local Research Ethics Committee (LREC 97/084). Samples of thoracic aorta from wild type male 21 to 28 days old Sprague-Dawley rats from a previously published study.41 All animal studies were approved by the Ethics Committee for animal experiments of Kings College London, United Kingdom, in compliance with the UK Home Office Guidance on the Operation of the Animals (Scientific Procedures) Act, 1986. Rats were purchased from Charles River Laboratories (United Kingdom) and fed 3 mg warfarin and 1.5 mg vitamin K1 per gram of food (Arie Blok diets, 4165.00) for 1 week or 4 weeks (rats used for immunohistochemistry samples) or 11 days (rats used for Western blotting samples). The control groups consisted of rats fed a normal laboratory diet (PicoLab rodent diet 20). These human and rat tissue samples were fixed in 10% neutral buffered formalin and were then embedded in paraffin wax blocks and cut down in 7-µm thick sections. Immunohistochemistry was performed in serial sections, as described before42 using VECTASTAIN ABC kit (PK6102; Vector Labs), anti-Grp78, and Grp94 (anti-KDEL, SPA-827, 1:500; Assay Designs) anti-ATF4 (sc-200, 1:250; Santa Cruz) antibodies. Sections were additionally stained for von Kossa and Alizarin Red S to visualize calcification. The stained tissue sections were quantified using scores from 1 to 5 and distributed between nuclear and extracellular expression of Grp 78 and 94 of each vessel. The calcification was scored using grades from 0 to 4 where calcified areas were qualitatively graded; 0: no calcification, 1: <25%, 2: <50%, 3: <75%, and 4: >75%. The collected data from human carotid and aortic samples have been pooled together for statistical analysis.
Apoptosis and Necrosis Analysis
Analysis of cell death was carried out using the Green FLICA Caspases 3&7 Assay Kit (ImmunoChemistry) according to the manufacturers’ protocols, using NucleoCounter NC-3000 (Chemometec). The percentage of healthy, apoptotic, and necrotic cells in each sample was determined based on the scatter plots generated by the NucleoCounter software.
Intracellular Reactive Oxygen Species Measurement
VSMCs were stained with 20 μM DCFDA (2’,7’ –dichlorofluorescein diacetate; Abcam) in phenol red-free DMEM with 0.5% FBS for 1 hour. Afterward, treatments were applied in KRPG (Krebs-Ringer phosphate glucose) buffer (145 mmol/L NaCl, 5.7 mmol/L NaH2PO4*2 H2O, 4.86 mmol/L KCl, 0.54 mmol/L CaCl2*2 H2O, 1.22 mmol/L MgSO4*7 H2O, 5.5 mmol/L glucose, pH 7.35) containing 1:1000 Hoechst dye for 4 hours. Fluorescence was measured and normalized to cell count using the Cytation 3 imaging reader (BioTek).
Western Blotting and Immunocytochemistry
Western blotting was performed, as described before.11 Concentration of protein in samples was measured using DC (detergent compatible) Protein assay (BioRad). The following antibodies were used: Grp78 and Grp94 (anti-KDEL, SPA-827, 1:2500; Assay Designs), GAPDH (sc-25778, 1:5000; Santa Cruz), CNN1 (ab46794, 1:5000; Abcam), SM22α (ab14106, 1:1000; Abcam), p-MLC (36755, 1:500; Cell Signalling), ALP (alkaline phosphatse; ab108337, 1:10 000; Abcam), CD63 (556019, 1:500; BD Bioscience), TSG101 (tumor susceptibility gene 101 protein; T5701, 1:500; Sigma), SERCA2 (sarco/endoplasmic reticulum Ca2+-ATPase; 564702, 1:500; Calbiochem) α-actinin-4 (ab108198, 1:500; Abcam), pPERK (ab192591,1:1500; Abcam) anti-mouse (926-32210, 1:10000; Li-Cor and p0447, 1:3000; Dako) and anti-rabbit (926-68071, 1:10000; Li-Cor and 7074S, 1:3000; Cell Signalling). Total protein was quantified in gels using Coomassie Brilliant Blue or Revert (Li-Cor). All blots shown in figures are from the same membrane, cropped for clarity of presentation. Immunocytochemistry was performed as described before,43 using the following antibodies: Grp78, Santa Cruz, 1:100, total MGP (matrix Gla-protein), VitaK, 1:75, carboxylated MGP, VitaK, 1:75, uncarboxylated MGP, VitaK, 1:250,44 anti-mouse–fluorescein isothiocyanate (Dako, F0205, 1:250), anti-rabbit FITC (115-095-068, 1:250; Jackson). Images were taken and signal quantified using the Cytation 3 imaging reader (BioTek).
Dot Blot Analysis
Dot blots were performed as described before.45 Briefly, EVs isolated by ultracentrifugation were absorbed onto nitrocellulose membranes, which were blocked with 5% nonfat milk or 3% bovine serum albumin (BSA; TSG101) in PBS in the absence or presence 0.1% (v/v) Tween-20. Antibodies were used as described above.
Quantitative Real-Time Polymerase Chain Reaction
Quantitative polymerase chain reaction (PCR) was performed as described before11 with primers listed in the Table. The quality and concentration of RNA were measured using a Nanodrop ND 1000, only samples with a 260/280 ratio above 1.8 were used.
Table.
QPCR Primers
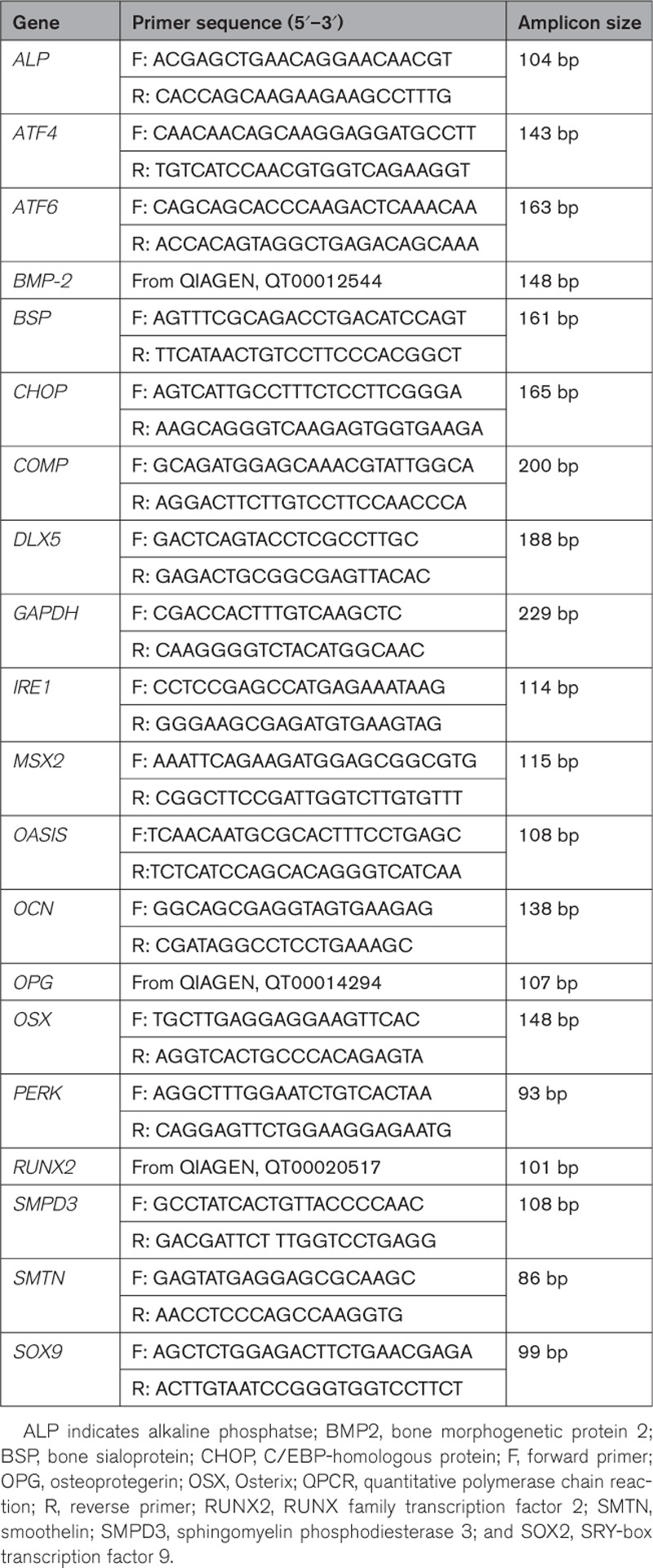
Calcification Assays
Cells were lysed with 0.1 M HCl. Ca2+ concentrations were determined colorimetrically using the o-cresolphthalein assay (adapted from Gitelman46) and normalized to protein concentrations. To visualize calcification, VSMCs were fixed and stained with Alizarin Red S (adapted from Bancroft and Stevens47).
Alkaline Phosphatase Activity Assay
ALP activity in cell lysates was determined colorimetrically using pNPP and normalized to protein concentration.
Extracellular Vesicle Isolation and Calcification
EVs were isolated by differential ultracentrifugation as described before.11 EVs where isolated from 2 different fractions (2000 g and 100 000 g pellet) and equal amounts of EVs from either fraction were used to study calcification in collagen-coated wells. Calcification was quantified using o-cresolphthalein, as described above.
Bead Capture Assay
Bead capture assays were performed as described before.11 Anti-human CD63 antibody was immobilized on beads. VSMC culture media was incubated with anti-CD63–coated. Beads were incubated with anti-CD81–PE (phycoerythrin) antibodies and analyzed by flow cytometry. Arbitrary Units were calculated as mean fluorescence units x percentage of positive beads and normalized to the number of VSMCs.
Electron Microscopy and Immunogold Labeling
For immunolabeling, VSMCs were fixed with 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M PHEM (60 mM 1,4 piperazine diethylsulfonic acid [PIPES], 25 mM N-2-hydroxyethylpiperazine Nl-2-ethanesulfonic acid [HEPES], 10 mM EGTA, and 2 mM MgCl2, pH 6.9) buffer followed by washing in PB containing 50 mmol/L glycine, scraped in 1% gelatin, embedded in 12% gelatin, and infused in 2.3 M sucrose. EVs were isolated by ultracentrifugation as described above and pellets were fixed with 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1M PHEM buffer. After washing with PB containing 50 mmol/L glycine, vesicles were embedded in 12% gelatin and infused in 2.3 M sucrose. For both cells and EVs mounted in gelatin, blocks were frozen in liquid nitrogen. Sections were prepared in an ultracryomicrotome (Leica EM Ultracut UC6/FC6, Leica Microsystems), collected with 2% methylcellulose in 2.3 M sucrose, and incubated at room temperature on drops of PBS for 60 minutes at 37 °C, followed by 50 mmol/L gelatin in PBS for 10 minutes and 1% BSA in PBS for 15 minutes at room temperature. Then, cell sections were incubated with anti-Grp78/Grp94 coupled to protein-A 6 nm diameter colloidal gold (1:50, conjugated by Aurion to anti-KDEL, SPA-827; Assay Designs) diluted in 1% BSA in PBS. This was followed by washes with drops of PBS for 10 minutes, and 2 washes with distilled water. Cryosections with EVs were incubated at room temperature on drops of PBS for 60 minutes at 37 °C, followed by 50 mmol/L gelatin in PBS for 10 minutes and 1% BSA in PBS for 15 minutes at room temperature. Then, they were incubated with mouse anti-CD63 (1:5, BD Biosciences, 556019), rabbit anti-TSG101 (1:5, Sigma, T5701) diluted in 1% BSA in PBS for 45 minutes. After 3 washes with drops of PBS for 10 minutes, sections were incubated for 20 minutes in protein-A coupled to 10 nm diameter colloidal gold particles (1:50, CMC Utrecht) diluted in 1% BSA. Sections were fixed with 1% glutaraldehyde in PBS for 5 minutes, followed by 50 mmol/L gelatin in PBS for 10 minutes, and 1% BSA in PBS for 15 minutes. Then, they were incubated with anti-Grp78/Grp94 coupled to protein-A 6 nm diameter colloidal gold (1:50, conjugated by Aurion to anti-KDEL, SPA-827; Assay Designs) diluted in 1% BSA in PBS. This was followed by washes with drops of PBS for 10 minutes, 2 washes of distilled water. Grids were embedded in a thin layer of 1.8% methylcellulose (25 cP) containing 0.4% uranyl acetate. As a control for nonspecific binding of the colloidal gold-conjugated antibody, the primary antibody was omitted. Sections were observed in an Electron Microscope Tecnai T12 Spirit (Thermo Fisher Scientific) equipped with an Eagle 4kx4k CCD (charge-coupled device) camera.
Data Analysis
All data represent 3 independent experiments, unless stated otherwise. Data analysis was performed with pooled data from male and female subjects, as we did not have sufficient material for large enough groups to derive a meaningful comparison between the sexes. Graphs show mean+SD with individual data points. Normality of all data was tested using the Shapiro-Wilks test. Variance was tested using the F test alongside a t test and Brown-Forsythe test alongside 1-way ANOVA. Where appropriate t tests, 1-way ANOVA with Tukey or Dunnett post hoc, and 2-way ANOVA with Sidak correction for multiple comparison tests were performed. When the data failed the equal variance or normality tests, the nonparametric Mann-Whitney, 1-sample Wilcoxon and Kruskal-Wallis tests were used, as indicated in figure legends. Statistical analysis was performed in GraphPad Prism 8.2.1. Statistical significance is indicated with asterisks: *P between 0.05 and 0.01, **P between 0.01 and 0.001, ***P<0.001.
Results
Increased Extracellular Grp78 and Grp94 Deposition in Calcified Human Vessels
We examined ER stress signaling in human arteries by quantitative PCR and immunocytochemistry comparing noncalcified with calcified arteries. The ER stress markers PERK, ATF6, ATF4, CHOP (C/EBP-homologous protein), and Grp78 were all expressed in human carotid and aortic samples. All markers showed a trend towards reduced expression in early disease (fatty streak lesions) and calcified arteries compared with healthy, with the lowest expression in calcified arteries (Table I in the Data Supplement; Figure 1A). This trend was statistically significant for PERK.
Figure 1.
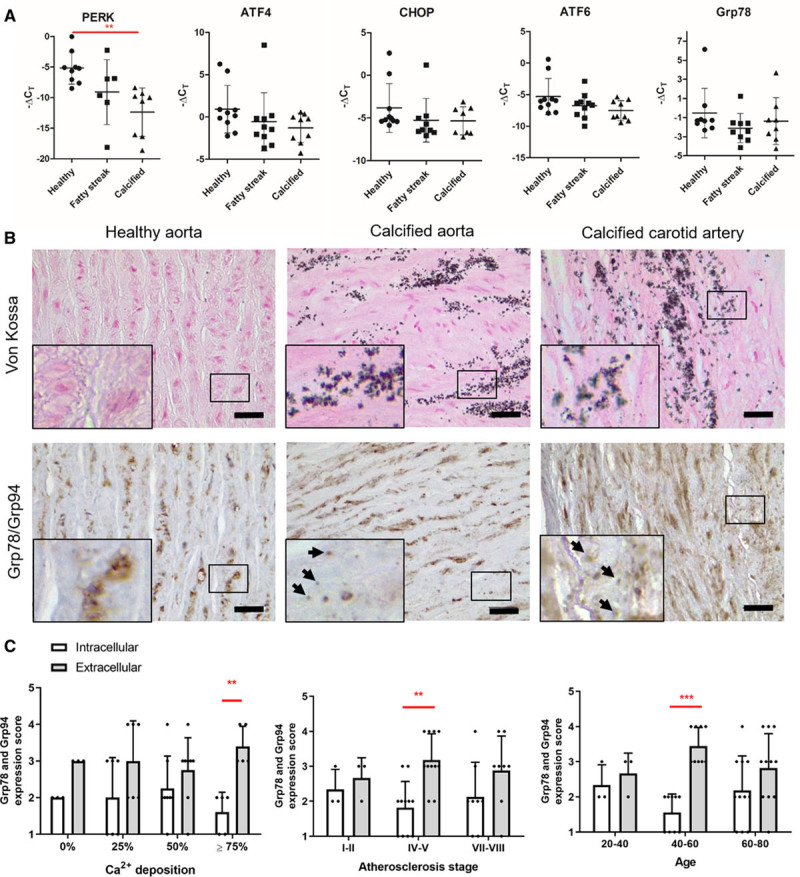
Vascular calcification is associated with changes in endoplasmic reticulum (ER) stress markers in human vasculature. A, Quantitative real-time polymerase chain reaction analysis of ER stress markers in healthy, fatty streak, and calcified human aortas. ΔCT values were calculated relative to GAPDH expression in corresponding samples. Graphs show individual data points and mean+SD, n=10 for each group, ANOVA with Tukey post hoc test (PERK [PKR (protein kinase RNA)-like ER kinase]) or Kruskal-Wallis test (ATF [activating transcription factor] 4, CHOP [C/EBP-homologous protein], ATF6, Grp78 [glucose-regulated protein, 78 kDa]) were performed. B, Immunohistochemical analysis of Grp78 and Grp94 expression in artery samples from human donors. Arrows indicate extracellular, punctate Grp78/Grp94 staining. Scale bars are 50 µm. C, Quantification of Grp78 and Grp94 expression scores. Calcification was scored based on von Kossa staining. Graphs show mean+SD and individual data points. Statistical significance between atherosclerosis plaque types and Ca2+ deposition groups was tested using 2-way ANOVA. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
As PCR analysis only gives a snapshot of gene expression at a single time point, we used immunohistochemistry to investigate deposition of downstream ER stress markers Grp78/Grp94 in normal and calcified vessels (carotid and aortic sections from 22 donors, Table II in the Data Supplement; Figure 1B). Grp78/Grp94 staining was detectable with 2 distinct localization patterns; either intracellular or deposited in a punctate pattern in the ECM in a pattern similar to previous observations of EVs in the ECM.43 Although smaller types of vesicles (exosomes) are too small to detect with regular light microscopy, these puncta might represent larger microvesicles or vesicle aggregates, as aggregation has recently been proposed to promote calcification.48 When Grp78/Grp94 deposition was scored for each section a clear increase in extracellular deposition of Grp78 and Grp94 was observed in donors aged 40 to 60 years, in sections with 75% calcification and in vessels with stages IV to V of atherosclerosis/plaque type (according to American Heart Association classification49; Figure 1C). These data suggest that calcification of both intima and media coincides with changes in ER stress-related gene expression and extracellular deposition of ER stress chaperones.
ER Stress Enhances Calcification of VSMCs In Vitro and Modifies VSMC Phenotype
To understand further the relationship between ER stress and VC, VSMCs in vitro were treated with 2 ER stress inducers, tunicamycin and thapsigargin. This resulted in comprehensive activation of all 3 unfolded protein response branches (IRE1, ATF6, PERK, and transcription factors downstream of PERK—ATF4 and CHOP, Figure 2A) and their target genes Grp78 and Grp94 (Figure 2B and 2C).
Figure 2.
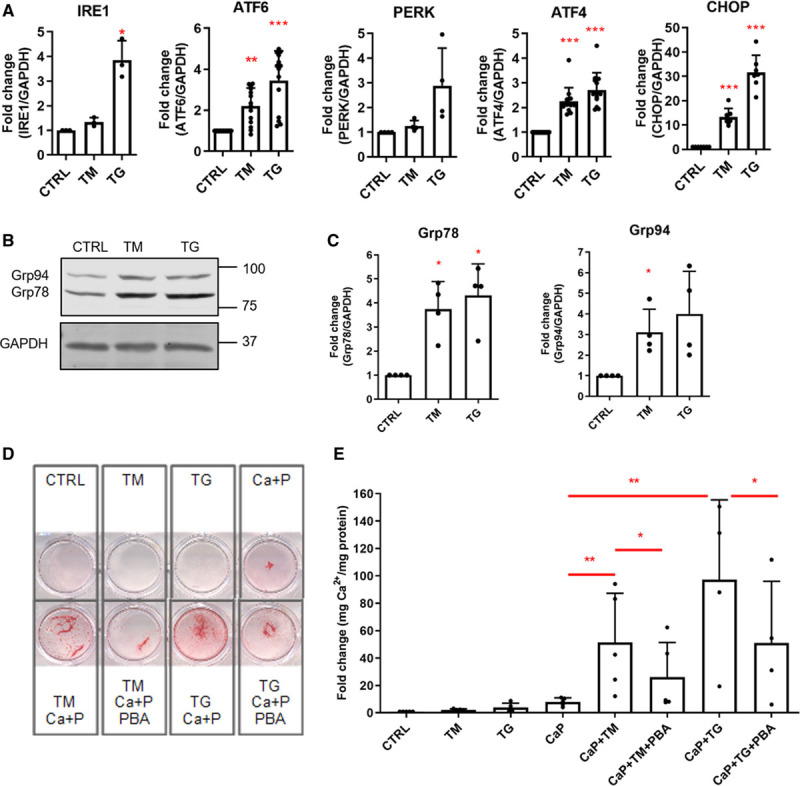
Endoplasmic reticulum (ER) stress increased calcification of human primary vascular smooth muscle cells (VSMCs) in vitro. Cells were treated with 0.4 μg/mL tunicamycin (TM) or 0.2 μg/mL thapsigargin (TG) for 24 h, in M199 with 0.5% FBS. A, Quantitative real-time polymerase chain reaction analysis of ER stress marker activation in VSMCs. Statistical significance was tested with 1-sample Wilcoxon (ATF [activating transcription factor] 4, ATF6) and t tests (IRE [inositol-requiring protein] 1, PERK [PKR (protein kinase RNA)-like ER kinase], CHOP [C/EBP-homologous protein]). B, Western blot and quantification showing increased levels of ER stress markers Grp78 (glucose-regulated protein, 78 kDa) and Grp94 and (C) quantification. Statistical significance was tested with t tests. D, Calcification assay; Alizarin Red S staining. Cells were treated with 2.7 mmol/L Ca2+, 2.5 mmol/L PO43- (CaP), 0.04 μg/mL TM or 0.01 μg/mL TG, 0.5 mmol/L 4-phenylbutyric acid (PBA) for 8 d in M199 with 5% FBS. E, Calcification quantified with the o-cresolphthalein assay. Graph shows mean+SD and individual data points, statistical significance was tested with ANOVA with Tukey post hoc tests. CTRL indicates control. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
To examine whether induction of ER stress influenced calcification, VSMCs were treated with calcifying media in the presence of tunicamycin or thapsigargin (Figure 2D and 2E). This resulted in increased calcification, which could be blocked by the ER stress inhibitor PBA, a chemical chaperone, which inhibits production of Grp78/94,50 suggesting ER stress can promote calcification.
To examine the mechanisms promoting VSMC calcification in response to ER stress, we first examined the proportion of cells undergoing death (Figure 3A through 3C). Tunicamycin and thapsigargin alone did not change cell viability; however, calcifying media significantly increased cell necrosis and this was significantly decreased by tunicamycin and thapsigargin. In the presence of Ca and P, tunicamycin did not increase apoptosis and cell death rates were the same in pairs of samples treated or not with ER stress pathway modifiers suggesting increased calcification in response to ER stress could not be attributed to increased apoptosis.
Figure 3.
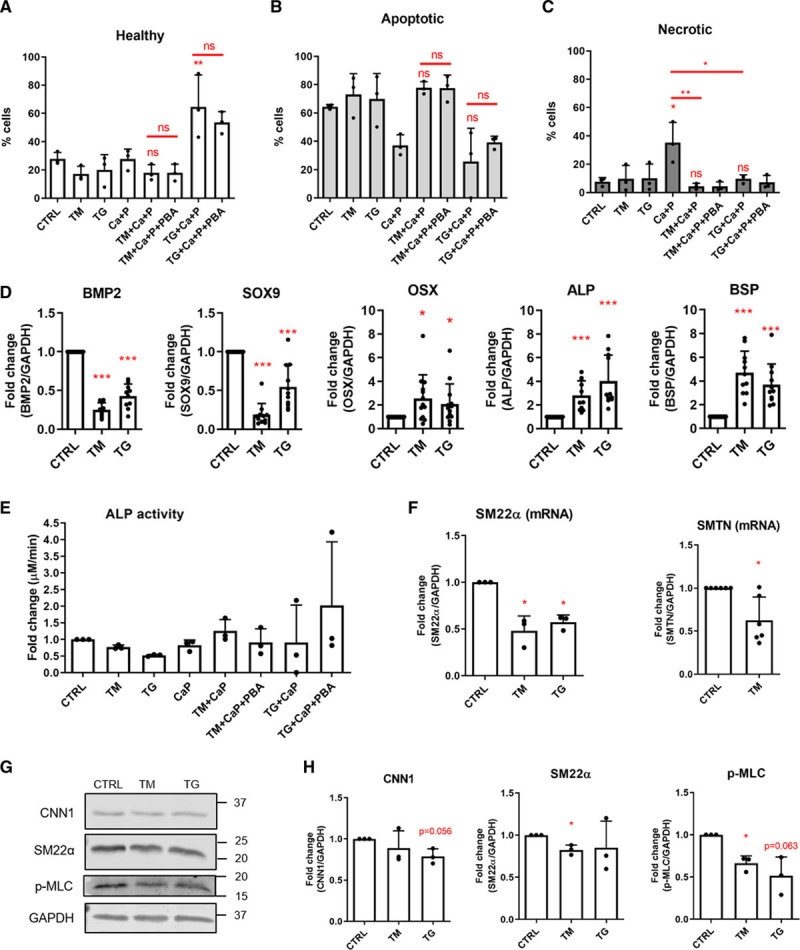
Endoplasmic reticulum (ER) stress modulated vascular smooth muscle cell (VSMC) phenotype. A–C, VSMCs were treated with 2.7 mmol/L Ca2+, 2.5 mmol/L PO43-, 0.04 μg/mL tunicamycin (TM), or 0.01 μg/mL thapsigargin (TG), 0.5 mmol/L 4-phenylbutyric acid (PBA) for 8 d in M199 with 5% FBS. Cell death was analyzed with the NucleoCounter NC-3000 using a FLICA caspase assay and propidium iodide staining, where cells are described as healthy, apoptotic, or necrotic. Graph shows mean+SD and individual data points, ANOVA with Tukey post hoc test was performed. Statistical significance is marked for Ca+P vs Ca+P and TM/TG or as indicated with brackets. D, VSMCs were treated with 0.4 μg/mL TM or 0.2 μg/mL TG for 24 h. Osteogenic gene expression in response to ER stress was analyzed by real-time polymerase chain reaction (PCR). Graphs show mean+SD, 1-sample t tests (BMP-2 [bone morphogenetic protein 2], SOX9 [SRY-box transcription factor 9], BSP [bone sialoprotein]), and 1-sample Wilcoxon tests (OSX [Osterix], ALP [alkaline phosphatase]) were performed. E, VSMCs were treated with 2.7 mmol/L Ca2+, 2.5 mmol/L PO43-, 0.04 μg/mL TM or 0.01 μg/mL TG, 0.5 mmol/L PBA for 8 d in M199 with 5% FBS. ALP activity was measured using a colorimetric assay. Graph shows mean+SD and individual data points, Kruskal-Wallis test was performed. F, Analysis of SM22α (smooth muscle protein 22α) and SMTN (smoothelin) expression by real-time PCR. One-sample t tests were performed. G, Representative western blots of CNN1 (calponin 1), SM22α, and p-MLC (phosphorylated myosin light chain) and H, quantifications. Graphs show mean+SD and individual data points, 1-sample t tests were performed. CTRL indicates control. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
Drawing on the similarities between bone formation and VSMC calcification, we examined whether ER stress is involved in regulating expression of osteo/chondrogenic genes in VSMCs. Quantitative real-time PCR revealed a complex pattern of gene expression. While some genes were downregulated (BMP-2 [bone morphogenetic protein 2], SOX9 [SRY-box transcription factor 9]), others were upregulated (OSX [Osterix], ALP, BSP [bone sialoprotein]) or unchanged (Figure 3D), whereas some showed different responses to tunicamycin and thapsigargin (Runx2 [RUNX family transcription factor 2], OPG [osteoprotegerin]; Figure IA in the Data Supplement). As ALP was consistently upregulated and is an early indicator of osteogenic differentiation, we examined its protein expression and activity. Both tunicamycin and thapsigargin increased protein expression, however, this did not translate into higher activity levels, and the induction could not be blocked by PBA. Taken together, these data suggest ER stress does not induce calcification via regulation of osteogenic processes (Figure 3E, Figure IB through ID in the Data Supplement).
However, examination of contractile protein expression showed that ER stress decreased SMC-specific contractile markers SM22α, SMTN (smoothelin), CNN1, and p-MLC (Figure 3F through 3H), indicative of VSMC phenotypic modulation to a synthetic phenotype. This is in line with a recent study showing a negative correlation between ER stress signaling and myocardin-driven phenotype regulation.51
ER Stress Increases EV Release and Influences Cargo Loading
Phenotypic modulation of VSMCs has been linked to the induction of EV release, an early event in calcification.11,52–54 Moreover, we previously reported the presence of ER-resident proteins in VSMC-derived EVs including chaperones (Hsp70 [heat shock protein, 70 kDa], Grp94, Grp78), and the punctate extracellular pattern of Grp78 deposition in calcified arteries was consistent with vesicle-mediated calcification.43 Therefore, we tested the effect of ER stress on VSMC EV release and cargo loading. Both tunicamycin and thapsigargin increased EV release up to 5-fold (Figure 4A), and this was accompanied by increased expression of SMPD3 (Figure 4B), a key regulator of EV release. Importantly, the SMPD3 inhibitor spiroepoxide blocked ER stress-enhanced VSMC calcification, suggesting EVs were mediating ER stress–induced calcification (Figure 4C).
Figure 4.
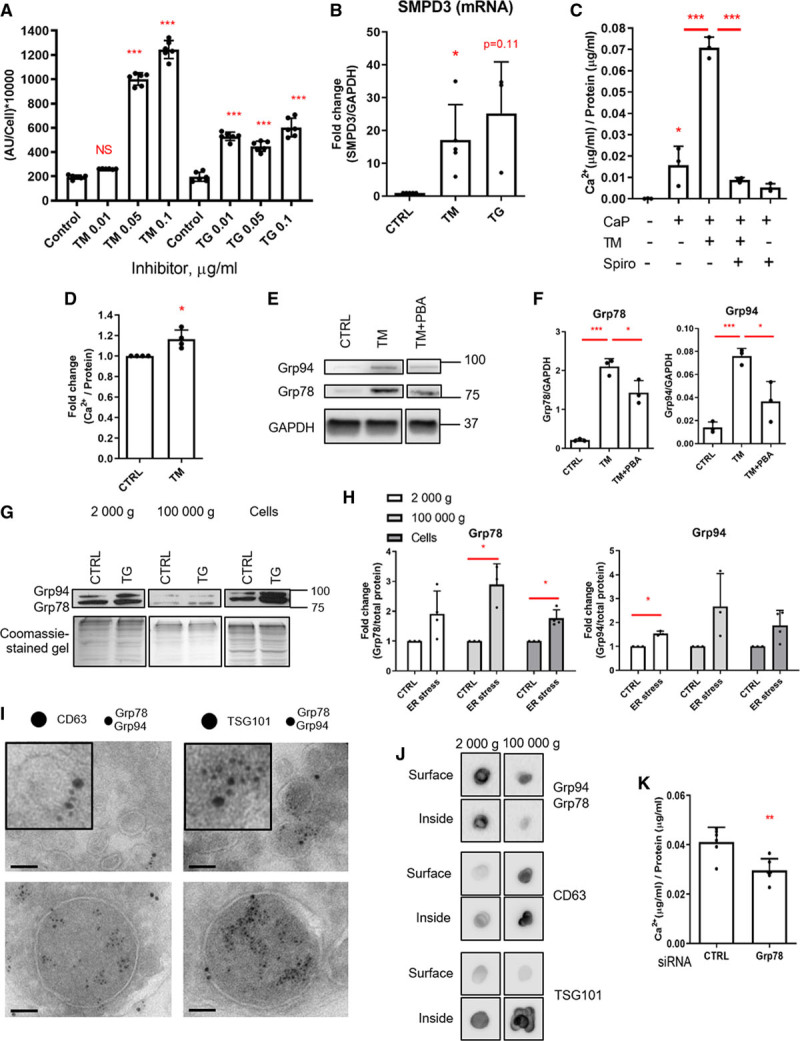
Endoplasmic reticulum (ER) stress–induced vascular smooth muscle cell (VSMC) calcification is mediated by extracellular vesicle (EV) release and Grp78 (glucose-regulated protein, 78 kDa). A, Quantification of EV release using a bead capture assay. VSMCs were treated 0.01 to 0.1 μg/mL tunicamycin (TM) or 0.01 to 0.1 μg/mL thapsigargin (TG) for 24 h in M199 with 0.5% FBS. Statistical significance was tested with ANOVA. B, SMPD3 (sphingomyelin phosphodiesterase 3) mRNA expression measured by real-time polymerase chain reaction. VSMCs were treated 0.1 μg/mL TM or 0.1 μg/mL TG for 24 h. Statistical significance was tested with 1-sample t tests. C, SMPD3 inhibitor spiroepoxide blocks ER stress-enhanced VSMC calcification. VSMCs were treated with 0.5 µmol/L spiroepoxide, 0.05 µg TM and 2.7 mmol/L Ca2+ and 2.5 mmol/L PO43- for 5 d in 5% FBS. Statistical significance was tested with ANOVA. D, EVs from VSMCs treated with TM have a higher propensity to calcify. Pellet (100 000g) EVs were isolated by ultracentrifugation and equal amounts (protein) were calcified on a collagen matrix with 2.7 mmol/L Ca2+ and 2.5 mmol/L PO43-. Statistical significance was tested with 1-sample t test. E and F, Western blot and quantification, showing that 4-phenylbutyric acid (PBA), which decreases ER stress-induced calcification, decreases Grp78 (glucose-regulated protein, 78 kDa) expression in VSMCs. VSMCs were treated with 0.1 μg/mL TM and 1 mmol/L PBA for 24 h. Statistical significance was tested with ANOVA. G and H, Western blot showing Grp78 is increased in EVs released from cells treated with ER stress inducers. VSMCs were treated with 0.2 μg/mL TM or TG in phenol red-free DMEM with 0.1% bovine serum albumin (FBS free); EVs were isolated from culture media by ultracentrifugation. Statistical significance was tested with 1-sample t tests. I, EVs were isolated by ultracentrifugation (100 000g), immunogold staining, and electron microscopy was carried out on sections of fixed EV pellets. Grp78/Grp94 (6 nm gold) are localized in EVs of various sizes expressing CD63 and TSG101 (tumor susceptibility gene 101 protein; both 10 nm gold). Scale bars are 100 nm. J, Dot blot showing Grp78/Grp94 is detected predominantly on the surface of EVs (samples without detergent). TSG101 and CD63 were used as positive and negative control, respectively. EVs were isolated by ultracentrifugation from primary VSMCs culture supernatants. Representative blots from n=3 experiments. K, siRNA (short interfering RNA) knock-down of Grp78 decreases calcification of VSMCs treated with 2.7 mmol/L Ca2+ and 2.5 mmol/L PO43- for 18 h. Cells were treated with Grp78 siRNA or control siRNA 24 h before applying calcification media. Calcification was quantified with o-cresolphthalein assay. Statistical significance was tested with t test. All graphs show mean+SD and individual data points. AU indicates arbitrary units; and CTRL, control. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
To assess whether EVs released in response to ER stress have increased calcification capacity, we performed in vitro calcification assays. The 100 000 g pellets of EVs (enriched for exosomes11) isolated from cells treated with tunicamycin and control cells were placed in collagen-coated wells in the presence of calcifying media. We found that EVs from tunicamycin-treated cells had a higher propensity to calcify compared with EVs from vehicle-treated cells (Figure 4D; Figure IIA through IIC in the Data Supplement). We also observed that PBA, which decreased calcification, also decreased Grp78 expression in VSMCs (Figure 4E and 4F). This suggested that Grp78, which was previously shown to nucleate mineral,55 could be responsible for the increased calcification capacity of EVs. Indeed, western blot showed that Gpr78 loading into both the 2000 g and 100 000 g pellets of EVs (apoptotic bodies and exosomes, respectively) was increased in response to ER stress (Figure 4G and 4H, Figure IID through IIF in the Data Supplement). Grp94 loading to EVs was also increased but not significantly.
Electron microscopy confirmed the specificity of Grp78/Grp94 loading into TSG101- and CD63-positive EVs from the 100 000 g pellet (Figure 4I). We found Grp78/Grp94 in small vesicles (consistent with exosomes, 20–100 nm) but predominantly in larger vesicles (microvesicles, 200–1000 nm). Of note, not all EVs were positive for TSG101, CD63, or Grp78/Grp94, which reflects their heterogeneity (Figure IIIA in the Data Supplement). Electron microscopy of VSMCs stained for Grp78/Grp94 shows that additional to the expected ER staining, Grp78/Grp94 were also present in multivesicular bodies, further supporting specific loading of the chaperones into EVs (Figure IIIA in the Data Supplement). Dot blot analysis of EVs with and without lysis suggested that Grp78/Grp94 are present both inside and on the surface of EVs, with possibly a larger amount on the surface (Figure 4J). As expected, we found more TSG101 inside EVs than on the surface, and no difference in CD63. To confirm a role for Grp78 in calcification, we used siRNA to knock-down Grp78 which resulted in decreased calcification of VSMCs (Figure 4K, Figure IIIB in the Data Supplement).
Warfarin Induces EV Release via a Specific ER Stress Pathway
Warfarin is known to be a potent inducer of calcification, as it inhibits γ-carboxylation of calcification inhibitors,56 which occurs within the ER. Therefore, we next tested whether warfarin could induce ER stress. We treated human primary VSMCs with warfarin and found that ATF4 mRNA was upregulated in response (Figure 5A). Western blot showed increased Grp78 but not Grp94 expression in response to warfarin (Figure 5B and 5C), as well as a transient increase in PERK phosphorylation (Figure 5D and 5E, Figure IVA in the Data Supplement). However, we observed no consistent increase of Grp78 loading into EVs (Figure IVB and IVC in the Data Supplement). Treating cells with PBA blocked warfarin-induced calcification of VSMCs cultured in calcifying conditions (Figure 5F). As we ruled out effects of warfarin on oxidative stress (Figure V in the Data Supplement), these data suggest ER stress mediates in part the procalcific effects of warfarin.
Figure 5.
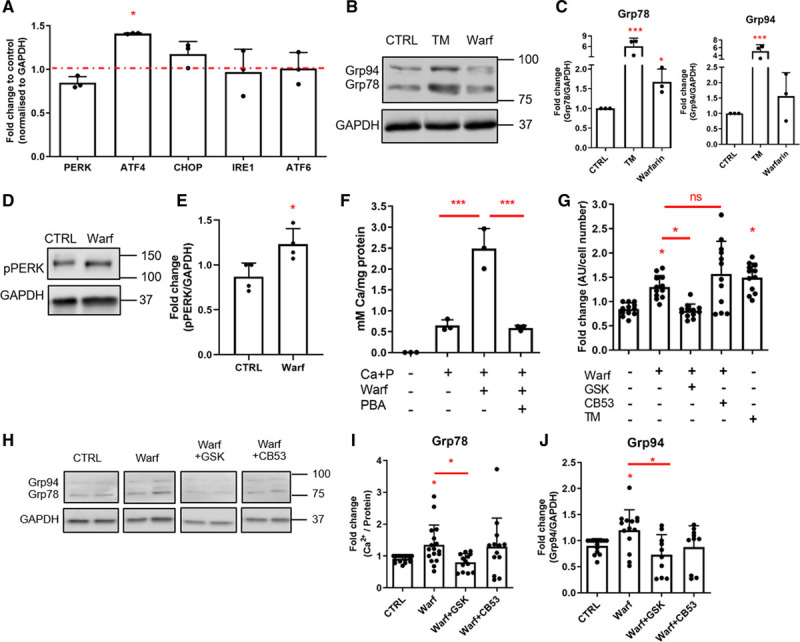
Warfarin-mediated endoplasmic reticulum (ER) stress induces extracellular vesicle (EV) release in a PERK (PKR [protein kinase RNA]-like ER kinase)-dependent manner. A, Real-time polymerase chain reaction analysis of ER stress markers in vascular smooth muscle cells (VSMCs) treated with 10 μM warfarin for 8 d in M119 with 5% FBS. Statistical significance was tested with 1-sample t tests. B and C, Western blot showing that warfarin induces Grp78 (glucose-regulated protein, 78 kDa) and Grp94 expression in human VSMCs in vitro. Cells were treated with 10 μM warfarin for 24 h in M199 with 0.5% FBS. Statistical significance was tested with ANOVA. D and E, Western blot showing that warfarin induces PERK phosphorylation in human VSMCs in vitro. Cells were treated with 50 μM warfarin for 1 h in M199 with 0.5% FBS. Statistical significance was tested with t test. F, ER stress inhibitor 4-phenylbutyric acid (PBA) decreased warfarin-enhanced calcification of VSMCs. Cells were treated with 2.7 mmol/L Ca2+, 2.5 mmol/L PO43-, 0.5 mmol/L PBA and 10 μM warfarin for 8 days in M199 with 5% FBS. Statistical significance was tested with ANOVA. G, Quantification of EV release using a bead capture assay. Cells were treated with 50 μM warfarin with or without ER stress inhibitors GSK (10 µmol/L GSK2656157) or CB53 (25 µmol/L CB5305630) for 24 h in M199 with 0.5% FBS. Statistical significance was tested with ANOVA. H–J, Western blot showing that GSK, but not CB53, can inhibit a warfarin-induced Grp78 increase. Statistical significance was tested with ANOVA. All graphs show mean+SD and individual data points. ATF indicates activating transcription factor; AU, arbitrary units; CHOP, C/EBP-homologous protein; CTRL, control; IRE, inositol-requiring protein; pPERK, phosphorylated PKR (protein kinase RNA)-like ER kinase; TM, tunicamycin; and Warf, warfarin. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
Next, we measured EV release in warfarin-treated VSMCs and its regulation by ER stress signaling. Warfarin increased EV release by VSMCs, and this was blocked by a PERK inhibitor, which acts upstream of ATF4 (Figure 5G). The PERK inhibitor also blocked the warfarin-induced increase in Grp78 expression (Figure 5H through 5J). In contrast, an IRE1 inhibitor had no effect on EV release or Grp78 levels (Figure 5H through 5J, Figure VI in the Data Supplement) suggesting specific involvement of the PERK-ATF4 ER stress pathway.
Next, we set out to assess by immunohistochemistry whether ER stress was induced in an animal model of warfarin-induced medial VC (Figure 6A). Aortas from control rats showed no calcification, expressed Grp78/Grp94 which showed an intracellular localization, and showed no ATF4 staining. Rats treated with warfarin for 1 week showed no calcification but had increased Grp78/Grp94 and markedly elevated nuclear ATF4 expression. At this stage, some deposition of Grp78/Grp94 in the ECM was also observed. After 4 weeks of warfarin treatment, aortas were calcified and rounded puncta of Grp78/Grp94 in the ECM were clearly apparent, with reduced intracellular staining at sites of calcification. Increased expression of Grp78/Grp94 and ATF4 was observed in calcified areas and increased expression of Grp78 (and not Grp94) in aortas of rats treated with warfarin was confirmed by Western blotting (Figure 6B and 6C).
Figure 6.
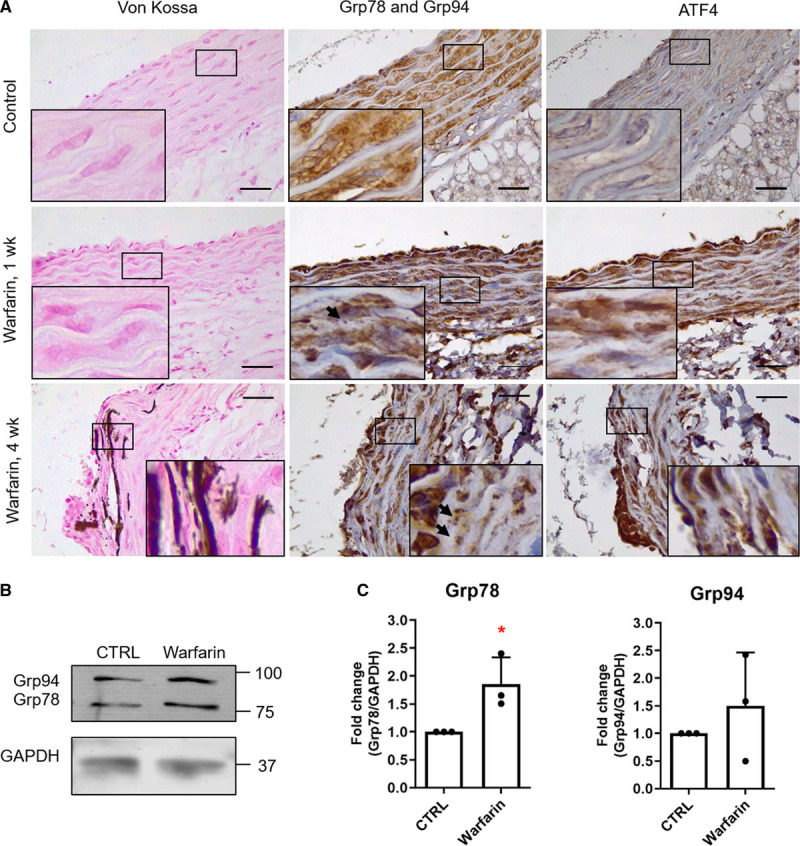
Warfarin induces endoplasmic reticulum stress in vivo. A, Warfarin increased calcification, Grp78 (glucose-regulated protein, 78 kDa)/Grp94, and ATF (activating transcription factor) 4 expression in rat aortic wall. Rats were fed 3 mg warfarin and 1.5 mg vitamin K1 per 1 g of food for 1 or 4 wk, n=3 was analyzed in each group, representative images shown. Arrows indicate extracellular, punctate Grp78/Grp94 staining. B and C, Western blot of Grp78 and Grp94 in aortas of rats fed with warfarin for 11 days, n=3. Statistical significance was tested with 1-sample t tests. CTRL indicates control. Dots denote individual data points, *P<0.05, **P<0.01, ***P<0.001.
Since we show that warfarin induces ER-mediated changes that lead to VSMC calcification, we also investigated whether ER stress mediates the well-known γ-carboxylation inhibiting effects of warfarin. Expression of total, carboxylated, and uncarboxylated MGP was measured in response to ER stress (Figure VII in the Data Supplement), and no changes were observed. This suggests that disruption of ER function does not translate into decreased protein carboxylation.
Discussion
Increased Deposition of Grp78 and Grp94 in Calcified Human Arteries
This study is the first to comprehensively examine expression of ER stress markers in human vascular tissue. We observed that mRNA expression of ER stress markers was decreased in fatty streak and calcified human aortic samples compared with healthy, suggesting there is dysregulation of homeostatic ER stress signaling during disease development. We also observed increased extracellular deposition of Grp78 and Grp94 at sites of calcification, consistent with the presence of Grp78 in EVs and its role in promoting calcification. To the best of our knowledge, only one other study has examined the expression of ER stress markers in human arteries57 showing upregulation of Grp78 and Grp94 in vessels from aneurysm patients supporting our conclusion that vascular disease is associated with changes in ER stress signaling components.
ER Stress Enhances VSMC Calcification In Vitro but Not via Apoptosis
We demonstrated that ER stress can be induced in human primary VSMCs in vitro by tunicamycin and thapsigargin and that all 3 branches of the UPR were activated.30,31,35 We also showed that the addition of ER stress inducers enhanced VSMC calcification, but consistent with previous studies in rodent models, tunicamycin, and thapsigargin alone did not induce calcification.30,32,33,36,37
ER stress-mediated apoptosis has been reported in some models of VC30,34–36; however, it was not involved in ER stress-enhanced calcification in response to elevated calcium and phosphate. On the contrary, thapsigargin decreased the rate of apoptosis and both tunicamycin and thapsigargin protected VSMCs from necrosis. These findings may be due to differences between in vivo and in vitro models or different ER stress inducers and suggests that ER stress can mediate both prosurvival and procalcification signaling, depending on the patho/physiological context. Indeed, several reports showed a protective role for ER stress in vascular disease, and this is consistent with the reduction in ER stress signaling we observed in diseased tissue samples.58–60
ER Stress Modulates VSMC Phenotype
Tunicamycin and thapsigargin treatments changed expression of almost all examined osteo/chondrogenic genes in VSMCs causing decreased as well as increased expression. However, ALP activity was not increased by ER stress despite increased mRNA and protein levels with activation required to form a functional enzyme.61 This finding is in contrast to osteo/chondrogenic differentiation in bone, where these proteins are expressed in a concerted manner, with many directly regulated by ER stress signaling.18,62 The apparently chaotic pattern of osteogenic gene expression observed in VSMCs in response to ER stress may reflect activation via aberrant signals and their expression is unlikely to play a role in the accelerated calcification observed in response to ER stress. This is in line with literature showing that osteogenic differentiation might not be the primary mechanism of calcification in all contexts.63
In contrast to osteogenic gene expression, VSMC contractile marker expression was uniformly downregulated by tunicamycin and thapsigargin treatments (SM22α and SMTN mRNA and p-MLC protein) supporting the notion that ER stress promotes de-differentiation of VSMCs to a synthetic phenotype.30
ER Stress Increases Release of Grp78-Loaded EVs
In our study, we focused on EVs isolated in the 2000 g and 100 000 g pellets, which correspond to apoptotic bodies and exosomes, respectively, as both were previously implicated in mediating calcification.11,38 We identified a novel role for ER stress in mediating increased EV release by VSMCs consistent with their transition to a synthetic phenotype (loss of contractile markers and increased SMPD3 expression). Our study confirms a link between EV release and ER stress, which was reported in other cell types64 and in VSMCs subjected to mechanical stress.65 Importantly, ER stress was also associated with increased levels of Grp78 in EVs. Grp78 has been shown to function on the cell surface in other cell types66 and to facilitate calcium phosphate crystal formation on a collagen matrix, due to its presence on the surface of osteoblasts.55,67 Our results suggest that it elicits the same effect when loaded into EVs. Because we did not observe Grp78 expression on the surface of VSMCs (data not shown), its procalcification properties demonstrated in the siRNA knock-down experiment are most likely due to it being secreted into the ECM and this is consistent with increased extracellular deposition of Grp78/Grp94 in calcified human arteries.
To exclude the possibility that Grp78 co-isolates with EVs during isolation by differential ultracentrifugation,68 we performed electron microscopy, which unequivocally shows Grp78/Grp94 staining associated with EVs and MVBs. Additionally, online EV database EVpedia69 lists 608 studies (on April 11, 2019), which identified Grp78 in EV samples derived from human material, confirming that this protein is indeed specifically loaded into vesicles.
In many experiments, we observed upregulation of Grp94 expression in response to ER stress induced by tunicamycin or thapsigargin and warfarin, alongside Grp78. We also found increased Grp94 loading into EVs of cells treated with ER stress but less consistent than for Grp78. However, we cannot exclude the possibility that some of the effects of ER stress that we observed were due to increased Grp94 expression, whose mineralization-promoting properties are unknown. Grp78 is hypothesized to nucleate mineralization via calcium and collagen I binding.55 Although Grp94 is also a calcium-binding protein70 found in mineralizing EVs,71 its ECM protein-binding capabilities have not been investigated.
Warfarin Induces VSMC Calcification via ER Stress-Dependent EV release
Warfarin is an oral anticoagulant commonly administered to patients that suffer from hypercoagulability.72 Warfarin inhibits VKORC1 (vitamin K epoxide reductase complex subunit 1) thereby diminishing available vitamin K, which is a required cofactor for protein carboxylation.73 Warfarin promotes VC as it inhibits carboxylation of the endogenous calcification inhibitor MGP.13 The link between warfarin and ER stress has not been examined before, even though it is known that protein carboxylation occurs within the ER.74 We show here that warfarin induces expression of Grp78, Grp94, and ATF4 in rat aortas and in human VSMCs in vitro. Additionally, warfarin increased VSMC calcification in vitro in an ER stress-dependent manner. We found that warfarin-induced EV release, an effect mediated by the PERK-ATF4 branch of the UPR. However, increased Grp78 loading into EVs was not observed as a result of warfarin treatment most likely due to warfarin acting specifically via the PERK-ATF4 branch of the UPR and only having a modest effect on upregulation of Grp78, in contrast to tunicamycin/thapsigargin, which strongly induce all 3 branches. Additionally, it is possible that the short time point of our in vitro experiments did not allow for sufficient Grp78 accumulation in EVs in vitro. However, the punctate pattern of Grp78/Grp94 staining in warfarin-fed rat aortas is consistent with Grp78 accumulation in EVs.
Conclusions
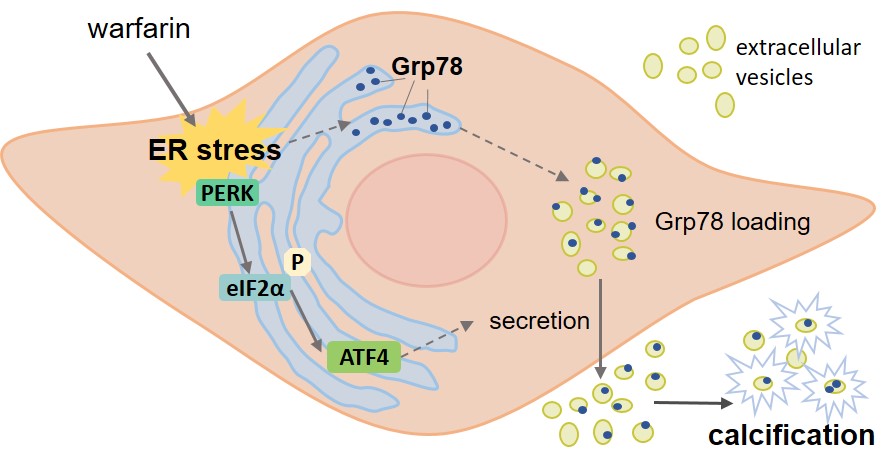
We provide evidence that ER stress-mediated EV release and Grp78 deposition in the ECM promote calcification (Figure 7). We show that ER stress induces changes in osteogenic gene expression and contractile VSMC phenotype loss. Both of these are known to be associated with increased EV release, and our results provide further evidence that ER stress is involved. We also show that warfarin induces the PERK-ATF4 branch of the UPR, which has previously been implicated in VC, and via this pathway increases EV release and VSMC calcification. This study is the first, to our knowledge, to show that warfarin induces ER stress and to link ER stress to cargo loading of EVs.
Figure 7.
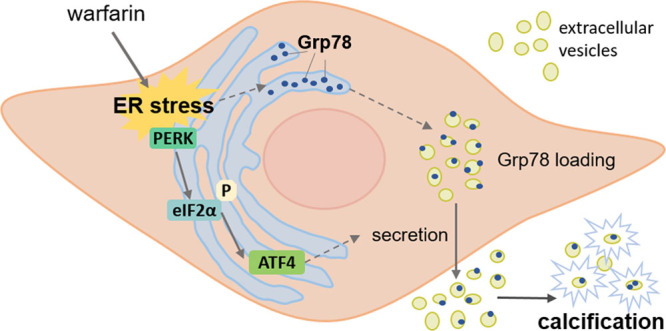
Endoplasmic reticulum (ER) stress-mediated extracellular vesicle (EV) release and Grp78 (glucose-regulated protein, 78 kDa) deposition in the ECM (extracellular matrix) promote vascular smooth muscle cell (VSMC) calcification. ER stress induces loading of Grp78 into EVs. Warfarin induces the PERK (PKR [protein kinase RNA]-like ER kinase)-ATF (activating transcription factor) 4 branch of the unfolded protein response, which via this pathway increases EV release. Increased EV release and the presence of Grp78 lead to VSMC calcification eIF2α indicates eukaryotic translation initiation factor 2α.
The next important steps would be investigating the molecular events that lead from ER stress activation to increased EV release and Grp78 loading into EVs. Because inhibiting ER stress has been recently described to attenuate VC in animal models,75–77 ER stress pathways might represent a new therapeutic possibility.
Acknowledgments
We would like to thank Dr Carmen López-Iglesias, Dr Kèvin Knoops, Willine van de Wetering, and Hans Duimel from the Microscopy Core Lab at Maastricht University for support with electron microscopy.
Sources of Funding
This work was funded by the British Heart Foundation, PhD Studentship FS/11/9/28695, and in part by the Trombose Stichting Nederland (grant 2014.02) and the Norwegian Research Council and NattoPharma ASA (Project 241584).
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- ATF
- activating transcription factor
- ECM
- extracellular matrix
- ER
- endoplasmic reticulum
- EV
- extracellular vesicle
- IRE
- inositol-requiring protein
- MGP
- matrix Gla-protein
- PCR
- polymerase chain reaction
- PERK
- PKR (protein kinase RNA)-like ER kinase
- UPR
- unfolded protein response
- VC
- vascular calcification
- VKORC1
- vitamin K epoxide reductase complex subunit 1
- VSMC
- vascular smooth muscle cell
These authors contributed equally to this article.
For Sources of Funding and Disclosures, see page 912.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.120.315506.
Contributor Information
Malgorzata Furmanik, Email: gosia.furmanik@maastrichtuniversity.nl.
Rick van Gorp, Email: rick.vangorp@maastrichtuniversity.nl.
Meredith Whitehead, Email: meredith.whitehead@kcl.ac.uk.
Jayanta Bordoloi, Email: jayanta.bordoloi@crick.ac.uk.
Alexander Kapustin, Email: Alexander.kapustin@kcl.ac.uk.
References
- 1.Shanahan CM. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat Rev Nephrol. 2013;9:661–670. doi: 10.1038/nrneph.2013.176 [DOI] [PubMed] [Google Scholar]
- 2.Karwowski W, Naumnik B, Szczepański M, Myśliwiec M. The mechanism of vascular calcification - a systematic review. Med Sci Monit. 2012;18:RA1–R11. doi: 10.12659/msm.882181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA. Vascular calcifications as a marker of increased cardiovascular risk: a meta-analysis. Vasc Health Risk Manag. 2009;5:185–197. doi: 10.2147/vhrm.s4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528–536. doi: 10.1038/nrcardio.2010.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Proudfoot D, Shanahan CM. Biology of calcification in vascular cells: intima versus media. Herz. 2001;26:245–251. doi: 10.1007/pl00002027 [DOI] [PubMed] [Google Scholar]
- 6.Ehara S, Kobayashi Y, Yoshiyama M, Shimada K, Shimada Y, Fukuda D, Nakamura Y, Yamashita H, Yamagishi H, Takeuchi K, et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: an intravascular ultrasound study. Circulation. 2004;110:3424–3429. doi: 10.1161/01.CIR.0000148131.41425.E9 [DOI] [PubMed] [Google Scholar]
- 7.Shanahan CM. Vascular calcification. Curr Opin Nephrol Hypertens. 2005;14:361–367. doi: 10.1097/01.mnh.0000172723.52499.38 [DOI] [PubMed] [Google Scholar]
- 8.Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711. doi: 10.1161/CIRCRESAHA.110.234914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol. 2013;24:179–189. doi: 10.1681/ASN.2011121191 [DOI] [PubMed] [Google Scholar]
- 10.Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification: In vitro evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. doi: 10.1161/01.res.87.11.1055 [DOI] [PubMed] [Google Scholar]
- 11.Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RT, Alvarez-Hernandez D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–1323. doi: 10.1161/CIRCRESAHA.116.305012 [DOI] [PubMed] [Google Scholar]
- 12.Krohn JB, Hutcheson JD, Martínez-Martínez E, Irvin WS, Bouten CV, Bertazzo S, Bendeck MP, Aikawa E. Discoidin domain receptor-1 regulates calcific extracellular vesicle release in vascular smooth muscle cell fibrocalcific response via transforming growth factor-β signaling. Arterioscler Thromb Vasc Biol. 2016;36:525–533. doi: 10.1161/ATVBAHA.115.307009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med. 2013;19:217–226. doi: 10.1016/j.molmed.2012.12.008 [DOI] [PubMed] [Google Scholar]
- 14.Jahnen-Dechent W, Schinke T, Trindl A, Müller-Esterl W, Sablitzky F, Kaiser S, Blessing M. Cloning and targeted deletion of the mouse fetuin gene. J Biol Chem. 1997;272:31496–31503. doi: 10.1074/jbc.272.50.31496 [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. 2013;112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543 [DOI] [PubMed] [Google Scholar]
- 16.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Mönckeberg’s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100:2168–2176. doi: 10.1161/01.cir.100.21.2168 [DOI] [PubMed] [Google Scholar]
- 18.Karsenty G, Kronenberg HM, Settembre C. Genetic control of bone formation. Annu Rev Cell Dev Biol. 2009;25:629–648. doi: 10.1146/annurev.cellbio.042308.113308 [DOI] [PubMed] [Google Scholar]
- 19.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 20.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 21.Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS. Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem. 2003;278:37375–37385. doi: 10.1074/jbc.M303619200 [DOI] [PubMed] [Google Scholar]
- 22.Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- 23.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clauss IM, Gravallese EM, Darling JM, Shapiro F, Glimcher MJ, Glimcher LH. In situ hybridization studies suggest a role for the basic region-leucine zipper protein hXBP-1 in exocrine gland and skeletal development during mouse embryogenesis. Dev Dyn. 1993;197:146–156. doi: 10.1002/aja.1001970207 [DOI] [PubMed] [Google Scholar]
- 25.Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, Morioka H, Nakamura M, Iwawaki T, Chiba K, et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011;12:451–457. doi: 10.1038/embor.2011.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han X, Zhou J, Zhang P, Song F, Jiang R, Li M, Xia F, Guo FJ. IRE1α dissociates with BiP and inhibits ER stress-mediated apoptosis in cartilage development. Cell Signal. 2013;25:2136–2146. doi: 10.1016/j.cellsig.2013.06.011 [DOI] [PubMed] [Google Scholar]
- 27.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/mcb.22.11.3864-3874.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, Li L, Brancorsini S, Sassone-Corsi P, Townes TM, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387–398. doi: 10.1016/s0092-8674(04)00344-7 [DOI] [PubMed] [Google Scholar]
- 29.Duan X, Zhou Y, Teng X, Tang C, Qi Y. Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun. 2009;387:694–699. doi: 10.1016/j.bbrc.2009.07.085 [DOI] [PubMed] [Google Scholar]
- 30.Duan XH, Chang JR, Zhang J, Zhang BH, Li YL, Teng X, Zhu Y, Du J, Tang CS, Qi YF. Activating transcription factor 4 is involved in endoplasmic reticulum stress-mediated apoptosis contributing to vascular calcification. Apoptosis. 2013;18:1132–1144. doi: 10.1007/s10495-013-0861-3 [DOI] [PubMed] [Google Scholar]
- 31.Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates nadph oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun. 2011;413:436–441. doi: 10.1016/j.bbrc.2011.08.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masuda M, Ting TC, Levi M, Saunders SJ, Miyazaki-Anzai S, Miyazaki M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J Lipid Res. 2012;53:1543–1552. doi: 10.1194/jlr.M025981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masuda M, Miyazaki-Anzai S, Levi M, Ting TC, Miyazaki M. PERK-eIF2α-ATF4-CHOP signaling contributes to TNFα-induced vascular calcification. J Am Heart Assoc. 2013;2:e000238 doi: 10.1161/JAHA.113.000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyazaki-Anzai S, Masuda M, Demos-Davies KM, Keenan AL, Saunders SJ, Masuda R, Jablonski K, Cavasin MA, Kendrick J, Chonchol M, et al. Endoplasmic reticulum stress effector CCAAT/enhancer-binding protein homologous protein (CHOP) regulates chronic kidney disease-induced vascular calcification. J Am Heart Assoc. 2014;3:e000949 doi: 10.1161/JAHA.114.000949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masuda M, Miyazaki-Anzai S, Keenan AL, Okamura K, Kendrick J, Chonchol M, Offermanns S, Ntambi JM, Kuro-O M, Miyazaki M. Saturated phosphatidic acids mediate saturated fatty acid-induced vascular calcification and lipotoxicity. J Clin Invest. 2015;125:4544–4558. doi: 10.1172/JCI82871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masuda M, Miyazaki-Anzai S, Keenan AL, Shiozaki Y, Okamura K, Chick WS, Williams K, Zhao X, Rahman SM, Tintut Y, et al. Activating transcription factor-4 promotes mineralization in vascular smooth muscle cells. JCI Insight. 2016;1:e88646 doi: 10.1172/jci.insight.88646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furmanik M, Shanahan CM. Endoplasmic reticulum stress in arterial smooth muscle cells: a novel regulator of vascular disease. Curr Cardiol Rev. 2017;13:94–105. doi: 10.2174/1573403X12666161014094738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen-Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28 [DOI] [PubMed] [Google Scholar]
- 39.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem. 2012;55:7193–7207. doi: 10.1021/jm300713s [DOI] [PubMed] [Google Scholar]
- 41.Bordoloi JK, Berry D, Khan IU, Sunassee K, de Rosales RT, Shanahan C, Blower PJ. Technetium-99m and rhenium-188 complexes with one and two pendant bisphosphonate groups for imaging arterial calcification. Dalton Trans. 2015;44:4963–4975. doi: 10.1039/c4dt02965h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, Hiorns M, Donald AE, Deanfield J, Rees L, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. doi: 10.1161/CIRCULATIONAHA.108.783738 [DOI] [PubMed] [Google Scholar]
- 43.Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, et al. Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–12. doi: 10.1161/CIRCRESAHA.110.238808 [DOI] [PubMed] [Google Scholar]
- 44.Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, van Diest R, Appels A, Reutelingsperger CP, Cleutjens JP, Vermeer C. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler Thromb Vasc Biol. 2005;25:1629–1633. doi: 10.1161/01.ATV.0000173313.46222.43 [DOI] [PubMed] [Google Scholar]
- 45.Sung BH, Weaver AM. Exosome secretion promotes chemotaxis of cancer cells. Cell Adh Migr. 2017;11:187–195. doi: 10.1080/19336918.2016.1273307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gitelman HJ. An improved automated procedure for the determination of calcium in biological specimens. Anal Biochem. 1967;18:521–531. [Google Scholar]
- 47.Bancroft JD, Stevens A. Theory and Practice of Histological Techniques. 1990. Churchill Livingstone [Google Scholar]
- 48.Blaser MC, Aikawa E. Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Front Cardiovasc Med. 2018;5:187 doi: 10.3389/fcvm.2018.00187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai JM, Hatsukami TS, Ferguson MS, Small R, Polissar NL, Yuan C. Classification of human carotid atherosclerotic lesions with in vivo multicontrast magnetic resonance imaging. Circulation. 2002;106:1368–1373. doi: 10.1161/01.cir.0000028591.44554.f9 [DOI] [PubMed] [Google Scholar]
- 50.Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones. 1996;1:109–115. doi: 10.1379/1466-1268(1996)001<0109:iomacc>2.3.co;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu B, Daoud F, Zeng S, Matic L, Hedin U, Uvelius B, Rippe C, Albinsson S, Swärd K. Antagonistic relationship between the unfolded protein response and myocardin-driven transcription in smooth muscle. J Cell Physiol. 2020;235:7370–7382. doi: 10.1002/jcp.29637 [DOI] [PubMed] [Google Scholar]
- 52.Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, van Eys G, Bochaton-Piallat ML, Proudfoot D, Biessen E, et al. Reactive oxygen-forming Nox5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res. 2020;127:911–927. doi: 10.1161/CIRCRESAHA.119.316159 [DOI] [PubMed] [Google Scholar]
- 53.Hutcheson JD, Goettsch C, Bertazzo S, Maldonado N, Ruiz JL, Goh W, Yabusaki K, Faits T, Bouten C, Franck G, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat Mater. 2016;15:335–343. doi: 10.1038/nmat4519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goettsch C, Hutcheson JD, Aikawa M, Iwata H, Pham T, Nykjaer A, Kjolby M, Rogers M, Michel T, Shibasaki M, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J Clin Invest. 2016;126:1323–1336. doi: 10.1172/JCI80851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ravindran S, Gao Q, Ramachandran A, Blond S, Predescu SA, George A. Stress chaperone GRP-78 functions in mineralized matrix formation. J Biol Chem. 2011;286:8729–8739. doi: 10.1074/jbc.M110.179341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, Vermeer C, Weissberg PL, Proudfoot D. Post-translational modifications regulate matrix Gla protein function: importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;5:2503–2511. doi: 10.1111/j.1538-7836.2007.02758.x [DOI] [PubMed] [Google Scholar]
- 57.Qin Y, Wang Y, Liu O, Jia L, Fang W, Du J, Wei Y. Tauroursodeoxycholic Acid Attenuates Angiotensin II Induced Abdominal Aortic Aneurysm Formation in Apolipoprotein E-deficient Mice by Inhibiting Endoplasmic Reticulum Stress. Eur J Vasc Endovasc Surg. 2017;53:337–345. doi: 10.1016/j.ejvs.2016.10.026 [DOI] [PubMed] [Google Scholar]
- 58.Matsushita E, Asai N, Enomoto A, Kawamoto Y, Kato T, Mii S, Maeda K, Shibata R, Hattori S, Hagikura M, et al. Protective role of Gipie, a Girdin family protein, in endoplasmic reticulum stress responses in endothelial cells. Mol Biol Cell. 2011;22:736–747. doi: 10.1091/mbc.E10-08-0724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Serrano RL, Yu W, Terkeltaub R. Mono-allelic and bi-allelic ENPP1 deficiency promote post-injury neointimal hyperplasia associated with increased C/EBP homologous protein expression. Atherosclerosis. 2014;233:493–502. doi: 10.1016/j.atherosclerosis.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noda T, Maeda K, Hayano S, Asai N, Enomoto A, Takahashi M, Murohara T. New endoplasmic reticulum stress regulator, Gipie, regulates the survival of vascular smooth muscle cells and the neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2015;35:1246–1253. doi: 10.1161/ATVBAHA.114.304923 [DOI] [PubMed] [Google Scholar]
- 61.Fukunaka A, Kurokawa Y, Teranishi F, Sekler I, Oda K, Ackland ML, Faundez V, Hiromura M, Masuda S, Nagao M, et al. Tissue nonspecific alkaline phosphatase is activated via a two-step mechanism by zinc transport complexes in the early secretory pathway. J Biol Chem. 2011;286:16363–16373. doi: 10.1074/jbc.M111.227173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakashima K, de Crombrugghe B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003;19:458–466. doi: 10.1016/S0168-9525(03)00176-8 [DOI] [PubMed] [Google Scholar]
- 63.O’Neill WC, Adams AL. Breast arterial calcification in chronic kidney disease: absence of smooth muscle apoptosis and osteogenic transdifferentiation. Kidney Int. 2014;85:668–676. doi: 10.1038/ki.2013.351 [DOI] [PubMed] [Google Scholar]
- 64.Kanemoto S, Nitani R, Murakami T, Kaneko M, Asada R, Matsuhisa K, Saito A, Imaizumi K. Multivesicular body formation enhancement and exosome release during endoplasmic reticulum stress. Biochem Biophys Res Commun. 2016;480:166–172. doi: 10.1016/j.bbrc.2016.10.019 [DOI] [PubMed] [Google Scholar]
- 65.Jia L, Zhang W-M, Li T-T, Liu Y, Piao C-M, Ma Y-C, Lu Y, Wang Y, Liu T-T, Qi Y-F, et al. Er-stress-dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin Sci. 2017;131:1287–1299. doi: 10.1042/CS20170252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Krieken R, Mehta N, Wang T, Zheng M, Li R, Gao B, Ayaub E, Ask K, Paton JC, Paton AW, et al. Cell surface expression of 78-kDa glucose-regulated protein (GRP78) mediates diabetic nephropathy. J Biol Chem. 2019;294:7755–7768. doi: 10.1074/jbc.RA118.006939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ravindran S, Gao Q, Ramachandran A, Sundivakkam P, Tiruppathi C, George A. Expression and distribution of grp-78/bip in mineralizing tissues and mesenchymal cells. Histochem Cell Biol. 2012;138:113–125. doi: 10.1007/s00418-012-0952-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913 doi: 10.3402/jev.v3.26913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim DK, Lee J, Kim SR, Choi DS, Yoon YJ, Kim JH, Go G, Nhung D, Hong K, Jang SC, et al. EVpedia: a community web portal for extracellular vesicles research. Bioinformatics. 2015;31:933–939. doi: 10.1093/bioinformatics/btu741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biswas C, Ostrovsky O, Makarewich CA, Wanderling S, Gidalevitz T, Argon Y. The peptide-binding activity of GRP94 is regulated by calcium. Biochem J. 2007;405:233–241. doi: 10.1042/BJ20061867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou X, Cui Y, Luan J, Zhou X, Zhang G, Zhang X, Han J. Label-free quantification proteomics reveals novel calcium binding proteins in matrix vesicles isolated from mineralizing Saos-2 cells. Biosci Trends. 2013;7:144–151. [PubMed] [Google Scholar]
- 72.van Gorp RH, Schurgers LJ. New insights into the pros and cons of the clinical use of vitamin K antagonists (VKAs) versus direct oral anticoagulants (DOACs). Nutrients. 2015;7:9538–9557. doi: 10.3390/nu7115479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Danziger J. Vitamin k-dependent proteins, warfarin, and vascular calcification. Clin J Am Soc Nephrol. 2008;3:1504–1510. doi: 10.2215/CJN.00770208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz HJ, Lappegard K, Seifried E, Scharrer I, Tuddenham EG, et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–541. doi: 10.1038/nature02214 [DOI] [PubMed] [Google Scholar]
- 75.Chang JR, Duan XH, Zhang BH, Teng X, Zhou YB, Liu Y, Yu YR, Zhu Y, Tang CS, Qi YF. Intermedin1-53 attenuates vascular smooth muscle cell calcification by inhibiting endoplasmic reticulum stress via cyclic adenosine monophosphate/protein kinase A pathway. Exp Biol Med (Maywood). 2013;238:1136–1146. doi: 10.1177/1535370213502619 [DOI] [PubMed] [Google Scholar]
- 76.Liu H, Li X, Qin F, Huang K. Selenium suppresses oxidative-stress-enhanced vascular smooth muscle cell calcification by inhibiting the activation of the PI3K/AKT and ERK signaling pathways and endoplasmic reticulum stress. J Biol Inorg Chem. 2014;19:375–388. doi: 10.1007/s00775-013-1078-1 [DOI] [PubMed] [Google Scholar]
- 77.Lu Y, Bian Y, Wang Y, Bai R, Wang J, Xiao C. Globular adiponectin reduces vascular calcification via inhibition of ER-stress-mediated smooth muscle cell apoptosis. Int J Clin Exp Pathol. 2015;8:2545–2554. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.