

SUMMARY
Our understanding of the genetic basis of host resistance to viral infection and disease has progressed significantly over the last century. Numerous genes coding for modifiers of immune functions have been identified which impact a variety of critical cellular processes, including signaling via lymphocyte receptors and their ligands, signal transduction, cytokine signaling, production and release of cytotoxic effectors, transcriptional regulation and proliferation. Genome-wide association studies implicate an important role for both highly polymorphic natural killer (NK) cell receptors and their major histocompatibility complex (MHC) class I ligands in modifying host resistance. These findings indicate NK cells are critical mediators of viral control with considerable potential to affect morbidity and mortality outcomes. They further suggest that both stimulatory and inhibitory NK receptor polymorphisms alter NK cell sensing of MHC I ligands on viral targets, which influences how NK cells respond to infection. In many cases, however, the underlying causes associated with host outcomes remain elusive. Herein, we discuss several modes of NK cell sensing of MHC I and MHC I-like molecules on viral targets, and the role of genetic diversity in this evolutionarily dynamic process. We further suggest that natural selection for paired NK receptors with opposing function, but shared MHC I ligands may give rise to rare, but highly effective MHC I-dependent modes of NK cell sensing of viral targets.
Keywords: genetic variation, host resistance to infection, Ly49 receptor, H-2, HLA, Killer-cell immunoglobulin-like receptor, natural killer cells, MHC, pathogen associated molecular pattern, polymorphism, viral immunity
Graphical Abstract
Graphical Summary: NK inhibitory and activating receptors undergo natural selection together with their MHC I ligands to provide optimal sensing of virus infected cells.
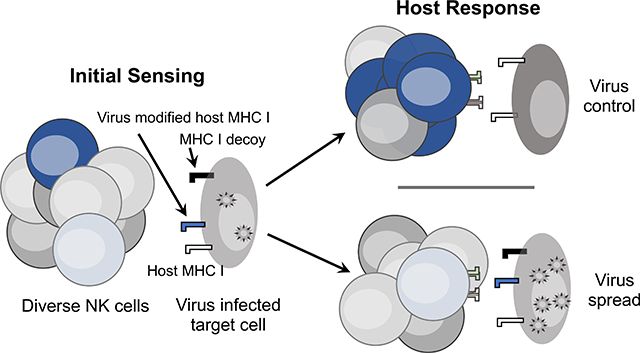
INTRODUCTION
Today, complications stemming from infectious disease account for ~25% of deaths globally [1]. Almost a century has passed since the advent of genetic approaches to explore the basis of mammalian susceptibility to viral infection [2–4]. Recent advances include identification of individual genes and the nucleotide variants within them that underpin disparity in host immune defenses and resultant impacts on viral growth and pathogenesis. A well-delineated inference of these studies focusing on the genetics of host resistance to viral infection (human and mouse) suggests that NK receptor polymorphisms are a frequent cause of variability in immune control and disease outcome. Moreover, genetic epistasis is typical of NK receptor and MHC I gene combinations affecting host morbidity and mortality outcomes [5]. These findings hint that critical interactions between NK receptors and their MHC I ligands are essential to modify NK cell control of viral spread and eventual health outcomes in infected hosts.
Genetic variation underlying polymorphic NK cell receptors for MHC I and their cognate MHC I ligands indicates that diversity favors host survival, at least at a population level. Indeed, NK cell deficiency is associated with severe host susceptibility to viral infection and premature death due to infectious causes in most affected persons [6, 7]. Whereas classical NK cell deficiency results from mutations in genes encoding GATA-2, MCM4 and IRF8 that are essential to develop mature NK cells, functional deficiency resulting in impaired NK cell activity is less well understood [6, 8, 9]. Moreover, both human killer-cell immunoglobulin-like receptor (KIR) and mouse Ly49 (killer lymphocyte receptor, subfamily a, Klra) genes are under natural selection and significant pressure to rapidly evolve [10]. While reproductive pressures exert a profound effect on gene selection, health-related impacts associated with increased fitness and host resistance to infection and pathogenesis have undoubtedly influenced these MHC I-binding NK receptors as well [11, 12].
NK cells display many different types of cell surface receptors that specifically bind classical or non-classical MHC I molecules and modify NK functional responses, including KIRs in human, Ly49 receptors in rodents, and NKG2A/C-CD94 receptors in both species that specifically bind classical and non-classical MHC I molecules. Although KIR and Ly49 receptors are structurally unrelated, both receptor types display variegated expression in NK cells, they serve analogous functional roles, and they either stimulate or inhibit NK activity upon engagement of MHC I or related cognate ligands [12, 13].
Inasmuch as the genes for these NK receptors and their MHC I ligands segregate independently in both human and mouse, NK receptor diversification should increase the likelihood that at least one can bind self-MHC I in a given host. NK cell education (licensing), which requires NK inhibitory receptor interaction with cognate MHC I, contributes to self-tolerance and endows NK cells with increased effector activity in response to stimulation through activation receptors [14, 15]. Hence, licensed NK cells are prevented from killing autologous tissues, yet they aggressively attack self-MHC I-deficient targets [16]. However, many viruses deliberately interfere with antigen processing and presentation pathways in infected cells, which results in altered or inadequate cell surface MHC I display and evasion of T cell immunity (reviewed in [17, 18]). MHC I-specific NK receptors therefore are well suited to detect viral pathogens striving to prevent host expression of self-antigens on the surface of infected cells.
Recent studies have shown that polymorphism affecting both MHC I-specific NK receptors and their cognate ligands has a synergistic effect on antiviral NK cell responses and host outcomes during infection. These findings suggest that natural selection for diversity in NK receptors and their ligands underlies the potential for highly discriminatory interactions which might assist NK cell sensing of virus modified self-antigens on infected targets. This review thus focuses primarily on human and mouse studies highlighting the importance of selection for polymorphic NK receptors and MHC I which jointly shape how NK cells sense and respond to viral infection (Figure 1).
Figure 1. A proposed model for NK cell sensing of MHC I on virus infected target cells.

Proposed NK cell sensing modes involving activation (AR) or inhibitory (IR) receptor detection of viral targets with consequent effects on antiviral NK cells and host virus control (VC) are depicted. Major NK receptor signaling pathways implicated in each mode are shown. The extent of VC may vary and, in most cases, has not been compared across sensing modes. VC associated with high expression KIR3DL1hi, however, exceeds mild HIV protection noted for low expression KIR3DL1lo. Expression-independent KIR3DL1–47V is associated with HIV elite control (EC) in HLA-B*57 people. Self MHC I-dependent IR enhancement of AR sensing is represented by increased expansion of activated NK cells that are able to mediate highly efficient lysis of infected target cells.
MHC AND NON-MHC GENETIC CONTROL OF HOST RESISTANCE TO VIRAL INFECTION
More than forty years ago, researchers found that outbred and different inbred strains of mice differed in their susceptibility to acute lethal MCMV infection [19]. Grundy (Chalmer) and colleagues predicted that mouse strain-specific susceptibility differences could be useful to explore the genetic basis of host resistance to viral infection [20]. They found that both MHC and non-MHC genetic factors contribute to host protection during experimental MCMV infection [20, 21]. Remarkably, the H-2k haplotype in C3H, B10.BR or BALB.K mice confers substantial host protection in comparison to congenic strains carrying H-2b, H-2d or H-2g haplotypes [20–23]. Indeed, BALB.K mice withstand ~10-fold higher dose MCMV infection than their H-2 congenic counterparts. Nonetheless, non-MHC effects are clearly at play since BALB.K and B10.BR also differ in the extent of host resistance to lethal infection [21].
Similar to strain-dependent variation in MCMV-induced lethality, spleen MCMV titers vary according to the level of host resistance within 3–4 days post-infection [23–25]. Scalzo and Shellam therefore measured MCMV titers in infected spleen and liver tissues to determine acute virus control phenotypes in different offspring obtained by crossing C57BL/6 (B6; MCMV resistant) to A/J or BALB/c (both MCMV susceptible) mice. They studied several different panels of recombinant inbred mouse strains, as well as large cohorts of intercross or backcross mice that were all typed with known genetic markers, including natural killer gene complex (NKC)-linked genetic markers [25–27]. A single autosomal dominant, NKC-linked (non-MHC) locus Cmv1, later identified as Ly49h [28–30], was found to confer robust host protection in B6 mice during acute MCMV infection.
NK CELL SENSING OF AN MHC I-RELATED VIRAL DECOY
The Ly49H activation receptor expressed in about one half of NK cells in B6 mice, but not in A/J or BALB/c mouse NK cells, binds the MCMV encoded m157 molecule which is structurally related to MHC I [31, 32]. Antigen-specific recognition of m157+ infected target cells causes rapid activation and expansion of Ly49H+ NK cells so that they exert profound control over viral spread during acute MCMV infection [33]. This initial expansion phase is followed closely by Ly49H+ NK cell contraction and eventual formation of NK cell memory which is marked by antigen recall responsiveness, increased effector activity and enhanced viral control in challenge experiments [34, 35].
Specific expansion of Ly49H+ NK cells during acute MCMV infection depends upon the coordinated action of several cytokines including type I interferon (IFN), IL-12, IL-15, IL-18 and IL-33 that regulate activation, proliferation, IFN-γ production, cytotoxicity and accordingly acute MCMV control [36–38]. Type I interferon or IL-12 cytokines elicited in response to MCMV infection boost granzyme B levels and Ly49H+ NK lysis of infected targets which requires both IFN-γ and perforin [39, 40]. Moreover, type I IFN α/β receptor signaling promotes expansion of Ly49H+ NK cells by protecting them from premature apoptotic death during MCMV infection [41]. Together these data establish the importance of various cytokines and effector molecules working together in support of the antiviral role of Ly49H+ NK cells.
In sharp contrast to Ly49H, MCMV m157 binds NK inhibitory receptors (e.g. Ly49I129) and dampens NK effector function, which suggests that it evolved primarily as a decoy MHC I to counter NK cell attack in favor of viral spread within the host [31, 42]. Although the single remaining Ly49IB6 receptor allele in inbred B6 mice lacks this specificity, selective pressure exerted by MCMV in a predecessor of C57-related strains might have given rise to m157-specific Ly49HB6 via alteration of an inhibitory receptor allele previously targeted for viral manipulation. Such a change would have required minor nucleotide sequence variants resulting in two amino acid adaptations, one in the transmembrane domain to enable interaction with a signaling adaptor (e.g. DAP-12), and another that alters a tyrosine in the immunoreceptor tyrosine-based inhibitory motif [10]. Inbreeding of B6 and 129 (one of Castle’s strains generated by crossing with C57-related mice) strains [43] thus could have resulted in the respective loss (B6 strain) or retention (129 strain) of an m157-specific Ly49I receptor allele, thereby further contributing to strain-specific differences in host resistance to MCMV infection.
In addition to m157-dependent effects, Ly49H+ NK sensing of the viral decoy is regulated by licensing receptors that recognize self-MHC I. mAb 5E6-reactive Ly49 receptors in B6 mice, for example, interfere with m157-dependent Ly49H+ NK cell expansion, and MCMV control upon adoptive transfer into Ly49H-deficient neonates due to sustained inhibitory interactions with self-MHC I [44]. This effect presumably is due to self-MHC I Db and Kb-licensed Ly49I receptors [45] since 5E6 does not readily bind Ly49C expressed on NK cells in B6 mice [46]. Ly49H-dependent sensing leading to NK cell expansion and virus control in MCMV-infected neonates is likewise thwarted by the NKR-P1B inhibitory NK receptor, which has Clr-b as its self-ligand [47]. Interestingly, the MCMV m12 protein, another viral decoy, binds NKR-P1B as a way to evade NK-mediated immunity [48]. The importance of licensed NK cells in viral control thus has been questioned. It remains unknown whether this supposed negative impact is restricted to NK sensing of stand-alone viral decoys. In any case, the Ly49H-m157 receptor-ligand pairing differs from other modes of NK cell sensing of viral infection that are primarily host MHC I-dependent.
HOST MHC I-DEPENDENT NK CELL SENSING OF VIRAL INFECTION
The H-2k haplotype imparts essential control of acute MCMV replication, viral spread and lethal infection [26, 49, 50]. As with B6 mice, depletion of NK cells in C57-related MA/My (H-2k) mice prior to infection fully eliminates MCMV control [26, 43, 49, 50]. A combination of genes within the NKC-Ly49 gene cluster and the MHC is essential to acute viral control in MA/My x BALB/c cross offspring [49], much like genetic epistasis relating defined pairs of KIR and HLA afford increased viral control and protection from disease progression in persons with chronic viral infection (discussed below). The MA/My Ly49P activation receptor which specifically binds MHC I Dk plus MCMV gp34 complexes in vitro, thus, is predicted to deliver antigen-specific NK cell control of MCMV [51]. The Ly49L activation receptor expressed on NK cells in BALB.K mice also binds MHC I Dk plus MCMV gp34 complexes in vitro. Although in vivo evidence of a role for Ly49P is limited, Ly49L+ NK cells expand in vivo during MCMV infection, and adoptive transfer of mature Ly49L+ NK cells protects neonates during infection [52]. These findings establish that antigen-specific activation receptors expressed by NK cells contribute to acute virus control and critical host resistance.
In separate work, genetic analysis of MA/My x C57L (MCMV resistant and susceptible, respectively) cross offspring identified MHC I Dk as a primary genetic factor influencing MCMV control [50, 53]. MHC I Dk-dependent MCMV resistance is equally abolished in these mice by depleting either total NK cells or a select subset expressing Ly49G2, a cognate inhibitory receptor for self-MHC I Dk [53, 54]. In bone marrow chimeric mice expressing MHC I Dk solely in hematopoietic or non-hematopoietic cells, Ly49G2+ NK cells display impaired stimulatory activity, less effective lysis of MHC I-deficient target cells and diminished MCMV control [54]. Both the frequency and numbers of resting Ly49G2+ NK cells in naïve mice are regulated by MHC I Dk [55], and they additionally undergo specific expansion and accumulation in response to MCMV in MHC I Dk mice, but not in mice lacking their cognate self-MHC I ligand [56]. Moreover, Ly49G2MA/My or C57L+ NK cell responsiveness to MCMV is specifically dependent on the MHC I Dk locus [55]. NK cells bearing certain Ly49G2 receptor allotypes become licensed when MHC I Dk is expressed in both hematopoietic and non-hematopoietic cells. Incomplete education due to absent or lineage-restricted self-MHC I exposure during development, however, results in inferior Ly49G2+ NK cells and less effective viral control. The importance of NK cell education in host resistance to infection, however, remains to be established in animals deficient for a single NK licensing receptor. These animals should provide an outstanding model system to explore how NK inhibitory receptor signaling contributes to the role of NK cells in viral immunity, which might further yield insights into the basis for elite virus control observed in HIV-infected people (discussed below).
Self-MHC regulated NK cell expansion in response to MCMV thus is not a common feature since Ly49G2B6+ NK cells expand irrespective of host H-2 haplotype in B6 and B10.D2 mice during Listeria or MCMV infection [57]. However, H-2-dependent expansion, tissue localization and virus control is characteristic of licensed NK cells responding to MCMV in B6 mice depleted of regulatory T cells, or in chimeric B6 hosts following hematopoietic stem cell transplantation [58, 59]. Licensed NK cells thus mediate critical virus control in different genetic backgrounds presumably due to rapid detection of viral targets and consequent expansion during infection. Unlicensed NK cells in these settings, in contrast, fail to respond similarly to infection, thereby delineating NK cell sensing modes based on detection of a viral decoy versus virus modified host MHC I molecules.
HUMAN KIR AND HLA GENE PAIRS IMPART FITNESS: HOST MHC I-DEPENDENT NK CELL SENSING OF VIRAL INFECTION INFERRED
More than a decade ago, Carrington and coworkers found that specific KIR and HLA genetic pairings or epistatic interactions correspond with delayed progression toward disease in HIV-infected persons [60]. Individuals with both KIR3DS1 and HLA-B Bw4 allotypes experienced delayed disease progression which suggests that KIR3DS1 NK activation receptor recognition of HIV-infected target cells might coincide with better NK cell clearance of viral infection and less disease.
HLA-B Bw4 defense against HIV intriguingly extends to persons carrying specific KIR3DL1 allotypes encoding inhibitory NK receptors [61]. When sorted by NK cell surface expression level, highly expressed KIR3DL1 allotypes best correspond with delayed disease progression and lower viral loads in persons carrying HLA-B*57 with an isoleucine at position 80 (80I) of the heavy chain. On the other hand, low expression KIR3DL1 allotypes are beneficial in HLA-B*27 persons with a threonine residue at position 80 (80T), albeit this pairing is less protective than KIR3DL1hi in HLA-B*57-80I persons. Although a basis of KIR3DL1 protection is tenuous, varied expression corresponds to inhibitory activity in NK cells [62, 63], which may selectively modify NK cell sensing of distinct HLA-B Bw4 ligands on infected targets. KIR3DL1 discernment of HLA-B*57 presented peptides [64] additionally suggests it may be sensitive to HIV-modified peptide cargo due to changes in the overall conformation of host MHC antigens in infected cells. In effect, less inhibitory KIR3DL1 signaling during infection should promote NK cell lysis of infected targets and HIV control via stimulation through a still unknown NK activation receptor. KIR3DS1 is an interesting candidate in this regard as it specifically binds HLA-B*57 presenting peptides with discrepant P8 residues, as well as two different HIV peptides [65].
Amongst HIV-infected persons, some that are serologically positive for anti-HIV antibodies exhibit slower progression towards clinical immunodeficiency while maintaining a relatively low viral burden. Many of these slow progressors eventually succumb to HIV infection without anti-retroviral treatment. A small fraction of this group referred to as ‘elite controllers’ maintain remarkably low to undetectable (plasma HIV RNA < 50–75 copies / ml) viral loads without therapy and do not progress to AIDS for still unknown reasons. These individuals represent < 1.0% of all HIV-infected persons [66, 67]. Elite control (EC) in these individuals appears to be highly durable and can extend beyond 25 years [66].
HLA-B*57 which confers significant HIV protection is highly represented amongst elite controllers. Carrington’s research team therefore recently sifted HLA-B*57+ controller and non-controller genome sequences for host genetic modifiers underpinning its effect in HIV-infected persons. Remarkably, only a single KIR3DL1 nucleotide variant (position 47 valine; 47V) was significantly associated with EC [68]. This KIR3DL147V effect was not strictly aligned with higher or lower KIR3DL1 expression, but it was HLA-B*57:01-dependent as EC was not evident in HLA-B*57:03 persons with two disparate amino acids at the base of the peptide binding groove [68]. Taken together, these data suggest that mere inhibitory NK receptor expression is not a key determinant of EC. Subtle structural variations in the NK receptor and its MHC I ligand might favor NK cell discernment of peptide cargo changes and resultant NK cell responses during viral infection.
Interactions between KIRs and their MHC I ligands differ from those between the T cell receptor and MHC I, which underlies KIR specificity for HLA alleles (e.g. position 80 in the alpha 1 helix of HLA-C) [12]. Nonetheless, KIRs display peptide-specific interactions with MHC I as in the case of KIR3DL2 which binds HLA-A3 or HLA-A11 and an EBNA3A peptide fragment from Epstein-Barr virus [69]. More recently, MHC I-restricted and peptide-specific recognition has been shown for the KIR2DS2 activation receptor that recognizes Hepatitis C Virus (HCV)-infected cells. Naiyer et al found that an HCV NS3 helicase 1a ‘LNP’ peptide fragment, conserved amongst the vast majority of sequenced HCV isolates, stimulates KIR2DS2 activity when presented by HCV-protective HLA-C*0102 molecules [70]. Though other flaviviruses lack expression of NS3 LNP peptides, 61 of 63 members of the genus flaviviruses instead express NS3 RNA helicases including a highly conserved KIR2DS2-binding peptide domain that fits the HLA-C*0102 peptide-binding groove [70]. Recent work from Hammer and colleagues further shows that human cytomegalovirus (HCMV)-responsive NKG2C+ adaptive NK cells expand and differentiate differently in response to HCMV UL40-derived peptides [71]. These findings together demonstrate that stimulatory NK receptors mediate antigen-specific recognition of viral infection, similar to other pattern recognition receptors (e.g. Toll-like receptors), via surveillance for conserved pathogen associated molecular patterns (PAMPs), which coincides with NK-mediated control of viral replication and host protection.
The importance of specific KIR and HLA genetic pairings in host protection from viral infection is not limited to HIV. Indeed, individuals carrying homozygous KIR2DL3 and HLA-C1 allotypes manifest significant resistance to Hepatitis C Virus (HCV)-induced disease [72]. Despite that an underlying mechanism for KIR2DL3 and HLA-C1 synergy during HCV infection remains elusive, it is possible that KIR2DL3 can detect subtle changes in virus-modified HLA-C1 since it displays some degree of peptide-selectivity in discriminating MHC I ligands [73, 74]. Moreover, KIR2DL3 is sensitive to variation in HCV core-derived peptides presented by HLA-C*03:04 which results in NK cell functional differences [75]. These data suggest that KIR2DL3 recognition of virus-modified HLA-C molecules favors NK-mediated HCV control, perhaps by promoting signaling through an activation receptor due to the loss of negative signaling through a licensing receptor. Notably, KIR2DS2 also binds HLA-C*03:04 although this interaction is not NS3 LNP peptide-specific [70]. An intriguing question is whether a combination of certain KIR2DL3 and KIR2DS2 alleles further augment the host response in HLA-C1 persons during HCV infection.
CONCLUDING REMARKS
NK cell surveillance for virus-induced changes in host cells during infection relies on polymorphic NK receptor sensing of MHC I and related antigens (Figure 1), and transduction of varied, and often opposing, functional signals in distinct, but overlapping NK cell subsets. Genetic analysis of host resistance to viral infection indicates that polymorphism in NK receptors and cognate MHC I significantly affect antiviral NK cell responses and ensuing outcomes in the infected host.
Increased fitness and host protection associated with MHC I-specific NK receptor and self-MHC I gene pairs suggests that specific receptor-ligand interactions synergistically modify NK cells, including target cell sensing, cytotoxic effector functions, activation, cytokine production, proliferation and/or antiviral activity. Inasmuch as both activation and inhibitory receptor genes show linkage to disease protection, opposing receptors working separately, or in tandem, apparently is essential to NK sensing of virus-modified host MHC I determinants and resultant antiviral NK effector functions.
A rare percentage of HIV infected people with antibody reactivity to HIV antigens intriguingly display overt and durable resistance (i.e. elite control) for years in the complete absence of anti-retroviral therapy. Recent work demonstrates that HLA-B*57+ persons carrying the KIR3DL147V allotype are significantly protected from HIV in comparison to KIR3DL147I persons. This finding suggests KIR3DL1–47V+ NK cells are equipped for specific sensing of HIV infected targets, although a basis of KIR3DL1–47V receptor protection remains unclear. Perhaps HIV-specific KIR3DS1 jointly expressed in KIR3DL1–47V+ NK cells promote sensing and clearance of HIV infected targets. Related to this, licensed NK cells displaying certain Ly49G2 allotypes provide essential MCMV control in MHC I Dk mice, which represents a novel and important model system to explore the role of licensed NK cells in viral immunity and possibly elite virus control.
These studies raise a number of important questions requiring further attention. Are the observed genetic associations in human studies related to NK cells directly, their effect on adaptive immunity, or some combination of the two? What’s the role of the inhibitory receptor? Are they sensitive to fine differences in peptide cargo (host v. viral) when carried by specific HLA alleles? Does inhibitory receptor sensing result in more pronounced effector NK cell stimulation? A better understanding of the role of both activation and inhibitory receptor sensing of viral infection should reveal if and how different receptors with divergent functions might partner to deliver optimal antiviral NK activities and increased host resistance during viral infection.
ACKNOWLEDGEMENTS
This work was supported by the Department of Medicine, Division of Nephrology, the Beirne Carter Center for Immunology Research, a Career Award from the AAI (MGB and AG), and PHS Grant R01-AI050072 (MGB). AG and JC received support on PHS Training Grant T32-AI7046. WTN received support on PHS training grant T32-DK072922. We thank past and current members of the Brown Laboratory. We also thank the many researchers who have generously contributed important tools, resources and creative thinking to help extend this work.
References.
- 1.Morens DM, Folkers GK, Fauci AS (2004) The challenge of emerging and re-emerging infectious diseases. Nature 430, 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theiler M (1930) Susceptibility of White Mice to the Virus of Yellow Fever. Science 71, 367. [DOI] [PubMed] [Google Scholar]
- 3.Lynch CJ and Hughes TP (1936) The Inheritance of Susceptibility to Yellow Fever Encephalitis in Mice. Genetics 21, 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sawyer WA and Lloyd W (1931) The Use of Mice in Tests of Immunity against Yellow Fever. J Exp Med 54, 533–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashirova AA, Thomas R, Carrington M (2011) HLA/KIR restraint of HIV: surviving the fittest. Annu Rev Immunol 29, 295–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orange JS (2012) Unraveling human natural killer cell deficiency. J Clin Invest 122, 798–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biron CA, Byron KS, Sullivan JL (1989) Severe herpesvirus infections in an adolescent without natural killer cells. The New England journal of medicine 320, 1731–1735. [DOI] [PubMed] [Google Scholar]
- 8.Mace EM, Bigley V, Gunesch JT, Chinn IK, Angelo LS, Care MA, Maisuria S, Keller MD, Togi S, Watkin LB, LaRosa DF, Jhangiani SN, Muzny DM, Stray-Pedersen A, Coban Akdemir Z, Smith JB, Hernandez-Sanabria M, Le DT, Hogg GD, Cao TN, Freud AG, Szymanski EP, Savic S, Collin M, Cant AJ, Gibbs RA, Holland SM, Caligiuri MA, Ozato K, Paust S, Doody GM, Lupski JR, Orange JS (2017) Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest 127, 306–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, Cohen JI, Frenkel EP, Bagwell JC, Sullivan JL, Biron CA, Spalding C, Zerbe CS, Uzel G, Holland SM, Orange JS (2013) Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood 121, 2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abi-Rached L and Parham P (2005) Natural selection drives recurrent formation of activating killer cell immunoglobulin-like receptor and Ly49 from inhibitory homologues. Journal of Experimental Medicine 201, 1319–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vilches C and Parham P (2002) KIR: Diverse, Rapidly Evolving Receptors of Innate and Adaptive Immunity. Annu. Rev. Immunol 20, 217–251. [DOI] [PubMed] [Google Scholar]
- 12.Parham P (2005) MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 5, 201–214. [DOI] [PubMed] [Google Scholar]
- 13.Trowsdale J, Barten R, Haude A, Stewart CA, Beck S, Wilson MJ (2001) The genomic context of natural killer receptor extended gene families. Immunol Rev 181, 20–38. [DOI] [PubMed] [Google Scholar]
- 14.Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song Y, Yang L, French AR, Sunwoo JB, Lemieux S, Hansen TH, Yokoyama WM (2005) Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature 436, 709–713. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez NC, Treiner E, Vance RE, Jamieson AM, Lemieux S, Raulet DH (2005) A subset of natural killer cells achieves self-tolerance without expressing inhibitory receptors specific for self-MHC molecules. Blood 105, 4416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S (2011) Innate or adaptive immunity? The example of natural killer cells. Science 331, 44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nash WT, Teoh J, Wei H, Gamache A, Brown MG (2014) Know Thyself: NK-Cell Inhibitory Receptors Prompt Self-Tolerance, Education, and Viral Control. Frontiers in immunology 5, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miletic A, Krmpotic A, Jonjic S (2013) The evolutionary arms race between NK cells and viruses: who gets the short end of the stick? Eur J Immunol 43, 867–77. [DOI] [PubMed] [Google Scholar]
- 19.Selgrade MK and Osborn JE (1974) Role of macrophages in resistance to murine cytomegalovirus. Infection and immunity 10, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalmer JE, Mackenzie JS, Stanley NF (1977) Resistance to murine cytomegalovirus linked to the major histocompatibility complex of the mouse. The Journal of general virology 37, 107–114. [DOI] [PubMed] [Google Scholar]
- 21.Grundy JE, Mackenzie JS, Stanley NF (1981) Influence of H-2 and non-H-2 genes on resistance to murine cytomegalovirus infection. Infection and immunity 32, 277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bancroft GJ, Shellam GR, Chalmer JE (1981) Genetic influences on the augmentation of natural killer (NK) cells during murine cytomegalovirus infection: correlation with patterns of resistance. J Immunol 126, 988–994. [PubMed] [Google Scholar]
- 23.Mercer JA and Spector DH (1986) Pathogenesis of acute murine cytomegalovirus infection in resistant and susceptible strains of mice. J Virol 57, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allan JE and Shellam GR (1984) Genetic control of murine cytomegalovirus infection: virus titres in resistant and susceptible strains of mice. Archives of virology 81, 139–150. [DOI] [PubMed] [Google Scholar]
- 25.Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR (1990) Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med 171, 1469–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scalzo AA, Lyons PA, Fitzgerald NA, Forbes CA, Yokoyama WM, Shellam GR (1995) Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics 27, 435–441. [DOI] [PubMed] [Google Scholar]
- 27.Scalzo AA, Fitzgerald NA, Wallace CR, Gibbons AE, Smart YC, Burton RC, Shellam GR (1992) The effect of the Cmv-1 resistance gene, which is linked to the natural killer cell gene complex, is mediated by natural killer cells. J Immunol 149, 581–589. [PubMed] [Google Scholar]
- 28.Brown MG, Dokun AO, Heusel JW, Smith HRC, Beckman DL, Blattenberger EA, Dubbelde CE, Stone LR, Scalzo AA, Yokoyama WM (2001) Vital Involvement of a natural killer cell activation receptor in resistance to viral infection. Science 292, 934–937. [DOI] [PubMed] [Google Scholar]
- 29.Lee SH, Girard S, Macina D, Busa M, Zafer A, Belouchi A, Gros P, Vidal SM (2001) Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nature Genetics 28, 42–45. [DOI] [PubMed] [Google Scholar]
- 30.Daniels KA, Devora G, Lai WC, O’Donnell CL, Bennett M, Welsh RM (2001) Murine Cytomegalovirus Is Regulated by a Discrete Subset of Natural Killer Cells Reactive with Monoclonal Antibody to Ly49H. J. Exp. Med 194, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL (2002) Direct Recognition of Cytomegalovirus by Activating and Inhibitory NK Cell Receptors. Science 296, 1323–1326. [DOI] [PubMed] [Google Scholar]
- 32.Smith HRC, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM (2002) Recognition of a virus-encoded ligand by a natural killer cell activation receptor. PNAS 99, 8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yokoyama WM, Kim S, French AR (2004) The dynamic life of natural killer cells. Annual Review of Immunology 22, 405–29. [DOI] [PubMed] [Google Scholar]
- 34.O’Sullivan TE, Sun JC, Lanier LL (2015) Natural Killer Cell Memory. Immunity 43, 634–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min-Oo G, Bezman NA, Madera S, Sun JC, Lanier LL (2014) Proapoptotic Bim regulates antigen-specific NK cell contraction and the generation of the memory NK cell pool after cytomegalovirus infection. J Exp Med 211, 1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nabekura T, Girard JP, Lanier LL (2015) IL-33 Receptor ST2 Amplifies the Expansion of NK Cells and Enhances Host Defense during Mouse Cytomegalovirus Infection. J Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madera S and Sun JC (2015) Cutting edge: stage-specific requirement of IL-18 for antiviral NK cell expansion. J Immunol 194, 1408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei X. q., Liew FY, Caligiuri MA, Durbin JE, Biron CA (2002) Coordinated and Distinct Roles for IFN-{alpha}{beta}, IL-12, and IL-15 Regulation of NK Cell Responses to Viral Infection. J Immunol 169, 4279–4287. [DOI] [PubMed] [Google Scholar]
- 39.Loh J, Chu DT, O’Guin AK, Yokoyama WM, Virgin H. W. t. (2005) Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol 79, 661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parikh BA, Piersma SJ, Pak-Wittel MA, Yang L, Schreiber RD, Yokoyama WM (2015) Dual Requirement of Cytokine and Activation Receptor Triggering for Cytotoxic Control of Murine Cytomegalovirus by NK Cells. PLoS Pathog 11, e1005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Madera S, Rapp M, Firth MA, Beilke JN, Lanier LL, Sun JC (2016) Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J Exp Med 213, 225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corbett AJ, Coudert JD, Forbes CA, Scalzo AA (2011) Functional Consequences of Natural Sequence Variation of Murine Cytomegalovirus m157 for Ly49 Receptor Specificity and NK Cell Activation. J Immunol 186, 1713–1722. [DOI] [PubMed] [Google Scholar]
- 43.Beck JA, Lloyd S, Hafezparast M, Lennon-Pierce M, Eppig JT, Festing MF, Fisher EM (2000) Genealogies of mouse inbred strains. Nature Genetics 24, 23–25. [DOI] [PubMed] [Google Scholar]
- 44.Orr MT, Murphy WJ, Lanier LL (2010) ‘Unlicensed’ natural killer cells dominate the response to cytomegalovirus infection. Nat Immunol 11, 321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brodin P, Lakshmikanth T, Johansson S, Karre K, Hoglund P (2009) The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 113, 2434–41. [DOI] [PubMed] [Google Scholar]
- 46.MacFarlane A. W. t., Yamazaki T, Fang M, Sigal LJ, Kurosaki T, Campbell KS (2008) Enhanced NK-cell development and function in BCAP-deficient mice. Blood 112, 131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rahim MM, Wight A, Mahmoud AB, Aguilar OA, Lee SH, Vidal SM, Carlyle JR, Makrigiannis AP (2016) Expansion and Protection by a Virus-Specific NK Cell Subset Lacking Expression of the Inhibitory NKR-P1B Receptor during Murine Cytomegalovirus Infection. J Immunol 197, 2325–37. [DOI] [PubMed] [Google Scholar]
- 48.Aguilar OA, Berry R, Rahim MMA, Reichel JJ, Popovic B, Tanaka M, Fu Z, Balaji GR, Lau TNH, Tu MM, Kirkham CL, Mahmoud AB, Mesci A, Krmpotic A, Allan DSJ, Makrigiannis AP, Jonjic S, Rossjohn J, Carlyle JR (2017) A Viral Immunoevasin Controls Innate Immunity by Targeting the Prototypical Natural Killer Cell Receptor Family. Cell 169, 58–71 e14. [DOI] [PubMed] [Google Scholar]
- 49.Desrosiers MP, Kielczewska A, Loredo-Osti JC, Adam SG, Makrigiannis AP, Lemieux S, Pham T, Lodoen MB, Morgan K, Lanier LL, Vidal SM (2005) Epistasis between mouse Klra and major histocompatibility complex class I loci is associated with a new mechanism of natural killer cell-mediated innate resistance to cytomegalovirus infection. Nat Genet 37, 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dighe A, Rodriguez M, Sabastian P, Xie X, McVoy M, Brown MG (2005) Requisite H2k role in NK cell-mediated resistance in acute murine cytomegalovirus-infected MA/My mice. J Immunol 175, 6820–8. [DOI] [PubMed] [Google Scholar]
- 51.Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, Hill AB, Koszinowski UH, Jonjic S, Lanier LL, Vidal SM (2009) Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med 206, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pyzik M, Charbonneau B, Gendron-Pontbriand EM, Babic M, Krmpotic A, Jonjic S, Vidal SM (2011) Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J Exp Med 208, 1105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie X, Stadnisky MD, Coats ER, Ahmed Rahim MM, Lundgren A, Xu W, Makrigiannis AP, Brown MG (2010) MHC class I D(k) expression in hematopoietic and nonhematopoietic cells confers natural killer cell resistance to murine cytomegalovirus. Proc Natl Acad Sci U S A 107, 8754–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei H, Nash WT, Makrigiannis AP, Brown MG (2014) Impaired NK-cell education diminishes resistance to murine CMV infection. Eur J Immunol 44, 3273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gillespie AL, Teoh J, Lee H, Prince J, Stadnisky MD, Anderson M, Nash W, Rival C, Wei H, Gamache A, Farber CR, Tung K, Brown MG (2016) Genomic Modifiers of Natural Killer Cells, Immune Responsiveness and Lymphoid Tissue Remodeling Together Increase Host Resistance to Viral Infection. PLoS Pathog 12, e1005419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prince J, Lundgren A, Stadnisky MD, Nash WT, Beeber A, Turner SD, Brown MG (2013) Multiparametric Analysis of Host Response to Murine Cytomegalovirus in MHC Class I-Disparate Mice Reveals Primacy of Dk-Licensed Ly49G2+ NK Cells in Viral Control. J Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barao I, Alvarez M, Ames E, Orr MT, Stefanski HE, Blazar BR, Lanier LL, Anderson SK, Redelman D, Murphy WJ (2011) Mouse Ly49G2+ NK cells dominate early responses during both immune reconstitution and activation independently of MHC. Blood 117, 7032–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sungur CM, Tang-Feldman YJ, Ames E, Alvarez M, Chen M, Longo DL, Pomeroy C, Murphy WJ (2013) Murine natural killer cell licensing and regulation by T regulatory cells in viral responses. Proc Natl Acad Sci U S A 110, 7401–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zamora AE, Aguilar EG, Sungur CM, Khuat LT, Dunai C, Lochhead GR, Du J, Pomeroy C, Blazar BR, Longo DL, Venstrom JM, Baumgarth N, Murphy WJ (2017) Licensing delineates helper and effector NK cell subsets during viral infection. JCI insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, Buchbinder S, Hoots K, Vlahov D, Trowsdale J, Wilson M, O’Brien SJ, Carrington M (2002) Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet 31, 429–34. [DOI] [PubMed] [Google Scholar]
- 61.Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, Colombo S, Brown EE, Shupert WL, Phair J, Goedert JJ, Buchbinder S, Kirk GD, Telenti A, Connors M, O’Brien SJ, Walker BD, Parham P, Deeks SG, McVicar DW, Carrington M (2007) Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet 39, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gardiner CM, Guethlein LA, Shilling HG, Pando M, Carr WH, Rajalingam R, Vilches C, Parham P (2001) Different NK cell surface phenotypes defined by the DX9 antibody are due to KIR3DL1 gene polymorphism. J Immunol 166, 2992–3001. [DOI] [PubMed] [Google Scholar]
- 63.Yawata M, Yawata N, Draghi M, Little AM, Partheniou F, Parham P (2006) Roles for HLA and KIR polymorphisms in natural killer cell repertoire selection and modulation of effector function. J Exp Med 203, 633–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vivian JP, Duncan RC, Berry R, O’Connor GM, Reid HH, Beddoe T, Gras S, Saunders PM, Olshina MA, Widjaja JM, Harpur CM, Lin J, Maloveste SM, Price DA, Lafont BA, McVicar DW, Clements CS, Brooks AG, Rossjohn J (2011) Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature 479, 401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Connor GM, Vivian JP, Gostick E, Pymm P, Lafont BA, Price DA, Rossjohn J, Brooks AG, McVicar DW (2015) Peptide-Dependent Recognition of HLA-B*57:01 by KIR3DS1. J Virol 89, 5213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Deeks SG and Walker BD (2007) Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27, 406–16. [DOI] [PubMed] [Google Scholar]
- 67.Hubert JB, Burgard M, Dussaix E, Tamalet C, Deveau C, Le Chenadec J, Chaix ML, Marchadier E, Vilde JL, Delfraissy JF, Meyer L, Rouzioux (2000) Natural history of serum HIV-1 RNA levels in 330 patients with a known date of infection. The SEROCO Study Group. Aids 14, 123–31. [DOI] [PubMed] [Google Scholar]
- 68.Martin MP, Naranbhai V, Shea PR, Qi Y, Ramsuran V, Vince N, Gao X, Thomas R, Brumme ZL, Carlson JM, Wolinsky SM, Goedert JJ, Walker BD, Segal FP, Deeks SG, Haas DW, Migueles SA, Connors M, Michael N, Fellay J, Gostick E, Llewellyn-Lacey S, Price DA, Lafont BA, Pymm P, Saunders PM, Widjaja J, Wong SC, Vivian JP, Rossjohn J, Brooks AG, Carrington M (2018) Killer cell immunoglobulin–like receptor 3DL1 variation modifies HLA-B*57 protection against HIV-1. The Journal of Clinical Investigation 128, 1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, Rowland-Jones S, Braud VM (2004) Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur J Immunol 34, 1673–9. [DOI] [PubMed] [Google Scholar]
- 70.Naiyer MM, Cassidy SA, Magri A, Cowton V, Chen K, Mansour S, Kranidioti H, Mbirbindi B, Rettman P, Harris S, Fanning LJ, Mulder A, Claas FHJ, Davidson AD, Patel AH, Purbhoo MA, Khakoo SI (2017) KIR2DS2 recognizes conserved peptides derived from viral helicases in the context of HLA-C. Science immunology 2. [DOI] [PubMed] [Google Scholar]
- 71.Hammer Q, Ruckert T, Borst EM, Dunst J, Haubner A, Durek P, Heinrich F, Gasparoni G, Babic M, Tomic A, Pietra G, Nienen M, Blau IW, Hofmann J, Na IK, Prinz I, Koenecke C, Hemmati P, Babel N, Arnold R, Walter J, Thurley K, Mashreghi MF, Messerle M, Romagnani C (2018) Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat Immunol 19, 453–463. [DOI] [PubMed] [Google Scholar]
- 72.Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, Cheng J, Goedert JJ, Vlahov D, Hilgartner M, Cox S, Little A, Alexander GJ, Cramp ME, O’Brien SJ, Rosenberg WMC, Thomas DL, Carrington M (2004) HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305, 872–874. [DOI] [PubMed] [Google Scholar]
- 73.Borhis G, Ahmed PS, Mbiribindi B, Naiyer MM, Davis DM, Purbhoo MA, Khakoo SI (2013) A peptide antagonist disrupts NK cell inhibitory synapse formation. J Immunol 190, 2924–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A, Stathopoulos S, Middleton D, Mulder A, Claas FH, Elliott T, Davis DM, Purbhoo MA, Khakoo SI (2010) Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A 107, 10160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lunemann S, Martrus G, Holzemer A, Chapel A, Ziegler M, Korner C, Garcia Beltran W, Carrington M, Wedemeyer H, Altfeld M (2016) Sequence variations in HCV core-derived epitopes alter binding of KIR2DL3 to HLA-C *03:04 and modulate NK cell function. J Hepatol 65, 252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]