

Abstract
Diabetic nephropathy (DN) is considered the primary causes of end-stage renal disease (ESRD) and is related to abnormal glycolipid metabolism, hemodynamic abnormalities, oxidative stress and chronic inflammation. Antagonism of vascular endothelial growth factor B (VEGF-B) could efficiently ameliorate DN by reducing renal lipotoxicity. However, this pharmacological strategy is far from satisfactory, as it ignores numerous pathogenic factors, including anomalous reactive oxygen species (ROS) generation and inflammatory responses. We found that the upregulation of VEGF-B and downregulation of interleukin-22 (IL-22) among DN patients were significantly associated with the progression of DN. Thus, we hypothesized that a combination of a VEGF-B antibody and IL-22 could protect against DN not only by regulating glycolipid metabolism but also by reducing the accumulation of inflammation and ROS. To meet these challenges, a novel anti-VEGFB/IL22 fusion protein was developed, and its therapeutic effects on DN were further studied. We found that the anti-VEGFB/IL22 fusion protein reduced renal lipid accumulation by inhibiting the expression of fatty acid transport proteins and ameliorated inflammatory responses via the inhibition of renal oxidative stress and mitochondrial dysfunction. Moreover, the fusion protein could also improve diabetic kidney disease by increasing insulin sensitivity. Collectively, our findings indicate that the bifunctional VEGF-B antibody and IL-22 fusion protein could improve the progression of DN, which highlighted a novel therapeutic approach to DN.
KEY WORDS: Diabetic nephropathy, Vascular endothelial growth factor B, Interleukin-22, Fusion protein
Abbreviations: ACR, urine albumin-to-creatinine ratio; ADFP, adipocyte differentiation-related protein; AGEs, advanced glycation end products; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; Ccr, creatinine clearance rate; DN, diabetic nephropathy; ECM, extracellular matrix; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; FA, fatty acid; FATPs, fatty acid transport proteins; GBM, glomerular basement membrane; GSEA, gene set enrichment analysis; H&E, hematoxylin & eosin; HbA1c%, glycosylated hemoglobin; IL-22, interleukin-22; KEGG, Kyoto Encyclopedia of Genes and Genomes; NAC, N-acetyl-l-cysteine; NLRP3, NOD-like receptor family pyrin domain-containing protein 3; NRP-1, neuropilin-1; PAS, periodic acid-Schiff; ROS, reactive oxygen species; SDS-PAGE, SDS-polyacrylamide gel electrophoresis; TEM, transmission electron microscopy; VEGF-B, vascular endothelial growth factor B; VEGFR, vascular endothelial growth factor receptor; β2-MG, β2 microglobulin
Graphical abstract
The expression of VEGF-B was increased and that of IL-22 was decreased in diabetic nephropathy (DN) patients. Anti-VEGFB/IL22 fusion protein combining VEGF-B antibody and IL22 ameliorated DN by alleviating ectopic lipid accumulation, inflammatory responses and insulin resistance.
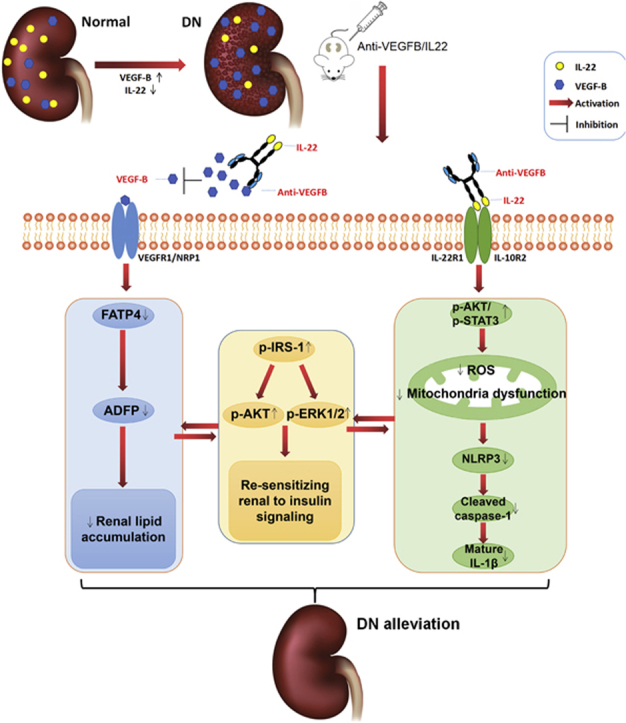
1. Introduction
Diabetic nephropathy (DN), one of the crucial microvascular complications of diabetes mellitus, is currently considered to be the major cause of end-stage renal disease (ESRD)1. DN is caused by a variety of factors, such as the production of advanced glycation end products (AGEs), hemodynamic abnormalities, ectopic lipid deposition and inflammatory injury, which ultimately lead to kidney damage and renal dysfunction2. Unfortunately, the therapeutic agents developed to date for DN that target renal pathogenesis are restricted. Patients can only depend on renal replacement therapy after DN progresses to ESRD, which imposes a heavy economic burden on individuals and families3. Therefore, it is necessary to further explore the corresponding potential therapeutic approaches of DN.
Vascular endothelial growth factor B (VEGF-B) is a crucial factor in promoting abnormal lipid metabolism as a part of the vascular endothelial growth factor family4. VEGF-B can specifically upregulate fatty acid transport proteins (FATPs) and increase ectopic lipid deposition, which ultimately leads to insulin resistance by binding to the membrane receptors neuropilin-1 (NRP-1) or vascular endothelial growth factor receptor (VEGFR)-15. Furthermore, recent studies6, 7, 8 have reported that VEGF-B can also promote lipid accumulation in podocytes and directly result in DN by reducing podocyte insulin sensitivity. Therefore, an anti-VEGF-B monoclonal antibody is expected to treat DN by simultaneously regulating lipid metabolism and improving insulin resistance. However, there is no definitive evidence that reducing VEGF-B signaling could alleviate oxidative stress and inflammatory responses, which are equally the leading causes of DN.
Multiple studies9,10 have shown that inflammatory responses are closely related to the development of DN; however, glucose-lowering agents and antihypertensive therapies cannot easily eliminate inflammatory responses. Therefore, further amelioration of inflammatory responses as well as the control of abnormal glycolipid metabolism is necessary. Interleukin-22 (IL-22) can be produced by CD4+ T helper subtypes (such as Th17 cells and Th22 cells) and innate lymphoid cells (ILCs, such as natural killer cells, dendritic cells, and macrophages) as one of the interleukin-10 cytokine superfamilies11. IL-22 mainly binds to the receptors IL-22R1 and IL-10R2, and then specifically activates the intracellular JAK/STAT3 signaling pathway, thereby mediating anti-inflammatory and other biological effects. We and others have reported that IL-22 can significantly reduce liver fibrosis, which is caused by acetaminophen, concanavalin A or overconsumption of a high-fat diet by preventing liver inflammatory responses and promoting hepatocyte regeneration12, 13, 14. Additionally, our colleagues have reported that IL-22 can significantly reduce inflammatory cytokines (such as IL-1β) and simultaneously protect renal function in DN mice15. Moreover, studies have also indicated that IL-22 can alleviate renal injury caused by renal ischemia-reperfusion16. Thus, we hypothesize that combining VEGF-B antibody and IL-22 can alleviate renal lipotoxicity and insulin resistance as well as ameliorate inflammatory responses.
Here we found that serum levels of VEGF-B were clearly upregulated and that IL-22 levels were obviously downregulated in DN patients, which might be related to the progression of DN. To meet these challenges, we successfully constructed and purified a novel anti-VEGFB/IL22 fusion protein, simultaneously combining a VEGF-B antibody and IL-22. We further demonstrated the beneficial effects of the anti-VEGFB/IL22 fusion protein on DN by ameliorating glomerular and tubular lipid accumulation while alleviating oxidative stress and inflammatory responses. Moreover, the bifunctional fusion protein could increase insulin sensitivity. Overall, these findings indicate that the anti-VEGFB/IL22 fusion protein might be expected to become a prospective therapeutic strategy for DN.
2. Materials and methods
2.1. DN patient selection and nondiabetic subject recruitment
Human serum samples were obtained from 18 nondiabetic subjects and 128 DN patients. The inclusion criteria for DN patients were as follows: diagnosis of DM with concomitant macroalbuminuria, diabetes duration >10 years with concomitant microalbuminuria, or diabetic retinopathy with concomitant microalbuminuria. In addition, human kidney tissues were obtained from DN patients requiring renal puncture biopsy and nondiabetic subjects undergoing radical nephrectomy or requiring renal puncture biopsy. The clinical characteristics of these subjects are shown in Supporting Information Table S1. Our study was given authorization by the Institutional Review Board at ChangHai Hospital (Shanghai, China), and all subjects signed an informed consent form.
2.2. mRNA-seq analysis
Total RNA was isolated from mouse kidney tissues. RNA integrity (agarose gel electrophoresis), purity (nanodrop) and concentration were analyzed. Then, the mRNA was concentrated using magnetic beads with Oligo (dT) and randomly broken into short fragments. Double-stranded cDNA was synthesized and purified from template mRNA with AMPure XP beads. The resulting cDNA end sequences of were completed via End Repair Mix, and poly A tails and sequencing connectors were added. Double-stranded DNA was digested to single-stranded DNA by USER enzyme, and 15 amplification cycles were performed to obtain the final cDNA library using PCR technology. Clusters were generated by bridge-type PCR amplification on cBot instrument. Finally, sequencing was performed using 2 × 151 sequencing mode.
2.3. Production of anti-VEGF-B antibody
The genes encoding the heavy chain and light chain of the anti-VEGF-B antibody (2H10, described by PD Scotney5) were successfully cloned and digested by EcoRI and BamHI (New England Biolabs Inc., Ipswich, MA, USA). Then, the final sequence was ligated into the directional pTT5 plasmid vector. Next, we produced an anti-VEGF-B antibody by transient gene expression in CHO-S cells. DNA transfection procedures were performed in strict accordance with Thermo Fisher's protocols. Finally, we collected cell supernatants and purified the VEGF-B antibody using a protein A affinity chromatography column.
2.4. Production of anti-VEGFB/IL22 fusion protein
The C-terminus of the gene encoding the anti-VEGF-B antibody heavy chain was fused to the murine IL22 moiety with a flexible linker sequence. Then, the genes described above and that encoding the anti-VEGF-B antibody light chain were digested concurrently by EcoRI and BamHI, and the final sequence was ligated into the directional pTT5 plasmid vector. The DNA transfection and purification protocol for the anti-VEGFB/IL22 fusion protein is the same as that described above for the anti-VEGF-B antibody.
2.5. Animals
db/m and db/db mice (C57BKS/Lepr, male, 5 weeks old) were obtained from the Model Animal Research Center of Nanjing University (Najing, China) and were kept under specific pathogen-free (SPF) conditions. db/m mice were fed chow-food, and db/db mice were kept a high-fat diet (HFD) for 5 weeks, followed by HFD feeding and treatment for 8 weeks. Mice were randomly divided into seven groups: lean controls (db/m, n = 7); DN model group (db/db, n = 9); anti-VEGF-B antibody-treated group (db/db, n = 7, 400 μg, twice weekly for 8 weeks, intraperitoneal injection); IL-22-Fc-treated group (db/db n = 7, 245 μg, twice weekly for 8 weeks, intraperitoneal injection); low-dose anti-VEGFB/IL22 fusion protein-treated group (db/db, n = 7, 100 μg, twice weekly for 8 weeks, intraperitoneal injection); high-dose anti-VEGFB/IL22 fusion protein-treated group (db/db, n = 7, 505 μg, twice weekly for 8 weeks, intraperitoneal injection); and losartan-treated group (db/db, n = 7, 20 mg/kg/day, irrigation). And anti-VEGF-B antibody-treated mice, IL-22-Fc-treated mice, high-dose anti-VEGFB/IL22 fusion protein-treated mice were treated with equimolar administration (molecular weight: anti-VEGF-B antibody: approximately 150 kD; IL-22-Fc: approximately 92 kD, IL-22: approximately 20 kD; anti-VEGFB/IL22 fusion protein: approximately 190 kD). Furthermore, losartan was used as positive control drug. Blood was sampled retro-orbitally and stored at −70 °C for experimental analyses. Urine was collected biweekly in 24-h metabolic cages. All animal experiments were conducted with the approval of the Ethics Committee of Fudan University School of Pharmacy (Shanghai, China).
2.6. Metabolic measurements
Body weight, serum triglyceride level, serum total cholesterol level, blood glucose level, serum creatinine level and blood urea nitrogen (BUN) level were measured with an assay kit (NanJingJianCheng, Nanjing, China). The levels of urine microalbumin were measured with a mouse microalbumin (mALB) ELISA kit (NanJingJianCheng). The urine creatinine level and 24-h urinary protein quantity were measured with an assay kit (NanJingJianCheng). The creatinine clearance rate (Ccr) was determined using Eq (1):
| Ccr = (Urine creatinine × Urine volume/24 h)/Serum creatinine | (1) |
2.7. Histopathological study
Mice were euthanized at the age of 18 weeks under 5% chloral hydrate, and kidney and liver tissues were immediately weighed and fixed with 4% formaldehyde. Then, we stained kidney tissues with H&E, Masson, PAS and oil red O for histopathological research. In addition, we processed and stained liver tissues with H&E and oil red O. Subsequently, we assessed the renal samples and liver samples under 100 × , 200 × and 400 × magnification, randomly obtained five images and analyzed the percentage of positive Masson, PAS, oil red O stained area in the whole image with ImageJ software (NIH, Bethesda, MA, USA).
2.8. Immunohistochemical study
Immunohistochemical (IHC) staining of VEGF-B (Abcam, Lon., UK, 1:200), with Hoechst 33342-labeling of the nuclei, was used to explore the correlation between the expression of VEGF-B and the progression of DN. IHC staining of NF-κB P65 (Servicebio, Wuhan, China, 1:200) and TNF-α (Servicebio, 1:200), counterstained with Hoechst 33342 to label the nuclei, was carried out to assess the effect of the anti-VEGFB/IL22 fusion protein on improving inflammatory responses. IHC staining of IRS-1 (Bioss, Beijing, China, 1:200), counterstained with Hoechst 33342 to label the nuclei, was carried out to assess the effect of the anti-VEGFB/IL22 fusion protein on resensitization to insulin signaling. We counted positively staining areas with ImageJ software and calculated the percentage of positively staining areas in the whole image.
2.9. Immunofluorescence study
Immunofluorescence analysis of NLRP3 (CST, Boston, Ma, USA, 1:200) and ADFP (Bioss, 1:200), with Hoechst 33342-labeling of the nuclei, was carried out to assess the expression of NLRP3 and ADFP in the kidney tissues of DN patients. Immunofluorescence analysis of FATP4 (Abcam, 1:200), ADFP (Bioss, 1:200), counterstained with Hoechst 33342 to label the nuclei, was carried out to assess the effect of the anti-VEGFB/IL22 fusion protein on reducing the transportation and accumulation of fatty acids. The immunofluorescence results were assessed and calculated using ImageJ software.
2.10. ROS and mitochondrial membrane potential study
SV40 MES13 cells were incubated in medium supplemented with 10% FBS in 6-well plates for 24 h. Then, cells were exposed to a high dose of glucose (HG: 30 mmol/L) or a low dose of glucose (LG: 5 mmol/L). Subsequently, cells were cultured and respectively added with anti-VEGF-B antibody (200 ng/mL), anti-VEGFB/IL22 fusion protein (252 ng/mL), IL-22-Fc (122 ng/mL), NAC (5 mmol/L) and PBS overnight. NAC was used as a positive control drug for antioxidant.
Kidney samples and SV40 MES13 cells were incubated for 20 min after adding Mitosox (10 μmol/L, Beyotime Biotechnology, Shanghai, China). Subsequently, we measured the fluorescence intensity of kidney samples and SV40 MES13 cells at 485 and 530 nm (excitation and emission wavelength). Furthermore, we measured the mitochondrial membrane potential with a JC-1 assay kit (Beyotime Biotechnology). Kidney samples and SV40 MES13 cells were exactly incubated for 30 min after adding JC-1 at 37 °C (10 μmol/L) and measured at 514 and 529 nm (excitation and emission wavelength of JC-1 monomer) and at 529 and 585 nm (excitation and emission wavelength of J-aggregates).
2.11. Electron microscopy
For analysis of the electron microscopy results, kidney samples were collected in 4% stationary liquid for electron microscopy overnight at 4 °C. Then, they were measured by using an electron microscope at 60 kV. The podocyte foot processes and glomerular basement membrane (GBM) thickness were measured, and we randomly examined three mice from each group.
2.12. Western blot analysis
Renal tissues and liver tissues were homogenized, and total proteins were collected and incubated with specific antibodies for NLRP3 (CST, 1:1000), cleaved caspase-1 (CST, 1:1000), mature IL-1β (CST, 1:1000), phospho-Ser 616 IRS-1 (Bioss, 1:1000), total IRS-1 (Bioss, 1:1000), p-AKT (CST, 1:1000), total AKT (CST, 1:1000), p-ERK1/2 (Abcam, 1:1000), total ERK1/1 (Abcam, 1:1000), collagen IV (Abcam, 1:1000), vimentin (CST, 1:1000), α-SMA (CST, 1:1000), GAPDH (CST, 1:1000) and β-actin (CST, 1:1000). Then, we visualized target proteins with an enhanced chemiluminescence substrate. Semiquantitative Western blot analysis was performed using ImageJ software.
We cultured SV40 MES13 cells in medium supplemented with 10% FBS at 1 × 105 cells/well in 6-well plates for 24 h. Anti-VEGF-B monoclonal antibody was added to each well at 200 ng/mL, and anti-VEGFB/IL22 fusion protein was added to each well at 250 ng/mL for 0.5, 1, 2, 4 or 8 h. Then, cells were collected and incubated with p-STAT3 (CST, 1:1000), STAT3 (CST, 1:1000), p-AKT (CST, 1:1000) and β-actin (CST, 1:1000). Finally, cells were visualized with enhanced chemiluminescence substrate.
Human umbilical vein endothelial cells (HUVECs) were cultured in medium (10% FBS) in 6-well plates for 24 h. Then, cells were starved in medium (0.5% FBS) for 6 h and were subsequently added 100 ng/mL of VEGF-B, treated with 1 μg/mL anti-VEGF-B antibody, 1.25 μg/mL anti-VEGFB/IL22 fusion protein, 0.25 μg/mL IL-22-Fc, or new medium for placebo therapy, and then incubated for 18 h. Additional cells were collected and incubated with FATP4 (Abcam, 1:1000) and GAPDH (CST, 1:1000). Then, the cells were visualized with enhanced chemiluminescence substrate.
2.13. Cell culture
ExpiCHO-S cells (Invitrogen, Carlsbad, CA, USA) were incubated in an incubator with 8% CO2 at 37 °C. CHO-S cells were transferred to 32 °C on the second day of gene expression. We used a Countstar IC 1000 cell counter to assess cell density and viability.
SV40 MES13 cells and HUVECs were obtained from Cell Bank of the Chinese Academy of Sciences, Shanghai Branch (Shanghai, China) and incubated in an incubator (5% CO2, 37 °C). Dulbecco's modified Eagle's medium (Invitrogen) containing 10% FBS (Invitrogen), 1% streptomycin and 1% penicillin (Beyotime Biotechnology) was used to culture all cells.
2.14. Statistical analysis
All data were expressed as the mean ± standard error of mean (SEM). Assessment of differences between groups was performed using Student's t-test or one-way ANOVA. Pearson correlation analysis or Pearson's correlation analysis was performed by SPSS version 20. Statistically significant differences were indicated as ∗P < 0.05, ∗∗P < 0.01 or ∗∗∗P < 0.001.
3. Results
3.1. VEGF-B expression was abnormally increased and IL-22 expression was visibly decreased in DN patients
Our study group included 128 DN patients and 18 nondiabetic subjects. The clinical characteristics of these nondiabetic subjects and DN patients are indicated in Table S1. Our data suggest that the circulating VEGF-B level was notably higher and the circulating IL-22 level was substantially lower in DN patients than in nondiabetic subjects (Fig. 1A). Furthermore, we found a positive correlation of VEGF-B level with the levels of urine albumin-to-creatinine ratio (ACR), serum creatinine, 24-h urinary protein quantity and glycosylated hemoglobin (HbA1c%) but not with β2 microglobulin (β2-MG) or estimated glomerular filtration rate (eGFR). We also observed a negative correlation of IL-22 level with the levels of ACR and β2-MG but not with 24-h urinary protein quantity, HbA1c% or eGFR in Fig. 1B and Supporting Information Fig. S1A–S1E. Renal pathology was diagnosed based on the Pathologic Classification of Diabetic Nephropathy17. To ulteriorly explore the effect of VEGF-B on the progression of DN, immunohistochemistry was conducted to detect the expression of VEGF-B in kidney tissues. Our data indicate that the expression of VEGF-B significantly increased in the renal tissues of DN patients (Fig. 1C and Fig. S1F). Furthermore, we found that the expression of adipocyte differentiation-related protein (ADFP) and the NOD-like receptor family pyrin domain-containing protein 3 (NLRP3) inflammasome significantly increased in DN patients, indicating that the development of DN was due to ectopic lipid accumulation and inflammatory responses (Fig. 1D and E). Taken together, our findings suggest that abnormal glucose metabolism, abnormal lipid metabolism and inflammation play a role in DN and that upregulation of VEGF-B and downregulation of IL-22 may be relevant to the progression of DN. Therefore, we hypothesized that DN could be improved by combined administration of the VEGF-B antibody and IL-22.
Figure 1.

Upregulation of VEGF-B and downregulation of IL22 in DN patients. (A) Serum levels of VEGF-B from healthy subjects (n = 17) and DN patients (n = 49) and serum levels of IL22 in healthy subjects (n = 17) and DN patients (n = 65). (B) Pearson correlation between VEGF-B and ACR (n = 38); Pearson correlation between IL22 and ACR (n = 38); Pearson correlation between VEGF-B and serum creatinine (n = 46); Pearson correlation between IL22 and serum creatinine (n = 65); Pearson correlation between VEGF-B and β2-MG (n = 30); Pearson correlation between IL22 and β2-MG (n = 33). (C) Representative PAS images of kidney samples and immunohistochemical staining of VEGF-B in kidney samples (scale bar: PAS: 50 μm; VEGF-B: 100 μm). (D) Immunofluorescence analysis of ADFP (scale bar: 50 μm). (E) Immunofluorescence analysis of NLRP3 (scale bar: 50 μm). ∗∗P < 0.01; ∗∗∗P < 0.001.
3.2. Expression, purification and bioactivity of anti-VEGF-B antibody and IL-22 fusion protein
In the present study, to develop an anti-VEGFB/IL22 fusion protein, the C-terminus of the anti-VEGF-B antibody heavy chain was fused to the murine IL22 moiety with a flexible linker sequence. The genes encoding the protein described above and the anti-VEGF-B antibody light chain were concurrently inserted into pTT5 plasmid vector and transfected into CHO-S cells (Fig. 2A). SDS-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and size-exclusion chromatography show the molecular weight of the anti-VEGF-B antibody and anti-VEGFB/IL22 fusion protein, and show that anti-VEGF-B antibody and anti-VEGFB/IL22 fusion protein had a high purity without forming polymer (Fig. 2B and D). The surface plasmon resonance results indicate that the anti-VEGF-B antibody and anti-VEGFB/IL22 fusion protein had a similar high-binding affinity to the VEGF-B antigen (Fig. 2C). Furthermore, anti-VEGFB/IL22 fusion protein could block the high expression of FATP4 caused by the VEGF-B antigen in HUVECs (Fig. 2I and J), proving that the fusion protein and anti-VEGF-B antibody could repress the interaction of VEGF-B and VEGFR1/NRP-1. In addition, representative immunoblots and quantitative analysis suggest that the anti-VEGFB/IL22 fusion protein as well as IL-22-Fc could significantly stimulate the phosphorylation of STAT3 and AKT, indicating that the fusion protein activated IL22-related signaling (Fig. 2H, Supporting Information Fig. S2A and S2B). More importantly, our data demonstrated that the fusion protein, IL-22-Fc and the ROS scavenger N-acetyl-l-cysteine (NAC) completely recovered high glucose-induced increase of oxidative stress level and the collapse of the mitochondrial membrane potential (Fig. 2E–G), further proving its biological effects. Meanwhile, our results illustrated that anti-VEGFB/IL22 fusion protein at different concentrations were almost nontoxic in various cell types including SV-40 MES13, HK-2, HEK-293T and C2C12 cells (Fig. S2C). Histological examination on major organs show no pathological damages after the fusion protein administration in 1 day and 7 days post-injection, indicating that the fusion protein could be perceived as a safe agent (Fig. S2D). Given these results, we suggest that an anti-VEGFB/IL22 fusion protein combining VEGF-B antibody and IL-22 was successfully expressed and bifunctional.
Figure 2.

Anti-VEGFB/IL22 fusion protein was expressed and purified. (A) Generation and purification of anti-VEGFB/IL22 fusion proteins (linker sequence: GGCGGCGGCGGCTCCGGAGGAGGAGGATCTGGTGGTGGTGGTTCG. (B) SDS-PAGE analysis of the anti-VEGFB/IL22 fusion protein. (C) Affinity of anti-VEGFB-hIL22 for human VEGF-B and mouse VEGF-B measured by surface plasmon resonance (Ka: association constant; Kd: dissociation constant; KD: affinity constant). (D) Size-exclusion chromatography profile of purified anti-VEGF-B antibody and anti-VEGFB/IL22 fusion protein. (E) SV40 MES13 cells were incubated with high-dose glucose (30 mmol/L) or low-dose glucose (5 mmol/L) for 1 h, followed by the addition of anti-VEGF-B antibody, IL22 or anti-VEGFB/IL22 fusion protein for 12 h. Then, the cells were stained with Hoechst 33342 and Mitosox to measure intracellular ROS (scale bar: 20 μm). (F) A JC-1 kit was used to visualize the mitochondrial membrane potential (scale bar: 50 μm). (G) Statistical assessment of the levels of ROS and JC-1 aggregates/monomers (n = 3–5). (H) Anti-VEGF-B antibody or anti-VEGFB/IL22 fusion protein was added at different time points. Anti-VEGFB/IL22 fusion protein induced STAT3 and AKT phosphorylation in SV40 MES13 cells. (I) Anti-VEGFB/IL22 fusion protein reduced the expression of FATP4 in HUVECs. (J) Densitometric values of FATP4 expression. ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.3. Anti-VEGFB/IL22 fusion protein ameliorated renal dysfunction and reduced kidney injury in db/db mice
The db/db mice are employed as a normal experimental model of type two diabetes for the analysis of DN. As shown in Fig. 3A and Supporting Information Fig. S7B, our data demonstrated that diabetes-induced increasing ACR level was almost completely recovered by anti-VEGFB/IL22 fusion protein injection. Anti-VEGFB antibody and IL-22-Fc treatment also significantly alleviated the level of ACR, but their efficacy was relatively lower than that of fusion protein. Apart from ACR level, we illustrated that the fusion protein treatment remarkably downregulated the levels of serum creatinine, blood urea nitrogen (BUN), 24-h urinary volume and 24-h urinary protein, and reversed Ccr levels (Fig. 3B, E, F and Supporting Information Figs. S3–S6). More importantly, these results suggest that anti-VEGFB/IL22 fusion protein treatment showed a better improvement in comparison with anti-VEGFB antibody, IL-22-Fc and losartan treatment to a certain degree, indicating the protective effects of the fusion protein in alleviating kidney injury and renal dysfunction. In addition, anti-VEGFB/IL22 fusion protein improved the survival rate of db/db mice and effectively reduced the diabetes-induced increase in the renal index, whereas anti-VEGFB antibody, IL-22-Fc and losartan only achieved marginal improvement (Fig. 3C and D). Renal pathological alterations were significantly improved in db/db mice as shown by hematoxylin & eosin (H&E) staining after administration of anti-VEGFB/IL22 fusion protein therapy for 8 weeks. Obviously, the effects of the fusion protein were better than anti-VEGFB antibody, IL-22-Fc and losartan (Fig. 3G). Consistent with histological observations, transmission electron microscopy (TEM) results show anti-VEGFB/IL22 fusion protein distinctly reduced glomerular basement membrane (GBM) thickness and reversed podocyte foot process effacement, whereas anti-VEGFB antibody and IL-22-Fc only showed moderate amelioration, including the protective effects of the fusion protein on the pathological ultramicrostructure of the kidney (Fig. 3H and I). In conclusion, these data indicate that the anti-VEGFB/IL22 fusion protein dramatically ameliorated renal function and reduced kidney injury in a diabetes-induced mouse model of DN.
Figure 3.

Anti-VEGFB/IL22 fusion protein therapy reduced renal injury and improved kidney function in db/db mice. (A) Measurements of ACR. (B) Measurements of serum creatinine, BUN and Ccr (n = 5–9). (C) Survival rate of db/db mice after anti-VEGFB/IL22 fusion protein therapy. ∗P < 0.05 compared with DN model mice; #P = 0.095 compared with DN model mice; &P = 0.097 compared with DN model mice. (D) Renal index indicated by kidney weight/body weight of db/db mice. (E)–(F) Measurements of 24-h urine volume and 24-h urinary protein quantity. (G) Representative H&E images of kidney samples from db/db mice (scale bar: 100 μm, 200 × ; 50 μm, 400 ×). (H)–(I) Representative electron microscopy images of a db/db mouse glomerulus and TEM quantification of GBM thickness (scale bar: 10 μm) (n = 3). ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.4. Anti-VEGFB/IL22 fusion protein reduced kidney oxidative stress, inhibited inflammatory responses and ameliorated renal fibrosis in diabetes-induced mouse model of DN
The NLRP3 inflammasome is an inflammation mediator18. And NLRP3 inflammasome has significant effects on the progression of DN. Our colleagues found that NLRP3 inflammasome activation was markedly upregulated in experimental mice with DN15. In the present study, our data suggest that the anti-VEGFB/IL22 fusion protein therapy had a stronger inhibitory effect on the activation of NLRP3 inflammasome, cleaved caspase-1 and mature IL-1β in kidney tissues compared with the anti-VEGF-B antibody and IL-22-Fc treatment (Fig. 4B and G). Similarly, diabetes induced an up-regulation of the TNF-α and NF-κB P65 levels in the kidney, which was completely reversed by anti-VEGFB/IL22 fusion protein therapy and partly alleviated by IL-22-Fc treatment and minimally relieved by anti-VEGFB antibody treatment, indicating that the fusion protein had a significant therapeutic effect in inhibiting inflammatory responses in vivo (Fig. 4F). In addition, anti-VEGFB/IL22 fusion protein administration prominently reduced diabetes-induced intracellular ROS accumulation as well as lowered mitochondrial dysfunction in comparison with anti-VEGFB antibody- and IL-22-Fc-treated groups (Fig. 4A and D).
Figure 4.

Anti-VEGFB/IL22 fusion protein inhibited NLRP3 inflammasome activation, alleviated ROS generation and reduced renal fibrosis in db/db mice. (A) The levels of ROS and mitochondrial membrane potential in the kidney sections were measured by Mitosox staining and JC-1 assay kit (scale bar: 100 μm). (B)–(C) The levels of NIRP3, cleaved caspase-1 and mature IL-1β and the levels of vimentin, α-SMA and collagen IV were measured by Western blotting. (D) The statistical assessment of the levels of ROS and JC-1 aggregates/monomers (n = 3–5). (E) and (H) Representative Masson images of mouse kidney samples and the assessment of Masson-positive staining (scale bar: 100 μm) (n = 3–5). (F) Immunohistochemical images of TNF-α and NF-κB P65 in kidney sections (scale bar: 50 μm). (G) and (I) Quantitative analysis of expression of NLRP3, cleaved caspase-1 and mature IL1-β, and the expression of vimentin, α-SMA and collagen IV (n = 3–5). ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
It is universally understand that excessive deposition of extracellular matrix (ECM) leads to renal fibrosis, which eventually causes renal failure in DN19. Thus, we next asked whether the anti-VEGFB/IL22 fusion protein could inhibit renal fibrosis. We found that renal collagen accumulation was considerably reduced as shown by Masson staining after anti-VEGFB antibody, IL-22-Fc, losartan, and anti-VEGFB/IL22 fusion protein therapy for 8 weeks. However, the anti-VEGFB/IL22 fusion protein almost recovered diabetes-induced renal collagen accumulation compared with anti-VEGFB antibody, IL-22-Fc and losartan (Fig. 4E and H). Concurrently, our results further demonstrated that administration of the anti-VEGFB/IL22 fusion protein completely reversed diabetes-induced high expression of proteins related to fibrosis (collagen IV, vimentin and α-SMA) in kidney tissues. The increase of above proteins could be obviously reduced by anti-VEGFB antibody and IL-22-Fc treatment, whereas the effects of which were partly inferior to the fusion protein (Fig. 4C and I). Overall, these results demonstrated that kidney fibrosis and inflammatory responses mediated by oxidative stress and mitochondrial dysfunction were effectively ameliorated after anti-VEGFB/IL22 fusion protein administration, indicating the combinational protective role of anti-VEGFB antibody and IL-22.
3.5. Anti-VEGFB/IL22 fusion protein reduced diabetes-induced renal lipid accumulation
Studies8,20,21 have found that lipid deposits as well as inflammatory responses have a considerable effect on the pathogenesis of DN and that kidney function was immensely improved by reducing renal lipotoxicity. In the present study, we found that the accumulation of neutral lipids in the kidney, as assessed by oil red O staining and ADFP expression, was completely rescued by anti-VEGFB/IL22 fusion protein therapy and partly relieved by IL-22-Fc treatment. Specifically, anti-VEGFB antibody treatment also elicited significant improvement, but the fusion protein displayed relatively better efficacy than anti-VEGFB antibody (Fig. 5A, C, E, and F). In addition, renal lipid accumulation was closely related to FATP4 expression22. We further confirmed that anti-VEGFB/IL22 fusion protein administration completely normalized the expression of FATP4, whereas anti-VEGFB antibody treatment showed partial recovery and IL-22-Fc treatment only exhibited minimal reduction. Above data suggest the combinational protective role of anti-VEGFB antibody and IL-22 in alleviating lipid droplets transported into podocytes and tubular epithelial cells via reducing FATP4 expression (Fig. 5B and F). Furthermore, we assessed whether dyslipidemia was improved after anti-VEGFB/IL22 fusion protein therapy. As shown in Fig. 5D, the fusion protein remarkably reduced triglycerides but not total cholesterol in diabetic mice, which might be due to anti-VEGFB/IL22 fusion treatment having targeted effects on neutral lipids. In summary, the beneficial effect of anti-VEGFB/IL22 fusion protein on DN was closely related with reduced glomerular and tubular lipid accumulation.
Figure 5.

Anti-VEGFB/IL22 fusion protein therapy prevented the accumulation of neutral lipids in blood circulation and kidney tissue. (A) Representative oil red O images of kidney sections (scale bar: 200 μm, 100 × ; 50 μm, 400 ×). (B) Immunofluorescence images of FATP4 in kidney sections (scale bar: 50 μm). (C) Immunofluorescence images of ADFP (adipophilin) in kidney sections (scale bar: 20 μm). (D) Measurements of serum triglyceride and serum total cholesterol. (E) Quantification of oil red O analysis of kidney sections (n = 3–5). (F) Quantification of FATP4 and ADFP expression in kidney sections (n = 3–5). ∗P < 0.05; ∗∗P < 0.01.
3.6. Anti-VEGFB/IL22 fusion protein therapy increased insulin sensitivity in diabetic mice
Hyperglycemia, lipid deposition and inflammatory responses jointly promote renal insulin resistance and kidney dysfunction23. To study whether anti-VEGFB/IL22 fusion protein therapy reduced insulin resistance, we monitored blood glucose fluctuation for 8 weeks and assessed insulin-related signaling by IHC staining of IRS-1. We found that anti-VEGFB/IL22 fusion protein and IL-22-Fc treatment could obviously abate blood glucose while anti-VEGFB antibody treatment did little to lower blood glucose (Fig. 6D and Supporting Information Fig. S7A. Our data further suggest that the fusion protein immensely up-regulated the expression of IRS-1 in the liver, the ameliorative effects of which were statistically more significant than anti-VEGFB antibody and IL-22-FFc (Fig. 6C and E). In addition, administration of anti-VEGFB/IL22 fusion protein markedly stimulated IRS-1 phosphorylation and AKT phosphorylation in kidney compared with anti-VEGFB antibody- and IL-22-FFc-treated mice. Similarly, the expression of signal transductions of insulin such as p-IRS-1 and p-AKT was also up-regulated in liver of mice administrated with the fusion protein, whereas anti-VEGFB antibody and IL-22-FFc treatment exhibited relatively lighter improvement. Specially, the phosphorylation of ERK1/2 was prominently triggered by the fusion protein and IL-22-Fc, but not by anti-VEGFB antibody (Fig. 6F–I). Furthermore, administration of anti-VEGFB/IL22 fusion protein profoundly decreased the area positive for PAS staining in comparison with anti-VEGFB antibody-, IL-22-Fc-, and losartan-treated groups, indicating glycogen deposition (Fig. 6A and B). Taken together, these data suggest a beneficial role of combination of VEGF-B antibody and IL-22 in alleviating insulin resistance and further ameliorating diabetic kidney diseases.
Figure 6.

Anti-VEGFB/IL22 fusion protein induced IRS-1 and AKT phosphorylation in db/db mice. (A) and (B) Representative PAS images and quantification of PAS analysis of kidney sections (scale bar: 100 μm) (n = 3–5). (C) and (E) Immunohistochemical images and the statistics of IRS-1 in liver sections (scale bar: 100 μm, 200 × ; 50 μm, 400 ×) (n = 3–5). (D) Measurements of blood glucose. (F)–(I) Western blotting analysis and quantitative analysis of the expression of p-IRS-1, p-ERK1/2 and p-AKT in the kidney and liver (n = 3–5). ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001.
3.7. Anti-VEGFB/IL22 fusion protein regulated glucose metabolism-related process, lipid metabolism-related process and inflammatory responses
To further validate that the therapeutic effect of the anti-VEGFB/IL22 fusion protein on DN was related to the amelioration of abnormal glucose metabolism, lipid metabolism and inflammatory responses, we sequenced the mRNA of kidney tissues from db/m mice and HFD-fed db/db mice (a diabetes-induced mouse model of DN) and anti-VEGFB/IL22 fusion protein-treated DN mice. We found 103 identical upregulated genes and 214 identical downregulated genes in anti-VEGFB/IL22 fusion protein-treated mouse and db/m mice compared with DN mice, shown in Supporting Information Fig. S8C. Moreover, the heat map result suggest that the genes prominently altered in DN mice included lipid metabolism process (such as Dgkq and Gpd1), glucose metabolism process (such as Pck1 and Creb3l3) and inflammatory responses (such as Chad and Wnt 11) compared with db/m mice (Fig. 7A). And the genes that were altered upon administration of anti-VEGFB/IL22 fusion protein in diabetes-induced DN mice also were related to lipid metabolism process (such as Dgkq and Gpd1), glucose metabolism process (such as Pck1 and Creb3l3) and inflammatory responses (such as Chad and Wnt11) (Fig. 7B). Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and gene set enrichment analysis (GSEA) results further confirmed that the most significantly altered pathways were lipid metabolism-, glucose metabolism-, and inflammatory responses-related pathway in db/m mice and anti-VEGFB/IL22 fusion protein-treated mice (Fig. 7C and D and Supporting Information Fig. S8A and S8B). More importantly, we have found JAK–STAT signaling is obviously enriched in anti-VEGFB/IL22 fusion protein-treated mice according to GSEA analysis, indicating that the fusion protein could activate IL-22 signaling pathway and further ameliorate inflammatory responses. Collectively, these findings suggest potential therapeutic effects of anti-VEGFB/IL22 fusion protein on DN via regulating glucose metabolism-related process, lipid metabolism-related process and inflammatory responses.
Figure 7.

mRNA-seq analysis results validate that anti-VEGFB/IL22 fusion protein regulated glucose metabolism-related process, lipid metabolism-related process and inflammatory responses. (A) Heat map of meaningfully altered lipid metabolism-related genes, glucose metabolism-related genes and inflammation-related genes in db/m mice versus DN mice. (B) Heat map of meaningfully altered lipid metabolism-related genes, glucose metabolism-related genes and inflammation-related genes in DN mice and anti-VEGFB/IL22 fusion protein-rescued DN mice. (C) KEGG pathway analysis of lipid metabolism-related genes, glucose metabolism-related genes or inflammation-related genes in diabetes-induced DN mice and db/m mice. (D) KEGG pathway analysis of lipid metabolism-related genes, glucose metabolism-related genes or inflammation-related genes after anti-VEGFB/IL22 fusion protein treatment in db/db mice.
3.8. Anti-VEGFB/IL22 fusion protein protected against diabetes-induced liver injury
There was a strong correlation between metabolic disorders and diabetes complications24. Moreover, the liver has been widely considered to have significant effects on metabolic diseases. Therefore, we asked whether diabetes-induced liver injury was improved after anti-VEGFB/IL22 fusion protein therapy, which might have further beneficial effects on diabetes complications. We found that anti-VEGFB/IL22 fusion protein and IL-22-Fc administration prominently reduced diabetes-induced increases in serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT), whereas anti-VEGFB antibody only displayed partial mitigation (Fig. 8D). Moreover, H&E staining analysis suggest that administration of anti-VEGFB/IL22 fusion protein obviously ameliorated diabetes-induced hepatocellular necrosis compared with anti-VEGFB antibody- and IL-22-Fc-treated mice. oil red O staining results demonstrated that diabetes could trigger lipid accumulation in the liver, which was meaningfully relieved by the fusion protein injection and partly reduced by anti-VEGFB antibody or IL-22-Fc treatment (Fig. 8A and C). Immunoblotting analysis indicates that anti-VEGFB/IL22 fusion protein therapy completely reversed diabetes-induced over expression of NLRP3 inflammasome and cleaved caspase-1 in comparison with anti-VEGFB antibody and IL-22-Fc treatment (Fig. 8B and E). These results indicate that the anti-VEGFB/IL22 fusion protein improved diabetes-induced liver injury, which further protected against the progression of DN.
Figure 8.

Reduced liver injury and inflammation generation in db/m mice after anti-VEGFB/IL22 fusion protein therapy. (A) Representative images of H&E staining and images of oil red O staining (scale bar: 100 μm, 200 × ; 50 μm, 400 ×). (B) and (E) Immunoblotting and quantitative analysis of the expression of NLRP3 and cleaved caspase-1 in the liver (n = 3–5). (C) Quantification of oil red O analysis of liver sections (n = 3). (D) Measurements of serum ALT and serum AST (n = 5–7). ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001; ∗∗∗∗P < 0.0001.
4. Discussion
Although DN is treated by strictly lowering blood glucose, blood pressure, and blood lipids and protecting renal function, it is still the most common cause of ESRD25. Therefore, novel treatment strategies that target the pathogenesis of DN and prevent progression towards ESRD are urgently needed. In our work, confirming a close correlation between the progression of DN and increased VEGF-B levels as well as decreased IL-22 levels, a bifunctional anti-VEGFB/IL22 fusion protein combining a VEGF-B antibody and IL-22 was first expressed and purified. We provided evidence that this fusion protein not only ameliorated glomerular and tubular lipid deposition by reducing the expression of FATP4 but also reduced oxidative stress, mitochondrial dysfunction and inflammatory responses, which further prevented the progression of DN.
Recent studies have indicated that abnormal lipid metabolism and ectopic neutral lipid accumulation play a potential role in the progression of DN. Accumulation of neutral lipids in the renal tubules might induce tubulointerstitial fibrosis, yet glomerular fatty acid (FA) uptake might lead to podocyte death and insulin resistance26, 27, 28, 29. Lowering VEGF-B signaling could improve glomerular lipid accumulation and re-sensitize podocytes to insulin signaling8. Moreover, we and others14,30 have suggested that IL-22 is beneficial for ameliorating lipid metabolic disorders. As expected, anti-VEGFB/IL22 fusion protein can play relatively better role in reducing glomerular and tubular lipid accumulation via combining VEGF-B signaling reduction and IL-22, indicating that anti-VEGFB/IL22 fusion protein might be a potential mechanism in reducing lipotoxicity, proteinuria and renal fibrosis in diabetic kidneys.
Inflammatory responses have also been widely considered to be a major factor in the progression of diabetic renal complications, indicating that the reduction in inflammatory responses could ameliorate diabetic-induced proteinuria and kidney damage31,32. Moreover, a recent study indicated that activation of the NLRP3 inflammasome contributes to inflammatory responses, further resulting in the progression of DN33, 34, 35, 36. Furthermore, we and others have suggested that IL-22 was beneficial for ameliorating inflammatory responses and accelerating kidney regeneration. IL-22 gene therapy could protect against the development of DN by inhibiting NLRP3 inflammasome activation and reducing renal fibrosis15,30,37,38. In the current investigation, we demonstrated that the anti-VEGFB/IL22 fusion protein noticeably reduced the activation of the NLRP3 inflammasome by lowering ROS generation and preventing mitochondrial dysfunction in the diabetic kidney, indicating that the beneficial effects of the fusion protein on DN might be due to ameliorating inflammatory responses. Notably, thus far, there is no definite evidence that VEGF-B is closely related to inflammatory responses. However, our data indicate that reducing VEGF-B signaling could alleviate NLRP3 inflammasome activation and further decrease IL-1β expression mediated by cleaved caspase-1, so the further relationship between VEGF-B signaling and inflammatory responses will be explored in the future studies.
Insulin resistance and insulin deficiency is well known to react on the progression of DN. A recent study suggested that decreased VEGF-B signaling could ameliorate glucose tolerance and restore insulin sensitivity5. Moreover, IL-22 is reported to improve metabolic disorders and insulin resistance in diabetes30. Our data suggest that the anti-VEGFB/IL22 fusion protein played a better role in ameliorating insulin resistance, which might be due to the beneficial effects of the fusion protein in protecting the liver. The liver is recognized as the main organ involved in metabolism and is closely related to metabolic disorders. Therefore, we hypothesized that in addition to its own effects in re-sensitizing the kidneys to insulin signaling, the anti-VEGFB/IL22 fusion protein could also reduce insulin resistance by improving liver function and alleviating liver injury, further protecting against DN. Moreover, considerable evidence proves that serum fatty acid levels could intensely promote insulin resistance; conversely, increased insulin resistance accelerates hepatic lipid accumulation by activating insulin receptor kinase, phosphorylating IRS-1/IRS-2 and upregulating p-AKT39, 40, 41, 42, 43. In addition, inflammatory responses had a crucial effect on the pathogenesis of insulin resistance and the development of diabetes by upregulating IL-1β and TGF-α levels44, 45, 46, 47. In the present study, the beneficial effects of the anti-VEGFB/IL22 fusion protein in improving glucose metabolism and insulin resistance might be exerted by reducing renal lipid deposition and inflammatory responses. Furthermore, the positive effects of the fusion protein in ameliorating diabetes-induced renal lipotoxicity and inflammatory responses might be due to increased insulin sensitivity. Thus, we suggest that ectopic lipid accumulation, inflammatory responses and increased insulin resistance were interrelated and interacted with each other, which ultimately promoted the development of DN.
5. Conclusions
A noteworthy increase in circulating VEGF-B levels and a remarkable decrease in circulating IL-22 in DN patients, which were strongly related to the progression of DN, were found. Furthermore, our data provide evidence that the anti-VEGFB/IL22 fusion protein could ameliorate renal dysfunction and alleviate kidney injury in diabetic mice. We suggest that the important effects of the fusion protein on DN are mediated by reducing ectopic lipid accumulation, inflammatory responses and insulin resistance in diabetic mice, indicating that the bifunctional anti-VEGFB/IL22 fusion protein might be an effective therapeutic strategy for DN.
Acknowledgments
The study was supported by grants from the National Natural Science Foundation of China (31872746 and 81773620) and Scientific Research Projects of Shanghai Municipal Commission of Health and Family Planning (201740140, China).
Author contributions
Xiaobin Mei and Dianwen Ju designed the study, Yilan Shen, Wei Chen, Lei Han, Qi Bian, Jiajun Fan and Zhonglian Cao carried out experiments; Zongshu Xian, Zhiyong guo and Wei Zhang analyzed the data; Xin Jin and Tao Ding made the figures; Yilan Shen, Wei Chen drafted and revised the paper; all authors approved the final version of the manuscript. The final version of the paper has been approved by all authors.
Conflicts of interests
All authors declare no competing interests.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2020.07.002.
Contributor Information
Dianwen Ju, Email: dianwenju@fudan.edu.cn.
Xiaobin Mei, Email: meixiaobin@smmu.edu.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Rask-Madsen C., King G.L. Diabetes: podocytes lose their footing. Nature. 2010;468:42–44. doi: 10.1038/468042a. [DOI] [PubMed] [Google Scholar]
- 2.Schrijvers B.F., De Vriese A.S., Flyvbjerg A. From hyperglycemia to diabetic kidney disease: the role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev. 2004;25:971–1010. doi: 10.1210/er.2003-0018. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Vargas P.A., Tong A., Howell M., Craig J.C. Educational interventions for patients with CKD: a systematic review. Am J Kidney Dis. 2016;68:353–370. doi: 10.1053/j.ajkd.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Bry M., Kivelä R., Leppänen V.M., Alitalo K. Vascular endothelial growth factor-B in physiology and disease. Physiol Rev. 2014;94:779–794. doi: 10.1152/physrev.00028.2013. [DOI] [PubMed] [Google Scholar]
- 5.Hagberg C.E., Mehlem A., Falkevall A., Muhl L., Fam B.C., Ortsäter H. Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature. 2012;490:426–430. doi: 10.1038/nature11464. [DOI] [PubMed] [Google Scholar]
- 6.Mehrotra D., Wu J., Papangeli I., Chun H.J. Endothelium as a gatekeeper of fatty acid transport. Trends Endocrinol Metab. 2014;25:99–106. doi: 10.1016/j.tem.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehlem A., Palombo I., Wang X., Hagberg C.E., Eriksson U., Falkevall A. PGC-1α coordinates mitochondrial respiratory capacity and muscular fatty acid uptake via regulation of VEGF-B. Diabetes. 2016;65:861–873. doi: 10.2337/db15-1231. [DOI] [PubMed] [Google Scholar]
- 8.Falkevall A., Mehlem A., Palombo I., Heller Sahlgren B., Ebarasi L., He L. Reducing VEGF-B signaling ameliorates renal lipotoxicity and protects against diabetic kidney disease. Cell Metab. 2017;25:713–726. doi: 10.1016/j.cmet.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Gurley S.B., Ghosh S., Johnson S.A., Azushima K., Sakban R.B., George S.E. Inflammation and immunity pathways regulate genetic susceptibility to diabetic nephropathy. Diabetes. 2018;67:2096–2106. doi: 10.2337/db17-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu J.D., Wu C.C., Hung C.N., Wang C.J., Huang H.P. Myrciaria cauliflora extract improves diabetic nephropathy via suppression of oxidative stress and inflammation in streptozotocin-nicotinamide mice. J Food Drug Anal. 2016;24:730–737. doi: 10.1016/j.jfda.2016.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabat R., Ouyang W., Wolk K. Therapeutic opportunities of the IL-22–IL-22R1 system. Nat Rev Drug Discov. 2014;13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 12.Chen W., Zhang X., Fan J., Zai W., Luan J., Li Y. Tethering interleukin-22 to apolipoprotein A-I ameliorates mice from acetaminophen-induced liver injury. Theranostics. 2017;7:4135–4148. doi: 10.7150/thno.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W., Luan J., Wei G., Zhang X., Fan J., Zai W. In vivo hepatocellular expression of interleukin-22 using penetratin-based hybrid nanoparticles as potential anti-hepatitis therapeutics. Biomaterials. 2019;187:66–80. doi: 10.1016/j.biomaterials.2018.09.046. [DOI] [PubMed] [Google Scholar]
- 14.Zai W., Chen W., Wu Z., Jin X., Fan J., Zhang X. Targeted interleukin-22 gene delivery in the liver by polymetformin and penetratin-based hybrid nanoparticles to treat nonalcoholic fatty liver disease. ACS Appl Mater Interfaces. 2019;11:4842–4857. doi: 10.1021/acsami.8b19717. [DOI] [PubMed] [Google Scholar]
- 15.Wang S., Li Y., Fan J., Zhang X., Luan J., Bian Q. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017;8:e2937. doi: 10.1038/cddis.2017.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu M.J., Feng D., Wang H., Guan Y., Yan X., Gao B. IL-22 ameliorates renal ischemia–reperfusion injury by targeting proximal tubule epithelium. J Am Soc Nephrol. 2014;25:967–977. doi: 10.1681/ASN.2013060611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tervaert T.W., Mooyaart A.L., Amann K., Cohen A.H., Cook H.T., Drachenberg C.B. Pathologic classification of diabetic nephropathy. J Am Soc Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z., Xu G., Gao Y., Zhan X., Qin N., Fu S. Cardamonin from a medicinal herb protects against LPS-induced septic shock by suppressing NLRP3 inflammasome. Acta Pharm Sin B. 2019;9:734–744. doi: 10.1016/j.apsb.2019.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ha H., Lee H.B. Reactive oxygen species and matrix remodeling in diabetic kidney. J Am Soc Nephrol. 2003;14:S246–S249. doi: 10.1097/01.asn.0000077411.98742.54. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z., Jiang T., Li J., Proctor G., McManaman J.L., Lucia S. etc. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVB db/db mice with type 2 diabetes. Diabetes. 2005;54:2328–2335. doi: 10.2337/diabetes.54.8.2328. [DOI] [PubMed] [Google Scholar]
- 21.Lennon R., Pons D., Sabin M.A., Wei C., Shield J.P., Coward R.J. Saturated fatty acids induce insulin resistance in human podocytes: implications for diabetic nephropathy. Nephrol. Dial. 2009;24:3288–3296. doi: 10.1093/ndt/gfp302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagberg C.E., Falkevall A., Wang X., Larsson E., Huusko J., Nilsson I. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- 23.Laville M., Rigalleau V., Riou J.P., Beylot M. Respective role of plasma nonesterified fatty acid oxidation and total lipid oxidation in lipid-induced insulin resistance. Metabolism. 1995;44:639–644. doi: 10.1016/0026-0495(95)90122-1. [DOI] [PubMed] [Google Scholar]
- 24.Guebre-Egziabher F., Alix P.M., Koppe L., Pelletier C.C., Kalbacher E., Fouque D. Ectopic lipid accumulation: a potential cause for metabolic disturbances and a contributor to the alteration of kidney function. Biochimie. 2013;95:1971–1979. doi: 10.1016/j.biochi.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 25.Molitch M.E., Adler A.I., Flyvbjerg A., Nelson R.G., So W.Y., Wanner C. Diabetic kidney disease: a clinical update from kidney disease: improving global outcomes. Kidney Int. 2015;87:20–30. doi: 10.1038/ki.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yiu W.H., Li R.X., Wong D.W.L., Wu H.J., Chan K.W., Chan L.Y.Y. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol Dial Transplant. 2018;33:1323–1332. doi: 10.1093/ndt/gfx336. [DOI] [PubMed] [Google Scholar]
- 27.Herman-Edelstein M., Scherzer P., Tobar A., Levi M., Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. 2014;55:561–572. doi: 10.1194/jlr.P040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang H.M., Ahn S.H., Choi P., Ko Y.A., Han S.H., Chinga F. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh D.K., Winocour P., Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- 30.Wang X., Ota N., Manzanillo P., Kates L., Zavala-Solorio J., Eidenschenk C. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature. 2014;514:237–241. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- 31.Wada J., Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 32.Liang G., Song L., Chen Z., Qian Y., Xie J., Zhao L. Fibroblast growth factor 1 ameliorates diabetic nephropathy by an anti-inflammatory mechanism. Kidney Int. 2018;93:95–109. doi: 10.1016/j.kint.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y., Xu X., Tang C., Gao P., Chen X., Xiong X. Reactive oxygen species promote tubular injury in diabetic nephropathy: the role of the mitochondrial ROS–TXNIP–NLRP3 biological axis. Redox Biol. 2018;16:32–46. doi: 10.1016/j.redox.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu R., Liu X., Yin J., Wu H., Cai X., Wang N. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism. 2018;83:18–24. doi: 10.1016/j.metabol.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Yi H., Peng R., Zhang L.Y., Sun Y., Peng H.M., Liu H.D. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017;8:e2583. doi: 10.1038/cddis.2016.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S.M., Ka S.M., Wu H.L., Yeh Y.C., Kuo C.H., Hua K.F. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia. 2014;57:424–434. doi: 10.1007/s00125-013-3115-6. [DOI] [PubMed] [Google Scholar]
- 37.Kulkarni O.P., Hartter I., Mulay S.R., Hagemann J., Darisipudi M.N., Kumar Vr S. Toll-like receptor 4-induced IL-22 accelerates kidney regeneration. J Am Soc Nephrol. 2014;25:978–989. doi: 10.1681/ASN.2013050528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weidenbusch M., Rodler S., Anders H.J. Interleukin-22 in kidney injury and regeneration. Am J Physiol Renal Physiol. 2015;308:F1041–F1046. doi: 10.1152/ajprenal.00005.2015. [DOI] [PubMed] [Google Scholar]
- 39.Meikle P.J., Summers S.A. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat Rev Endocrinol. 2017;13:79–91. doi: 10.1038/nrendo.2016.169. [DOI] [PubMed] [Google Scholar]
- 40.Petersen M.C., Shulman G.I. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38:649–665. doi: 10.1016/j.tips.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 42.Petersen K.F., Dufour S., Befroy D., Lehrke M., Hendler R.E., Shulman G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petersen K.F., Oral E.A., Dufour S., Befroy D., Ariyan C., Yu C. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin W., Cui B., Li P., Hua F., Lv X., Zhou J. 1,25-Dihydroxyvitamin D3 protects pbese rats from metabolic syndrome via promoting regulatory T cell-mediated resolution of inflammation. Acta Pharm Sin B. 2018;8:178–187. doi: 10.1016/j.apsb.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Olefsky J.M., Glass C.K. Macrophages, inflammation and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 46.Shoelson S.E., Lee J., Goldfine A.B. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang S., Wang B., Humphries F., Hogan A.E., O'Shea D., Moynagh P.N. The E3 ubiquitin ligase pellino3 protects against obesity-induced inflammation and insulin resistance. Immunity. 2014;41:973–987. doi: 10.1016/j.immuni.2014.11.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.