

Visual Abstract

Key Words: appetite, blood pressure, cardiac metabolism, heart failure, MC4R
Abbreviations and Acronyms: AMPK, adenosine monophosphate–activated protein kinase; BP, blood pressure; CNS, central nervous system; HF, heart failure; HR, heart rate; ICV, intracerebroventricular; LV, left ventricular; MC4R, melanocortin-4 receptor; MI, myocardial infarction; MTII, melanotan II; mTOR, mechanistic target of rapamycin
Highlights
-
•
Leptin protects against progression to heart failure after myocardial infarction.
-
•
This beneficial effect requires activation of the brain melanocortin system.
-
•
Stimulation of brain MC4R recapitulates the cardiac protective effects of leptin.
-
•
Leptin-MC4R activation improves cardiac substrate oxidation and mitochondrial function.
-
•
It also improves Ca2+ coupling and contractile function in viable cardiomyocytes after MI.
Summary
Heart failure has a high mortality rate, and current therapies offer limited benefits. The authors demonstrate that activation of the central nervous system leptin-melanocortin pathway confers remarkable protection against progressive heart failure following severe myocardial infarction. The beneficial cardiac-protective actions of leptin require activation of brain melanocortin-4 receptors and elicit improvements in cardiac substrate oxidation, cardiomyocyte contractility, Ca2+ coupling, and mitochondrial efficiency. These findings highlight a potentially novel therapeutic approach for myocardial infarction and heart failure.
Heart failure (HF) is a major contributor to the high mortality rate from cardiovascular diseases in the United States and worldwide (1). Although the pathophysiology of HF is heterogeneous, the common outcome is the inability of the heart to pump the required blood flow to meet the metabolic demands of the body at rest or during increased metabolic needs associated with exercise or even normal daily activities. Most patients with HF caused by myocardial infarction (MI) have reduced left ventricular (LV) pumping ability (2). Despite efforts to treat HF, current therapies offer only limited benefits to patients who develop HF post-MI, especially those who have not been reperfused within a short period of time and experience extensive cardiac injury (3,4). Therefore, new, more effective therapies are needed to protect the heart and improve its function following MI.
Leptin, an adipocyte-derived peptide, regulates body weight by reducing food intake and increasing energy expenditure and stimulates glucose use and fatty acid metabolism in peripheral tissues (5). Previous studies that investigated the effects of leptin on cardiac function provided conflicting results, with some suggesting a deleterious effect of hyperleptinemia on cardiac inflammation and fibrosis (6, 7, 8) and others showing a beneficial impact of leptin replacement therapy to improve heart function in leptin-deficient animals (9,10). The majority of the studies showing detrimental cardiac effects of leptin are confounded by the fact that subjects with hyperleptinemia are usually obese, making it difficult to control for other effects of obesity on cardiac structure and function. Conversely, studies that support a protective action of leptin on the heart have examined the effect of raising peripheral leptin levels in animals with normal or reduced baseline leptin levels (9). Although leptin appears to have rapid direct effects to increase glucose and fatty acid uptake and use in peripheral tissues, including the heart (11, 12, 13), these actions appear to be short lived, and the majority of leptin’s long-term effects on overall metabolism and energy balance are mediated by activation of leptin receptors in the central nervous system (CNS) (14,15).
Previous studies have also shown that long-term intravenous leptin administration may reduce inflammatory responses and accelerate repair of ischemic tissues (16). Conversely, disruption of leptin signaling because of mutations of leptin receptors or the leptin gene is associated with cardiac lipid accumulation, cardiac dysfunction, and myocardial apoptosis (17). In addition, cardiomyocyte-specific deletion of leptin receptors in adult mice caused lethal HF in a Cre-recombinase-mediated cardiotoxicity model through actions that impaired cardiomyocyte energy metabolism (18). The mechanisms by which leptin exerts these protective effects are still unclear and may involve direct activation of cardiac leptin receptors or indirect effects on the heart via activation of leptin receptors in the CNS.
We previously demonstrated that leptin has powerful CNS-mediated antidiabetic effects capable of normalizing blood glucose levels even in insulin-deficient diabetic animals (19,20). We also demonstrated that intracerebroventricular (ICV) leptin administration reversed bradycardia and normalized intrinsic heart rate (HR) in this model of diabetes (19). Leptin also markedly improves cardiac glucose metabolism via its CNS actions in insulin-deficient animals. Although these effects of leptin could be beneficial in the setting of HF, whether leptin protects the heart and improves its function after MI through its CNS actions has, to our knowledge, not been previously tested.
In preliminary studies we discovered that leptin administration elicited impressive recovery of cardiac function after MI. However, the mechanisms involved in mediating this recovery were unknown. Our previous studies also indicated that a major part of leptin’s effects on blood pressure (BP) and glucose metabolism required activation of the CNS melanocortin system (21,22). Leptin activates proopiomelanocortin neurons to stimulate their release of α-melanocyte stimulating hormone, an endogenous agonist of melanocortin-4 receptors (MC4Rs). α-Melanocyte stimulating hormone exhibits immunomodulatory and anti-inflammatory activities and facilitates regeneration of injured tissues (23,24). However, the role of the CNS proopiomelanocortin-MC4R in mediating leptin’s effects on cardiac function has, to our knowledge, not been previously determined.
In the present study we tested the hypothesis that leptin improves cardiac metabolism and attenuates cardiac dysfunction after MI via CNS actions. We also tested whether these beneficial cardiac actions of leptin require activation of CNS MC4Rs using genetic and pharmacological approaches. As long-term leptin infusion and MC4R activation reduce appetite and promote weight loss, we also determined if reductions in food intake and body weight contribute to the beneficial cardiac effects of leptin and MC4R activation. Finally, we investigated the metabolic mechanisms by which CNS leptin-MC4R activation improves recovery of cardiac function and attenuates adverse cardiac remodeling after MI.
Methods
All experimental procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center. (Detailed methods can be found in the Supplemental Appendix).
Animals
Experiments were performed in 12- to 14-week-old male Sprague-Dawley rats purchased from Harlan/Envigo (Houston, Texas), and MC4R-knockout rats from our colony maintained at the animal facilities at the University of Mississippi Medical Center.
Animal surgery
The rats were anesthetized with isoflurane and telemetry BP transmitters (model HSD10, Data Sciences International, New Brighton, Minnesota) were implanted in the abdominal aorta distal to the renal arteries under sterile conditions, as previously described (19,25). After stable control measurements for food intake and hemodynamic status, the rats were submitted to baseline echocardiography followed by left anterior descending coronary artery ligation surgery, as previously described (26).
Experimental protocols
Food intake and hemodynamic measurements
The rats were allowed to recover for 10 days after telemetry transmitters and ICV cannulas were implanted before control measurements were recorded. Daily food intake was measured, and BP and HR were recorded 24 h/day and analyzed using Dataquest ART software (Data Science International), as previously described (27).
Long-term central infusion of leptin, MC4R agonist, or vehicle after MI
Approximately 15 to 20 min after left anterior descending coronary artery ligation surgery, an osmotic minipump (model 2002, Durect, Cupertino, California) was implanted subcutaneously in the scapular region and connected to the ICV cannula using Tygon tubing (0.38-mm inside diameter; Cole Parmer, Vernon Hills, Illinois) to deliver leptin (0.62 μg/h), vehicle (saline, 0.5 μl/h), or the MC4R agonist melanotan II (MTII) (10 ng/h) for 28 consecutive days. The rates of leptin and MTII infusion were based on previous studies showing that these doses effectively decrease food intake and promote weight loss (20,28). On the 14th day of infusion, the rats were briefly anesthetized with isoflurane for osmotic minipump replacement. On day 28, the rats were euthanized and hearts were collected.
Echocardiography
Transthoracic echocardiography was performed at baseline before left anterior descending coronary artery ligation and at weeks 1, 2, 3, and 4 after ligation using the VEVO 3100 system (VisualSonics, Toronto, Ontario, Canada) equipped with a 21-MHz scan-head probe (MX250) at 100 frames/s. Rats were anesthetized with 1.5% isoflurane and placed on a heating table, and their extremities were fixed to 4 electrocardiographic leads on the table. After removal of all hair in the chest area, warm Aquasonic 100 gel (Parker Laboratories, Fairfield, New Jersey) was applied to optimize visibility of the heart chambers. A parasternal long-axis B-mode image was acquired at the maximum LV length. Three short-axis B-mode videos of the left ventricle were also acquired at the midventricular level for strain analysis. The left atrium/aorta ratio was calculated using a B-mode image from the short-axis view to measure the left atrial and aortic diameters. The long-axis image with the maximal and minimal cross-sectional areas in a heart cycle and LV end-systolic and end-diastolic volumes were calculated as previously described (29). LV ejection fraction was calculated as: [(LV end-diastolic volume − LV end-systolic volume)/LV end-diastolic volume] ⋅ 100. The myocardium at the midventricular level was divided into 6 segments (average radial strain), including the infarcted areas, and regional wall motion in each segment was measured using speckle-tracking analysis. We also removed segments 1 and 6 (area of infarction) and examined overall radial strain in the remaining areas (radial strain only).
Glucose and fatty acid oxidation in perfused working hearts
Glucose and fatty acid oxidation were measured in hearts from saline-treated (n = 5), leptin-treated (n = 6), and MTII-treated (n = 5) rats using a working heart system at the end of week 2 of ICV infusion. We used nonrecirculating Krebs-Henseleit buffer supplemented with 5.5 mmol/l glucose and 0.4 mmol/l oleate as the long-chain fatty acid bound to 0.5% (w/v) bovine serum albumin in the perfusion medium and the radiolabeled tracers [U-14C]glucose (0.1 μCi/ml) and [9,3-3H]oleate (0.1 μCi/ml) to determine myocardial rates of glucose and long-chain fatty acid oxidation every 5 min by quantitative collection of [14C]O2 and [3H]2O released in the coronary effluent, as previously described (30). At the end of the 20-min mark (samples were collected at 0, 5, 10, 15, and 20 min), hearts were quickly removed from the working heart apparatus and snap-frozen for determination of wet and dry weights. Coronary blood flow was calculated by measuring the amount of perfusate collected in the perfusion chamber in 1 min. Total cardiac output was calculated by measuring the amount of perfusate pumped against an afterload of 90 mm Hg in 1 min plus the coronary blood flow. Cardiac power was also calculated using cardiac output and the afterload pressure. Cardiac O2 consumption was measured using the difference of O2 content in the perfusate before passing through the heart and in the perfusate collected from the coronary blood flow. These measurements were corrected by dry heart weights.
Cardiomyocyte isolation and measurement of contractility and calcium transient
Rats were injected intravenously with 1,000 U/kg of heparin for anticoagulation before anesthesia with 2% to 3% isoflurane. The heart was excised, cannulated, and connected to a heart perfusion apparatus (Radnoti, Covina, California), and perfusion was initiated in the Langendorff mode. The heart was perfused with a calcium-free-based buffer (pH 7.2, 37°C) containing 135 mmol/l NaCl, 4 mmol/l KCl, 1 mmol/l MgCl2, 10 mmol/l HEPES, 0.33 mmol/l NaH2PO4, 10 mmol/l glucose, 10 mmol/l 2,3-butanedione monoxime (B0753, Sigma-Aldrich, St. Louis, Missouri), and 5 mmol/l taurine (T0625, Sigma-Aldrich) and bubbled with 95% O2/5% CO2. After 3 to 5 min of perfusion, the buffer was replaced with a similar buffer containing 0.6 mg/g body weight collagenase D (11088858001, Roche, Basel, Switzerland), 0.8 mg/g body weight collagenase B (11088807001, Roche), and 0.1 mg/g body weight protease type XIV (P5147, Sigma-Aldrich) dissolved in 50 ml perfusion buffer (31). After complete digestion (6 to 8 min), the heart was removed and minced. Extracellular Ca2+ was gradually added back to the cells, starting at 0.06 mmol/l to a final concentration of 1.2 mmol/l. The interval of each calcium concentration was 15 min (32).
Cardiomyocyte contractility was assessed using a SoftEdge MyoCam system (IonOptix, Westwood, Massachusetts). Cardiomyocytes were placed in a chamber and stimulated with a suprathreshold voltage at a frequency of 1 Hz (33,34). IonOptix SoftEdge software was used to record the changes in sarcomere length and duration of shortening and relengthening. Intracellular Ca2+ transient was measured using a dual-excitation, single-emission photomultiplier system (IonOptix). Cardiomyocytes were treated with Fura 2-AM (2 μmol/l; Thermo Fisher Scientific, Waltham, Massachusetts) at 37°C for 20 min and then exposed to light emitted by a 75-W halogen lamp through either a 340- or 380-nm filter while being stimulated to contract at a frequency of 1 Hz.
Histological analysis and infarct size
Heart samples were sectioned (5 μm thick) and stained using picrosirius red for quantification of interstitial collagen. Stained cross sections were captured using light microscopy (Eclipse 50i, Nikon, Tokyo, Japan) at 40× magnification. To estimate the fraction area (percentage) of collagen using picrosirius red–stained sections, 15 to 20 images of the septum were randomly captured for posterior analysis using ImageJ software (National Institutes of Health, Bethesda, Maryland). LV infarct size was measured using picrosirius red–stained sections and calculated by dividing the length of the infarcted area by the total circumference of the left ventricle (expressed as a percentage). Only cells with well-defined cell membranes and visible cell nuclei at midwall depth were selected to measure cardiomyocyte diameter on sections stained with Masson’s trichrome.
Statistical methods
Results are expressed as mean ± SEM. One-way analysis of variance was used for comparisons of single time points among 3 or more groups, and 1-way analysis of variance with repeated measures followed by Dunnett’s post hoc test was used for comparisons between control (baseline) and experimental values (treatment) within each group as appropriate. Comparisons between different groups over time were made using 2-way analysis of variance followed by the Bonferroni post hoc test. Single time points between 2 groups were compared using Student’s t-test. A p value <0.05 was considered to indicate statistical significance. The repeated-measures analyses were completed in GraphPad Prism (GraphPad Software, La Jolla, California), thereby using the default compound symmetrical (exchangeable) covariance structures. Time point and treatment group were the only fixed effects. There were no missing data, and all treatment groups were equivalent at baseline, thus no baseline adjustment was necessary.
Results
Blood glucose, tissue weight, and infarct size responses to long-term ICV leptin or MTII infusion after MI
Blood glucose concentration and heart weight were not different in rats with MI that were treated for 28 days with ICV leptin, MTII, or vehicle. However, ratio of heart weight to tibia length, liver weight, and lung wet and dry weights were significantly lower in ICV leptin–treated rats with MI compared with vehicle treatment (Table 1). ICV MTII infusion also reduced liver weight compared with vehicle-treated rats (Table 1). Infarct size was similar in all 3 groups (Table 1 and Supplemental Figure S1).
Table 1.
Blood Glucose, Tissue Weight, and Infarct Area in ICV Leptin–Treated, ICV MTII–Treated, and ICV Vehicle–Treated Rats 28 Days After Myocardial Infarction
| Blood Glucose (mg/dl) | Heart (g) | Heart/Tibia Length | Liver (g) | Lung (Wet, g) | Lung (Dry, g) | Infarcted Area (%) | |
|---|---|---|---|---|---|---|---|
| Vehicle | 80 ± 6 | 1.5 ± 0.1 | 0.33 ± 0.02 | 12.8 ± 0.4 | 1.94 ± 0.04 | 0.39 ± 0.01 | 29 ± 4 |
| Leptin | 76 ± 6 | 1.3 ± 0.1 | 0.27 ± 0.01∗ | 8.8 ± 1.2∗ | 1.56 ± 0.09∗ | 0.31 ± 0.02∗ | 30 ± 3 |
| MTII | 70 ± 3 | 1.6 ± 0.1 | 0.34 ± 0.01 | 10.9 ± 0.6∗ | 1.89 ± 0.06 | 0.38 ± 0.01 | 32 ± 4 |
Values are mean ± SEM obtained on day 28 of chronic ICV infusion.
ICV = intracerebroventricular; MTII = melanotan II.
p < 0.05 compared with vehicle (n = 6 to 8 per group).
Food intake and body weight responses to long-term ICV leptin or MTII infusion after MI
After MI, food intake decreased transiently in vehicle-, leptin-, and MTII-infused rats. However, long-term ICV leptin or MTII infusion for 28 consecutive days further reduced food intake during the first few days after MI (Figure 1A). The impact of long-term ICV leptin or MTII treatment to decrease food intake compared with saline vehicle infusion is more evident when analyzing the net cumulative deficit food intake during the entire 28 days of treatment (Figure 1B). This decrease in food intake was associated with significantly greater weight loss in leptin-treated rats at weeks 1 (−39 ± 5 g vs. 6 ± 6 g), 2 (−30 ± 5 g vs. 18 ± 6 g), and 3 (−18 ± 8 g vs. 38 ± 7 g) and reduced weight regain at week 4 (5 ± 11 g vs. 53 ± 5 g) compared with vehicle treatment (Figure 1C). Similarly, long-term ICV MTII infusion was associated with a significant decrease in body weight at weeks 1 (−45 ± 4 g vs. 6 ± 6 g) and 2 (−27 ± 3 g vs. 18 ± 6 g) and reduced weight regain at weeks 3 (−3 ± 6 g vs. 38 ± 7 g) and 4 (−7 ± 5 g vs. 53 ± 5 g) compared with vehicle treatment (Figure 1C).
Figure 1.
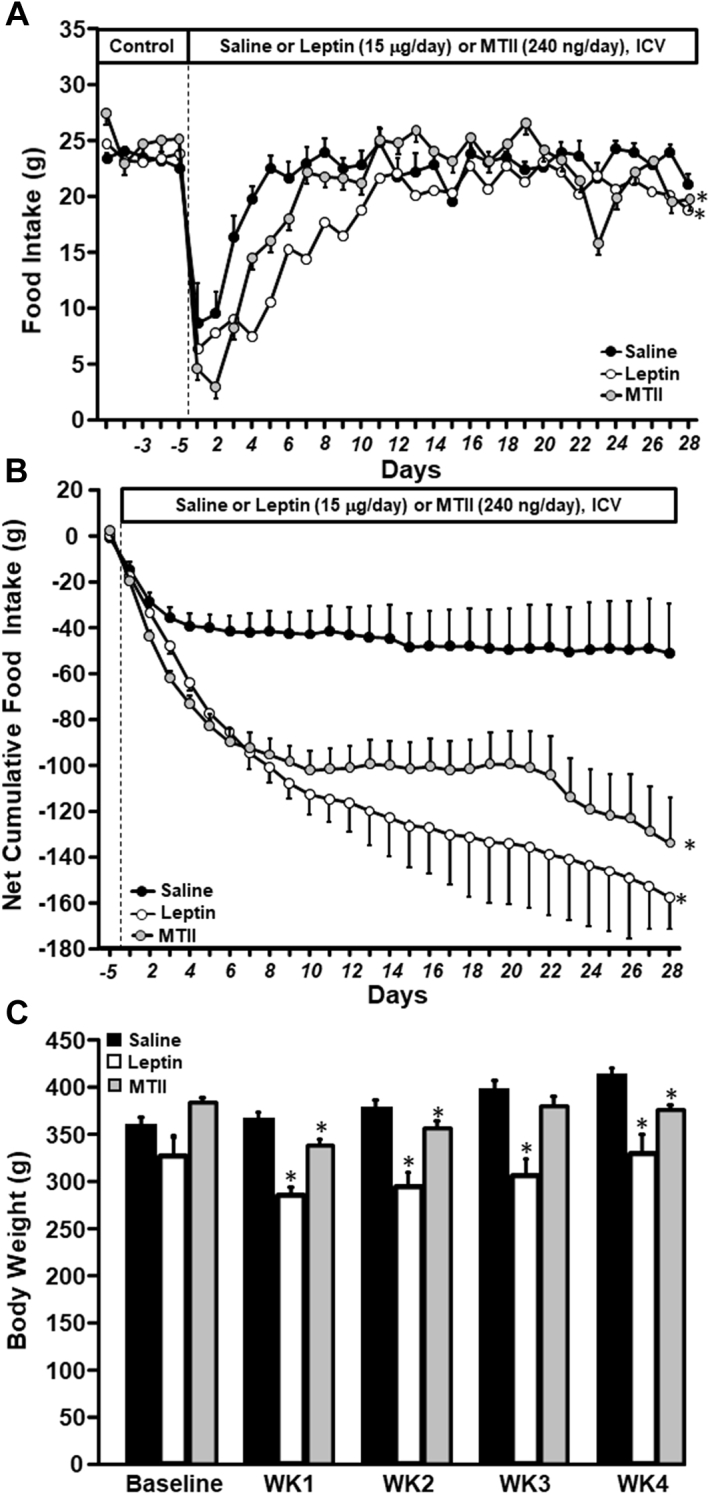
Long-Term ICV Leptin and MTII Infusion Reduced Food Intake and Body Weight in Rats With Myocardial Infarction
(A) Food intake, (B) net cumulative food intake, and (C) body weight in Sprague-Dawley rats with myocardial infarction and intracerebroventricular (ICV) infusion of vehicle (n = 6), leptin (n = 8), or melanotan II (MTII) (n = 8) for 4 weeks. ∗p < 0.05 compared with vehicle-treated group.
Bp, HR, and cardiac function responses to long-term ICV leptin or MTII infusion after MI
Long-term ICV leptin infusion for 28 days attenuated the fall in BP observed at week 1 post-MI in vehicle-treated rats, although mean arterial pressure during weeks 2 to 4 of treatment was not different compared with baseline for ICV vehicle or leptin treatment (Figure 2A). HR in vehicle-treated rats did not change at week 1 post-MI but gradually decreased, reaching the lowest value at week 4 (Figure 2B). In leptin-treated rats, HR spiked at week 1 post-MI but also gradually decreased, returning to baseline values by week 3 post-MI (Figure 2B).
Figure 2.

Chronic ICV Leptin or MTII Infusion Attenuated Cardiac Dysfunction in Rats With Myocardial Infarction
(A) Mean arterial pressure (MAP), (B) heart rate (HR), (C) ejection fraction (EF), (D) fractional shortening (FS), (E) cardiac output (CO), (F) average radial strain (segments 1 to 6), (G) radial strain (segments 2 to 5), (H) left atrium/aorta diameter ratio, (I) collagen content, and (J) myocyte diameter in Sprague-Dawley rats with myocardial infarction and ICV infusion with vehicle (n = 6), leptin (n = 8), or MTII (n = 8) for 4 weeks. ∗p < 0.05 compared with vehicle-treated group; #p < 0.05 compared with baseline. Abbreviations in Figure 1.
ICV leptin infusion post-MI significantly improved ejection fraction and fractional shortening compared with vehicle treatment (Figures 2C and 2D). In addition, leptin infusion returned cardiac output back to baseline levels before MI (Figure 2E) and markedly improved systolic function after MI, as evidenced by increased average LV radial strain and radial strain from segments 2 to 5 of the noninfarcted LV area (Figures 2F and 2G). In fact, leptin infusion restored radial strain in the noninfarcted area to values that were not significantly different from control values measured prior to MI. In vehicle-treated rats, however, the average radial strain and radial strain in the noninfarcted area remained significantly reduced 4 weeks after MI compared with baseline values before MI (Figures 2F and 2G). Leptin infusion also attenuated cardiac congestion, as indicated by reduced left atrium/aorta diameter ratio (Figure 2H) and lung weight compared with vehicle treatment (Table 1). These results indicate that leptin acts on the CNS to markedly improve cardiac function and attenuate the progression of HF after MI.
To determine if long-term CNS activation of MC4R recapitulates the protective effects of leptin on cardiac function post-MI, we infused MTII for 4 weeks beginning 15 to 20 min after left anterior descending coronary artery ligation. ICV MTII infusion prevented the decreases in BP and HR that were observed in vehicle-treated rats after MI (Figures 2A and 2B), and similar to our observations with ICV leptin infusion, MTII also markedly improved ejection fraction and fractional shortening compared with vehicle treatment (Figures 2C and 2D). In addition, ICV MTII infusion returned cardiac output back to baseline levels (Figure 2E), increased average radial strain and radial strain from segments 2 to 5 in noninfarcted areas (Figures 2F and 2G), and prevented cardiac congestion, as indicated by reduced left atrium/aorta diameter ratio (Figure 2H) compared with vehicle treatment. These results indicate that activation of CNS MC4R with MTII significantly improved cardiac function and attenuated progression of HF after MI in a similar fashion as observed in ICV leptin-treated rats.
Reduced food intake and weight loss do not mediate leptin’s beneficial effect on cardiac function after MI
As leptin treatment caused significant reductions in food intake and promoted weight loss in rats with MI, we examined if reduction in food intake contributes to the cardiac-protective effect of ICV leptin treatment. For this purpose, we included a group of rats with MI and infused ICV with saline vehicle that were pair-fed the same amount of food consumed by rats infused ICV with leptin. We found that pair feeding did not attenuate the progression of HF after MI, suggesting that leptin’s effects on appetite and body weight do not contribute importantly to leptin’s beneficial cardiac effects in HF (Supplemental Figure S2).
Cardiac collagen deposition and cardiomyocyte diameter in rats with MI treated with vehicle, leptin, or MTII
To assess the impact of long-term ICV leptin infusion and CNS MC4R activation on cardiac remodeling and collagen deposition after MI, we quantified cardiomyocyte size and estimated collagen content using picrosirius red–stained heart sections. Chronic ICV leptin infusion post-MI significantly reduced collagen deposition by 21% in the noninfarcted area compared with similar areas in hearts from vehicle-treated rats with MI (Figure 2I). We also observed an 11% reduction in cardiomyocyte diameter in ICV leptin–treated rats compared with vehicle treatment 4 weeks after MI (Figure 2J), which is consistent with our finding of reduced cardiac weight and suggests that ICV leptin treatment attenuates cardiomyocyte hypertrophy post-MI. In rats treated with ICV MTII after MI, we found no major effect in collagen deposition (Figure 2I), but cardiomyocyte diameter was significantly reduced in MTII-treated compared with vehicle-treated rats (Figure 2J).
Long-term ICV leptin or MTII infusion after MI alters cardiac metabolism
We investigated if improvements in cardiac function 4 weeks post-MI in ICV leptin–treated rats were associated with alterations in markers of cardiomyocyte energy metabolism including phosphorylated adenosine monophosphate–activated protein kinase (p-AMPK) and mechanistic target of rapamycin (mTOR). At the end of the 4-week treatment post-MI, p-AMPK/AMPK and p-mTOR/mTOR ratios were significantly lower in noninfarcted LV areas from ICV leptin– or MTII-treated rats compared with vehicle treatment (Supplemental Figures S3A and S3B). As AMPK is a cellular metabolic sensor that can be activated during energy deprivation, the lower p-AMPK/AMPK ratio in hearts from ICV leptin–treated rats may suggest improved myocardial bioenergetics compared with hearts from vehicle-treated animals 4 weeks after MI, when functional echocardiographic data show stable improved heart function (Figures 2C to 2G).
We also examined gene expression of factors involved in glucose metabolism, including pyruvate dehydrogenase kinase 4, hypoxia-inducible factor–alpha, and glucose transporter 4 (Supplemental Figures S3C to S3E), and fatty acid metabolism such as fatty acid transport protein 1, fatty acid translocase, and carnitine palmitoyltransferase 1b (Supplemental Figures S3F to S3H) in hearts from the vehicle- and leptin-treated groups. We found no significant differences in gene expression at the end of 4 weeks of treatment between ICV leptin and vehicle infusion.
We also found that p-AMPK/AMPK and p-mTOR/mTOR ratios were lower in hearts from ICV MTII–treated rats compared with vehicle treatment (Supplemental Figures S3A and S3B). This may suggest that, similar to ICV leptin treatment, activation of CNS MC4R led to improved myocardial bioenergetics in these hearts 4 weeks post-MI. We also examined expression of genes involved in glucose (pyruvate dehydrogenase kinase 4, hypoxia-inducible factor–alpha, and glucose transporter 4 (Supplemental Figures S3C to S3E) and fatty acid metabolism (fatty acid transport protein 1, fatty acid translocase, and carnitine palmitoyltransferase 1b) (Supplemental Figures S3F to S3H) in hearts from rats treated with MTII and found no significant differences compared with vehicle treatment.
Long-term ICV leptin or MTII infusion after MI increases cardiac substrate oxidation
To test the possibility that long-term activation of the CNS leptin-MC4R axis in rats with MI-induced HF induced time-dependent changes in cardiac energy metabolism, we also quantified p-AMPK/AMPK and p-mTOR/mTOR ratios at the end of the second week post-MI. This period was chosen because it corresponds to the interval immediately before significant improvements in cardiac function begin to be detectable on echocardiography in rats infused with leptin or MTII (Figures 2C to 2G). Contrary to what we observed at the end of 4 weeks of treatment, ICV leptin or MTII infusion significantly increased cardiac p-AMPK/AMPK ratio (Figure 3A). Conversely, p-mTOR/mTOR ratio remained reduced in ICV leptin– or MTII-treated rats compared with vehicle treatment 2 weeks post-MI (Figure 3B), which was similar to what we observed at 4 weeks post-MI.
Figure 3.

Long-Term ICV Leptin or MTII Infusion for 2 Weeks Increased Cardiac Substrate Oxidation, p-AMPK/AMPK Ratio, and Mitochondrial Efficiency in Rats With Myocardial Infarction
(A) Phosphorylated adenosine monophosphate–activated protein kinase (p-AMPK)/AMPK ratio, (B) phosphorylated mechanistic target of rapamycin (p-mTOR)/mTOR ratio, (C) glucose oxidation, (D) fatty acid oxidation, and (E) cardiac fiber mitochondria adenosine triphosphate (ATP)–linked respiration in hearts from rats with myocardial infarction treated with ICV vehicle, leptin, or MTII at the end of the second week of treatment. ∗p < 0.05 compared with the vehicle-treated group. Abbreviations as in Figure 1.
We then hypothesized that the increased p-AMPK/AMPK ratio would be associated with increased substrate oxidation at 2 weeks post-MI, which would lead to a new steady state of nutrient surplus resulting in adaptive reduction in p-AMPK/AMPK ratio at 4 weeks of treatment. Therefore, we analyzed glucose and fatty acid oxidation in isolated perfused hearts from rats treated with ICV leptin, MTII, or vehicle 2 weeks after MI and found marked increases in glucose oxidation and fatty acid oxidation, respectively, for ICV leptin and MTII infusion compared with ICV vehicle treatment (Figures 3C and 3D). This improvement in cardiac energy metabolism observed during the initial stage of treatment at 2 weeks post-MI may be necessary for the consequent amelioration of cardiac function observed by echocardiographic measurements that are noticeable at weeks 3 and 4 post-MI compared with vehicle treatment. Then, AMPK phosphorylation is suppressed in response to the new state of improved metabolism and cardiac function in rats infused with leptin or MTII.
Perfused hearts from MTII-treated rats had higher cardiac output and cardiac power compared with vehicle and leptin treatment groups (Supplemental Table S1). Leptin and MTII-treated groups also exhibited increased O2 consumption compared with vehicle treatment (Supplemental Table S1). No differences in coronary blood flow were observed among the 3 groups.
Long-term ICV leptin infusion after MI improves cardiac mitochondrial function and cardiomyocyte contractility
To further investigate potential mechanisms contributing to leptin’s amelioration of cardiac function in rats with MI-induced HF, we instrumented additional groups of male Sprague-Dawley rats divided into 4 groups: 1) sham controls; 2) MI plus ICV saline vehicle treatment for 2 weeks; 3) MI plus ICV leptin infusion for 2 weeks; and 4) MI plus ICV MTII infusion for 2 weeks. On day 14 of treatment, hearts were excised, and pieces of the left ventricle (from healthy areas around the infarct) were used to assess mitochondrial function, while remaining portions of the heart were digested, and cardiomyocytes isolated from healthy areas, away from the infarct, were used to examine cardiomyocyte contractile function and calcium transients.
We found that long-term ICV leptin or MTII administration significantly improved mitochondrial function, as evidenced by augmented adenosine triphosphate–linked respiration compared with ICV saline treatment (Figure 3E), indicating improved cardiomyocyte mitochondrial efficiency. We also observed impaired sarcomere contractile function in cardiomyocytes isolated from noninfarcted areas of the hearts of saline-treated rats after MI compared with sham controls and that ICV leptin or MTII administration fully restored cardiomyocyte contractile function, including sarcomere shortening and velocity of contraction (Figures 4A and 4D). No differences were observed in sarcomere length at rest among all 4 groups (Figure 4B).
Figure 4.

Long-Term ICV Leptin or MTII Infusion Increased Cardiomyocyte Contractility in Melanocortin-4 Receptor–Knockout Rats With Myocardial Infarction
(A) Representative curves of isolated cardiomyocyte sarcomere shortening, (B) baseline sarcomere length, (C) sarcomere shortening (index of cardiomyocyte contraction), (D) sarcomere shortening velocity (contraction velocity), (E) resting calcium signal, and (F) calcium transient in isolated cardiomyocytes from hearts of rats with myocardial infarction treated with ICV vehicle or leptin at the end of the second week of treatment and sham controls. ∗p < 0.05 compared with vehicle-treated group; ‡p < 0.05 compared with sham controls. Abbreviations as in Figure 1.
ICV leptin treatment not only normalized cardiomyocyte contractility but also increased baseline calcium signal in cardiomyocytes (Figure 4E) and improved calcium-contractility coupling, which was impaired in saline-treated rats with MI; saline vehicle–treated rats with MI required markedly greater calcium transients for cardiomyocyte contraction than leptin-treated rats after MI or sham rats that did not receive MI (Figure 4F). Long-term ICV MTII infusion also improved cardiomyocyte calcium-contractility coupling (Figure 4F); however, MTII did not significantly increase resting baseline calcium signal (Figure 4E).
ICV leptin or MTII infusion in rats with MI did not alter expression of genes involved in regulation of transcription, remodeling, inflammation, and neovascularization of cardiac tissue
We further investigated if ICV leptin infusion up-regulates the expression of several genes known to be involved in transcription, neovascularization, and cardiac remodeling while reducing expression of genes involved in inflammation in noninfarcted areas of the left ventricle. However, we found no major differences in interleukin-1-beta, tumor necrosis factor–alpha, vascular endothelial growth factor–alpha, peroxisome proliferator–activated receptor–gamma coactivator 1–alpha, peroxisome proliferator–activated receptor–alpha, or sarcoendoplasmic reticulum calcium transport adenosine triphosphatase 2 (Supplemental Figures S3I to S3N) messenger ribonucleic acid expression in hearts from ICV leptin– or vehicle-treated groups after 4 weeks of treatment. Similar results were observed in rats treated with MTII (Supplemental Figures S3I to S3N).
Cardiac function responses to long-term ICV leptin infusion post-MI in MC4R-deficient rats
As long-term CNS MC4R activation with MTII recapitulated the improvement in cardiac function observed in rats infused with leptin, and our previous studies supported a critical role of brain MC4R in mediating leptin’s effects on BP and overall glucose regulation, we tested whether genetic disruption of MC4R attenuates leptin’s CNS-mediated cardiac-protective effects in rats with MI-induced HF. Male MC4R-knockout rats from our colony (MC4R-knockout rats originally developed by Transposagen, Lexington, Kentucky [35]) were instrumented with ICV cannulas, and left anterior descending coronary artery ligation was performed followed by implantation of osmotic pumps to deliver vehicle or leptin via ICV infusion for 4 weeks according to the same protocols used in Sprague-Dawley rats described previously. As we hypothesized, ICV leptin infusion failed to improve cardiac function in MC4R-knockout rats post-MI, and we observed similar reductions in cardiac output, ejection fraction, fractional shortening, and radial strain in ICV leptin–treated rats compared with vehicle treatment (Figures 5A to 5D). These observations suggest that leptin’s powerful CNS-mediated effects to improve cardiac function require functional brain MC4R.
Figure 5.
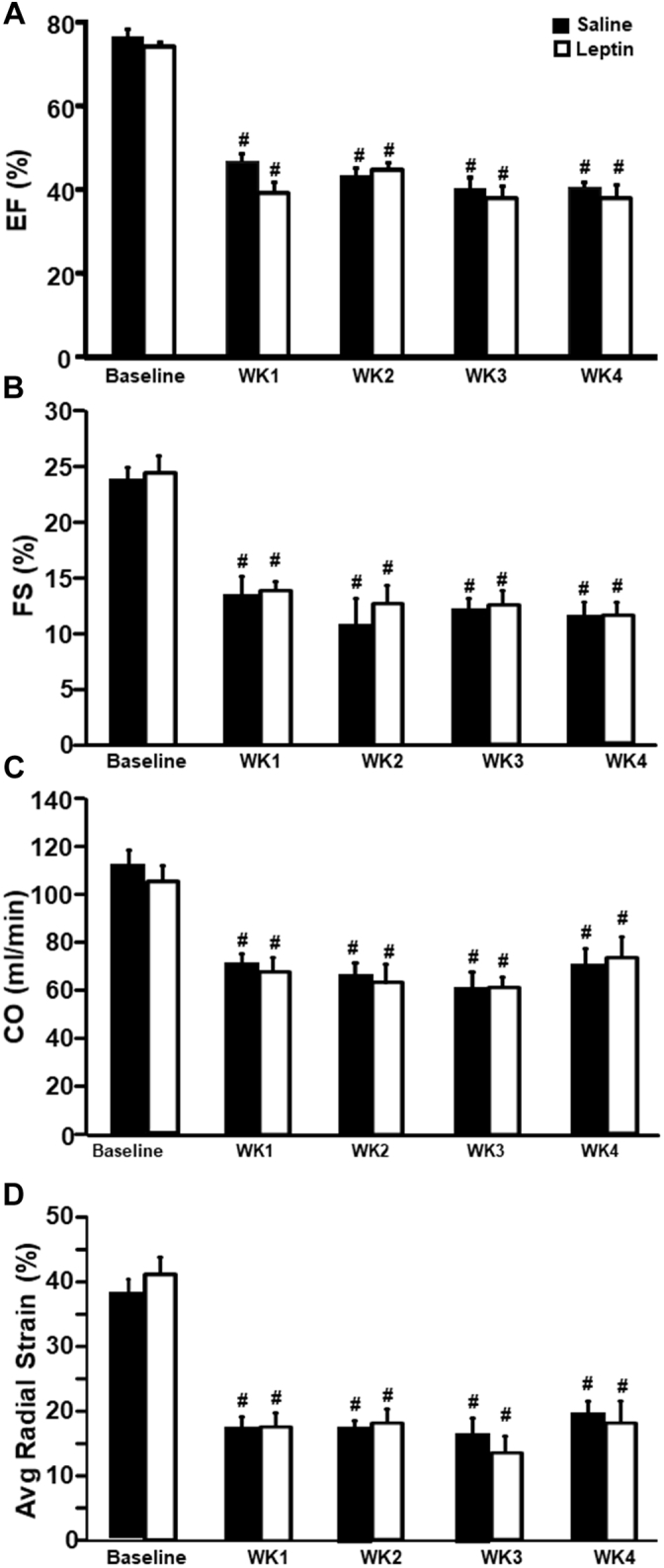
Long-Term ICV Leptin Infusion Failed to Attenuate Cardiac Dysfunction in Melanocortin-4 Receptor–Knockout Rats With Myocardial Infarction
(A) Ejection fraction (EF), (B) fractional shorting (FS), (C) cardiac output (CO), and (D) average (Avg) radial strain in melanocortin-4 receptor–knockout rats with myocardial infarction and ICV treatment with vehicle (n = 4) or leptin (n = 5) for 4 weeks.
Discussion
A major goal of this study was to test the hypothesis that pharmacological activation of the brain leptin-MC4R axis attenuates cardiac dysfunction and adverse cardiac remodeling after severe MI and to determine the mechanisms involved. Our results indicate that ICV leptin infusion for 4 weeks, beginning about 15 to 20 min after MI at rates that have no major effects on plasma leptin concentrations, elicited remarkable improvements in cardiac function, as evidenced by the return of cardiac output and radial strain back to baseline values, increased ejection fraction and fractional shortening, and attenuation of cardiac congestion as indicated by reduced left atrium/aorta diameter and wet and dry lung weights. We also found that leptin, via its actions on the CNS, attenuated cardiac hypertrophy, cardiomyocyte diameter, and cardiac collagen deposition after MI. Similar protective effects on cardiac structure and function, with few exceptions, were observed with CNS MC4R activation by ICV infusion of MTII for 4 weeks after MI. Moreover, the beneficial effects of ICV leptin administration were completely abrogated in rats with genetic deficiency of MC4R. These findings strongly support the hypothesis that leptin-induced stimulation of the CNS melanocortin system, particularly MC4R, is necessary for leptin’s impressive effects to improve cardiac function post-MI.
Our findings also provide important insights on the metabolic mechanisms by which CNS leptin-MC4R activation improves cardiac function after MI. Long-term ICV leptin infusion or CNS MC4R activation with MTII increased expression of global metabolic energy sensors, such as p-AMPK, in hearts at 2 weeks post-MI, which was associated with increased cardiac glucose and fatty acid oxidation. This transient decrease was followed by a new energy status at 4 weeks post-MI whereby metabolic energy sensors were reduced, likely because of restoration of adequate energy supply to the heart. Whether the reduction in p-AMPK observed at 4 weeks of treatment is a consequence of a new steady state in which the heart has adequate energy supply and thus does not require activation of energy sensors is still unclear and would require additional studies. Furthermore, we demonstrated that the improvement in cardiac function post-MI in ICV leptin–treated rats was mediated, at least in part, by increased mitochondrial efficiency, normalization of cardiomyocyte contractile function, and attenuation of calcium transient–contractility uncoupling compared with vehicle treatment.
Food intake and body weight responses to long-term activation of the CNS leptin-melanocortin axis post-MI
Long-term ICV leptin or MTII infusion significantly reduced food intake during the first 2 weeks of treatment, resulting in weight loss compared with ICV vehicle treatment. de Lucia et al. (36) reported that caloric restriction after established HF reduced cardiac dysfunction and improved inotropic reserve. In the present study, however, reduction in caloric intake alone in vehicle-treated rats with MI did not attenuate the impairment of cardiac function following MI. In addition, ICV leptin or MTII infusion caused only transient reductions in food intake, and body weight returned to baseline values by the end of treatment. We also conducted studies in rats that were pair-fed the same amount of food as ingested by rats infused with leptin after MI and demonstrated that this did not significantly improve recovery of cardiac function after MI. Thus, it is unlikely that the beneficial impact of activating the brain leptin-MC4R axis on cardiac function post-MI can be explained by reduced food intake and weight loss.
Changes in Bp, HR, and cardiac function in response to long-term activation of the CNS leptin-melanocortin axis post-MI
We previously demonstrated that leptin’s long-term CNS-mediated BP effects are mediated by activation of the brain melanocortin system, ultimately requiring stimulation of MC4R (27). For example, blockade of the proopiomelanocortin-MC4R pathway using pharmacological agents or transgenic mice that do not have functional MC4R or leptin receptors in proopiomelanocortin neurons completely prevented leptin-induced sympathetic activation as well as leptin’s ability to modulate BP and HR (27,37). We also showed that long-term ICV leptin infusion reverses bradycardia, restores cardiac sympathetic tone, reduces HR variability, and normalizes intrinsic HR in insulin-deficient diabetic rats (19). Although the precise mechanisms by which leptin alters pacemaker activity and autonomic outflow to the heart in this model of diabetes are still unknown, they appear to require activation of CNS MC4R, as no improvement in HR was observed when leptin was infused after pharmacological blockade of MC4R (37). In the present study, ICV leptin infusion attenuated the fall in HR observed in vehicle-treated rats with MI. Similar attenuation was observed in rats treated with the MC4R agonist MTII.
Although leptin administration greatly improved recovery of cardiac function after MI, many patients with MI are overweight or obese and may be resistant to the CNS effects of leptin (7). In contrast, obesity does not appear to be associated with resistance to the CNS actions of MC4R agonists (38). Therefore, we examined whether activation of the brain MC4R would mimic the cardioprotective effects of leptin in a model of MI-induced HF. As we hypothesized, long-term activation of brain MC4R recapitulated most of the effects of leptin on cardiac function post-MI. These findings highlight an important and novel role for the brain melanocortin system in attenuating the adverse cardiac effects that accompany MI and in mediating the beneficial cardiac effects of leptin after MI.
These results have potential clinical relevance, as MC4R agonists that cross the blood-brain barrier have been developed and tested in clinical trials for treatment of patients with proopiomelanocortin gene mutations (39) and some other genetic forms of severe obesity (40,41). MC4R agonists also appear to have minimal adverse side effects when used to treat premenopausal women with acquired, generalized hypoactive sexual desire disorder (42,43). If our results in rodents can be extrapolated to humans, they suggest that MC4R agonists may be a promising new therapeutic approach for treating patients who experience severe MI. However, clinical studies are needed to test the potential therapeutic value of MC4R agonists in patients with MI, HF, and other associated pathologies.
Long-term activation of the CNS leptin-melanocortin axis improves cardiac metabolism, mitochondrial efficiency, and cardiomyocyte contractile function post-MI
LV dysfunction post-MI is usually accompanied by decreased overall cardiomyocyte energy production (44). Although the mechanisms by which activation of the CNS leptin-melanocortin axis improves systolic function post-MI have not been fully elucidated, they may involve improved cardiomyocyte energy metabolism and contractility in the noninfarcted portions of the heart after MI. Administration of leptin or MTII for 2 weeks after MI markedly increased cardiac substrate oxidation in perfused hearts from rats with MI. Also, ICV leptin or MTII administration fully reversed impaired cardiomyocyte contractility and uncoupling of calcium transient–contractility observed in noninfarcted regions of the heart in vehicle-treated rats 2 weeks post-MI. The improvements in cardiomyocyte contractility observed during activation of the CNS leptin-MC4R pathway were accompanied by increased mitochondrial efficiency, as shown by higher percentage of adenosine triphosphate–linked respiration compared with hearts of vehicle-treated rats. These results indicate that activation of the CNS leptin-MC4R axis slows progression of HF post-MI by improving cardiac mitochondrial function and energy metabolism that translates into better contractile function.
Although our results support a key role for brain MC4R in contributing to leptin’s cardiac-protective actions in HF, leptin and MC4R activation may not have the same quantitative effects on cardiac metabolism. Leptin administration markedly increased glucose oxidation, while MTII infusion mainly increased fatty acid oxidation and had minimal impact on glucose oxidation in perfused hearts 2 weeks after MI. These observations suggest that leptin and MC4R activation may trigger other mechanisms that contribute to improved cardiac function in addition to differentially modulating cardiac glucose and fatty acid oxidation.
We also observed increased AMPK phosphorylation in cardiac tissue of rats treated with leptin or MTII compared with vehicle treatment 2 weeks post-MI. At 4 weeks of treatment, however, AMPK phosphorylation was reduced in ICV leptin– or MTII-treated rats. One interpretation of these findings is that activation of the brain leptin-MC4R axis attenuated the impairment in metabolic function induced by MI, stimulated cardiac AMPK phosphorylation, and increased glucose and fatty acid uptake and oxidation as previously demonstrated in normal animals without MI (45,46). This improvement in cardiac energy metabolism observed after 2 weeks of treatment post-MI was likely necessary for the consequent improvement in cardiac function observed by echocardiographic measurements that began to be significant compared with vehicle treatment at week 3 post-MI. Then, AMPK phosphorylation was suppressed in response to the new state of improved metabolism and cardiac function in rats infused with leptin or MTII.
Although ICV leptin treatment attenuated cardiac fibrosis in rats with MI, we observed no major changes in gene expression of key regulators of cardiac inflammation, remodeling, and neovascularization after 4 weeks of treatment. For example, we found no significant changes in expression of genes associated with remodeling and hypertrophy (sarcoendoplasmic reticulum calcium transport adenosine triphosphatase 2), inflammation (interleukin-1-beta and tumor necrosis factor–alpha), and neovascularization (vascular endothelial growth factor–alpha) or transcriptional factors such as peroxisome proliferator–activated receptor–gamma coactivator 1–alpha or peroxisome proliferator–activated receptor–alpha in hearts from leptin- or MTII-treated animals compared with vehicle-treatment. These results suggest that any reduction in pro-inflammatory processes elicited by ICV leptin or MTII infusion during treatment may have already subsided by week 4 of treatment, coinciding with the marked improvements in cardiac function at that time. It is also possible that the beneficial effects of the leptin-brain MC4R on cardiac function post-MI may be independent of alterations in inflammatory processes that occur in this model. Additional studies are needed to test these possibilities.
Conclusions
Our results provide strong evidence for an important beneficial effect of leptin, via its actions on the CNS, to improve cardiac function in rats with HF post-MI. The CNS actions of leptin restored ejection fraction, cardiac output, LV muscle strain, and left atrium/aorta diameter ratio almost all the way back to baseline values before MI. These cardiac-protective actions of leptin are mimicked by long-term activation of CNS MC4R and are completely abolished in rats with genetic deficiency of MC4R. Furthermore, our study demonstrated that the CNS actions of leptin-MC4R activation enhance cardiac substrate oxidation, improve mitochondrial efficiency, and increase cardiomyocyte contractile function, thus halting progression of HF following severe MI. Whether an initial transient increase in p-AMPK levels and improved cardiac substrate oxidation and mitochondrial efficiency observed at 2 weeks of ICV leptin or MTII administration are directly responsible for the functional improvements observed at weeks 3 and 4 post-MI is still unclear and represents a limitation of our studies. Thus, additional studies are needed to unravel the link between the brain leptin-MC4R pathway and improved cardiac function in HF.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: HF is a serious health condition associated with high mortality rates and significant morbidity for which current therapies offer only modest benefits. Thus, development of new, more effective therapies is urgently needed. Our results demonstrate an important and novel beneficial effect of activating the brain leptin-MC4R pathway to improve cardiac performance and attenuate progression of HF post-MI. These findings represent a shift in the current paradigm of focusing on therapies that directly target the heart and show remarkable CNS-mediated effects that are capable of ameliorating cardiac dysfunction and protecting against progressive HF following severe MI.
TRANSLATIONAL OUTLOOK: Our findings highlight a promising new therapeutic approach for treatment of MI and HF. A limitation of our study is that we used ICV administration of leptin and MC4R agonist, which is impractical in clinical settings. However, new MC4R agonists that cross the blood-brain barrier have been developed and tested to treat rare genetic disorders in humans and may represent an alternative approach to target brain MC4R and to overcome the challenge of leptin resistance in obese patients with MI. Future studies designed to test the therapeutic value of leptin or MC4R agonist administration for MI in large animal models and in humans are needed.
Author Disclosures
This research was supported by the National Heart, Lung, and Blood Institute (grants P01 HL51971, R01HL136438, and RO1 HL136438-01A1), the National Institute of General Medical Sciences (grants P20 GM104357 and U54 GM115428), and the National Institute of Diabetes and Digestive and Kidney Diseases (grant R01 DK121411). The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
Appendix
For supplemental methods, figures, and a table, please see the online version of this paper.
Appendix
References
- 1.Benjamin E.J., Blaha M.J., Chiuve S.E. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cikes M., Solomon S.D. Beyond ejection fraction: an integrative approach for assessment of cardiac structure and function in heart failure. Eur Heart J. 2016;37:1642–1650. doi: 10.1093/eurheartj/ehv510. [DOI] [PubMed] [Google Scholar]
- 3.Roger V.L. Epidemiology of heart failure. Circ Res. 2013;113:646–659. doi: 10.1161/CIRCRESAHA.113.300268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Go A.S., Mozaffarian D., Roger V.L. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortes V.A., Cautivo K.M., Rong S., Garg A., Horton J.D., Agarwal A.K. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2−/− lipodystrophic mice independent of hepatocyte leptin receptors. J Lipid Res. 2014;55:276–288. doi: 10.1194/jlr.M045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leifheit-Nestler M., Wagner N.M., Gogiraju R. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J Transl Med. 2013;11:170. doi: 10.1186/1479-5876-11-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khafaji H.A., Bener A.B., Rizk N.M., Al Suwaidi J. Elevated serum leptin levels in patients with acute myocardial infarction; correlation with coronary angiographic and echocardiographic findings. BMC Res Notes. 2012;5:262. doi: 10.1186/1756-0500-5-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soderberg S., Stegmayr B., Ahlbeck-Glader C., Slunga-Birgander L., Ahren B., Olsson T. High leptin levels are associated with stroke. Cerebrovasc Dis. 2003;15:63–69. doi: 10.1159/000067128. [DOI] [PubMed] [Google Scholar]
- 9.McGaffin K.R., Sun C.K., Rager J.J. Leptin signalling reduces the severity of cardiac dysfunction and remodelling after chronic ischaemic injury. Cardiovasc Res. 2008;77:54–63. doi: 10.1093/cvr/cvm023. [DOI] [PubMed] [Google Scholar]
- 10.McGaffin K.R., Witham W.G., Yester K.A. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc Res. 2011;89:60–71. doi: 10.1093/cvr/cvq288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denroche H.C., Huynh F.K., Kieffer T.J. The role of leptin in glucose homeostasis. J Diabetes Investig. 2012;3:115–129. doi: 10.1111/j.2040-1124.2012.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamohara S., Burcelin R., Halaas J.L., Friedman J.M., Charron M.J. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. doi: 10.1038/38717. [DOI] [PubMed] [Google Scholar]
- 13.Atkinson L.L., Fischer M.A., Lopaschuk G.D. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis. J Biol Chem. 2002;277:29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 14.Li X., Wu X., Camacho R., Schwartz G.J., LeRoith D. Intracerebroventricular leptin infusion improves glucose homeostasis in lean type 2 diabetic MKR mice via hepatic vagal and non-vagal mechanisms. PLoS ONE. 2011;6 doi: 10.1371/journal.pone.0017058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morton G.J., Schwartz M.W. Leptin and the central nervous system control of glucose metabolism. Physiol Rev. 2011;91:389–411. doi: 10.1152/physrev.00007.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warzecha Z., Dembinski A., Ceranowicz P. Influence of leptin administration on the course of acute ischemic pancreatitis. J Physiol Pharmacol. 2002;53:775–790. [PubMed] [Google Scholar]
- 17.Hall M.E., Maready M.W., Hall J.E., Stec D.E. Rescue of cardiac leptin receptors in db/db mice prevents myocardial triglyceride accumulation. Am J Physiol Endocrinol Metab. 2014;307:E316–E325. doi: 10.1152/ajpendo.00005.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall M.E., Smith G., Hall J.E., Stec D.E. Cardiomyocyte-specific deletion of leptin receptors causes lethal heart failure in Cre-recombinase-mediated cardiotoxicity. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1241–R1250. doi: 10.1152/ajpregu.00292.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.do Carmo J.M., Hall J.E., da Silva A.A. Chronic central leptin infusion restores cardiac sympathetic-vagal balance and baroreflex sensitivity in diabetic rats. Am J Physiol Heart Circ Physiol. 2008;295:H1974–H1981. doi: 10.1152/ajpheart.00265.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Silva A.A., Tallam L.S., Liu J., Hall J.E. Chronic antidiabetic and cardiovascular actions of leptin: role of CNS and increased adrenergic activity. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1275–R1282. doi: 10.1152/ajpregu.00187.2006. [DOI] [PubMed] [Google Scholar]
- 21.do Carmo J.M., da Silva A.A., Cai Z., Lin S., Dubinion J.H., Hall J.E. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57:918–926. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parton L.E., Ye C.P., Coppari R. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 23.Singh M., Mukhopadhyay K. Alpha-melanocyte stimulating hormone: an emerging anti-inflammatory antimicrobial peptide. Biomed Res Int. 2014;2014:874610. doi: 10.1155/2014/874610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dores R.M. Adrenocorticotropic hormone, melanocyte-stimulating hormone, and the melanocortin receptors: revisiting the work of Robert Schwyzer: a thirty-year retrospective. Ann N Y Acad Sci. 2009;1163:93–100. doi: 10.1111/j.1749-6632.2009.04434.x. [DOI] [PubMed] [Google Scholar]
- 25.do Carmo J.M., da Silva A.A., Rushing J.S., Hall J.E. Activation of the central melanocortin system contributes to the increased arterial pressure in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2012;302:R561–R567. doi: 10.1152/ajpregu.00392.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaprielian R., Sah R., Nguyen T., Wickenden A.D., Backx P.H. Myocardial infarction in rat eliminates regional heterogeneity of AP profiles, I(to) K(+) currents, and [Ca(2+)](i) transients. Am J Physiol Heart Circ Physiol. 2002;283:H1157–H1168. doi: 10.1152/ajpheart.00518.2001. [DOI] [PubMed] [Google Scholar]
- 27.da Silva A.A., Spradley F.T., Granger J.P., Hall J.E., do Carmo J.M. Brain-mediated antidiabetic, anorexic, and cardiovascular actions of leptin require melanocortin-4 receptor signaling. J Neurophysiol. 2015;113:2786–2791. doi: 10.1152/jn.00911.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.do Carmo J.M., Bassi M., da Silva A.A., Hall J.E. Systemic but not central nervous system nitric oxide synthase inhibition exacerbates the hypertensive effects of chronic melanocortin-3/4 receptor activation. Hypertension. 2011;57:428–434. doi: 10.1161/HYPERTENSIONAHA.110.163931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wahr D.W., Wang Y.S., Schiller N.B. Left ventricular volumes determined by two-dimensional echocardiography in a normal adult population. J Am Coll Cardiol. 1983;1:863–868. doi: 10.1016/s0735-1097(83)80200-9. [DOI] [PubMed] [Google Scholar]
- 30.Goodwin G.W., Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. 2000;279:H1490–H1501. doi: 10.1152/ajpheart.2000.279.4.H1490. [DOI] [PubMed] [Google Scholar]
- 31.Jain R.L.M. Isolation, culture, and functional analysis of adult mouse cardiomyocytes. Methods Mol Med. 2007;139:251–262. doi: 10.1007/978-1-59745-571-8_16. [DOI] [PubMed] [Google Scholar]
- 32.Li X., Liu J., Hu H. Dichloroacetate ameliorates cardiac dysfunction caused by ischemic insults through AMPK signal pathway—not only shifts metabolism. Toxicol Sci. 2019;167:604–617. doi: 10.1093/toxsci/kfy272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moshal K.S., Kumar M., Tyagi N. Restoration of contractility in hyperhomocysteinemia by cardiac-specific deletion of NMDA-R1. Am J Physiol Heart Circ Physiol. 2009;296:H887–H892. doi: 10.1152/ajpheart.00750.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moshal K.S., Tipparaju S.M., Vacek T.P. Mitochondrial matrix metalloproteinase activation decreases myocyte contractility in hyperhomocysteinemia. Am J Physiol Heart Circ Physiol. 2008;295:H890–H897. doi: 10.1152/ajpheart.00099.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spradley F.T., Palei A.C., Granger J.P. Differential body weight, blood pressure and placental inflammatory responses to normal versus high-fat diet in melanocortin-4 receptor-deficient pregnant rats. J Hypertens. 2016;34:1998–2007. doi: 10.1097/HJH.0000000000001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Lucia C., Gambino G., Petraglia L. Long-term caloric restriction improves cardiac function, remodeling, adrenergic responsiveness, and sympathetic innervation in a model of postischemic heart failure. Circ Heart Fail. 2018;11 doi: 10.1161/CIRCHEARTFAILURE.117.004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tallam L.S., da Silva A.A., Hall J.E. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48:58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 38.Goncalves J.P.L., Palmer D., Meldal M. MC4R Agonists: structural overview on antiobesity therapeutics. Trends Pharmacol Sci. 2018;39:402–423. doi: 10.1016/j.tips.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Ayers K.L., Glicksberg B.S., Garfield A.S. Melanocortin 4 receptor pathway dysfunction in obesity: patient stratification aimed at MC4R agonist treatment. J Clin Endocrinol Metab. 2018;103:2601–2612. doi: 10.1210/jc.2018-00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhnen P., Clement K., Wiegand S. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. N Engl J Med. 2016;375:240–246. doi: 10.1056/NEJMoa1512693. [DOI] [PubMed] [Google Scholar]
- 41.Clement K., Biebermann H., Farooqi I.S. MC4R agonism promotes durable weight loss in patients with leptin receptor deficiency. Nat Med. 2018;24:551–555. doi: 10.1038/s41591-018-0015-9. [DOI] [PubMed] [Google Scholar]
- 42.Simon J.A., Kingsberg S.A., Portman D. Long-term safety and efficacy of bremelanotide for hypoactive sexual desire disorder. Obstet Gynecol. 2019;134:909–917. doi: 10.1097/AOG.0000000000003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhillon S., Keam S.J. Bremelanotide: first approval. Drugs. 2019;79:1599–1606. doi: 10.1007/s40265-019-01187-w. [DOI] [PubMed] [Google Scholar]
- 44.van der Velden J., Merkus D., Klarenbeek B.R. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- 45.Sloan C., Tuinei J., Nemetz K. Central leptin signaling is required to normalize myocardial fatty acid oxidation rates in caloric-restricted ob/ob mice. Diabetes. 2011;60:1424–1434. doi: 10.2337/db10-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keung W., Cadete V.J., Palaniyappan A., Jablonski A., Fischer M., Lopaschuk G.D. Intracerebroventricular leptin administration differentially alters cardiac energy metabolism in mice fed a low-fat and high-fat diet. J Cardiovasc Pharmacol. 2011;57:103–113. doi: 10.1097/FJC.0b013e31820014f9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.