

SUMMARY
Immune cells can metabolize glucose, amino acids, and fatty acids (FAs) to generate energy. The roles of different FA species and their impacts on humoral immunity remain poorly understood. Here, we report that proliferating B cells require monounsaturated FAs (MUFAs) to maintain mitochondrial metabolism and mTOR activity and to prevent excessive autophagy and endoplasmic reticulum (ER) stress. Furthermore, B cell-extrinsic stearoyl-CoA desaturase (SCD) activity generates MUFA to support early B cell development and germinal center (GC) formation in vivo during immunization and influenza infection. Thus, SCD-mediated MUFA production is critical for humoral immunity.
Graphical Abstract
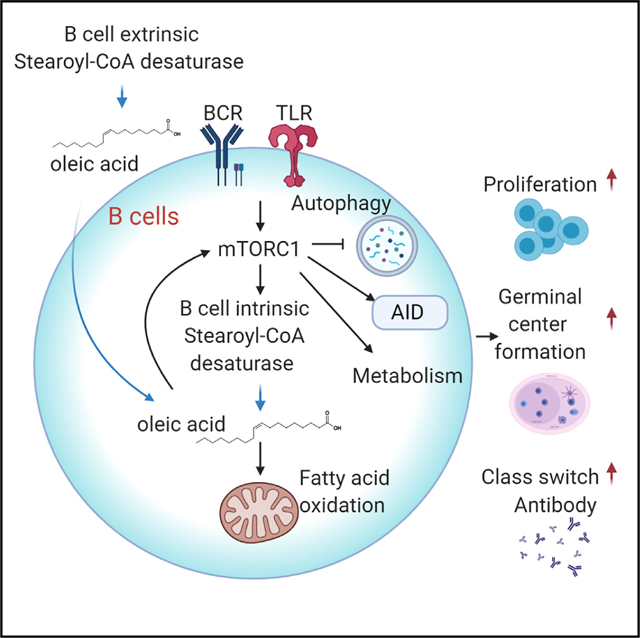
In Brief
Zhou et al. show that monounsaturated fatty acids (MUFAs), generated by stearoyl-CoA desaturase (SCD), support B cell mitochondrial metabolism and mTOR activity and promote B cell development and humoral immune responses. These data establish MUFA availability as a key regulator for humoral immunity and a potential therapeutic target.
INTRODUCTION
There is growing evidence that B cell development and activation are regulated by metabolic processes (Boothby and Rickert, 2017). In particular, glucose metabolism was shown to be activated by B cell receptor (BCR) or Toll-like receptor (TLR) ligand stimulation and support B cell function (Caro-Maldonado et al., 2014; Cho et al., 2011; Dufort et al., 2007). Activation of mitochondrial oxidative phosphorylation (OXPHOS) by TLR ligand or CD40L, and supported by glutamine, is required for B cell survival (Akkaya et al., 2018; Le et al., 2012; Waters et al., 2018). The central metabolic regulator, mammalian target of rapamycin (mTOR), is key to support B cell development and humoral response by mediating organelle biogenesis and various anabolic processes (Iwata et al., 2016; Jones et al., 2016; Raybuck et al., 2018; Zeng et al., 2018). However, a recent study suggested that activated B cells use glucose mainly for ribonucleotide and fatty acid (FA) biosynthesis but not for lactate production or feeding into TCA cycle (Waters et al., 2018). Thus, biomass accumulation appears to be the main feature of early B cell activation (Dufort et al., 2014). Glucose, glutamine, and FAs are the three major carbon sources. It has been recognized that nutrient availability is a key regulatory mechanism controlling immune responses (Kedia-Mehta and Finlay, 2019). Although the importance of glucose and glutamine has been evaluated, as described above, the functions of different FAs in B cells are not fully understood, partly because of the sheer diversity of FA species. Thus, it is critical to investigate how individual FA species contribute to B cell functions.
Both malnutrition and obesity impair humoral immunity (Alwarawrah et al., 2018; Rytter et al., 2014). Because a plethora of metabolites are altered in general malnutrition or obesity, it is challenging to parse how each metabolite affects humoral immunity, and thus exact mechanisms linking immunity to systemic metabolism remain obscure. This is reflected in many studies that focused on the relationship between diets with varying FA contents and systemic inflammation (Fritsche, 2015). A recent study showed that germinal center (GC) B cells mainly use FA β-oxidation (FAO), rather than glycolysis, to meet their energetic needs, highlighting the importance of FA availability for humoral immunity (Weisel et al., 2020). However, most immunometabolism studies do not distinguish different FAs. Several earlier studies have probed the contribution of polyunsaturated FAs (PUFAs), and their derivatives, to B cell functions (Gurzell et al., 2013; Kosaraju et al., 2017; Ramon et al., 2012, 2014; Roper et al., 1995), yet little is known about the contribution of endogenously derived MUFAs to humoral immunity.
Stearoyl-CoA desaturase (SCD) is a rate-limiting enzyme in de novo FA biosynthesis. It converts saturated FAs (SFAs) into monounsaturated FAs (MUFAs), including oleic acid (OA) and palmitoleic acid (PO). SCD plays a central role in fuel metabolism and constitutes a potential therapeutic target for treatment of obesity and cancer (ALJohani et al., 2017). SCD1-deficient mice are protected from diet-induced obesity and hepatic steatosis (Miyazaki et al., 2007; Ntambi et al., 2002). Interestingly, some of the metabolic defects in SCD1-deficient mice persisted even when they were fed a diet containing a high level of OA (Miyazaki et al., 2001a, 2001b; Ntambi et al., 2002), highlighting the importance of endogenously synthesized MUFAs for proper cellular function.
Here, we use a reductionist approach to investigate the impacts of SFA and MUFA on B cells. We present mechanistic evidence that B cell development and activation require SCD-generated MUFAs, particularly OA, which maintains B cell metabolic fitness partly by supporting OXPHOS and mTORC1 activity and preventing excessive autophagy and ER stress. In vivo, B cells can rely on B cell-extrinsic SCD activity to provide MUFAs. In response to immune challenges, the host enhances MUFA availability, partly through SCD activity, which is required to sustain antibody production. Suppression of SCD reduces humoral immune response to immunization and weakens immune defense against respiratory influenza infection. Thus, our results provide a novel link between endogenous biosynthesis of MUFAs and humoral immunity.
RESULTS
SCD-Mediated MUFA Biosynthesis Is Induced during B Cell Activation
We first sought to determine the FA biosynthesis gene expression program during B cell activation. RNA sequencing (RNA-seq) was performed using fresh murine B cells and lipopolysaccharide (LPS)/IL-4-activated B cells. Expression of major FA biosynthesis genes, including Acaca, Elovl1, Elovl5, Elovl6, Fasn, and Scd2, increased upon B cell activation (Figure 1A). Among the four murine Scd genes, Scd3 and Scd4 were below the detection limit. Scd1 expression was slightly reduced, while Scd2 expression, both mRNA and protein, was substantially increased (Figures 1B and 1C). Real-time PCR analysis confirmed the gene expression kinetics, in which Scd2 expression increased the most after 24 h stimulation (Figure S1A). Multiple stimuli promoted SCD2 expression, including LPS/IL-4, anti-IgM, anti-CD40, and CpG, but not IL-4 alone (Figure S1B). Furthermore, human B cell activation also induced robust SCD expression (Figure 1D). Finally, the upregulation of Fasn and Scd2 genes was dependent on mTORC1 signaling, as rapamycin and Torin 1 treatment blocked their induction (Figure 1E). Thus, antigenic stimulation of B cells activates FA biosynthetic pathway in an mTORC1 dependent manner.
Figure 1. SCD-Mediated MUFA Biosynthesis during B Cell Activation In Vitro.
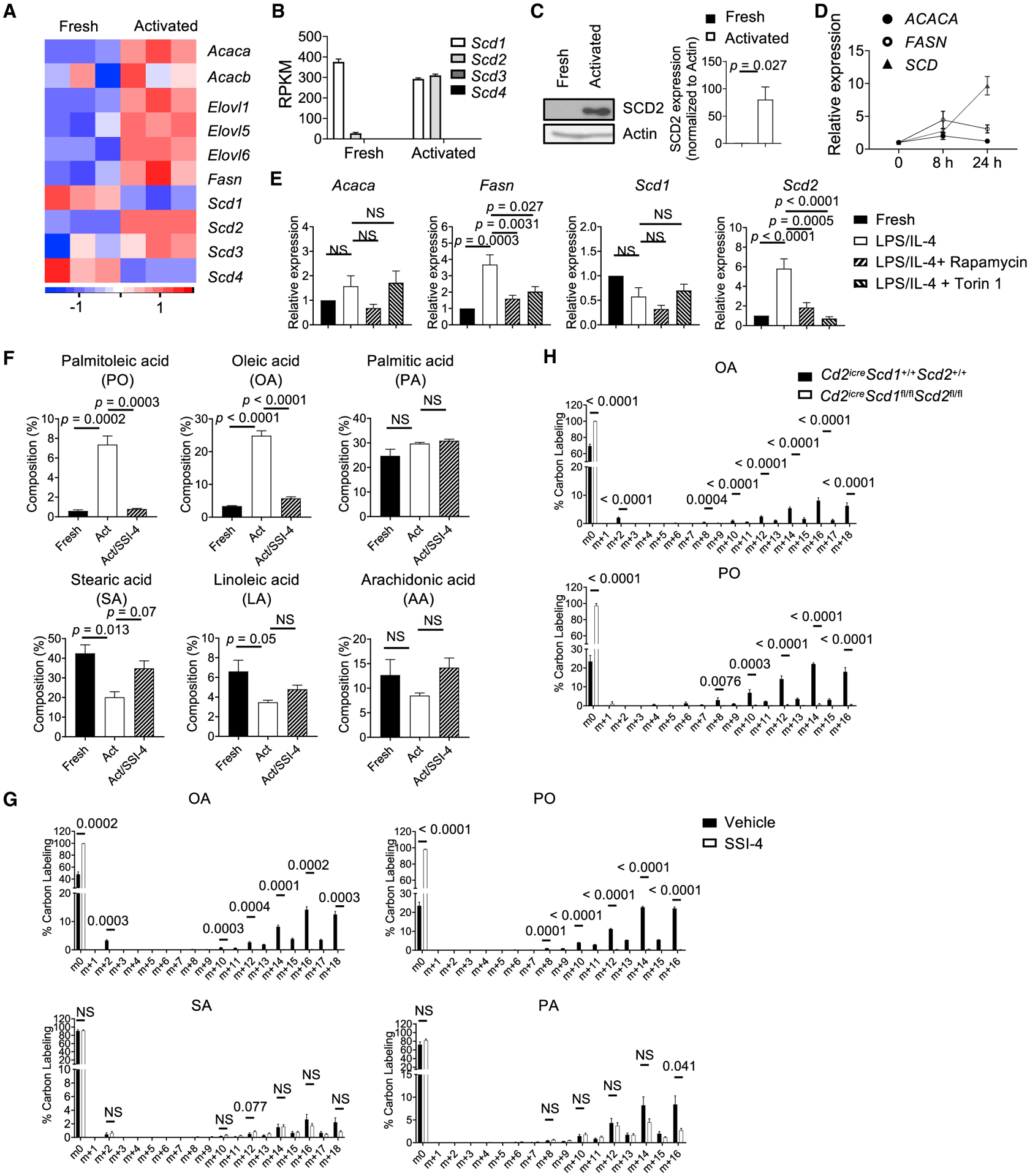
(A) Heatmap of fatty acid (FA) synthesis gene expression in fresh and LPS/IL-4-activated murine B cells.
(B) mRNA expression of four Scd isoforms in freshly and activated B cells, extracted from RNA-seq data. ND, not detected.
(C) Immunoblot analysis of SCD2 in fresh and activated murine B cells. Right: summary of SCD2 expression.
(D) Reverse transcription polymerase chain reaction (RT-PCR) analysis of ACACA, FASN, and SCD expression in human B cells at 0, 8, and 24 h after CpG/anti-CD40/IL-15/IL-10/IL-2 activation.
(E) RT-PCR analysis of Acaca, Fasn, Scd1, and Scd2 expression in fresh and activated murine B cells treated with rapamycin (10 nM) or Torin 1 (100 nM).
(F) Composition (among all cellular FAs) of palmitoleic acid (PO), oleic acid (OA), palmitic acid (PA), stearic acid (SA), linoleic acid (LA), and arachidonic acid (AA) in fresh and activated murine B cells treated with vehicle or the SCD inhibitor SSI-4 for 48 h (n = 3).
(G) Isotopomer distribution of U-13C-glucose incorporation in OA, SA, PO, and PA in activated B cell treated with vehicle or SSI-4 at 48 h (n = 3).
(H) Isotopomer distribution of U-13C-glucose incorporation in OA and PO in activated B cells from WT or Cd2iCreScd1fl/flScd2fl/fl mice (n = 3).
p values were calculated using Student’s t test (C, G, and H) and one-way ANOVA (E and F). Data are representative of at least three (C–E) or two (F–H) independent experiments. Error bars represent SEM. See also Figure S1.
To examine the functional outcome of increased FA biosynthesis gene expression, we used triple-quadrupole liquid chromatography-tandem mass spectrometry (LC-MS/MS). Our data showed that PO and OA, the main MUFAs produced by SCD, exhibited the highest increases of their relative contents in activated B cells compared with unstimulated B cells. The composition (among all cellular FAs) of palmitic acid (PA), the precursor of PO, did not change significantly, while the composition of stearic acid (SA), the precursor of OA, and the PUFAs, including linoleic acid (LA) and arachidonic acid (AA), modestly reduced after activation (Figure 1F). This dramatic accumulation of MUFAs was dependent on SCD activity, because treatment with an SCD-specific inhibitor, SSI-4 (von Roemeling et al., 2017), largely reversed it (Figure 1F). Furthermore, the de novo biosynthesis of FAs from glucose was confirmed in a U-13C-6-glucose tracing experiment. When B cells were activated in medium containing 13C-labeled glucose, LC-MS/MS assay detected substantial 13C incorporation in OA and PO, which were abolished upon SSI-4 treatment. However, the incorporation of 13C into SA and PA was largely unaffected, or only modestly affected, by SSI-4 treatment (Figure 1G). Finally, we confirmed these observations using a genetic model in which Scd1 and Scd2 were efficiently deleted in B cells through Cd2iCre (Figure S1C), an optimized variant of Cre recombinase under human CD2 promoter, which mediates efficient recombination in lymphocytes (de Boer et al., 2003; Siegemund et al., 2015). SCD1/SCD2 deficiency completely eliminated 13C-glucose incorporation into OA and PO, but not PA or SA (Figure 1H; Figure S1D). Incorporation of 13C into AA, LA, and α-linolenic acid was not detected in our assays (data not shown), suggesting that B cells are unable to generate these PUFAs from glucose de novo. Therefore, these results showed that B cell activation is associated with activation of SCD activity and increased SCD-generated MUFA content.
SCD-Generated MUFA Supports B Cell Proliferation and Class Switch In Vitro
To investigate the impact of SCD-generated MUFA on B cell functions, we measured the proliferation of mouse B cells treated with different SCD inhibitors, including SSI-4, MF438, and A939572. All were capable of inhibiting B cell proliferation (Figure 2A). The inhibitory effect of SSI-4 on cell proliferation was dose dependent (Figure S2A). SSI-4 did not significantly affect B cell survival at 48 h (Figure S2B), but it reduced cell viability at 72 h (Figure S2C). Importantly, exogenous OA could rescue the proliferation and viability defects caused by SCD inhibition, demonstrating that SCD-derived MUFA is required for B cell proliferation and survival (Figures 2B, 2C, and S2A–S2C). Similar phenotypes were observed when we stimulated B cells with anti-IgM/anti-CD40 or TLR9 ligand, CpG (Figure 2B). LPS/IL-4 also stimulates class switch to IgG1. SCD inhibition strongly suppressed IgG1 class switch (Figure 2D). OA alone further enhanced B cell proliferation and IgG1 expression, and it could restore both parameters upon SSI-4 treatment (Figures 2C and 2D). In contrast, PO alone did not improve class switch and had substantial, but incomplete, rescue effects on proliferation and IgG1 expression upon SSI-4 treatment. PA and SA showed no rescue effects (Figure 2D). Moreover, PA or SA alone was unable to promote B cell proliferation and class switch (Figure S2D). Of note, the concentration of exogenous FAs used in these in vitro assays was physiological on the basis of the levels of serum non-esterified FAs (NEFAs) in wild-type (WT) mice (Table S1). Moreover, SCD1/2-deficient B cells proliferated poorly and had highly reduced IgG1 expression (Figure 2E), which were fully and partially restored by OA and PO, respectively (Figure 2E). Last, we made similar observations using human B cells. Exogenous OA promoted human B cell proliferation (Figure 2F) and activation, measured by expression of costimulatory ligands CD80 and CD86 (Figure 2G). It also restored all these parameters upon SSI-4 treatment (Figures 2F and 2G). Thus, proliferating B cells preferentially use MUFA, especially OA, rather than SFAs, to promote their proliferation and class switch in vitro. The fact that exogenous OA can promote B cell activation and rescue SCD inhibition indicates that B cells can reply on either intrinsically synthesized MUFA or extrinsically provisioned MUFA.
Figure 2. SCD-Generated MUFA Is Required for B Cell Proliferation and Class Switch In Vitro.
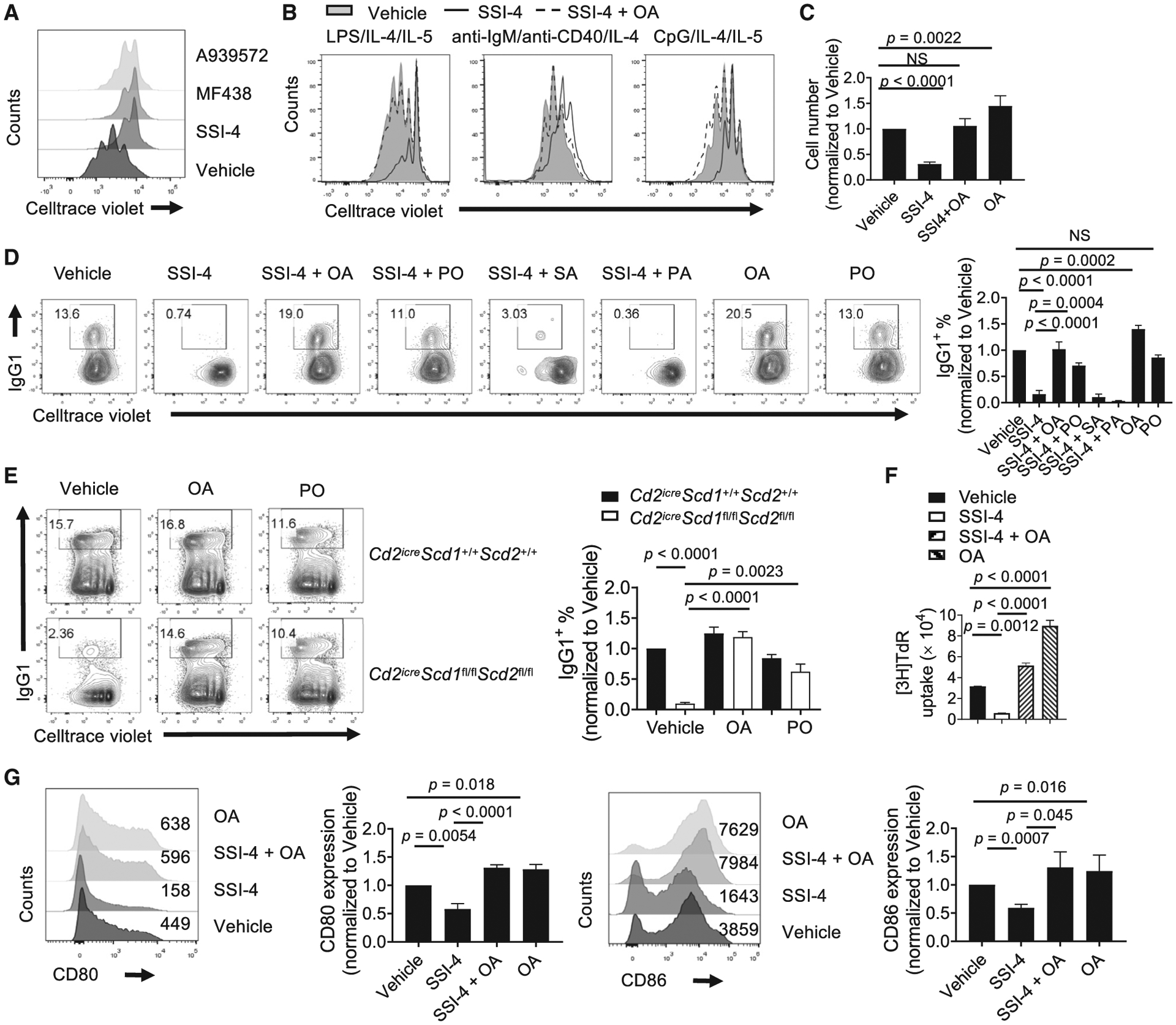
(A) Cell proliferation measured by dilution of CellTrace Violet (CTV) dye in LPS/IL-4-activated murine B cells treated with the SCD inhibitors A939572 (1 μM), MF438 (1 μM), and SSI-4 (1 μM).
(B) Cell proliferation of murine B cells stimulated with indicated stimuli in presence of vehicle, SSI-4, or SSI-4 plus OA for 3 days.
(C) Murine B cells were stimulated with LPS/IL-4/IL-5 for 3 days in the presence of indicated inhibitor and/or fatty acids. Numbers of live B cells were summarized and normalized to vehicle group.
(D) Murine B cell proliferation and IgG1 expression upon LPS/IL-4 stimulation in the presence of vehicle, SSI-4, SSI-4 with OA (100 μM), or PO (25 μM), or SA (25 μM), or PA (100 μM), OA alone, and PO alone. Right: summary of IgG1+ percentages normalized against vehicle group.
(E) Cell proliferation and IgG1 expression in activated B cells from WT or Cd2iCreScd1fl/flScd2fl/fl mice. Right: summary of IgG1+ percentage normalized to vehicle-treated WT cells.
(F) Human B cell proliferation was measured by thymidine incorporation after activation in the presence of vehicle, SSI-4, SSI-4 plus OA, and OA alone.
(G) Expression of CD80 and CD86 on activated human B cells in the presence of vehicle, SSI-4, SSI-4 plus OA, or OA alone. Numbers indicate the mean fluorescence intensity (MFI). Right: the summary of CD80 and CD86 expression.
p values were calculated using one-way ANOVA (C–G). NS, not significant. Data are representative of three (A and F) or pooled from four (C–E and G) independent experiments. Error bars represent SEM. See also Figure S2.
MUFA Maintains B Cell Metabolic Fitness
B cell activation is accompanied by increased OXPHOS, which is important to support humoral immunity (Akkaya et al., 2018; Caro-Maldonado et al., 2014; Waters et al., 2018). FAs can be used to fuel OXPHOS, but it is unclear how different FAs contribute to B cell metabolism. The inhibitor etomoxir, which blocks carnitine palmitoyltransferase I (CPT1)-mediated FA import to mitochondria, reduced B cell class switch and negated the effects of OA treatment at 40 μM, a dose selective to CPT1 inhibition (Weisel et al., 2020) (significantly lower than OA treatment alone, but not to the level of etomoxir treatment alone) (Figure 3A), suggesting that B cells oxidize OA to promote cell activation. FAO leads to the incorporation of FA-derived hydrogen and carbon into H2O and citrate (Koutsari et al., 2013). Indeed, we verified that OA was oxidized in mitochondrial by measuring 3H2O generation from [9,10-3H]-OA, which was greatly suppressed by etomoxir (Figure 3B). Furthermore, when B cells were cultured with increasing doses of exogenous OA, they exhibited an increasing FAO rate (Figure 3B). Similarly, stable isotope tracing using U-13C-OA showed that 13C incorporation into citrate was significantly blocked by etomoxir (Figures S3A and S3B). Increased FAO should result in an elevated cell oxygen consumption rate (OCR). To analyze the metabolic impact of SFAs and MUFAs, we measured OCR of CpG/IL-4/IL-5-stimulated B cells with different exogenous FA treatment. Addition of OA, but not SFA, enhanced B cell OCR (Figure 3C). Similar findings were observed using LPS/IL-4 and anti-IgM/IL-4 stimulation (Figures S3C and S3D). Importantly, the OA-augmented respiration was also observed in human B cells (Figure 3D). The effects of PO on B cell metabolism appeared to be variable. Although it did not promote mouse B cell OCR upon CpG and anti-IgM/anti-CD40 stimulation (Figures 3C and S3D), it enhanced basal OCR in LPS/IL-4-stimulated mouse B cells (Figure S3C) and in CpG/anti-CD40/IL-10/IL-15/IL-4/IL-2-stimulated human B cells (Figure 3D), suggesting a context-dependent metabolic impact of PO. Moreover, OA and, to a lesser degree, PO increased the glycolytic capacity of activated murine and human B cells, while SFA had either a negative or no effect on glycolysis (Figures 3E and 3F). Conversely, inhibition of SCD activity strongly suppressed B cell respiration and glycolysis (Figure 3G). Overall, these data suggest that provision of OA through SCD activity is critical for B cell metabolic fitness.
Figure 3. MUFA Supports B Cell Metabolic Fitness.
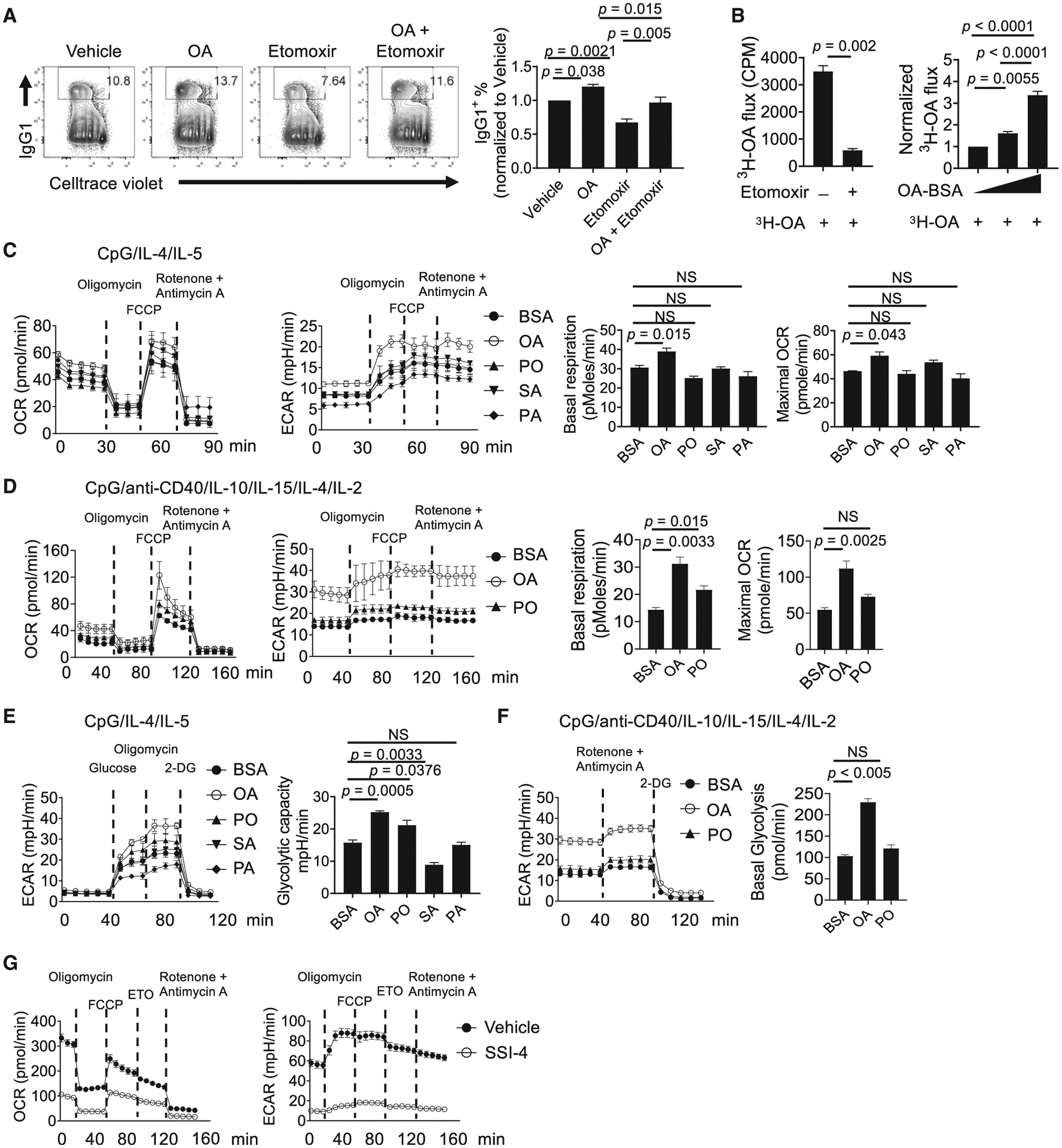
(A) Flow cytometry analysis of murine B cell proliferation and IgG1 expression. B cells were activated in the presence of vehicle (BSA), OA alone, etomoxir, and OA plus etomoxir at 72 h. Right: summary of IgG1+ B cell percentage normalized to vehicle treatment.
(B) The OA oxidation measurement by [9,10-3H]-OA flux assay. Left: activated B cells were treated with vehicle or etomoxir for 1 h before being incubated with 3 μCi [9,10-3H]-OA for another 4 h. Right: B cells were activated in the presence of increasing dose of OA, from 0 μM (BSA control) to 20 and 50 μM for 48 h, followed by incubation with 3 μCi [9,10-3H]-OA for another 4 h. OA oxidation was determined by measuring the detritiation of [9,10-3H]-OA.
(C) Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse XFe96 analyzer using murine B cells activated in the presence of BSA, OA, PO, SA, and PA for 48 h with MitoStress Test. Right: summary of basal respiration and maximal OCR.
(D) Measurement of OCR and ECAR in human B cells activated in the presence of BSA, OA, and PO for 72 h. Right: summary of basal respiration and maximal OCR.
(E) Glycolytic capacity of murine B cells activated in presence of BSA, OA, PO, SA, and PA at 48 h was measured using a glycolysis stress test. Right: summary of glycolytic capacity.
(F) ECAR was measured in activated human B cells in the presence of BSA, OA, and PO for 72 h. Right: summary of basal glycolysis rate.
(G) Measurement of OCR and ECAR in the activated murine B cells treated with vehicle or SSI-4 for 48 h.
p values were calculated using Student’s t test (B) and one-way ANOVA (A–F). NS, not significant. Data are representative of at least three (B–G) independent experiments or pooled from four (A) independent experiments. Error bars represent SEM. See also Figure S3.
SCD-Mediated MUFA Supports mTORC1 Activity and Prevents Excessive Autophagy
SCD-mediated FA metabolism has been linked to autophagy induction in non-immune cells, but there are contradictory findings (Huang et al., 2015; Ogasawara et al., 2014; Tan et al., 2014). It is unclear how FA metabolism may link to autophagy in lymphocytes. To examine how SCD inhibition affects subcellular organelles, we performed transmission electron microscopy on SSI-4-treated B cells and SCD-deficient B cells. We observed increased structures with double-layer membrane encompassing various organelles or multi-lamellar membrane structures, which are signatures of autophagosomes (Mizushima et al., 2011) (Figures 4A and S4A). The conversion of the soluble form of LC3 (LC3-I) to the autophagic vesicle-associated form (LC3-II) is a hallmark of autophagy. Immunoblot analysis showed that SSI-4-treated B cells (Figure 4B) and SCD-deficient B cells (Figure 4C) had significant increase of LC3-II/LC3-I ratio, which was completely rescued by exogenous OA. Consistent with these data, B cells isolated from GFP-LC3 reporter mouse had higher LC3 expression upon SCD inhibition (Figure S4B). mTORC1 is known to control autophagy (Kim and Guan, 2015). We observed reduced mTORC1 activation, measured by phosphorylation of its downstream targets S6K and S6, in SSI-4-treated B cells or SCD1/2-deficient B cells, which were restored by exogenous OA (Figures 4B and 4C). Reduced expression of activation-induced cytidine deaminase (AID) was observed upon SSI-4 treatment or in the absence of SCD1/2, consistent with the positive effect of mTORC1 on AID induction (Raybuck et al., 2018) (Figures 4B and 4C). Furthermore, OA treatment alone enhanced mTORC1 activity (Figure 4C). SCD inhibition also suppressed B cell mTORC1 activity in vivo, when mice were fed with SSI-4 chow or control chow followed by immunization with hapten 4-hydroxy-3-nitrophenylacetyl conjugated with ovalbumin (NP-OVA) (Figure 4D). Finally, the classic autophagy inhibitors 3-methyladenine (3-MA) and wortmannin successfully rescued B cell proliferation and IgG1 expression in the presence of SSI-4 (Figure 4E). Increased SFA content is associated with enhanced ER stress (Wei et al., 2006). Because SCD inhibition increases the SFA/MUFA ratio (Figure 1F), it could also induce ER stress. We found that expression of ER stress-related genes, including Chop, Atf4, and Sqstm1, increased after SSI-4 treatment, whereas exogenous OA reversed them (Figure S4C). Taken together, these data showed that SCD-mediated MUFA availability suppresses ER stress and maintains mTORC1 activity, which prevents overactivation of autophagy.
Figure 4. Inhibition of SCD Activity Induces the Formation of Autophagosomes and Suppresses mTORC1 Activity.
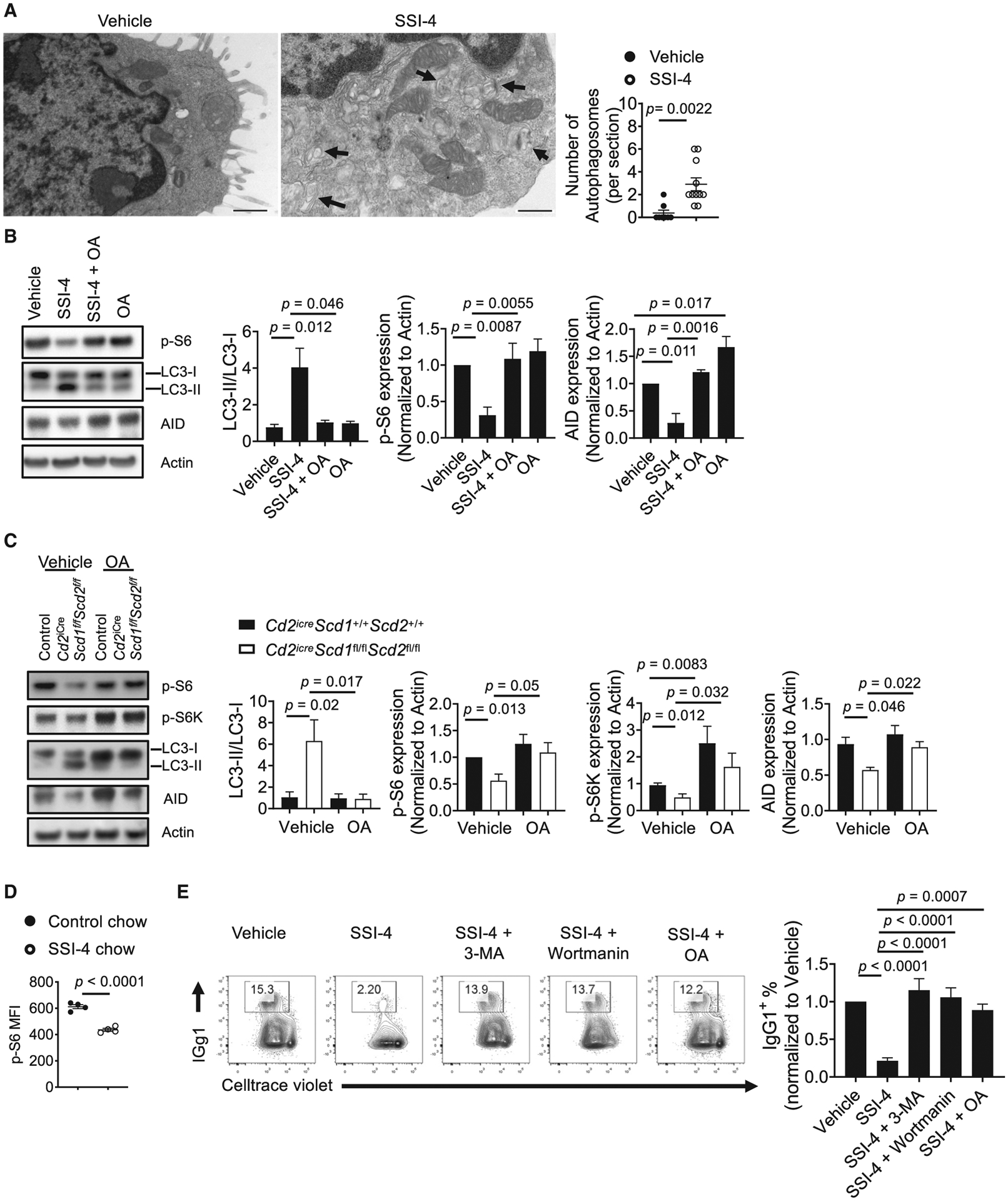
(A) Transmission electron microscopic image of activated mouse B cell treated with vehicle or SSI-4. Arrows indicate autophagosome structures. Right: numbers of autophagosomes (per section) found in vehicle (n = 8) or SSI-4 (n = 11) treated cells. Scale bars: 0.5 μm.
(B) Immunoblot analysis of LC3-I/LC3-II, p-S6, and AID in activated murine B cells in presence of vehicle, SSI-4, SSI-4 plus OA, and OA alone. Right: summary of the LC-II/LC-I ratio and the relative expression of p-S6 and AID (normalized to that in vehicle-treated samples).
(C) Immunoblot analysis of LC3-I/LC3-II, p-S6, p-S6K, and AID in activated B cells from control and Cd2iCreScd1fl/flScd2fl/fl mice with or without exogenous OA.
Right: summary of the LC-II/LC-I ratio and the relative expression of p-S6, p-S6K, and AID (normalized to that in vehicle-treated WT samples).
(D) Mice were fed with control chow and SSI-4 chow, followed by NP-OVA immunization for 1 week. p-S6 in splenic B cells was measured using flow cytometry.
(E) Cell proliferation and IgG1 expression in activated murine B cells treated with SSI-4 in the presence of 3-methyadenine (3-MA) (1 mM) and wortmannin (1 μM) or OA. Right: summary of the IgG1+ percentages.
p values were calculated using Student’s t test (A and D) and one-way ANOVA (B–C and E). Data were pooled from four (B and C) and five (E) independent experiments. Error bars represent SEM. See also Figure S4.
Systemic SCD Activity Supports Early B Cell Development and Humoral Immunity In Vivo
Next, we investigated how SCD activity contributes to B cell development and function in vivo. Feeding mice with SSI-4 chow for 1 week reduced the frequencies of immature B cells and B cell precursors (CD19+B220int) (Figure S5A) and CD25+ pre-B cells (Figure S5B) in bone marrow (BM) but did not affect the composition of thymocytes (Figure S5C), and peripheral B cells and CD4+ T cells (Figures S5D–S5F), with a slight increase of mature B cell frequency (Figure S5G). Thus, short-term suppression of SCD activity reduces early B cell development but does not affect peripheral mature B and T cells. However, there was a modest reduction of GC percentage and IgA expression in Peyer’s patches, where gut microbiota drives constitutive GC generation and IgA class switch (Fagarasan et al., 2010) (Figures S5H and S5I), suggesting that SCD activity may be required for these processes. We next evaluated the impact of SCD inhibition on foreign antigen-induced humoral immunity. Notably, NP-OVA immunization significantly increased serum OA, but not PO content (Figure 5A), indicating that immune challenge is associated with increased systemic MUFA content in vivo. SSI-4 treatment substantially reduced both OA and PO contents in immunized mice as expected (Figure 5A). Importantly, SCD inhibition significantly suppressed GC formation (Figure 5B) and reduced NP+ GC B cells (Figure 5C), but not B220intCD138+ plasmablast generation (Figure 5D), suggesting that GC formation, but not plasmablast differentiation, relies more on MUFA availability. Furthermore, the percentage of follicular helper T (Tfh) cells remained normal, suggesting that B cell activation is more sensitive to SCD inhibition (Figure 5E). Consequently, the production of anti-NP-specific IgG and IgG1, but not IgM, antibodies was significantly reduced upon SCD inhibition (Figure 5F). Therefore, SCD activity supports BM B cell development and promotes GC formation and antibody production upon immunization.
Figure 5. Systemic SCD Activity Is Essential for Humoral Immune Response In Vivo.
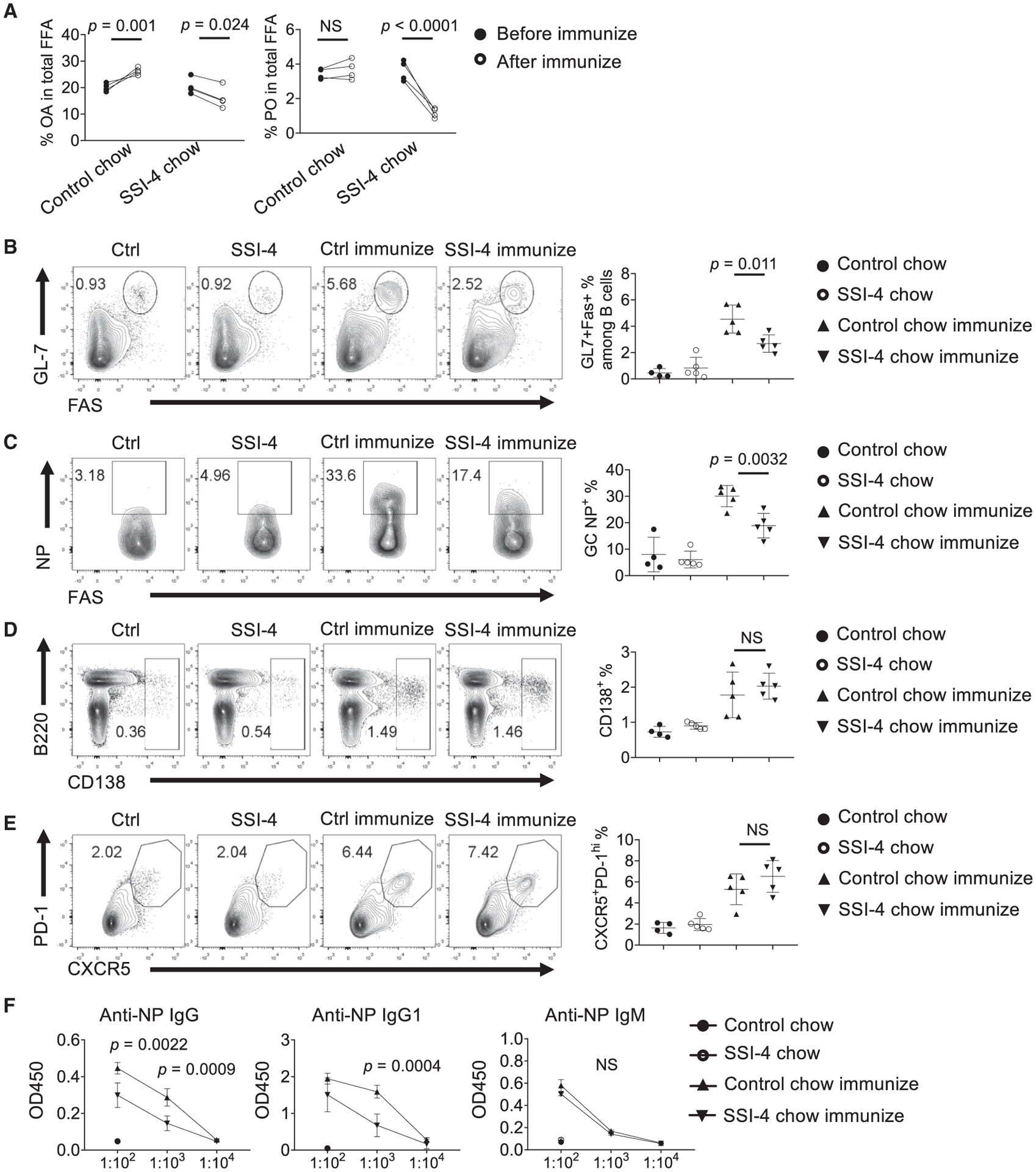
(A) Composition of OA and PO among total free FAs in sera (n = 4).
(B–E) Mice were divided into four groups. They were fed with control chow or SSI-4 chow, followed with or without NP-OVA immunization. Flow cytometry of germinal center (GC) (B), antigen-specific NP+ expression among GC B cells (C), B220intCD138+ expression (D), and expression of PD-1 and CXCR5 among CD4+ T cell (E). Right: the frequencies of GC B cells (B), NP+ GC B cells (C), B220intCD138+ plasmablasts (D), and PD-1+CXCR5+ Tfh cells (E).
(F) ELISA measurements of serum anti-NP immunoglobulins from unimmunized and immunized mice fed with control chow or SSI-4 chow, presented as absorbance at 450 nM (A450).
p values were calculated using Student’s t test (A–F). NS, not significant. Data are representative of two (A) and three (B–F) independent experiments. Error bars represent SEM. See also Figure S5.
SCD Activity Is Required for Humoral Immunity against Influenza Infection
Humoral immunity plays a critical role against influenza infection (Rangel-Moreno et al., 2008). How FA metabolism regulates anti-influenza immunity remains incompletely understood. A previous study showed that deficiency of FABP5 increased anti-influenza antibody production (Gally et al., 2013). Deficiencies of FABP family molecules are associated with increased MUFA content, suggesting a link between MUFA availability and anti-influenza humoral immunity (Hertzel et al., 2006; Hotamisligil and Bernlohr, 2015). Indeed, we observed increased serum OA and PO contents after H1N1 influenza A infection, indicating that influenza infection enhances MUFA availability (Figure 6A). Feeding with SSI-4 chow led to more severe weight loss compared with control chow (Figure 6B). SCD inhibition significantly reduced the frequency of GC B cells (Figure 6C) and the expression of Bcl6, a key transcription factor for GC B cell differentiation (Figure 6D). However, it did not affect CD138+ plasmablast formation (Figure 6E), nor did it affect CXCR5+Bcl6hi Tfh cell differentiation (Figure 6F). Consequently, SCD inhibition significantly dampened the production of serum anti-influenza IgG, IgG1, and IgG2c, but not IgM (Figure 6G). Altogether, these data suggested that SCD activity is essential for antigen specific GC B cell formation and antibody production upon respiratory viral infection in vivo.
Figure 6. SCD Activity Is Required for Humoral Immunity against Influenza Infection.
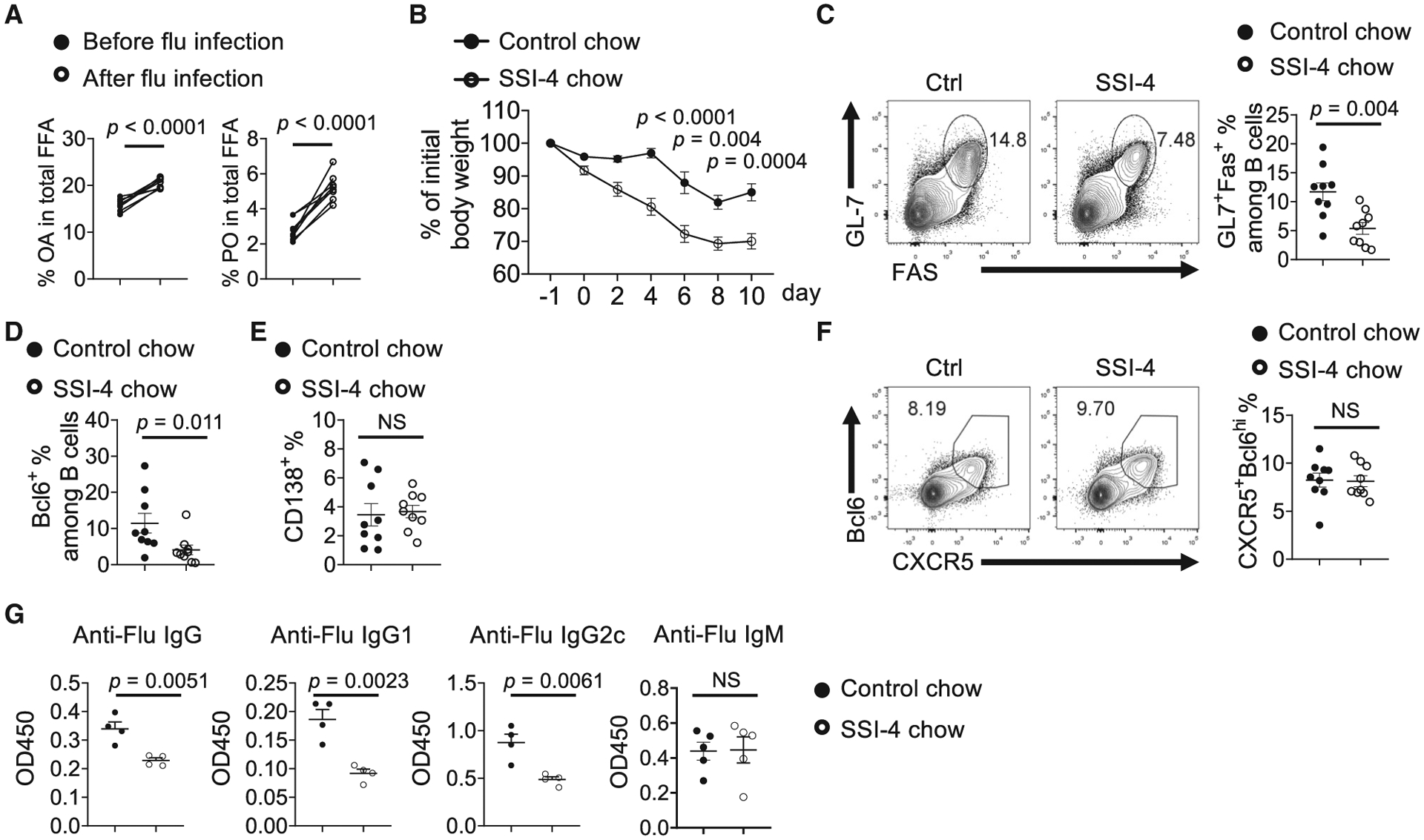
(A) Composition of OA and PO among total free FAs in sera before and after influenza virus infection (n = 8).
(B) Body weight change in mice fed with control chow or SSI-4 chow for 1 week following influenza infection.
(C) Flow cytometry analysis of GC B cells in mediastinal lymph nodes at day 11 following infection. Right: the frequencies of GC B cells.
(D) The frequencies of Bcl6+ expression among B cells in mediastinal lymph nodes.
(E) The frequencies of B220intCD138+ plasmablasts.
(F) Flow cytometry analysis of Tfh cells in mediastinal lymph nodes. Right: the frequencies of CXCR5+Bcl6hi Tfh among CD4+ T cells in mediastinal lymph nodes.
(G) Influenza virus-specific antibodies IgG, IgG1, IgG2c, and IgM in sera were measured using ELISA.
p values were calculated using Student’s t test (A–G). Results are representative of three (B–G) independent experiments. Error bars represent SEM.
Intrinsic SCD Activity Is Not Required for B Cell Development and B Cell Response In Vivo
Although our above data clearly demonstrated that SCD-generated MUFAs were critically required for B cell development and function, the sources of the SCD activity remained unknown. Because our in vitro experiments demonstrated that exogenous OA was able to rescue the SCD-deficient B cells, it is possible that B cell-extrinsic SCD activity could compensate the loss of SCD in B cells. To address this question, we first examined B cell development in Cd2iCreScd1fl/flScd2fl/fl mice, in which Scd1 and Scd2 were deleted in all lymphocytes. We did not observe significant alteration in early B cell development, indicating that B cell-intrinsic SCD activity was dispensable for early B cell development (Figure 7A). Moreover, we examined Cd19creScd1fl/flScd2fl/fl mice, in which Scd1 and Scd2 were deleted specifically and efficiently in B cells at a later stage (Hobeika et al., 2006) (Figure S6A). Cd19creScd1fl/flScd2fl/fl mice had normal GC formation upon NP-OVA immunization (Figure 7B). To eliminate any possible secondary effects elicited by chronic SCD deficiency during B cell development, we constructed chimera mice by transferring purified SCD-deficient B cells from tamoxifen-treated CreERScd1fl/flScd2fl/fl mice, together with CD4 T cells from OT-II transgenic mice and WT mice, into Rag1−/− mice, followed by immunization with NP-OVA (Lee et al., 2013). Again, we did not observe any differences in terms of GC formation (Figure 7C) and antibody production (Figure 7D). Altogether, these data indicate that B cell-intrinsic SCD activity is not essential for B cell development and function, or alternatively, B cell-extrinsic SCD activity can compensate for the loss of SCD activity in B cells to support humoral immunity.
Figure 7. B Cell-Intrinsic SCD Activity Is Not Required for B Cell Development and Humoral Response In Vivo.
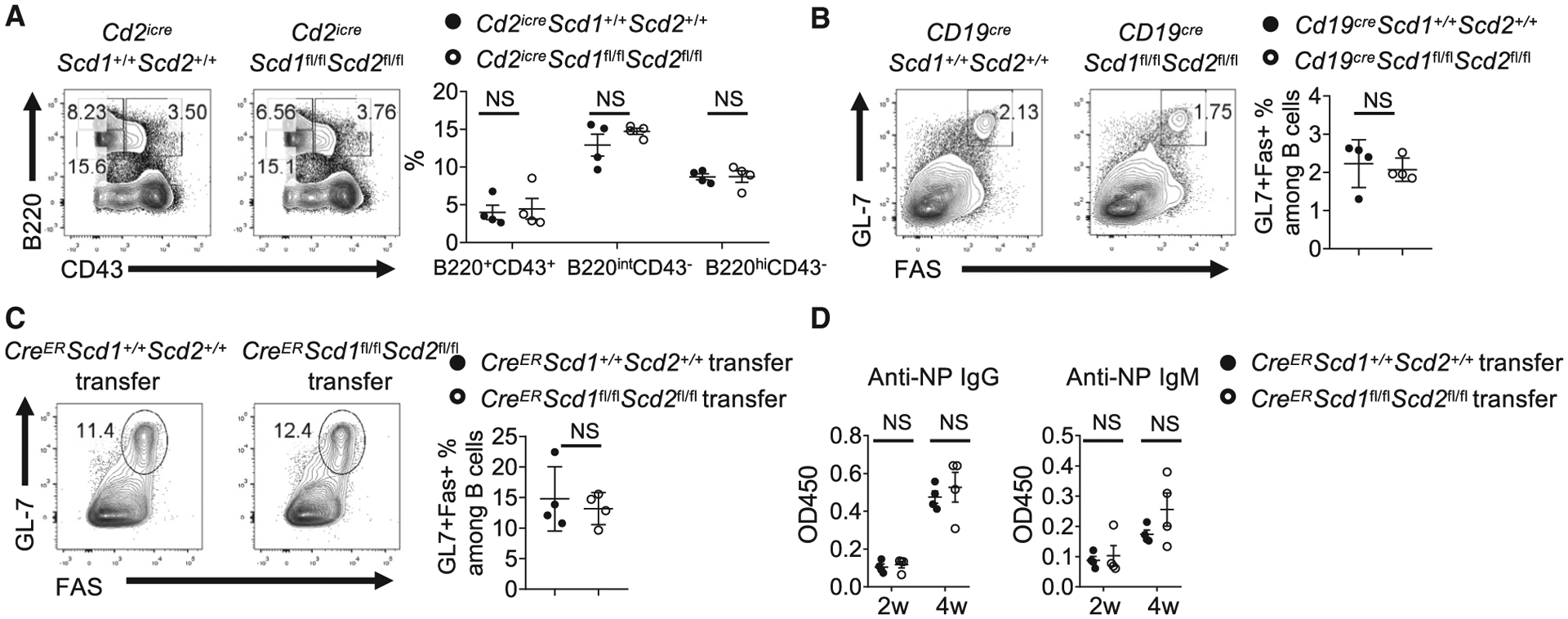
(A) Flow cytometry analysis of B cell development in bone marrow from WT and Cd2iCreScd1fl/flScd2fl/fl mice. Right: the frequencies of B220+CD43+ pro-B cells, B220intCD43− pre-B/immature B cells, and B220hiCD43− circulating mature B cells.
(B) WT and Cd19CreScd1fl/fl Scd2fl/fl mice were immunized with NP-OVA. Flow cytometry analysis of GC B cells. Right: the frequencies of GC B cells.
(C and D) B cells were purified from tamoxifen-treated CreERScd1+/+Scd2+/+ and CreERScd1fl/flScd2fl/fl mice. They were mixed with CD4 T cells purified from OT-II mice and WT C57BL/6 mice and transferred into Rag1−/− mice. The recipient mice were immunized with NP-OVA.
(C) Flow cytometry analysis of GC B cells in spleen. Right: the frequencies of GC B cells.
(D) ELISA measurements of serum anti-NP immunoglobulins at 2 and 4 weeks after first immunization, presented as absorbance at 450 nM (A450).
p values were calculated using Student’s t test (A–D). NS, not significant. Data were pooled from four (A and B) and represent two (C and D) independent experiments. Error bars represent SEM. See also Figure S6.
DISCUSSION
Despite the rapid progress in the immunometabolism field, our understanding of FA metabolism in adaptive immunity remains highly limited. Most previous research has focused on PUFA and its derivatives, including n-3 PUFA (Crouch et al., 2019; Gurzell et al., 2013; Kosaraju et al., 2017), prostaglandin E2 (Roper et al., 1995), and the pro-resolving mediator 17-hydroxydosa-hexaenoic acid (Ramon et al., 2012, 2014). The impact of other FA species, especially endogenously generated FAs, remains poorly defined. Our work offers a comprehensive study of the relationship between MUFA availability and humoral immunity. It reveals that murine and human B cells preferentially rely on MUFA to maintain their metabolic fitness and promote antibody production. SCD, which catalyzes endogenous MUFA generation, critically contributes to B cell development and activation. These observations are consistent with a recent finding that GC B cells preferentially use FAO rather than glycolysis (Weisel et al., 2020). However, we show that all FAs are not created equal for B cells. The quality of FA matters, and proliferating B cells preferentially use MUFA rather than SFA. Thus, our data suggest that the SCD may constitute a potential target for B cell-mediated diseases. Modulation of MUFA availability, through either dietary intervention or pharmaceutical targeting of SCD, may benefit B cell-dependent immunological diseases, including viral infection and certain autoimmunity.
Our results demonstrated that SCD-generated OA is required to sustain mTORC1 activity and metabolic fitness of B cells. mTORC1 activity is critical for B cell isotype switch (Raybuck et al., 2018), and mitochondrial metabolism supports B cell survival (Akkaya et al., 2018). Thus, our results link MUFA availability to B cell antibody generation and metabolism for the first time. In contrast to a recent study, we found no major defects in CD4+ T cell differentiation upon SCD inhibition, at least within our experimental schemes. The discrepancy could reflect the use of different SCD inhibitors and different immune challenge models (primary intranasal infection using PR8 strain in our study versus intraperitoneal immunization using X31 strain) (Son et al., 2020). Nevertheless, our results indicate that CD4+ T cells are less sensitive to SCD inhibition, and they may preferentially use different fuel sources, which warrant further investigation.
Notably, our data demonstrate that even though B cells are able to synthesize MUFA, loss of B cell-intrinsic SCDs can be compensated by extrinsic SCD activity, indicating that humoral immunity is supported by non-lymphoid cell-derived MUFAs. Although the source of the B cell-extrinsic SCD activity that sustains humoral immunity awaits future investigation, major metabolic organs expressing high levels of SCD, such as liver and adipose tissues, are potential candidates (Kaestner et al., 1989). Alternatively, stromal cells in lymphoid organs may supply MUFA to support B cells at the local microenvironment. Previous studies indicated that memory CD8+ T cells rely on intrinsically produced FAs to maintain their functions (O’Sullivan et al., 2014). In contrast, we show that B cells can use extrinsic source of MUFA to support their functions. Thus, B cells are nurtured by cell-extrinsic FA and targeting non-immune cells may be required to modulating MUFA availability and thus humoral immunity.
There are some major limitations of our study. MUFA likely contributes to a multitude of cellular functions, including membrane structure, ER stress, metabolism, proliferation, and survival. How MUFA coordinates these functions requires further investigation. Another limitation is that most of our metabolic assays were performed using in vitro activated bulk B cells because of current technical constraint even with a state-of-the-art metabolomics platform. In vitro activated B cells may not necessarily recapitulate their in vivo metabolic features. Future studies with more sensitive metabolomics technologies are required to tease out the metabolic requirement of FAs in B cell subsets in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author Hu Zeng (zeng.hu1@mayo.edu).
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated during this study are available at the Gene Expression Omnibus (accession number GSE162460).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human B cell
This study was conducted with approval from the institutional review boards of Mayo Clinic, Rochester. Peripheral blood mononuclear cells were isolated from apheresis collection of platelets. Briefly, blood was diluted 1:10 using PBS. The diluted blood was then overlaid with Ficoll-Paque PLUS density gradient medium. The gradient was centrifuged at 400 g with no brake for 25 min at room temperature. The interphase layer was collected, washed with PBS with 0.1% BSA/2 mM of EDTA. Naive B cells were enriched using Human naive B cell negative selection kit.
Mice
Scd floxed mice were from Dr. Makoto Miyazaki, University of Colorado School of Medicine. Mice were crossed with Cre-ERT2 (Zeng et al., 2013), Cd2-iCre (Zeng et al., 2018) or Cd19-Cre (Rickert et al., 1997) transgenic mice (Jackson Laboratory). C57BL/6 and Rag1−/− mice were purchased from the Jackson Laboratory. Spleen cells from GFP-LC3 reporter mice were a gift from Dr. Douglas Green, St. Jude Children’s Research Hospital. All mice were used at 2–4 months old. Both male and female mice were used. Mice of the same sex were used in a single experiment. For in vivo SCD inhibition experiments, animals were fed on chow containing SSI-4 (30 mg/kg) or otherwise equivalent control chow, which were gifts from Dr. John A. Copland, III. One day prior to immunization, mice were fed with SSI-4 and control chow for 1 week. Antigen for immunization was prepared by mixing 100 μg NP-OVA, 10% KAL(SO4)2 dissolved in PBS at a ratio of 1:1, together with 10 μg LPS at pH 7 (Zeng et al., 2016). Mice were immunized intraperitoneally. Transfer model were generated by transferring 5 × 106 B cells isolated from CreERScd1fl/flScd2fl/fl or CreERScd1+/+Scd2+/+ treated with tamoxifen for 4 consecutive days, mixed with 4 × 106 CD4 T and 1 × 106 OT-II transgenic T cells, into Rag1−/− mice. Three weeks after first immunization with NP-OVA/Alum, mice were boosted with NP-OVA/Alum. The mice were sacrificed one week after second immunization.
For influenza virus infection, influenza A/PR8 strain (60 pfu/mouse) were diluted in FBS-free DMEM media on ice and inoculated in anesthetized mice through intranasal route. Sera were collected before and two weeks after infection for fatty acid component measurement. The mice were fed with control chow or SSI-4 chow for 1 week following influenza infection before switching to regular diet. The mediastinal lymph nodes were analyzed for GC B cell formation and Tfh differentiation. Mice were bred and maintained under specific pathogen-free conditions in Department of Comparative Medicine of Mayo Clinic with Institutional Animal Care and Use Committee approval.
METHOD DETAILS
RNA-seq
RNA from isolated fresh B cells and activated B cells was extracted using a RNeasy Micro kit. After quality control, high quality total RNA was used to generate the RNA sequencing library. Paired-end RNA-seq reads were aligned to the mouse reference genome (mm10) using a spliced-aware reads mapper Tophat2 (v2.0.6) (Kim et al., 2013). Pre- and post-alignment quality controls, gene level raw read count and normalized read count (i.e., FPKM) were performed using RSeQC package (v2.3.6) with NCBI mouse RefSeq gene model (Wang et al., 2012). Differential gene expression analyses were conducted using edgeR (version 3.6.8) and the builtin “TMM” (trimmed mean of M-values) normalization method were used (Robinson et al., 2010). The criteria for selection of significant differentially expressed genes were: | log2 fold change | > = 1.0 and p value ≤ 0.001.
Immune cell Purification and Culture
Mouse B cells were isolated from spleen and peripheral lymph nodes using CD19 Microbeads or Mouse B cell Isolation Kit. B cells were stimulated with LPS (10 μg/mL) and recombinant IL-4 (10 ng/mL) with or without SSI-4, and proliferation was measured by dilution of CellTrace Violet proliferation dye. B cells were also stimulated with anti-IgM (10 μg/mL), anti-CD40 (10 μg/mL) and IL-4, or TLR ligand CpG (2.5 μM), recombinant IL-4 and IL-5 (10 ng/mL). To test the function of monounsaturated and saturated fatty acids, B cells were activated in presence of exogenous fatty acid-BSA conjugate. Palmitoleic acid, palmitic acid, and stearic acid were conjugated with fatty acid free BSA as previously described (Wang et al., 2011). Human B cells were activated with CpG OND2006 (2.5 μM), recombinant human cytokine IL-10 (50 μg/mL), IL-15 (10 ng/mL), IL-4 (10 ng/mL), IL-2 (10 unit/mL) and anti-human CD40 (1 μg/mL). Human B cell proliferation was measured by 3[H]-thymidine incorporation (1 μCi/mL).
Non-esterified free fatty acids (NEFAs) and Total Fatty Acid Composition
Fatty acids and total fatty acid composition were measured against a standard curve on a triple quadrupole mass spectrometer coupled with an Ultra Pressure Liquid Chromatography system (LC/MS) as previously described. Briefly, the cell pellets were spiked with internal standard prior to extraction with tert-Butyl Methyl Ether (MTBE). Roughly 25% of the sample was dried down, hydrolyzed, re-extracted and brought up in running buffer for the analysis of total fatty acid composition. The remaining portion of the extract was dried down and brought up in running buffer prior to injecting on the LC/MS for the NEFA measurement. Data acquisition was performed under negative electrospray ionization condition.
Mass spectrometer measurement of serum NEFA using liquid chromatography system
Fatty acids were measured against a standard curve on a Thermo Quantum Ultra triple quadrupole connected to a Waters Acquity Ultra high-pressure liquid chromatography system (LC/MS) as previously described. Briefly, 50 μL of serum was spiked with internal standard prior to extraction. The extracts were dried down and brought up in running buffer prior to injecting on the LC/MS. Data acquisition was performed under negative electrospray ionization condition (Persson et al., 2010).
NEFA isotopomer analysis
5 × 106 activated B cells were washed with PBS and cultured in glucose-free medium RPMI medium containing 10% dialyzed FBS and uniformly labeled [U-13C]-glucose (2 g/L) for 48 h. Cell pellets were lysed in 1 × PBS prior to lipid extraction. The extract was dried down and brought up in running buffer before underwent analysis on an Agilent 6550 iFunnel Q-TOF mass spectrometer/1290 Infinity liquid chromatographic system. A mixed standard containing 14 fatty acids was also run at the beginning and at the end of the sequence to generate retention time lock as well as unlabeled mass spectrum for each fatty acid. The mass spec was operating in negative electrospray ionization. Data was acquired in scan mode from 50–1700 m/z range. Data analysis was performed on Agilent Technologies software including Profinder, Mass Profiler Professional (MPP) and Vista Flux. The features extracted from the data files were aligned using Profinder and converted to a compound exchange file (CEF) format to import in the MPP. The list was filtered for frequency and abundance to identify features that are present in all samples and with high abundance. A library was created using mass, molecular formula, and retention time of these features and used in conjunction with the Vista Flux software to determine the presence of isotopologue in individual feature. Fatty acids displaying a presence of isotopic pattern were annotated using retention time lock, accurate mass, and the METLIN database with an error of 5 ppm.
[9,10-3H]-OA flux assay
OA oxidation was determined by measuring the detritiation of [9,10-3H]-OA. Briefly, Isolated B cells were activated with LPS/IL-4 for 48 h with or without exogenous OA, and then washed with fresh medium. The activated B cells were treated with 40 μM etomoxir for 1 h, and then incubated in 3 μCi of [9,10-3H]-OA for 4 h. The whole cell culture was mixed with 50 mL of 5 N HCL to stop the reaction in a microcentrifuge tube. The tube was then placed in 20 mL scintillation vials containing 0.5 mL water with the vial capped and sealed. 3H2O was separated from unmetabolized [9,10-3H]-OA by evaporation diffusion for 24 h at room temperature. A control sample with [9,10-3H]-OA containing medium alone (without B cells) was used to calculate background, which was subtracted from eventual scintillation reading.
[U-13C]-OA oxidation assay
OA oxidation activity was measured by monitoring the intracellular conversion rate of uniformly labeled 13C OA to [M+2, 4, 6]-labeled 13C citrate. B cells were activated with LPS/IL-4 for 48 h. The activated B cells were treated with 40 μM etomoxir for 1 h, and then incubated in 100 μM [U-13C]-OA conjugated to BSA for other 6 h. The extent of isotopic 13C labeling was calculated by summing the percentage intensities of isotopologues of [M + 2, 4, 6]-labeled citrate after natural-abundance correction as previously performed (Kurmi et al., 2018). The extent of isotopic 13C labeling in citrate was further divided by percent isotopic enrichment of intracellular [U-13C]-OA to determine the conversion rate of [U-13C]-oleic acid to [M + 2, 4, 6]-labeled citrate in cells. OA and citrate were detected by GC-MS as TBDMS derivatives: oleic acid (m/z 339), [U-13C]-OA (m/z 357), and citrate (m/z 459–465).
Metabolic Assays
The bioenergetic activities were measured using a Seahorse XFe96 Analyzer. Briefly, equal number of live B cells were seeded at 200,000 cells/well on Cell-Tak (Corning) coated XFe96 plate with fresh XF media (Seahorse XF RPMI medium containing 10 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate, PH 7.4). OCR was measured in the presence of Oligomycin (1.5 μM), FCCP (1.5 μM), and Rotenone (1 μM)/ Antimycin A (1 μM) in Mito Stress assay. For ECAR measurement, B cells were seeded in XFe96 plate with fresh Seahorse XF RPMI medium and treated with glucose (10 mM), Oligomycin (1.5 μM), and 2-DG (50 mM) in Glycolysis Stress assay.
Electron microscopy
Cells were fixed in Trumps fixative (pH 7.2) at 4°C overnight, spun down and the supernatant removed. They were re-suspended in 2% agarose which was allowed to cool and solidify. The cells in agarose were then post-fixed in 1% OsO4, dehydrated through a graded series of ethanol and embedded in Spurr resin. One hundred nm ultra-thin sections were mounted on 200-mesh copper grids, post-stained with lead citrate, and observed under a JEOL JEM-1400 transmission electron microscope at 80kV.
Immunoblotting
Immunoblot was performed as previously described (Zeng et al., 2013). The membrane was blocked with TBST (0.1% Tween 20) containing 5% BSA for 1 h at room temperature. Primary antibodies included: anti-p-S6 (Ser235/Ser236, D57.2.2E), anti-p-p70 S6 kinase (Thr389, 108D2), anti-LC3B; anti-AID (EK2 5G9), anti-β-actin (13E5), and anti-SCD2 (H-12). The band intensity of all the immunoblot was analyzed by ImageJ software.
Quantitative Real-time PCR
For mRNA analysis, total mRNA was isolated from mouse and human B cells by RNeasy Micro kit (QIAGEN), reverse transcribed from mRNA to cDNA for subsequent real-time PCR analysis. Scd1, Scd2, Fasn, Acaca, Chop, Atf4, Sqstm1 in mouse B cells and Scd, Fasn, Acaca expression in human B cells were with a Bio-Rad Realtime PCR system. β-actin expression was used as control. The primers information was provided in Table S2.
Flow Cytometry
Cells were stained in PBS containing 1% (w/v) bovine serum albumin on ice for 30 min, with anti-IgG1 (RMG1–1), anti-CD19 (ID3), anti-B220 (RA3–6B2), anti-CD4 (GK1.5), anti-CD8α (53–6.7), anti-CD25 (PC16), anti-GL7 (GL-7), anti-CD138 (281–2), anti-IgD (11–26c.2a), anti-CD95 (Jo2), anti-PD-1 (J43), anti-IgM (II/41), and anti-CXCR5 (2G8). Antigen specific GC response was detected with PE-conjugated anti-NP tetramer. Cell viability was assessed using the Fixable Dye Ghost 510. Phosflow staining was performed using Phosflow Fix/Perm kit (BD Biosciences). Flow cytometry was performed on the LSR II, LSR Fortessa (BD Biosciences) or Attune NxT (Thermo Fisher) cytometers, and analysis was performed using FlowJo software.
ELISA
For NP specific antibodies detection in sera, 96-well plates were coated with 2 μg/mL NP23-BSA in PBS overnight. Plates were washed twice (0.05% Tween 20 in PBS), blocked with 5% blocking protein for 1 h, and washed twice, and serially diluted serum samples were added for 1.5 h at 37°C. Plates were washed and horseradish peroxidase (HRP)-conjugated detection antibodies for IgG, IgM and IgG1 were added for 1 h, followed by reaction with tetramethylbenzidine (TMB) substrate. Similarly, antibodies specific to influenza A/PR8 strain in sera were measured with influenza virus coated plate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism (version 8). P values were calculated with Student’s t test, or one-way ANOVA (Tukey’s Post Hoc test) for multiple groups comparison. p < 0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgM | Jackson ImmunoResearch | Cat.#115-006-075; RRID:AB_2338474 |
| InVivoMab anti-mouse CD40 antibody | Bio × Cell | Cat.#BE0016–2; RRID:AB_1107647 |
| InVivoMab anti-human CD40 antibody | Bio × Cell | Cat.#BE0189; RRID: AB_10950314 |
| Rabbit polyclonal anti-p-S6 (Ser235/Ser236, D57.2.2E) | Cell Signaling | Cat.#4858S; RRID: AB_916156 |
| Rabbit polyclonal anti-p-p70 S6 kinase (Thr389, 108D2) | Cell Signaling | Cat.#9205S; RRID: AB_330944 |
| Rat anti-AID (EK2 5G9) | Cell Signaling | Cat.#4959; RRID: AB_10692771 |
| Rabbit polyclonal anti-LC3B | NOVUS | Cat.#NB600–1384 ; RRID: AB_669581 |
| Mouse anti-b-actin (13E5), | Sigma-Aldrich | Cat.#A5441; RRID: AB_476744 |
| Anti-SCD2 Antibody (H-12) | Santa Cruz | Cat.#sc-518034 |
| Alexa 488 anti-mouse IgG1 (RMG1–1) | Biolegend | Cat.# 406625 ; RRID: AB_2715988 |
| PE/Cy7 anti-mouse CD19 (ID3) | Biolegend | Cat.#115520 ; RRID: AB_313655 |
| Anti-mouse B220-BV605 (RA3–6B2) | Biolegend | Cat.#103244; RRID: AB_2563312 |
| FITC anti-mouse CD4 (GK1.5) | Biolegend | Cat.#100510; RRID: AB_312713 |
| APC-Cyanine7 Anti-Mouse CD8a (53–6.7) | Tonbo Biosciences | Cat.# 25–0081; RRID:AB_2621623 |
| FITC anti-mouse CD25 (PC61) | Tonbo Biosciences | Cat.# 35–0251; RRID:AB_2621685 |
| AF647 Anti-mouse CD95 (Jo2) | BD Biosciences | Cat.# 563647; RRID: AB_2738346 |
| BV711 Anti-mouse CD138 (Syndecan-1) | Biolegend | Cat.#142519; RRID:AB_2562571 |
| APC Anti-mouse IgD (11–26c.2a) | Biolegend | Cat.# 405714 ; RRID:AB_10643423 |
| PE-Cy7 anti-mouse PD1 (J43) | ThermoFisher | Cat.# 25-9985-82; RRID:AB_10853805 |
| eFluor 450 anti-mouse IgM (II/41) | ThermoFisher | Cat.# 48-5790-82; RRID: AB_2574073 |
| Pacific Blue anti-mouse/human GL-7 (GL-7) | Biolegend | Cat.# 144614; RRID:AB_2563292 |
| BV421 Anti-Bcl6 (K112–91) | BD Biosciences | Cat.# 563363; RRID:AB_2738159 |
| BV605 Anti-mouse CD25 (PC61) | Biolegend | Cat.#102036; RRID:AB_2563059 |
| FITC Anti-mouse IgA (mA-6E1) | ThermoFisher | Cat.#11-4204-82; RRID: AB_465221 |
| PE Anti-mouse CD43 (S7) | BD Biosciences | Cat.#553271; RRID: AB_394748 |
| APC anti-human CD80 (B7–1) (2D10.4) | Tonbo Biosciences | Cat.#20–0809; RRID: AB_2076147 |
| PE anti-human CD86 (B7–2) (IT2.2) | Tonbo Biosciences | Cat.#50–0869; RRID: AB_2621776 |
| Biotinylated anti-mouse CXCR5 (2G8) | BD Biosciences | Cat.#551960; RRID: AB_394301 |
| PE Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) | Cell Signaling | Cat.#5316S ; RRID: AB_10694989 |
| Mouse IgG-heavy and light chain Antibody HRP conjugated | Bethyl Laboratories | Cat.#A90–137P; RRID: AB_1211460 |
| Mouse IgM Antibody HRP conjugated | Bethyl Laboratories | Cat.#A90–101P; RRID: AB_67189 |
| Rat Anti-Mouse IgG1-HRP | SouthernBiotech | Cat.#1144–05; RRID:AB_2734757 |
| Goat Anti-Mouse IgG2c, Human/Bovine/Horse SP ads-HRP | SouthernBiotech | Cat.#1077–05; RRID:AB_2794452 |
| Bacterial and Virus Strains | ||
| influenza A/PR8 strain | Gift from Jie Sun, Mayo Clinic Rochester, USA | N/A |
| Biological Samples | ||
| PBMCs from healthy subjects | Mayo Clinic Blood Donor | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NP-OVAL (Ovalbumin) | BioSearch Technologies | Cat.#N-5051 |
| Aluminum potassium sulfate dodecahydrate (KAL(SO4)2.12H2O) | Sigma | Cat.#A6435 |
| Ficoll-Paque PLUS density gradient medium | GE Healthcare | Cat.#45-001-749 |
| SSI-4 and control diet | Gift from John A. Copland, III, Mayo Clinic Florida, USA | N/A |
| SSI-4, SCD inhibitor | Gift from John A. Copland, III, Mayo Clinic Florida, USA | N/A |
| A939572 | Sigma-Aldrich | Cat.#SML2356 |
| Stearoyl-CoA Desaturase 1 Inhibitor, MF438 | Sigma-Aldrich | Cat.# 569406 |
| LPS (Escherichia coli strain 055: B5) | Sigma-Aldrich | Cat.#L2637 |
| recombinant mouse IL-4 cytokine | Tonbo Bioscience | Cat.# 404-ML-010 |
| Recombinant Mouse IL-5 (Carrier-free) | Tonbo Bioscience | Cat.#21–8051 |
| recombinant human cytokine IL-10 | Peprotech | Cat.# 200–10 |
| recombinant human cytokine IL-4 | Biolegend | Cat.#574004 |
| recombinant human cytokine IL-15 | Peprotech | Cat.# 200–15 |
| recombinant human cytokine IL-2 | TECIN | Cat.#23–6019 |
| Oleic acid-Albumin from bovine serum | Sigma-Aldrich | Cat.#O3008 |
| Palmitoleic acid | NU-CHEK PREP, INC | Cat.#U-40-A |
| Palmitic acid | NU-CHEK PREP, INC | Cat.#N-16-A |
| Stearic acid | NU-CHEK PREP, INC | Cat.#N-18-A |
| Bovine Serum Albumin Fraction V, heat shock, fatty acid free | Sigma-Aldrich | Cat.#3117057001 |
| Thymidine, [methyl-3H] | PerkinElmer | Cat.#NET027W001MC |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories | Cat.#CLM-1396–2 |
| Oleic Acid, [9,10–3H(N)] | Perkin Elmer | Cat.# NET289001MC |
| [U-13C]-oleic acid | Larodan | Cat.# 78–1811 |
| (+)-Etomoxir (sodium salt) | Cayman | Cat.# 11969 |
| Cell-Tak Cell and Tissue Adhesive | Fisher Scientific | Cat.#354240 |
| Oligomycin | Sigma-Aldrich | Cat.#495455 |
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Sigma-Aldrich | Cat.#C2920 |
| Rotenone | Sigma-Aldrich | Cat.#R8875 |
| Antimycin A | Sigma-Aldrich | Cat.#A8674 |
| 2-deoxy-D-Glucose (2-DG) | Cayman Chemical | Cat.#14325 |
| Seahorse XF 1M Glucose solution | Agilent Technologies | Cat.#103577 |
| 7AAD | ThermoFisher | Cat.# A1310 |
| Fixable Dye Ghost 510 | Tonbo Bioscience | Cat.#13–0870 |
| CellTrace Violet proliferation dye | Thermo Fisher Scientific | Cat.# C34557 |
| Torin 1 | Cayman | Cat.#10997 |
| rapamycin (mTOR inhibitor) | LC Laboratories | Cat.#553210 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Fisher Scientific | Cat.#34580 |
| NP-PE (Phycoerythrin) | BioSearch Technologies | Cat.#N-5070–1 |
| NP(23)-BSA | BioSearch Technologies | Cat.#N-5050H |
| RT All-in-One Master Mix, 5X | Lamda Biotech | Cat.# G208–100 |
| 3-Methyladenine | Cayman | Cat.#13242 |
| Wortmannin | Cayman | Cat.# 10010591 |
| Tamoxifen | Cayman chemical | Cat.# 13258 |
| Critical Commercial Assays | ||
| RNeasy Micro Kit | QIAGEN | Cat.#74004 |
| Mouse CD19 Microbeads | Miltenyi | Cat.#130-052-201 |
| EasySep Mouse B Cell Isolation Kit | StemCell Technologies, Inc. | Cat.#19854 |
| EasySep Human naive B cell negative selection kit | StemCell Technologies, Inc. | Cat.# 17254 |
| Deposited Data | ||
| Raw and analyzed RNAseq data | This paper | GEO: GSE162460 |
| Experimental Models: Organisms/Strains | ||
| Mouse:C57BL/6 | Jackson Laboratory | Stock No:000664 |
| Mouse:Rag1−/− | Jackson Laboratory | Stock No:002216 |
| Mouse:Cre-ERT2 | Gift from Thomas Ludwig, Stanford University, USA | N/A |
| Mouse:Cd2-iCre | Jackson Laboratory | Stock No: 008520 |
| Mouse:Cd19-Cre | Jackson Laboratory | Stock No: 004126 |
| Mouse: Scd1flox/floxScd2flox/flox | Gift from Makoto Miyazaki, University of Colorado, USA | N/A |
| Mouse:OT-II transgenic | Gift from Jie Sun, Mayo Clinic Rochester, USA | N/A |
| Mouse:GFP-LC3 | Gift from Douglas Green, St. Jude Children’s Research Hospital, USA | N/A |
| Oligonucleotides | ||
| CpG ODN2006 5′-TCGTCGTTTTGTCG TTTTGTCGTT-3′ | IDT | N/A |
| All primers listed in Table S2 | This paper | N/A |
| Software and Algorithms | ||
| Rstudio | Rstudio | https://rstudio.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Flowjo v10 | FlowJo | https://www.flowjo.com/ |
| Prism 8 | GraphPad | https://www.graphpad.com |
| Wave | Agilent Technologies | https://www.agilent.com/en/product/cell-analysis/real-time-cell-metabolic-analysis/xf-software/seahorse-wave-desktop-software-740897 |
Highlights.
B cell activation increases SCD activity and MUFA content
MUFA maintains B cell metabolic fitness and promotes proliferation and class switch
SCD activity is required for anti-viral humoral immunity in vivo
B cell-extrinsic SCD activity can compensate intrinsic B cell SCD loss
ACKNOWLEDGMENTS
We thank Dr. Douglas Green at St. Jude Children’s Research Hospital for spleen cells from GFP-LC3 mice. We thank Dr. Michael Jensen for his expert advice on FA metabolism. This work was partly supported by National Institutes of Health (NIH) grants R01 CA225680 (to T.H.), R01 AG047156, R01 AI112844, and R01 AI147394 (to J.S.) and a Discovery Science Award (93059065) from the Center for Biomedical Discovery at Mayo Clinic (to H.Z.). The Mayo Clinic Metabolomics Core is supported by NIH grant U24DK100469.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108601.
DECLARATION OF INTERESTS
H.Z. and J.A.C. have a pending patent application regarding use of the SCD inhibitor SSI-4.
REFERENCES
- Akkaya M, Traba J, Roesler AS, Miozzo P, Akkaya B, Theall BP, Sohn H, Pena M, Smelkinson M, Kabat J, et al. (2018). Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol 19, 871–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALJohani AM, Syed DN, and Ntambi JM (2017). Insights into stearoyl-CoA desaturase-1 regulation of systemic metabolism. Trends Endocrinol. Metab 28, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alwarawrah Y, Kiernan K, and MacIver NJ (2018). Changes in nutritional status impact immune cell metabolism and function. Front. Immunol 9, 1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothby M, and Rickert RC (2017). Metabolic regulation of the immune humoral response. Immunity 46, 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR, and Rathmell JC (2014). Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J. Immunol 192, 3626–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SH, Ahn AK, Bhargava P, Lee CH, Eischen CM, McGuinness O, and Boothby M (2011). Glycolytic rate and lymphomagenesis depend on PARP14, an ADP ribosyltransferase of the B aggressive lymphoma (BAL) family. Proc. Natl. Acad. Sci. U S A 108, 15972–15977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch MJ, Kosaraju R, Guesdon W, Armstrong M, Reisdorph N, Jain R, Fenton J, and Shaikh SR (2019). Frontline science: a reduction in DHA-derived mediators in male obesity contributes toward defects in select B cell subsets and circulating antibody. J. Leukoc. Biol 106, 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, and Kioussis D (2003). Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur. J. Immunol 33, 314–325. [DOI] [PubMed] [Google Scholar]
- Dufort FJ, Bleiman BF, Gumina MR, Blair D, Wagner DJ, Roberts MF, Abu-Amer Y, and Chiles TC (2007). Cutting edge: IL-4-mediated protection of primary B lymphocytes from apoptosis via Stat6-dependent regulation of glycolytic metabolism. J. Immunol 179, 4953–4957. [DOI] [PubMed] [Google Scholar]
- Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, Richardson AD, Seyfried TN, and Chiles TC (2014). Glucose-dependent de novo lipogenesis in B lymphocytes: a requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J. Biol. Chem 289, 7011–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagarasan S, Kawamoto S, Kanagawa O, and Suzuki K (2010). Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol 28, 243–273. [DOI] [PubMed] [Google Scholar]
- Fritsche KL (2015). The science of fatty acids and inflammation. Adv. Nutr 6, 293S–301S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gally F, Kosmider B, Weaver MR, Pate KM, Hartshorn KL, and Oberley-Deegan RE (2013). FABP5 deficiency enhances susceptibility to H1N1 influenza A virus-induced lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol 305, L64–L72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurzell EA, Teague H, Harris M, Clinthorne J, Shaikh SR, and Fenton JI (2013). DHA-enriched fish oil targets B cell lipid microdomains and enhances ex vivo and in vivo B cell function. J. Leukoc. Biol 93, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertzel AV, Smith LA, Berg AH, Cline GW, Shulman GI, Scherer PE, and Bernlohr DA (2006). Lipid metabolism and adipokine levels in fatty acid-binding protein null and transgenic mice. Am. J. Physiol. Endocrinol. Metab 290, E814–E823. [DOI] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. U S A 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, and Bernlohr DA (2015). Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat. Rev. Endocrinol 11, 592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GM, Jiang QH, Cai C, Qu M, and Shen W (2015). SCD1 negatively regulates autophagy-induced cell death in human hepatocellular carcinoma through inactivation of the AMPK signaling pathway. Cancer Lett. 358, 180–190. [DOI] [PubMed] [Google Scholar]
- Iwata TN, Ramírez JA, Tsang M, Park H, Margineantu DH, Hockenbery DM, and Iritani BM (2016). Conditional disruption of raptor reveals an essential role for mTORC1 in B cell development, survival, and metabolism. J. Immunol 197, 2250–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DD, Gaudette BT, Wilmore JR, Chernova I, Bortnick A, Weiss BM, and Allman D (2016). mTOR has distinct functions in generating versus sustaining humoral immunity. J. Clin. Invest 126, 4250–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner KH, Ntambi JM, Kelly TJ Jr., and Lane MD (1989). Differentiation-induced gene expression in 3T3-L1 preadipocytes. A second differentially expressed gene encoding stearoyl-CoA desaturase. J. Biol. Chem 264, 14755–14761. [PubMed] [Google Scholar]
- Kedia-Mehta N, and Finlay DK (2019). Competition for nutrients and its role in controlling immune responses. Nat. Commun 10, 2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, and Guan KL (2015). mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest 125, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaraju R, Guesdon W, Crouch MJ, Teague HL, Sullivan EM, Karlsson EA, Schultz-Cherry S, Gowdy K, Bridges LC, Reese LR, et al. (2017). B cell activity is impaired in human and mouse obesity and is responsive to an essential fatty acid upon murine influenza infection. J. Immunol 198, 4738–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsari C, Ali AH, Mundi MS, and Jensen MD (2013). Measuring plasma fatty acid oxidation with intravenous bolus injection of 3H- and 14C-fatty acid. J. Lipid Res 54, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurmi K, Hitosugi S, Wiese EK, Boakye-Agyeman F, Gonsalves WI, Lou Z, Karnitz LM, Goetz MP, and Hitosugi T (2018). Carnitine palmitoyltransferase 1A has a lysine succinyltransferase activity. Cell Rep. 22, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Heffington L, Jellusova J, Nam KT, Raybuck A, Cho SH, Thomas JW, Rickert RC, and Boothby M (2013). Requirement for Rictor in homeostasis and function of mature B lymphoid cells. Blood 122, 2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Kim YC, and Ntambi JM (2001a). A lipogenic diet in mice with a disruption of the stearoyl-CoA desaturase 1 gene reveals a stringent requirement of endogenous monounsaturated fatty acids for triglyceride synthesis. J. Lipid Res 42, 1018–1024. [PubMed] [Google Scholar]
- Miyazaki M, Man WC, and Ntambi JM (2001b). Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J. Nutr 131, 2260–2268. [DOI] [PubMed] [Google Scholar]
- Miyazaki M, Flowers MT, Sampath H, Chu K, Otzelberger C, Liu X, and Ntambi JM (2007). Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 6, 484–496. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132. [DOI] [PubMed] [Google Scholar]
- Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, Song Y, Cohen P, Friedman JM, and Attie AD (2002). Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. U S A 99, 11482–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, et al. (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara Y, Itakura E, Kono N, Mizushima N, Arai H, Nara A, Mizukami T, and Yamamoto A (2014). Stearoyl-CoA desaturase 1 activity is required for autophagosome formation. J. Biol. Chem 289, 23938–23950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson XM, Blachnio-Zabielska AU, and Jensen MD (2010). Rapid measurement of plasma free fatty acid concentration and isotopic enrichment using LC/MS. J. Lipid Res 51, 2761–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon S, Gao F, Serhan CN, and Phipps RP (2012). Specialized proresolving mediators enhance human B cell differentiation to antibody-secreting cells. J. Immunol 189, 1036–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon S, Baker SF, Sahler JM, Kim N, Feldsott EA, Serhan CN, Martínez-Sobrido L, Topham DJ, and Phipps RP (2014). The specialized proresolving mediator 17-HDHA enhances the antibody-mediated immune response against influenza virus: a new class of adjuvant? J. Immunol 193, 6031–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel-Moreno J, Carragher DM, Misra RS, Kusser K, Hartson L, Moquin A, Lund FE, and Randall TD (2008). B cells promote resistance to heterosubtypic strains of influenza via multiple mechanisms. J. Immunol 180, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybuck AL, Cho SH, Li J, Rogers MC, Lee K, Williams CL, Shlomchik M, Thomas JW, Chen J, Williams JV, and Boothby MR (2018). B cell-intrinsic mTORC1 promotes germinal center-defining transcription factor gene expression, somatic hypermutation, and memory B cell generation in humoral immunity. J. Immunol 200, 2627–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickert RC, Roes J, and Rajewsky K (1997). B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25, 1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RL, Brown DM, and Phipps RP (1995). Prostaglandin E2 promotes B lymphocyte Ig isotype switching to IgE. J. Immunol 154, 162–170. [PubMed] [Google Scholar]
- Rytter MJ, Kolte L, Briend A, Friis H, and Christensen VB (2014). The immune system in children with malnutrition-a systematic review. PLoS ONE 9, e105017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegemund S, Shepherd J, Xiao C, and Sauer K (2015). hCD2-iCre and Vav-iCre mediated gene recombination patterns in murine hematopoietic cells. PLoS ONE 10, e0124661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YM, Cheon IS, Goplen NP, Dent AL, and Sun J (2020). Inhibition of stearoyl-CoA desaturases suppresses follicular help T- and germinal center B-cell responses. Eur. J. Immunol 50, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SH, Shui G, Zhou J, Shi Y, Huang J, Xia D, Wenk MR, and Shen HM (2014). Critical role of SCD1 in autophagy regulation via lipogenesis and lipid rafts-coupled AKT-FOXO1 signaling pathway. Autophagy 10, 226–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Roemeling CA, Caulfield TR, Marlow L, Bok I, Wen J, Miller JL, Hughes R, Hazlehurst L, Pinkerton AB, Radisky DC, et al. (2017). Accelerated bottom-up drug design platform enables the discovery of novel stearoyl-CoA desaturase 1 inhibitors for cancer therapy. Oncotarget 9, 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Waters LR, Ahsan FM, Wolf DM, Shirihai O, and Teitell MA (2018). Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience 5, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Wang D, Topczewski F, and Pagliassotti MJ (2006). Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab 291, E275–E281. [DOI] [PubMed] [Google Scholar]
- Weisel FJ, Mullett SJ, Elsner RA, Menk AV, Trivedi N, Luo W, Wikenheiser D, Hawse WF, Chikina M, Smita S, et al. (2020). Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol 21, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Yang K, Cloer C, Neale G, Vogel P, and Chi H (2013). mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature 499, 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, Cloer C, Kishton RJ, Gao X, Youngblood B, et al. (2016). mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity 45, 540–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Yu M, Tan H, Li Y, Su W, Shi H, Dhungana Y, Guy C, Neale G, Cloer C, et al. (2018). Discrete roles and bifurcation of PTEN signaling and mTORC1-mediated anabolic metabolism underlie IL-7-driven B lymphopoiesis. Sci. Adv 4, eaar5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at the Gene Expression Omnibus (accession number GSE162460).