

SUMMARY
Congenital hydrocephalus (CH), featuring markedly enlarged brain ventricles, is thought to arise from failed cerebrospinal fluid (CSF) homeostasis and is treated with lifelong surgical CSF shunting with substantial morbidity. CH pathogenesis is poorly understood. Exome sequencing of 125 CH trios and 52 additional probands identified three genes with significant burden of rare damaging de novo or transmitted mutations: TRIM71 (p = 2.15 × 10−7), SMARCC1 (p = 8.15 × 10−10), and PTCH1 (p = 1.06 × 10−6). Additionally, two de novo duplications were identified at the SHH locus, encoding the PTCH1 ligand (p = 1.2 × 10−4). Together, these probands account for ~10% of studied cases. Strikingly, all four genes are required for neural tube development and regulate ventricular zone neural stem cell fate. These results implicate impaired neurogenesis (rather than active CSF accumulation) in the pathogenesis of a subset of CH patients, with potential diagnostic, prognostic, and therapeutic ramifications.
In Brief
Congenital hydrocephalus (CH) is a major cause of childhood morbidity and mortality, affecting 1 in 1,000 live births and representing up to 3% of all pediatric hospital charges. Using data from the largest CH exome sequencing study to date, Furey et al. identify four genes (TRIM71, SMARCC1, PTCH1, and SHH) not previously implicated in CH. Remarkably, all four genes regulate ventricular zone neural stem cell fate and, together, explain ~10% of CH cases. These findings implicate impaired neurogenesis in pathogenesis of a significant number of CH patients, with potential diagnostic, prognostic, and therapeutic ramifications.
INTRODUCTION
Hydrocephalus has been defined as the active, progressive distension of the cerebral ventricular system resulting from inadequate passage of cerebrospinal fluid (CSF) from its point of production to its point of absorption (Rekate, 2008). This mechanism is evident in secondary hydrocephalus associated with brain tumors, infection, or hemorrhage, in which intracranial pressure is often elevated (Kahle et al., 2016). However, when infantile hydrocephalus occurs without a known antecedent, it is classified as primary or congenital hydrocephalus (CH) (Tully and Dobyns, 2014). CH can occur in the absence of obstruction to CSF flow (communicating hydrocephalus) or with complete/partial intraventricular obstruction (non-communicating hydrocephalus), most often due to aqueductal stenosis. Cases of communicating CH with normal or low intracranial pressure (Bret and Chazal, 1995; Tully and Dobyns, 2014) raise the question of whether the development of ventriculomegaly is a primary active process or a secondary passive process.
CH is a major cause of childhood morbidity and mortality, affecting 1 in 1,000 live births (Munch et al., 2012; Simon et al., 2008) and costing the U.S. health care system alone $2 billion annually (Shannon et al., 2011; Simon et al., 2008). Current therapy consists of catheter-based CSF shunting and endoscopic approaches, surgeries with high rates of failure and morbidity (Kahle et al., 2016). Significant gaps in our understanding of the molecular pathophysiology of CH impede the development of preventive, diagnostic, and therapeutic measures (McAllister et al., 2015).
Few bona fide human CH-causing genes have been identified (Kousi and Katsanis, 2016). These include L1CAM mutation in X-linked hydrocephalus and aqueductal stenosis (OMIM# 307000; Rosenthal et al., 1992), which constitutes up to 3% of CH, and rare recessive mutations in MPDZ (OMIM# 603785; Al-Dosari et al., 2013), CCDC88C (OMIM# 236600; Ekici et al., 2010), EML1, and WDR81 (Shaheen et al., 2017). These genes affect multiple cellular processes, confounding efforts to formulate a uniform paradigm of CH pathophysiology (Kousi and Katsanis, 2016). While ~40% of all CH cases are predicted to have a genetic etiology (Zhang et al., 2006), mutations in currently identified genes account for less than 5% of primary CH cases (Adle-Biassette et al., 2013; Haverkamp et al., 1999).
The sporadic nature of most CH cases limits the utility of traditional genetic approaches. This has motivated whole-exome sequencing (WES) of large patient cohorts, searching for genes mutated in affected subjects more often than expected by chance. This approach has proven powerful in other neurodevelopmental disorders (Deciphering Developmental Disorders Study, 2015, 2017), including brain malformations (Barak et al., 2011; Bilgüvar et al., 2010), epilepsy (Allen et al., 2013), craniosynostosis (Timberlake et al., 2016, 2017), autism (Awadalla et al., 2010; O’Roak et al., 2011; Neale et al., 2012; Sanders et al., 2012; Krumm et al., 2015), and others. We hypothesized that the apparent sporadic occurrence of CH may reflect, in some cases, damaging de novo mutations and/or transmitted mutations with incomplete penetrance.
RESULTS
Cohort Characteristics and Whole-Exome Sequencing
We recruited 177 probands with non-L1CAM-mutated CH in which hydrocephalus was the predominant phenotypic feature (see STAR Methods). All probands had undergone surgery for CSF diversion. The cohort included 125 parent-offspring sporadic CH trios, 47 singleton cases, and 5 multiplex kindreds (see STAR Methods; Table S1). 88 probands had communicating hydrocephalus and 89 had aqueductal stenosis. All study protocols were approved by the Yale Human Research Protection Program (HRPP) Institutional Review Board.
DNA was isolated, and WES was performed (Timberlake et al., 2016). 95.9% of targeted bases had 8 or more independent reads, and 92.7% had 15 or more (Table S2; Figure S1). 1,789 control trios that comprised unaffected siblings from the Simons simplex autism cohort were sequenced on the same platform and analyzed in parallel (Krumm et al., 2015). Variants were called using the Genome Analysis Toolkit (GATK) Haplotype Caller (McKenna et al., 2010; Van der Auwera et al., 2013), and allele frequencies were annotated in the Exome Aggregation Consortium (ExAC) and gnomAD databases (Lek et al., 2016; see STAR Methods). TrioDeNovo was used to identify de novo mutations, and MetaSVM was used to infer the impact of missense mutations (Dong et al., 2015). Direct Sanger sequencing of PCR amplicons containing the mutation verified variants in genes of interest.
We identified mutations in known CH genes in four probands, including one proband with compound heterozygous mutations in MPDZ, the cause of autosomal recessive (AR) non-syndromic hydrocephalus type 2 (OMIM# 615219; Al-Dosari et al., 2013); two male probands with transmitted D-mis mutations in FLNA, the cause of X-linked dominant (XLD) periventricular heterotopia (OMIM# 300049; Kamuro and Tenokuchi, 1993; Sheen et al., 2004); and one proband with compound heterozygous mutation in CRB2 (Hydro122), the cause of a AR ventriculomegaly with cystic kidney disease (OMIM# 219730; Slavotinek et al., 2015).
Global Analysis of Burden of De Novo Mutation
We determined a de novo mutation rate of 1.4 × 10−8 per base pair, with 1.09 de novo coding region mutations per proband (Table 1), consistent with expectation and previous work (Homsy et al., 2015; Timberlake et al., 2017; Ware et al., 2015). The burden of de novo mutations in the control cohort was similar (Table S3; Figure S2).
Table 1.
Observed and Expected De Novo Mutation Rates in 125 Probands with Congenital Hydrocephalus and CH Cases with Aqueductal Stenosis for Variant Classes by Phenotype and High Brain Expression
| Observed | Expected | |||||
|---|---|---|---|---|---|---|
| n | Rate | n | Rate | Enrichment | P | |
| Hydro (125) | ||||||
| All | 137 | 1.10 | 139.7 | 1.12 | 0.98 | 0.60 |
| Synonymous | 32 | 0.26 | 39.7 | 0.32 | 0.81 | 0.91 |
| Missense | 89 | 0.71 | 87.8 | 0.70 | 1.01 | 0.46 |
| D-Mis | 16 | 0.13 | 16.5 | 0.13 | 0.97 | 0.58 |
| LOF | 16 | 0.13 | 12.3 | 0.10 | 1.30 | 0.18 |
| Protein-Altering | 105 | 0.84 | 100.0 | 0.80 | 1.05 | 0.32 |
| Damaging | 32 | 0.26 | 28.7 | 0.23 | 1.11 | 0.30 |
| Hydro (125) - High Brain Expressed | ||||||
| All | 48 | 0.38 | 36.9 | 0.30 | 1.30 | 0.04a |
| Synonymous | 12 | 0.10 | 10.3 | 0.08 | 1.16 | 0.34 |
| Missense | 30 | 0.24 | 23.2 | 0.19 | 1.29 | 0.10 |
| D-Mis | 5 | 0.04 | 4.4 | 0.04 | 1.14 | 0.45 |
| LOF | 6 | 0.05 | 3.4 | 0.03 | 1.78 | 0.13 |
| Protein-Altering | 36 | 0.29 | 26.5 | 0.21 | 1.36 | 0.04a |
| Damaging | 11 | 0.09 | 7.8 | 0.06 | 1.42 | 0.16 |
| Hydro + AS (66) | ||||||
| All | 78 | 1.18 | 73.8 | 1.12 | 1.06 | 0.33 |
| Synonymous | 22 | 0.33 | 20.9 | 0.32 | 1.05 | 0.44 |
| Missense | 47 | 0.71 | 46.3 | 0.70 | 1.01 | 0.48 |
| D-Mis | 12 | 0.18 | 8.7 | 0.13 | 1.38 | 0.17 |
| LOF | 9 | 0.14 | 6.5 | 0.10 | 1.39 | 0.21 |
| Protein-Altering | 56 | 0.85 | 52.8 | 0.80 | 1.06 | 0.35 |
| Damaging | 21 | 0.32 | 15.2 | 0.23 | 1.38 | 0.09 |
| Hydro + AS (66) - High Brain Expressed | ||||||
| All | 30 | 0.45 | 19.5 | 0.30 | 1.54 | 0.01a |
| Synonymous | 9 | 0.14 | 5.5 | 0.08 | 1.65 | 0.10 |
| Missense | 17 | 0.26 | 12.2 | 0.19 | 1.39 | 0.11 |
| D-Mis | 4 | 0.06 | 2.3 | 0.04 | 1.72 | 0.21 |
| LOF | 4 | 0.06 | 1.8 | 0.03 | 2.25 | 0.11 |
| Protein-Altering | 21 | 0.32 | 14.0 | 0.21 | 1.50 | 0.04a |
| Damaging | 8 | 0.12 | 4.1 | 0.06 | 1.95 | 0.05a |
n, the number of de novo mutations; Rate, the number of de novo mutations per individual; Enrichment, ratio of observed to expected number of mutations; Hydro, congenital hydrocephalus; High Brain Expression, top quartile of expression; AS, aqueductal stenosis; Missense, tolerated missense mutations as predicted by MetaSVM; D-Mis, damaging missense mutations as predicted by MetaSVM; LOF, loss of function denotes premature termination, frameshift, or splice site mutations; Damaging, D-miss and LOF mutations.
p value ≤ 0.05
We compared the observed and expected number of de novo mutations in all genes and among genes in the top quartile of brain expression at embryonic day 9.5 (E9.5; see STAR Methods). Protein-altering de novo mutations were significantly enriched over expectation in the latter group, contributing to an estimated 8% of cases (Table 1). Controls showed no enrichment of de novo mutations in any gene class (Table S3).
Five genes contained two or more protein-altering de novo mutations: TRIM71, SMARCC1, PTCH1, PLOD2, and SGSM3 (Table S4). The first three genes have a probability of loss-of-function (LOF) intolerance (pLI) score ≥ 0.99; the latter two genes have pLIs of zero (Lek et al., 2016). The probability of finding five genes with two or more protein-altering de novo mutations in a cohort this size by chance is very low (p = 1.1 × 10−4). Four of these genes were in the upper quartile of expression in the developing brain. The probability of finding two or more protein-altering de novo mutations in four such genes in our cohort is very low (p = 1.1 × 10−4; Table S5).
Recurrent De Novo and Transmitted Mutations in TRIM71
LIN41/TRIM71, encoding lineage variant 41 (LIN41)/tripartite motif 71 (“TRIM71”) (Reinhart et al., 2000; Slack et al., 2000), harbored three novel de novo mutations, including the identical heterozygous de novo p.Arg608His mutation in two unrelated probands with prenatally diagnosed communicating hydrocephalus (Hydro101–1 and Hydro102–1) (Figure 1; Table S6). During the course of this study, Hydro102–1 became pregnant and transmitted the p.Arg608His mutation to her son, who was prenatally diagnosed with communicating hydrocephalus (Figure 1; Figure S5). A third de novo TRIM71 mutation (p.Arg796His) was identified in Hydro100–1, who had prenatally diagnosed communicating CH (Hydro100–1). All CH patients harboring TRIM71 mutations underwent surgical CSF shunting at birth because of extreme ventriculomegaly. The probability of finding three or more protein-altering de novo mutations in TRIM71 by chance in a cohort of this size is 2.15 × 10−7, surpassing genome-wide significance (Table 2) (Ware et al., 2015). TRIM71 is highly intolerant to LOF mutation (pLI = 0.99) and missense variation (Z score = 5.69) (Lek et al., 2016). With 105 protein-altering de novo missense mutations in our cohort, the probability of seeing any instances of the identical mutation at any position in the coding region by chance is very remote (p = 5.24 × 10−4).
Figure 1. Recurrent, Identical De Novo Mutations in LIN-41/TRIM71 Encoding the let-7 miRNA Target TRIM71.

(A) Representative sagittal (left) and axial (right) brain magnetic resonance images of CH probands Hydro100–1 and Hydro102–5 show communicating hydrocephalus.
(B) Pedigrees with Sanger-verified mutated bases (red) and the corresponding wild-type bases marked on the chromatograms.
(C) Structural modeling of TRIM71 mutation impact. The positively charged guanidinium side chain of p.Arg796 interacts with the negatively charged sugar-phosphate backbone of target RNAs to aid in maintaining the spatial position of the nucleic acid. Mutation of this p.Arg796 residue to the imidazole ring of histidine (ΔΔG = 2.0 kcal/mol) is predicted to disrupt this interaction (right). The side chain of p.Arg608 makes hydrogen bonds with uracil in target RNAs. The p.Arg608His mutation (ΔΔG = 1.6 kcal/mol) is predicted to disrupt these hydrogen bonds (left).
(D) Mapping of TRIM71 mutations. p.Arg608His and p.Arg796His affect conserved residues in the 16th position of the respective 1st and 5th blades of TRIM71’s NHL domain, which mediates the binding of target RNA.
(E) In situ hybridization of wild-type E12.5 and adult mouse brains for Trim71 showing signals in the ciliated neuroepithelium and ventricular zone (V, ventricle; arrow, neuroepithelium at E12.5 and ependymal layer in adulthood; 2.5× and 10× magnification).
Table 2.
Meta-analysis of Protein-Altering De Novo Mutations and Loss-of-Function Heterozygous Mutations in Probands for Genes with Multiple De Novo Mutations
| Gene | De Novo Mutations | Transmitted Loss-of-Function (LOF) | Meta p value | pLI | High Brain Expression Rank | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| # observed | # expected | enrichment | p value | # observed | # expected | enrichment | Binomial p value | ||||
| SMARCC1 | 2 | 2.3 × 10−3 | 861.7 | 2.7 × 10−6a | 3 | 4.3 × 10−2 | 70.5 | 1.2 × 10−5 | 8.2 × 10−10a | 1.0 | 97.4 |
| PTCH1 | 2 | 1.5 × 10−3 | 1374.3 | 1.1 × 10−6a | 1 | 3.8 × 10−2 | 26.1 | 3.8 × 10−2 | 7.2 × 10−7a | 1.0 | 93.2 |
| TRIM71 | 3 | 1.1 × 10−2 | 274.8 | 2.2 × 10−7a | 0 | 2.5 × 10−2 | 0 | 1 | 3.5 × 10−6 | 1.0 | 74.6 |
| PLOD2 | 2 | 3.5 × 10−3 | 580.2 | 5.9 × 10−6 | 0 | 7.8 × 10−2 | 0 | 1 | 7.7 × 10−5 | 0 | 82.8 |
| SGSM3 | 2 | 8.6 × 10−3 | 232.2 | 3.7 × 10−5 | 0 | 6.9 × 10−2 | 0 | 1 | 4.1 × 10−4 | 0 | 67.4 |
Meta-analysis performed by combining the p values from protein-altering de novo mutations and LOF heterozygous mutations using the Fisher’s method with 4 degrees of freedom.
p values surpassing the Bonferroni multiple testing correction (2.6 × 10−6, 0.05/18,989) for p values tabulated by either de novo, heterozygous, or meta-analysis. Of note, the denovolyzeR p value of damaging SMARCC1 mutations was calculated using 126 case-parent trios given the sporadic de novo mutation occurs in the proband’s father (Hydro106–3).
TRIM71 is the homolog of C. elegans Lin-41, a target of the let-7 (lethal 7) microRNA (miRNA) in the heterochronic pathway that regulates stem cell fate (Ecsedi and Grosshans, 2013). TRIM71, like Lin-41, mediates post-transcriptional silencing of mRNAs via direct interactions of its NHL domain and the 5′ or 3′ UTRs of target genes (Aeschimann et al., 2017; Ecsedi and Grosshans, 2013; Slack and Ruvkun, 1998; Vella et al., 2004). TRIM71 also contains ubiquitin ligase activity conferred by its RING domain (Nguyen et al., 2017). The NHL domain in TRIM71 consists of six repeats, each 40–50 residues long, that jointly comprise a barrel-like six-bladed β-propeller (Loedige et al., 2015). The de novo p.Arg608His and p.Arg796His mutations are at homologous positions in different blades of NHL domains. Arginines at these positions are conserved among orthologs from H. sapiens to C. elegans and reside in RPQGV motifs (Figure 1). The NHL domain of TRIM71 is the closest homolog of the NHL domain of D. melanogaster Brat (Loedige et al., 2013, 2014), which has been co-crystallized bound to a target RNA (Loedige et al., 2015). In this structure, the amino groups of side chains of the mutated arginines form hydrogen bonds with either the phosphate backbone (p.Arg796) or a uracil base (p.Arg608) of target RNA (Figure 1; Figure S3). These interactions are predicted to be altered by histidine substitution. While these mutations were predicted as tolerated by MetaSVM, they are predicted as damaging by 43 of 45 other predicting algorithms, including the very conservative MPC-D algorithm (see STAR Methods; Samocha et al., 2017). The recurrence of the identical de novo mutation and the fact that the third de novo mutation occurs at a homologous position in a different NHL domain suggest that TRIM71 mutations may not be simple LOF mutations.
In situ hybridization in E12.5 mouse brain (Figure 1; Figure S4) revealed abundant Trim71 expression in the neuroepithelium and ventricular zone (Cuevas et al., 2015; Maller Schulman et al., 2008). Analogous to its heterochronic expression in C. elegans (Kanamoto et al., 2006; Slack et al., 2000), Trim71 expression significantly decreases during development (Figure 1) (Schulman et al., 2005; Yu et al., 2010). Trim71 is highly expressed in neural progenitor cells (NPCs) of early mouse embryos (until E11.5) but declines secondary to increased expression of let-7 and mir-125 miRNAs as neural differentiation proceeds (Chen et al., 2012; Schulman et al., 2005). Trim71 deletion in mice results in exencephaly and embryonic lethality (failure of closure of the cephalic end of the neural tube) by E10 (Maller Schulman et al., 2008). Trim71 maintains NPC pluripotency NPCs by regulating the balance between self-renewal and differentiation via the post-transcriptional silencing of its target mRNAs (Chang et al., 2012; Ecsedi and Grosshans, 2013; Mitschka et al., 2015; Worringer et al., 2014). The neural tube closure defect in Trim71 knockout mice results from decreased proliferation and precocious differentiation of NPCs (Chen et al., 2012).
Multiple De Novo and Transmitted Mutations in SMARCC1
Two novel, damaging de novo mutations were identified in SMARCC1, encoding BAF155, a 155-kD subunit of the BRG1/BRM-associated factor (BAF; S. cerevisiae SWI/SNF) chromatin remodeling complex (Wang et al., 1996). These mutations included a p.Lys891fs*4 mutation (in Hydro106–3) and a predicted damaging missense p.His526Pro mutation (in Hydro105–1) located in a conserved position in the SWIRM domain, which mediates BAF complex subunit interactions (Da et al., 2006) (Figure 2). p.His526Pro occurred in a proband with obstructive CH with aqueductal stenosis and no other affected family members. p.Lys891fs*4 was de novo in an unaffected father (Hydro106–3) of three children with prenatally diagnosed CH with aqueductal stenosis. Two of these offsprings died in utero from obstructive hydrocephalus, while the surviving child inherited p.Lys891fs*4. In this cohort, the probability of seeing two damaging de novo mutations in SMARCC1 was 2.69 × 10−6 (Table 2).
Figure 2. De Novo and Transmitted Mutations in SMARCC1 Encoding the SWI/SNF Chromatin Modifier BAF155.
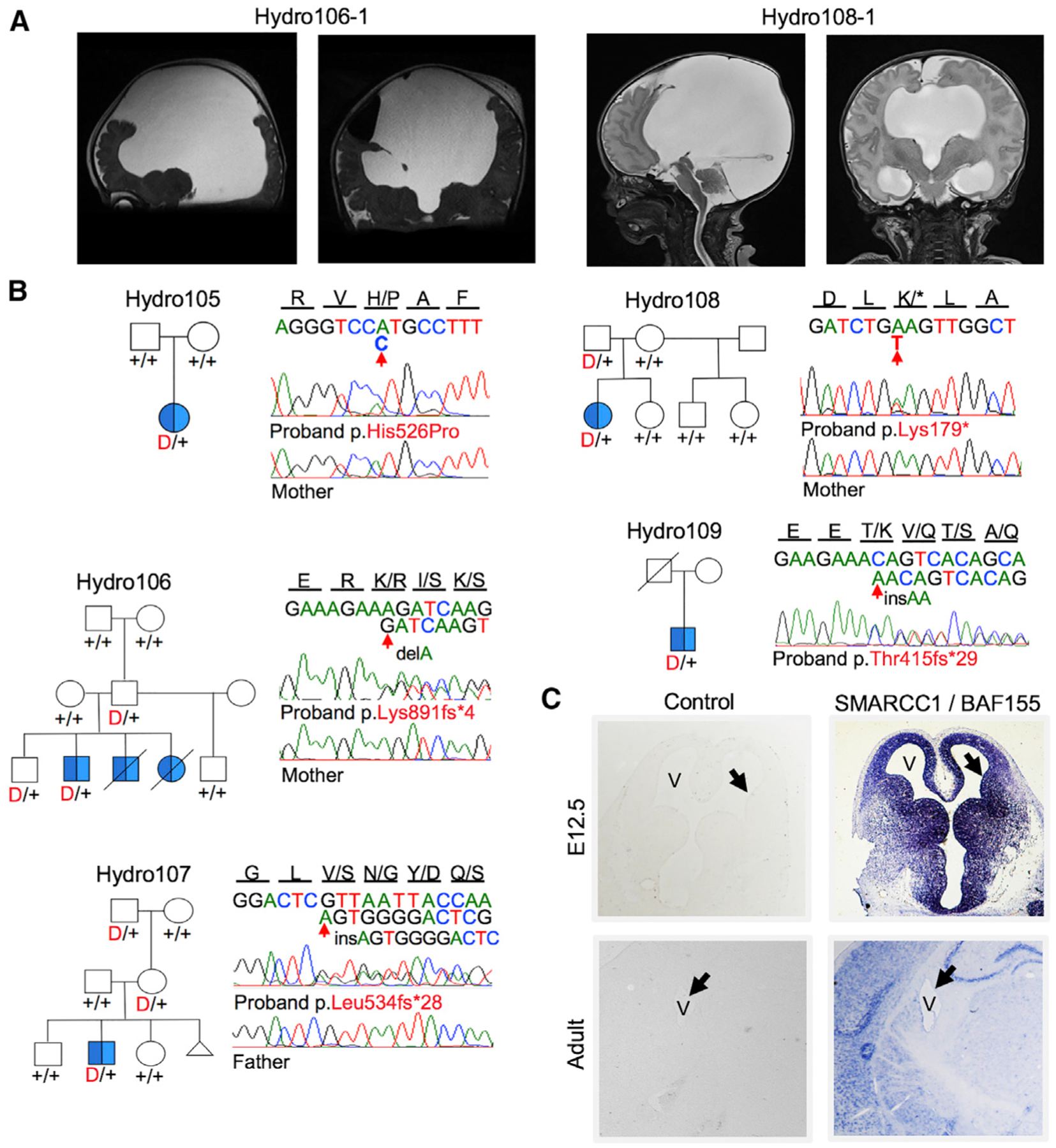
(A) Representative sagittal (left) and coronal (right) brain magnetic resonance images of CH probands Hydro106–1 and Hydro108–1 with obstructive hydrocephalus..
(B) Pedigrees with Sanger-verified mutated bases and the corresponding normal alleles marked on the chromatograms..
(C) In situ hybridization of wild-type E12.5 and adult mouse brains for Smarcc1 showing signals in the ciliated neuroepithelium and ventricular zone (V, ventricle; arrow, neuroepithelium at E12.5, 2.5×, and ependymal layer in adulthood; 10× magnification).
Three additional, previously unidentified, transmitted LOF mutations in SMARCC1 were found in three other CH probands with severe obstructive CH with aqueductal stenosis. In each of these kindreds, the proband was the sole affected member with mutation transmission from an unaffected parent (Figure 2; Figure S6; Table S7). The probability of two de novo damaging mutations and three rare transmitted LOFs occurring in SMARCC1 in this cohort was 8.15 × 10−10; this is a conservative estimate since SMARCC1 is highly intolerant to LOF mutation (pLI of 1), with far fewer LOF mutations than expected (3 LOF SMARCC1 mutations among 60,706 individuals in the ExAC database, and 2 among 3,578 autism controls).
In situ hybridization demonstrated that Smarcc1, like Trim71, is highly expressed in the neuroepithelium and ventricular zone but reduced in later brain development (Ho et al., 2009; Tuoc et al., 2013a; Yan et al., 2008) (Figure 2; Figure S4). Smarcc1 knockout causes embryonic lethality in mice (Kim et al., 2001). Roughly 20% of Smarcc1+/− mice (Kim et al., 2001) and ~80% of mice homozygous for a missense allele (Baf155msp/msp) (Harmacek et al., 2014) exhibit exencephaly similar to that of Trim71 knockout mice (Harmacek et al., 2014; Narayanan et al., 2015; Nguyen et al., 2016). The exencephaly phenotype has been attributed to inappropriate proliferation and increased apoptosis of NPCs in the neural tube (Harmacek et al., 2014; Kim et al., 2001). Neuronal specific knockout of Smarca4, encoding a Smarcc1 binding partner in the BAF complex, causes hydrocephalus and aqueductal stenosis (Cao and Wu, 2015). NPC-specific BAF complexes regulate the proliferation, differentiation, and survival of mouse NPCs via transcriptional regulation of genes critical for telencephalon development (Narayanan et al., 2015).
De Novo and Transmitted Mutations in PTCH1
Two previously unidentified de novo LOF mutations were identified in PTCH1, encoding the Sonic Hedgehog (SHH) receptor (Eggenschwiler and Anderson, 2007) Patched-1 (Figure 3). Heterozygous LOF PTCH1 mutations have been previously implicated in Gorlin syndrome (OMIM# 109400), featuring basal cell carcinomas and mandibular tumors, along with variable expressivity of other skeletal and non-skeletal phenotypes (Hahn et al., 1996). De novo PTCH1 mutations included a start-loss mutation (p.Met152fs*1) and a splice donor site mutation (c.1503+3T>C) in unrelated CH probands with aqueductal stenosis (Figure 3; Figure S7; Table S8). Both mutations were heterozygous and occurred in probands of uniplex CH kindreds. A transmitted frameshift mutation in PTCH1 (p.Leu664fs*12) was also found in two brothers with CH (Figure 3; Figure S7; Table S8). This mutation was transmitted from a mother who did not have hydrocephalus but had been diagnosed with Gorlin syndrome on the basis of numerous basal cell carcinomas. The probability of seeing at least two damaging de novo mutations by chance in a cohort of this size is 1.06 × 10−6, surpassing genome-wide significance (Table 2) (Ware et al., 2015).
Figure 3. De Novo and Transmitted Mutations in PTCH1 Encoding the Sonic Hedgehog Receptor Patched-1.
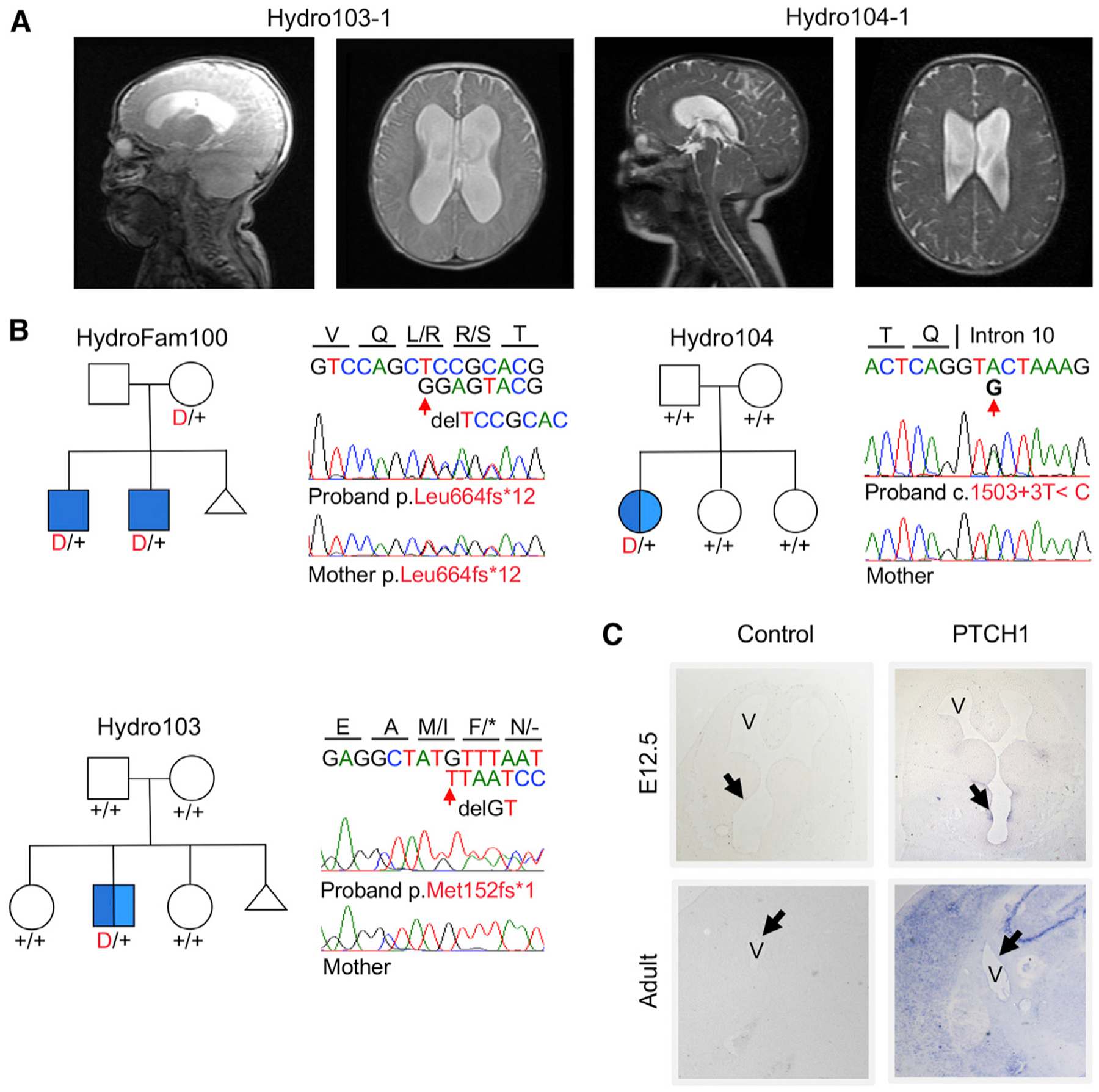
(A) Representative sagittal (left) and axial (right) brain magnetic resonance images of CH probands Hydro103–1 and Hydro104–1 with obstructive hydrocephalus..
(B) Pedigrees with Sanger-verified mutated bases and the corresponding normal alleles marked on the chromatograms..
(C) In situ hybridization of wild-type E12.5 and adult mouse brains for Ptch1 demonstrates expression in the hindbrain neuroepithelium (V, ventricle; arrow, neuroepithelium at E12.5, 2.5×, and ependymal layer in adulthood; 10× magnification).
Consistent with previous results (Eggenschwiler et al., 2001; Goodrich et al., 1996, 1997; Takahashi and Osumi, 2002), in situ hybridization showed Ptch1 expression in hindbrain neuroepithelium (Figure 3; Figure S4). Ptch1 knockout in mice results in exencephaly, neural tube overgrowth, and lethality between E9.0 and E10.5 (Ellis et al., 2003; Goodrich et al., 1997; Milenkovic et al., 1999). A significant fraction of Ptch1+/− mice develop hydrocephalus in two different genetic backgrounds (Svärd et al., 2009; Wetmore et al., 2000), similar to the variable expressivity seen in one of the multiplex kindreds. Penetrance of hydrocephalus in these mice increases to 100% for the homozygous quaking viable mutation (Ptch1+/−; qkv/v) (Gavino and Richard, 2011). Primary cilia in neuroepithelial cells sense gradients of SHH via PTCH1 and transduce these signals to regulate NPC proliferation, differentiation, and fate specification (Palma et al., 2005; Palma and Ruiz i Altaba, 2004).
De Novo Mutations in PLOD2
Two heterozygous de novo D-mis mutations were identified in the highly brain expressed (HBE) gene PLOD2 (Table S9). PLOD2 did not surpass thresholds for genome-wide significance. PLOD2 encodes a lysyl hydroxylase previously implicated in AR Bruck syndrome type 2 (OMIM# 609220), a variant of osteogenesis imperfecta (McPherson and Clemens, 1997). Further work will be required to assess the significance of these mutations in CH.
De Novo Mutations in Neural Tube Closure and Formation
Gene ontology (GO) pathway analysis identified our significant CH gene set to be highly enriched for genes involved in “neural tube closure and formation.” We searched for additional de novo mutations in genes in this GO pathway (Table S10). We found a single de novo missense mutation in CELSR2 (p.Arg2812Trp), encoding cadherin EGF LAG seven-pass G-type receptor 2, and a single de novo missense mutation in LRP6 (p.Val1415Phe), encoding a co-receptor with frizzled proteins in the WNT signaling pathway. Celsr2 and Lrp6 are highly expressed in ciliated neuroepithelia and regulate neurogenesis, and knockout of each gene results in lethal hydrocephalus in mice (Allache et al., 2014; Cuevas et al., 2015; Gavino and Richard, 2011; Harmacek et al., 2014; Maller Schulman et al., 2008; Tissir et al., 2010).
De Novo Duplications at the SHH Locus
Using the XHMM algorithm (Fromer and Purcell, 2001), we identified seven putative de novo copy number variants (CNVs) (see STAR Methods). Six of these, including five duplications and one deletion, were validated by independent tests (distortion of allelic ratios of heterozygous SNPs in implicated intervals or qPCR; Table S11). Two of these were duplications at the SHH locus, encoding SHH, the canonical ligand for PTCH1 that regulates neurogenesis by conferring positional information to ventral NPCs in the neural tube (Jessell and Sanes, 2000; Lupo et al., 2006). The probability of finding two duplications at the same locus among five duplication events with these gene compositions is very low (p = 1.22 × 10−4) (see STAR Methods).
DISCUSSION
Using exome sequencing, we implicate four new genes in human CH: TRIM71, SMARCC1, PTCH1, and SHH (Figure 4). High enrichment of mutations in these genes suggests their large effects on phenotypic risk. The genes showing heterozygous protein-altering de novo mutations are all highly intolerant to LOF mutation (pLI ≥ 0.99), and murine knockouts of each gene produce neural tube defects, including exencephaly (Feng et al., 2013; Gavino and Richard, 2011; Goodrich et al., 1997; Hahn et al., 1996; Harmacek et al., 2014; Maller Schulman et al., 2008). SHH encodes the PTCH1 ligand. Ptch1-deficient mice develop fatal hydrocephalus (Celen et al., 2017; Gavino and Richard, 2011). All four genes are expressed in the ciliated neuroepithelium and regulate NPC fate. Mutations in these four new genes account for 8.5% of cases; another 2.3% of cases are explained by previously identified genes.
Figure 4. Meta-analysis of Rare De Novo and Transmitted Mutations in Mutation-Intolerant Genes.

Quantile-quantile plot of observed versus expected p values from meta-analysis of protein-altering de novo and LOF transmitted heterozygous variants comparing the burden of rare variants in genes intolerant to LOF mutation (pLI ≥ 0.90, MAF ≤ 2 × 10−5).
From the inferred contribution of de novo point mutations to 8% of CH probands in our cohort and the number of genes with more than one de novo mutation, we estimate that additional genes contribute to CH by de novo point mutation (maximum likelihood estimate ~8 genes, with wide confidence interval) (Figures S8 and S9). This estimate is substantially smaller than autism and congenital heart disease (De Rubeis et al., 2014; Homsy et al., 2015; Iossifov et al., 2014; Sanders et al., 2012; Zaidi et al., 2013), consistent with the identification of multiple disease-causing genes from this relatively small cohort. Variants in non-coding elements of these genes might add to their contribution to CH. Sequencing of additional trios and isolated probands has high potential to detect additional rare mutations with large effect on disease risk.
All four cases of CH with TRIM71 mutation had communicating hydrocephalus, whereas all five cases with SMARCC1 mutation and all three cases with PTCH1 mutation had aqueductal stenosis. From the prevalence of these two sub-phenotypes in our cohort (50.2% communicating versus 49.8% aqueductal, respectively) we calculate that this correlation was highly unlikely to occur by chance (p = 0.002). These observations support the pathogenicity of the mutations and suggest phenotypic subsets of CH are influenced by genetic determinants.
Our results suggest incomplete penetrance and variable expressivity of mutations in two CH genes. Four first-degree relatives of probands with the same mutation in SMARCC1 and one relative of a PTCH1 mutation proband had no evidence of hydrocephalus. Moreover, heterozygous LOF mutations in PTCH1 are a well-described cause of Gorlin syndrome (Gorlin, 2004; Gorlin and Goltz, 1960), which can be associated with obstructive hydrocephalus due to medulloblastoma (Amlashi et al., 2003). Similar to murine hydrocephalus with PTCH1 mutation (Wang et al., 2010a; Wetmore et al., 2015), our patients had hydrocephalus without CSF obstruction from medulloblastoma. In addition, a mutation-bearing parent of one of our probands had been diagnosed with Gorlin syndrome owing to numerous basal cell carcinomas but did not have hydrocephalus. This suggests that LOF mutations in our probands are functionally equivalent to those causing Gorlin syndrome and that these probands may be at risk of developing basal cell carcinomas. The determinants of incomplete penetrance and variable expressivity remain obscure but may be explained by common genetic (Timberlake et al., 2016) or environmental (Stuart et al., 2015) modifiers or by stochastic elements.
Similar to other Mendelian forms of CH (e.g., L1CAM) (Rosenthal et al., 1992), several CH patients harboring TRIM71 and SMARCC1 mutations exhibited other neurological findings and structural brain abnormalities in addition to hydrocephalus. All CH patients with TRIM71 mutations and 3 of 5 CH patients with SMARCC1 mutations exhibited neurodevelopmental delay and epilepsy. A mother and son with mutant TRIM71 and ventriculomegaly had similar open schizencephalic clefts (Figure 1). Two patients with SMARCC1 mutations exhibited schizencephalic clefts and possibly other structural brain abnormalities (Figure 2). These findings are consistent with TRIM71 and SMARCC1 regulating key aspects of brain development (Cuevas et al., 2015; Harmacek et al., 2014; Kim et al., 2001; Maller Schulman et al., 2008; Narayanan et al., 2015; Nguyen et al., 2016).
SMARCC1 and PTCH1 in CH are LOF mutations. TRIM71 exhibits a highly specific mutation spectrum involving either of two homologous arginines in the NHL domain that directly binds mRNAs (Chang et al., 2012; Loedige et al., 2013, 2014, 2015). While these mutations likely impair binding to normal target mRNAs, other gene functions, such as its putative ubiquitin ligase activity, may be preserved. Alternatively, these mutations could impart neomorphic effects that cause binding to new mRNAs. Similarly, the observed duplications of SHH suggest that these mutations are gain of function. Dosage loss of PTCH1, which inhibits downstream signaling via effect on the SMO receptor, and dosage gain of SHH would be similarly expected to increase downstream Hedgehog signaling (Briscoe and Thérond, 2013).
Ciliated neuroepithelial stem cells in the neural tube ventricular zone undergo proliferation accompanied by minimal differentiation during early embryogenesis. These cells become differentiated neurons (Ever and Gaiano, 2005; Merkle et al., 2004) in mice between E8 and E14 (Chen et al., 2006; Noctor et al., 2004), a transformation driven by significant alterations in gene expression (Yao et al., 2016). TRIM71, SMARCC1, and PTCH1 are all highly expressed in the ciliated neuroepithelium (Cuevas et al., 2015; Gavino and Richard, 2011; Tuoc et al., 2013b). SHH engages its receptor PTCH1 on neuroepithelial cilia. All five genes regulate the proliferation, fate specification, and/or differentiation of NPCs the development of the neural tube (Kim et al., 2001; Nguyen et al., 2017; Shikata et al., 2011).
The gene mutations reported here suggest the importance of de novo or transmitted mutations that alter the balance between NPC proliferation and differentiation in the pathogenic mechanism of CH. In support of this human genetic data, Dusp16 deficiency in mice causes brain overgrowth and associated obstructive hydrocephalus due to hyperproliferation and expansion of ventricular zone NPCs at the level of the aqueduct (Zega et al., 2017). In contrast, the communicating hydrocephalus phenotype in a mouse model of the human ciliopathy Bardet-Biedl syndrome results from decreased proliferation and survival of NPCs (Carter et al., 2012). Interestingly, intraventricular hemorrhage in neonates, the most common cause of infantile secondary hydrocephalus (Mazzola et al., 2014; Robinson, 2012), is also associated with decreased NPC viability and associated cortical thinning (McAllister et al., 2017; Yung et al., 2011). A “neural stem cell” paradigm of hydrocephalus could provide a unifying mechanism to explain multiple forms of infantile hydrocephalus.
These findings have potential implications for CH treatment. Multiple non-neural mechanisms have been proposed to account for communicating hydrocephalus, including elevated venous pressure, arachnoid granulation immaturity, and lymphatic dysplasia (Govaert et al., 1991). However, given the new genetic data, a primary driver of some communicating forms of CH may not be attributable to active CSF accumulation and associated ventricular distension but rather impaired neurogenesis. In neonatal CH cases with normal or low intracranial pressure (Tully et al., 2016), it is unclear whether surgical CSF shunting addresses a pathologic feature of CH. Instead, it may expose CH patients to surgical morbidity without improving neurodevelopmental outcomes. The implications for families with CH children and their neurosurgeons will stimulate discussion and catalyze further investigation into CH pathogenesis and treatment.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Kristopher T. Kahle (kristopher.kahle@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Subjects and Samples
All study procedures and protocols comply with Yale University’s Human Investigation Committee and Human Research Protection Program. Written informed consent for genetic studies was obtained from all participants. Inclusion criteria included patients with primary congenital hydrocephalus without known genetic causes such as L1CAM or any other large chromosomal deletions or rearrangements. Hydrocephalus cases with secondarily acquired etiologies such as intraventricular hemorrhage (IVH), meningitis, obstruction due to tumors or cysts, and stroke were excluded. Children with hydranencephaly, large cysts and cephaloceles, posterior fossa crowding, myelomeningocele (Chiari II syndrome), or benign extra-axial CSF accumulation (i.e., benign external hydrocephalus) were also excluded. Sequenced trios were composed of two unaffected parents and one affected child with primary congenital hydrocephalus. All probands had undergone surgery for therapeutic CSF diversion (shunt placement and/or endoscopic third ventriculostomy). Patients and participating family members provided buccal swab samples (Isohelix SK-2S DNA buccal swab kits), medical records, radiological imaging studies, operative reports, and congenital hydrocephalus phenotype data.
The control cohort was composed of 1,789 previously whole-exome sequenced and analyzed families from the Simons Foundation Autism Research Initiative Simplex Collection (Fischbach and Lord, 2010; Iossifov et al., 2014; Krumm et al., 2015; O’Roak et al., 2011; Sanders et al., 2012). Sequenced families were comprised of two unaffected parents, one affected child with autism, and one unaffected sibling. Only the unaffected sibling and parents, as designated by the Simons Simplex Collection, were analyzed and served as controls for this study (Krumm et al., 2015).
Whole-Exome Sequencing and Variant Calling
DNA was isolated from buccal swab samples in accordance with manufacturer protocol. Whole-exome sequencing was performed using the IDT xGen capture reagent followed by 99 base paired-end sequencing on the Illumina HiSeq 2000 instrument at the Yale Center for Genome Analysis as previously described (Timberlake et al., 2016). Exome sequencing quality metrics are shown in Table S2 and Figures S1 and S2.
Sequence reads were mapped and aligned to the GRCh37/hg19 human reference genome using Burrows-Wheeler Aligner-MEM (Li and Durbin, 2009). In accordance with GATK Best Practices recommendations, the data were further processed using Genome Analysis Toolkit (GATK) base quality score recalibration (McKenna et al., 2010), indel realignment, duplication marking and removal, and base quality score recalibration (DePristo et al., 2011; Van der Auwera et al., 2013). Single nucleotide variants and small insertions and deletion were called using GATK Haplotype Caller and annotated using ANNOVAR (Wang et al., 2010b), NHLBI exome variant server (Fu et al., 2013), 1000 Genomes (Auton et al., 2015), DbSNP (Sherry et al., 2001), and gnomAD and ExAC databases (Lek et al., 2016).
The average depth of coverage of the whole-exome sequencing data was 54.3x, with greater than 8x coverage in 95.9% of the target region for exome capture (Table S2).
The sporadic or autosomal recessive mode of inheritance exhibited in our cohort pedigrees led us to prioritize de novo, compound heterozygous, and homozygous variants. Variants were filtered for predicted deleteriousness and conservation using a series of in silico prediction algorithms, including Meta-analytic support vector machine (MetaSVM) and Meta-analytic logistic regression (MetaLR) (Dong et al., 2015), Polymorphism Phenotyping (PolyPhen) (Adzhubei et al., 2010), Combined Annotation-Dependent Depletion (CADD) (Kircher et al., 2014), Sorting Intolerant From Tolerant (SIFT) (Kumar et al., 2009; Ng and Henikoff, 2001, 2002, 2003, 2006), conservation across 46 orthologs (cons46diff) (Cromer et al., 2012; Stuart et al., 2015), Likelihood Ratio Test (LRT) (Chun and Fay, 2009), MutationTaster (Schwarz et al., 2010), Mutation Assessor (Reva et al., 2011), Functional Analysis Through Hidden Markov Models (FATHMM) (Shihab et al., 2013), FATHMM–Multiple Kernel Learning (FATHMM-MKL) (Shihab et al., 2015), FATHMM-Coding (Shihab et al., 2014), Protein Variant Effect Analyzer (PROVEAN) (Choi et al., 2012), Variant Effect Scoring Tool (VEST3) (Carter et al., 2013), Mendelian Clinically Applicable Pathogenicity (M-CAP) (Jagadeesh et al., 2016), deleterious annotation of genetic variants using neural networks (DANN) (Quang et al., 2015), Eigen-PC (Ionita-Laza et al., 2016), Genomic Evolutionary Rate Profiling (GERP++ and GERPP++ GT2) (Davydov et al., 2010), phylogenetic p values (phyloP100way and phyloP20way) (Pollard et al., 2010), phastCons100way and phastCons20way (Siepel et al., 2005), Site-specific Phylogenetic analysis (SiPhy) (Garber et al., 2009; Lindblad-Toh et al., 2011), REVEL (Ioannidis et al., 2016), and MPC (Samocha et al., 2017).
METHOD DETAILS
Kinship Analysis
Pedigree information and participant relationships were confirmed utilizing pairwise PLINK identity-by-descent (IBD) calculation (Purcell et al., 2007). The IBD sharing between the probands and parents in all trios is between 45% and 55%. Pairwise individual relatedness was corroborated using KING (Manichaikul et al., 2010).
Haplotype Phasing and Analysis of Inbreeding
Haplotype phasing, inbreeding coefficient, and the longest homozygosity-by-descent (HBD) fragment were estimated using Beagle v3.3.2 (Browning and Browning, 2007) as described previously (Jin et al., 2017). The criteria of consanguinity are defined as runs of homozygosity in segments of 2cM or greater length that collectively comprise at least 0.35% of the genome.
Principal Component Analysis
In order to determine the ethnicity of each participant, we utilized the EIGENSTRAT software (Price et al., 2006) to analyze SNPs in cases, controls, and HapMap subjects as previously described (Jin et al., 2017; Timberlake et al., 2016).
De Novo and Inherited Variant Analysis and Filtering
De novo mutations were called in parent-offspring trios, each consisting of an affected child with primary congenital hydrocephalus and his or her unaffected biological parents, using the Bayesian framework TrioDeNovo (Dong et al., 2015). Candidate de novo variants were filtered based on the following criteria: (1) minor allele frequency (MAF) ≤ 5 × 10−3 in ExAC, 1000 Genomes, and EVS, (2) GATK variant quality score recalibration (VQSR) of ‘pass’, (3) minimum sequencing depth of 8 reads in the proband and each parent, (4) genotype quality (GQ) score ≥ 20 and alternate allele ratio ≥ 40%, (5) TrioDeNovo data quality (DQ) score ≥ 7, and (6) exonic or splice-site variant.
Transmitted dominant variants were filtered by similar criteria of rareness and quality: (1) MAF ≤ 2 × 10−5 in ExAC, (2) GQ ≥ 20 and alternate allele ratio ≥ 40%, (3) GATK VQSR of ‘pass’, and (4) minimum sequencing depth of 8 reads in each participant. Recessive variants were also filtered for rare (MAF ≤ 10−3 in ExAC) bi-allelic events (homozygous and compound heterozygous mutations) that met read quality criteria as above (GQ ≥ 20, alternate allele ratio ≥ 40%, ‘pass’ GATK VQSR, and minimum sequencing depth ≥ 8). Hemizygous recessive variants were filtered for rare events (MAF ≤ 2 × 10−5) using the same quality criteria described above.
The impact of nonsynonymous single nucleotide variants on protein function was predicted using the MetaSVM algorithm (Dong et al., 2015), identifying mutations with rank scores greater than 0.83357 as deleterious (‘D-mis’). D-Mis and loss-of function mutations (nonsense, frameshift insertions and deletions, and splice-site) were considered potentially damaging to protein function.
All de novo and transmitted calls were verified by in silico visualization of aligned reads using the BLAT search (Kent, 2002) and Integrative Genomics Viewer (IGV) (Robinson et al., 2011). Salient de novo and compound heterozygous calls were then verified in all participants that provided DNA by direct Sanger sequencing of PCR amplicons containing the mutation.
Burden of De Novo Mutations
The burden of de novo mutations in congenital hydrocephalus cases and unaffected autism controls was determined using the denovolyzeR package (Ware et al., 2015) as previously described The probability of observing a de novo mutation in each gene was calculated as illustrated previously (Jin et al., 2017), with the exception that the coverage adjustment factor was based on the full set of 125 case trios (or 126 case trios in the SMARCC1 analysis given the inclusion of a de novo in parent Hydro106–3) and 1,789 control trios (separate probability tables for each set). The expected number of de novo mutations across variant classes in case and control cohorts was calculated and compared to the observed number of de novo mutations in each cohort using the Poisson test (Samocha et al., 2014). Gene-set enrichment analyses and statistical tests considered only mutations observed or expected in genes within the specified set (i.e., high brain-expressed, aqueductal stenosis).
Contribution of De Novo Mutation to Congenital Hydrocephalus
The number of de novo mutations in risk genes that contribute to congenital hydrocephalus was calculated based on the observed count of protein-altering de novo mutations compared to expectation ( = Nx((M1–M2)/M1), where N is the total number of trios and M1 and M2 are the observed and expected count of protein-altering de novo mutations per trio, respectively)
Enrichment Analysis for the Dominant and Recessive Variants
To quantify the enrichment of LOF heterozygous variants, we calculated the expectation for a gene using the following formula:
where ‘j’ denotes the ‘jth’ gene and ‘L’ denotes the total number of LOF heterozygous mutations. A one-tailed binomial test was conducted to compare the observed number of heterozygous variants to expectation.
For damaging recessive genotypes (RGs) in a specific gene in cases, we conducted a one-tailed binomial test to evaluate enrichment as described previously (Jin et al., 2017). RG can also be modeled separately as compound heterozygotes or homozygotes. The expected number of compound heterozygotes for each gene is derived from distributing the observed number of RGs, N, across all genes according to the ratio of the squared de novo probabilities:
The expected number of homozygotes is derived similarly, but using the linear ratio of de novo probabilities:
The total number of expected RG for each gene is the sum of the derived expected compound heterozygous and homozygous values.
Gene Ontology Enrichment Analysis
Three genome-wide significant genes PTCH1, SMARCC1, and TRIM71 were input into GOrilla (Eden et al., 2009) to identify enriched GO terms compared to the background set of genes (M = 18,715). For gene-set enrichment analyses, each statistical test considered observed or expected mutations in genes within the specified gene set.
In Silico Splice-Site Prediction
In order to assess the impact of a missense mutation at the splice donor site of intron 10 of PTCH1 that changed the donor site sequenced from GTA to GTC, we utilized Human Splicing Finder (Desmet et al., 2009) and MaxEntScan (Yeo and Burge, 2004). Both programs predicted that this mutation is likely to affect splicing. MaxEntScan assigned a MaxEnt score of 7.64 to the wild-type canonical splice donor site; however, after mutation of the wild-type sequence to GTC, the alternate splice donor bases upstream was assigned a score of −0.18. The difference in MaxEnt scores between the mutant and the reference sequence is −7.82, which provides strong support for this mutation being a potential 5′ donor splice site.
Copy Number Variation Analysis
XHMM was run to call CNVs from WES as previously described (Ruderfer et al., 2016). GATK DepthOfCoverage was used to calculate mean read depth per targets from the alignment files. The data were normalized by removing the highest variance principal components (variance > 70%) and z scores were calculated from the mean read depths. CNVs were called using the Viterbi Hidden Markov model (HMM) and the quality scores were calculated using the forward-backward HMM. After filtering out common CNVs present at allele frequencies greater than 0.1% in 1000 Genomes (Sudmant et al., 2015) and 10% in the cohort, high quality CNVs (SQ > 60 where SQ indicates the phred-scaled quality score for the presence of a CNV event within the interval) were subjected to visual inspection. De novo CNVs were assessed using PLINK/Seq command-line tools (Fromer et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
De Novo CNV Permutation Test
The probability of finding one exon covered by multiple de novo CNVs was calculated by comparing the observed distribution to an empirical distribution derived from 1 million permutations. For each permutation, five de novo duplications were randomly distributed across the genome; each duplication contained the same number of exons as predicted from XHMM with adjustment if the CNV is partially validated by experiment. In each permutation, the experiment was considered a success if at least one locus contained exactly one exon was covered by multiple duplications. The number of successes was tallied and the p value was obtained by dividing the number of successes by the number of iterations.
Estimating the Number of Risk Genes
A maximum likelihood approach was used to estimate the number of genes contributing to congenital hydrocephalus via de novo events as described previously (Jin et al., 2017). We defined K to be the number of observed protein-altering de novo mutations in high brain-expressed (HBE) genes among cases. R1 indicates the number of HBE genes mutated exactly twice in cases and R2 indicates the number of high HBE mutated three times or more. We set the proportion (E) of protein-altering mutations in risk genes based on point estimate of enrichment in cases compared to expectation (E = (M1–M2)/M1, where M1 and M2 are the observed and expected count of protein-altering de novo mutations per trio, respectively). We then simulated the likelihood function as follows: First, we randomly selected G risk genes from the HBE gene set. Next, we simulated the number of contributing protein-altering mutations in risk genes, i.e., C, by sampling once from Binomial(K,E) distribution. Then, we simulated C contributing protein-altering mutations in G risk genes and K-C non-contributing protein-altering mutations in the complete HBE gene set, using each gene’s protein-altering mutability score as probability weights. We performed 20,000 simulations for G from 2 to 100, and calculated the likelihood function L(G) as the proportion of simulations in which the number of genes with two protein-altering de novo mutations equals to R1 and the number of genes with three or more protein-altering de novo mutations equals to R2. We then estimated the number of risk genes using the maximum likelihood estimate (MLE). Based on the likelihood function, we calculated the Fisher information and constructed the confidence interval based on the MLE and estimated Fisher information using the following equation
In Situ Hybridization
Mouse brains and embryos were fixed in 4% paraformaldehyde by overnight immersion and sectioned (10–15 μm, cryostat sections for digoxigenin [DIG] probes). Antisense RNA probes corresponding to murine Ptch1, Smarcc1 and Trim71 (approx. 200bp for DIG-labeled probes respectively) were synthesized to detect Ptch1, Smarcc1 and Trim71 transcripts in murine tissue, using methods previously described (Duncan et al., 1994; Petersen et al., 2002; Schaeren-Wiemers and Gerfin-Moser, 1993).
DATA AND SOFTWARE AVAILABILITY
The sequencing data for congenital hydrocephalus case-parent trios reported in this manuscript have been deposited in the NCBI database of Genotypes and Phenotypes (dbGaP). The accession number for the data reported in this paper is dbGaP: phs000744.
Supplementary Material
KEY RESOURCES TABLE
Highlights.
Exome sequencing identifies novel genetic drivers of congenital hydrocephalus (CH)
De novo and inherited rare variants in four genes explain ~10% of CH cases
All four CH genes (TRIM71, SMARCC1, PTCH1, and SHH) regulate neural stem cell fate
These data implicate aberrant neurogenesis in the pathogenesis of a subset of CH
ACKNOWLEDGMENTS
We are grateful to the patients and families who participated in this research. We would also like to thank Jenna Koschnitzky, Amanda Garzón, and the entire Hydrocephalus Association (HA); Jeannine Rockefeller and Jeannette Votto (Yale); and Diego Morales (WashU). This work is supported by the Yale-NIH Center for Mendelian Genomics (5U54HG006504), NIH T32GM007205, NIH TL1 TR001864, Hydrocephalus Foundation, Howard Hughes Medical Institute, and Rudi Schulte Research Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and eleven tables and can be found with this article online at https://doi.org/10.1016/j.neuron.2018.06.019.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adle-Biassette H, Saugier-Veber P, Fallet-Bianco C, Delezoide A-L, Razavi F, Drouot N, Bazin A, Beaufrère A-M, Bessières B, Blesson S, et al. (2013). Neuropathological review of 138 cases genetically tested for X-linked hydrocephalus: evidence for closely related clinical entities of unknown molecular bases. Acta Neuropathol. 126, 427–442. [DOI] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, and Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschimann F, Kumari P, Bartake H, Gaidatzis D, Xu L, Ciosk R, and Großhans H (2017). LIN41 post-transcriptionally silences mRNAs by two distinct and position-dependent mechanisms. Mol. Cell 65, 476–489.e4. [DOI] [PubMed] [Google Scholar]
- Al-Dosari MS, Al-Owain M, Tulbah M, Kurdi W, Adly N, Al-Hemidan A, Masoodi TA, Albash B, and Alkuraya FS (2013). Mutation in MPDZ causes severe congenital hydrocephalus. J. Med. Genet 50, 54–58. [DOI] [PubMed] [Google Scholar]
- Allache R, Lachance S, Guyot MC, De Marco P, Merello E, Justice MJ, Capra V, and Kibar Z (2014). Novel mutations in Lrp6 orthologs in mouse and human neural tube defects affect a highly dosage-sensitive Wnt non-canonical planar cell polarity pathway. Hum. Mol. Genet 23, 1687–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, et al. ; Epi4K Consortium; Epilepsy Phenome/Genome Project (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlashi SF, Riffaud L, Brassier G, and Morandi X (2003). Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer 98, 618–624. [DOI] [PubMed] [Google Scholar]
- Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, and Abecasis GR; 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awadalla P, Gauthier J, Myers RA, Casals F, Hamdan FF, Griffing AR, Côté M, Henrion E, Spiegelman D, Tarabeux J, et al. (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet 87, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak T, Kwan KY, Louvi A, Demirbilek V, Saygı S, Tüysüz B, Choi M, Boyacı H, Doerschner K, Zhu Y, et al. (2011). Recessive LAMC3 mutations cause malformations of occipital cortical development. Nat. Genet 43, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilgüvar K, Oztürk AK, Louvi A, Kwan KY, Choi M, Tatli B, Yalnizoğlu D, Tüysüz B, Cağlayan AO, Gökben S, et al. (2010). Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 467, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bret P, and Chazal J (1995). Chronic (“normal pressure”) hydrocephalus in childhood and adolescence. A review of 16 cases and reappraisal of the syndrome. Childs Nerv. Syst 11, 687–691. [DOI] [PubMed] [Google Scholar]
- Briscoe J, and Thérond PP (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol 14, 416–429. [DOI] [PubMed] [Google Scholar]
- Browning SR, and Browning BL (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet 81, 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M, and Wu JI (2015). Camk2a-Cre-mediated conditional deletion of chromatin remodeler Brg1 causes perinatal hydrocephalus. Neurosci. Lett 597, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CS, Vogel TW, Zhang Q, Seo S, Swiderski RE, Moninger TO, Cassell MD, Thedens DR, Keppler-Noreuil KM, Nopoulos P, et al. (2012). Abnormal development of NG2+PDGFR-α+ neural progenitor cells leads to neonatal hydrocephalus in a ciliopathy mouse model. Nat. Med 18, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Douville C, Stenson PD, Cooper DN, and Karchin R (2013). Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14 (Suppl 3), S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celen C, Chuang JC, Luo X, Nijem N, Walker AK, Chen F, Zhang S, Chung AS, Nguyen LH, Nassour I, et al. (2017). Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. eLife 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Martinez NJ, Thornton JE, Hagan JP, Nguyen KD, and Gregory RI (2012). Trim71 cooperates with microRNAs to repress Cdkn1a expression and promote embryonic stem cell proliferation. Nat. Commun 3, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CP, Lin SP, Wang TH, Chen YJ, Chen M, and Wang W (2006). Perinatal findings and molecular cytogenetic analyses of de novo interstitial deletion of 9q (9q22.3–>q31.3) associated with Gorlin syndrome. Prenat. Diagn 26, 725–729. [DOI] [PubMed] [Google Scholar]
- Chen J, Lai F, and Niswander L (2012). The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev. 26, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, and Chan AP (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, and Fay JC (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer MK, Starker LF, Choi M, Udelsman R, Nelson-Williams C, Lifton RP, and Carling T (2012). Identification of somatic mutations in parathyroid tumors using whole-exome sequencing. J. Clin. Endocrinol. Metab 97, E1774–E1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas E, Rybak-Wolf A, Rohde AM, Nguyen DTT, and Wulczyn FG (2015). Lin41/Trim71 is essential for mouse development and specifically expressed in postnatal ependymal cells of the brain. Front. Cell Dev. Biol 3, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da G, Lenkart J, Zhao K, Shiekhattar R, Cairns BR, and Marmorstein R (2006). Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc. Natl. Acad. Sci. USA 103, 2057–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, and Batzoglou S (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol 6, e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. ; DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, and Béroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, and Liu X (2015). Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet 24, 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SA, Manova K, Chen WS, Hoodless P, Weinstein DC, Bachvarova RF, and Darnell JE Jr. (1994). Expression of transcription factor HNF-4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: HNF-4 is a marker for primary endoderm in the implanting blastocyst. Proc. Natl. Acad. Sci. USA 91, 7598–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecsedi M, and Grosshans H (2013). LIN-41/TRIM71: emancipation of a miRNA target. Genes Dev. 27, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, and Anderson KV (2007). Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol 23, 345–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, Espinoza E, and Anderson KV (2001). Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature 412, 194–198. [DOI] [PubMed] [Google Scholar]
- Ekici AB, Hilfinger D, Jatzwauk M, Thiel CT, Wenzel D, Lorenz I, Boltshauser E, Goecke TW, Staatz G, Morris-Rosendahl DJ, et al. (2010). Disturbed Wnt Signalling due to a Mutation in CCDC88C Causes an Autosomal Recessive Non-Syndromic Hydrocephalus with Medial Diverticulum. Mol. Syndromol 1, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T, Smyth I, Riley E, Graham S, Elliot K, Narang M, Kay GF, Wicking C, and Wainwright B (2003). Patched 1 conditional null allele in mice. Genesis 36, 158–161. [DOI] [PubMed] [Google Scholar]
- Ever L, and Gaiano N (2005). Radial ‘glial’ progenitors: neurogenesis and signaling. Curr. Opin. Neurobiol 15, 29–33. [DOI] [PubMed] [Google Scholar]
- Feng W, Choi I, Clouthier DE, Niswander L, and Williams T (2013). The Ptch1(DL) mouse: a new model to study lambdoid craniosynostosis and basal cell nevus syndrome-associated skeletal defects. Genesis 51, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, and Lord C (2010). The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68, 192–195. [DOI] [PubMed] [Google Scholar]
- Fromer M, and Purcell SM (2001). Using XHMM software to detect copy number variation in whole-exome sequencing data. Curr. Protoc. Hum. Genet 81, 7.23.1–7.23.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O’Donovan MC, Owen MJ, et al. (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet 91, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E, Palta P, Ruderfer DM, et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, et al. ; NHLBI Exome Sequencing Project (2013). Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Guttman M, Clamp M, Zody MC, Friedman N, and Xie X (2009). Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25, i54–i62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavino C, and Richard S (2011). Patched1 haploinsufficiency impairs ependymal cilia function of the quaking viable mice, leading to fatal hydrocephalus. Mol. Cell. Neurosci 47, 100–107. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, and Scott MP (1996). Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 10, 301–312. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, and Scott MP (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ (2004). Nevoid basal cell carcinoma (Gorlin) syndrome. Genet. Med 6, 530–539. [DOI] [PubMed] [Google Scholar]
- Gorlin RJ, and Goltz RW (1960). Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N. Engl. J. Med 262, 908–912. [DOI] [PubMed] [Google Scholar]
- Govaert P, Oostra A, Matthys D, Vanhaesebrouck P, and Leroy J (1991). How idiopathic is idiopathic external hydrocephalus? Dev. Med. Child Neurol 33, 274–276. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. (1996). Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851. [DOI] [PubMed] [Google Scholar]
- Harmacek L, Watkins-Chow DE, Chen J, Jones KL, Pavan WJ, Salbaum JM, and Niswander L (2014). A unique missense allele of BAF155, a core BAF chromatin remodeling complex protein, causes neural tube closure defects in mice. Dev. Neurobiol 74, 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp F, Wölfle J, Aretz M, Krämer A, Höhmann B, Fahnenstich H, and Zerres K (1999). Congenital hydrocephalus internus and aqueduct stenosis: aetiology and implications for genetic counselling. Eur. J. Pediatr 158, 474–478. [DOI] [PubMed] [Google Scholar]
- Ho L, Ronan JL, Wu J, Staahl BT, Chen L, Kuo A, Lessard J, Nesvizhskii AI, Ranish J, and Crabtree GR (2009). An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 106, 5181–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. (2015). De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. (2016). REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, McCallum K, Xu B, and Buxbaum JD (2016). A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet 48, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, Bernstein JA, and Bejerano G (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet 48, 1581–1586. [DOI] [PubMed] [Google Scholar]
- Jessell TM, and Sanes JR (2000). Development. The decade of the developing brain. Curr. Opin. Neurobiol 10, 599–611. [DOI] [PubMed] [Google Scholar]
- Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. (2017). Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet 49, 1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle KT, Kulkarni AV, Limbrick DD Jr., and Warf BC (2016). Hydrocephalus in children. Lancet 387, 788–799. [DOI] [PubMed] [Google Scholar]
- Kamuro K, and Tenokuchi Y (1993). Familial periventricular nodular heterotopia. Brain Dev. 15, 237–241. [DOI] [PubMed] [Google Scholar]
- Kanamoto T, Terada K, Yoshikawa H, and Furukawa T (2006). Cloning and regulation of the vertebrate homologue of lin-41 that functions as a heterochronic gene in Caenorhabditis elegans. Dev. Dyn 235, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Kent WJ (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, et al. (2001). Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol. Cell. Biol 21, 7787–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, and Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousi M, and Katsanis N (2016). The genetic basis of hydrocephalus. Annu. Rev. Neurosci 39, 409–435. [DOI] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He Z-X, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, and Ng PC (2009). Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. ; Broad Institute Sequencing Platform and Whole Genome Assembly Team; Baylor College of Medicine Human Genome Sequencing Center Sequencing Team; Genome Institute at Washington University (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loedige I, Gaidatzis D, Sack R, Meister G, and Filipowicz W (2013). The mammalian TRIM-NHL protein TRIM71/LIN-41 is a repressor of mRNA function. Nucleic Acids Res. 41, 518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loedige I, Stotz M, Qamar S, Kramer K, Hennig J, Schubert T, Löffler P, Längst G, Merkl R, Urlaub H, and Meister G (2014). The NHL domain of BRAT is an RNA-binding domain that directly contacts the hunchback mRNA for regulation. Genes Dev. 28, 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loedige I, Jakob L, Treiber T, Ray D, Stotz M, Treiber N, Hennig J, Cook KB, Morris Q, Hughes TR, et al. (2015). The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of Drosophila brain-tumor-mediated gene regulation. Cell Rep. 13, 1206–1220. [DOI] [PubMed] [Google Scholar]
- Lupo G, Harris WA, and Lewis KE (2006). Mechanisms of ventral patterning in the vertebrate nervous system. Nat. Rev. Neurosci 7, 103–114. [DOI] [PubMed] [Google Scholar]
- Maller Schulman BR, Liang X, Stahlhut C, DelConte C, Stefani G, and Slack FJ (2008). The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 7, 3935–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, and Chen WM (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzola CA, Choudhri AF, Auguste KI, Limbrick DD Jr., Rogido M, Mitchell L, and Flannery AM; Pediatric Hydrocephalus Systematic Review and Evidence-Based Guidelines Task Force (2014). Pediatric hydrocephalus: systematic literature review and evidence-based guidelines. Part 2: management of posthemorrhagic hydrocephalus in premature infants. J. Neurosurg. Pediatr 14 (Suppl 1), 8–23. [DOI] [PubMed] [Google Scholar]
- McAllister JP 2nd, Williams MA, Walker ML, Kestle JRW, Relkin NR, Anderson AM, Gross PH, and Browd SR; Hydrocephalus Symposium Expert Panel (2015). An update on research priorities in hydrocephalus: overview of the third National Institutes of Health-sponsored symposium “Opportunities for Hydrocephalus Research: Pathways to Better Outcomes”. J. Neurosurg 123, 1427–1438. [DOI] [PubMed] [Google Scholar]
- McAllister JP, Guerra MM, Ruiz LC, Jimenez AJ, Dominguez-Pinos D, Sival D, den Dunnen W, Morales DM, Schmidt RE, Rodriguez EM, and Limbrick DD (2017). Ventricular zone disruption in human neonates with intraventricular hemorrhage. J. Neuropathol. Exp. Neurol 76, 358–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson E, and Clemens M (1997). Bruck syndrome (osteogenesis imperfecta with congenital joint contractures): review and report on the first North American case. Am. J. Med. Genet 70, 28–31. [PubMed] [Google Scholar]
- Merkle FT, Tramontin AD, García-Verdugo JM, and Alvarez-Buylla A (2004). Radial glia give rise to adult neural stem cells in the subventricular zone. Proc. Natl. Acad. Sci. USA 101, 17528–17532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, and Scott MP (1999). Mouse patched1 controls body size determination and limb patterning. Development 126, 4431–4440. [DOI] [PubMed] [Google Scholar]
- Mitschka S, Ulas T, Goller T, Schneider K, Egert A, Mertens J, Brüstle O, Schorle H, Beyer M, Klee K, et al. (2015). Co-existence of intact stemness and priming of neural differentiation programs in mES cells lacking Trim71. Sci. Rep 5, 11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch TN, Rostgaard K, Rasmussen ML, Wohlfahrt J, Juhler M, and Melbye M (2012). Familial aggregation of congenital hydrocephalus in a nationwide cohort. Brain 135, 2409–2415. [DOI] [PubMed] [Google Scholar]
- Narayanan R, Pirouz M, Kerimoglu C, Pham L, Wagener RJ, Kiszka KA, Rosenbusch J, Seong RH, Kessel M, Fischer A, et al. (2015). Loss of BAF (mSWI/SNF) complexes causes global transcriptional and chromatin state changes in forebrain development. Cell Rep. 13, 1842–1854. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S (2001). Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S (2002). Accounting for human polymorphisms predicted to affect protein function. Genome Res. 12, 436–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, and Henikoff S (2006). Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet 7, 61–80. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Sokpor G, Pham L, Rosenbusch J, Stoykova A, Staiger JF, and Tuoc T (2016). Epigenetic regulation by BAF (mSWI/SNF) chromatin remodeling complexes is indispensable for embryonic development. Cell Cycle 15, 1317–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DTT, Richter D, Michel G, Mitschka S, Kolanus W, Cuevas E, and Wulczyn FG (2017). The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ. 24, 1063–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Martínez-Cerdeño V, Ivic L, and Kriegstein AR (2004). Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat. Neurosci 7, 136–144. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet 43, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma V, and Ruiz i Altaba A (2004). Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 131, 337–345. [DOI] [PubMed] [Google Scholar]
- Palma V, Lim DA, Dahmane N, Sánchez P, Brionne TC, Herzberg CD, Gitton Y, Carleton A, Álvarez-Buylla A, and Ruiz i Altaba A (2005). Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 132, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen PH, Zou K, Hwang JK, Jan YN, and Zhong W (2002). Progenitor cell maintenance requires numb and numblike during mouse neurogenesis. Nature 419, 929–934. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, and Reich D (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, and Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang D, Chen Y, and Xie X (2015). DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, and Ruvkun G (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403, 901–906. [DOI] [PubMed] [Google Scholar]
- Rekate HL (2008). The definition and classification of hydrocephalus: a personal recommendation to stimulate debate. Cerebrospinal Fluid Res. 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, and Sander C (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S (2012). Neonatal posthemorrhagic hydrocephalus from prematurity: pathophysiology and current treatment concepts. J. Neurosurg. Pediatr 9, 242–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal A, Jouet M, and Kenwrick S (1992). Aberrant splicing of neural cell adhesion molecule L1 mRNA in a family with X-linked hydrocephalus. Nat. Genet 2, 107–112. [DOI] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Daly MJ, MacArthur DG, Fromer M, and Purcell SM; Exome Aggregation Consortium (2016). Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat. Genet 48, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, Kosmicki JA, Rehnström K, Mallick S, Kirby A, et al. (2014). A framework for the interpretation of de novo mutation in human disease. Nat. Genet 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, and Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. bioRxiv 10.1101/148353. [DOI] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, and Gerfin-Moser A (1993). A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100, 431–440. [DOI] [PubMed] [Google Scholar]
- Schulman BR, Esquela-Kerscher A, and Slack FJ (2005). Reciprocal expression of lin-41 and the microRNAs let-7 and mir-125 during mouse embryogenesis. Dev. Dyn 234, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, and Seelow D (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Sebai MA, Patel N, Ewida N, Kurdi W, Altweijri I, Sogaty S, Almardawi E, Seidahmed MZ, Alnemri A, et al. (2017). The genetic landscape of familial congenital hydrocephalus. Ann. Neurol 81, 890–897. [DOI] [PubMed] [Google Scholar]
- Shannon CN, Simon TD, Reed GT, Franklin FA, Kirby RS, Kilgore ML, and Wellons JC 3rd (2011). The economic impact of ventriculoperitoneal shunt failure. J. Neurosurg. Pediatr 8, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen VL, Basel-Vanagaite L, Goodman JR, Scheffer IE, Bodell A, Ganesh VS, Ravenscroft R, Hill RS, Cherry TJ, Shugart YY, et al. (2004). Etiological heterogeneity of familial periventricular heterotopia and hydrocephalus. Brain Dev. 26, 326–334. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, and Sirotkin K (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, Day IN, and Gaunt TR (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat 34, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Gough J, Mort M, Cooper DN, Day IN, and Gaunt TR (2014). Ranking non-synonymous single nucleotide polymorphisms based on disease concepts. Hum. Genomics 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day INM, Gaunt TR, and Campbell C (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikata Y, Okada T, Hashimoto M, Ellis T, Matsumaru D, Shiroishi T, Ogawa M, Wainwright B, and Motoyama J (2011). Ptch1-mediated dosage-dependent action of Shh signaling regulates neural progenitor development at late gestational stages. Dev. Biol 349, 147–159. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon TD, Riva-Cambrin J, Srivastava R, Bratton SL, Dean JM, and Kestle JR; Hydrocephalus Clinical Research Network (2008). Hospital care for children with hydrocephalus in the United States: utilization, charges, comorbidities, and deaths. J. Neurosurg. Pediatr 1, 131–137. [DOI] [PubMed] [Google Scholar]
- Slack FJ, and Ruvkun G (1998). A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem. Sci 23, 474–475. [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, and Ruvkun G (2000). The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell 5, 659–669. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Kaylor J, Pierce H, Cahr M, DeWard SJ, Schneidman-Duhovny D, Alsadah A, Salem F, Schmajuk G, and Mehta L (2015). CRB2 mutations produce a phenotype resembling congenital nephrosis, Finnish type, with cerebral ventriculomegaly and raised alpha-fetoprotein. Am. J. Hum. Genet 96, 162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart BD, Choi J, Zaidi S, Xing C, Holohan B, Chen R, Choi M, Dharwadkar P, Torres F, Girod CE, et al. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet 47, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH-Y, et al. ; 1000 Genomes Project Consortium (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd J, Rozell B, Toftgård R, and Teglund S (2009). Tumor suppressor gene co-operativity in compound Patched1 and suppressor of fused heterozygous mutant mice. Mol. Carcinog 48, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, and Osumi N (2002). Pax6 regulates specification of ventral neurone subtypes in the hindbrain by establishing progenitor domains. Development 129, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Timberlake AT, Choi J, Zaidi S, Lu Q, Nelson-Williams C, Brooks ED, Bilguvar K, Tikhonova I, Mane S, Yang JF, et al. (2016). Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. eLife 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timberlake AT, Furey CG, Choi J, Nelson-Williams C, Loring E, Galm A, Kahle KT, Steinbacher DM, Larysz D, Persing JA, and Lifton RP; Yale Center for Genome Analysis (2017). De novo mutations in inhibitors of Wnt, BMP, and Ras/ERK signaling pathways in non-syndromic midline craniosynostosis. Proc. Natl. Acad. Sci. USA 114, E7341–E7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Qu Y, Montcouquiol M, Zhou L, Komatsu K, Shi D, Fujimori T, Labeau J, Tyteca D, Courtoy P, et al. (2010). Lack of cadherins Celsr2 and Celsr3 impairs ependymal ciliogenesis, leading to fatal hydrocephalus. Nat. Neurosci 13, 700–707. [DOI] [PubMed] [Google Scholar]
- Tully HM, and Dobyns WB (2014). Infantile hydrocephalus: a review of epidemiology, classification and causes. Eur. J. Med. Genet 57, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully HM, Ishak GE, Rue TC, Dempsey JC, Browd SR, Millen KJ, Doherty D, and Dobyns WB (2016). Two hundred thirty-six children with developmental hydrocephalus: causes and clinical consequences. J. Child Neurol 31, 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuoc TC, Boretius S, Sansom SN, Pitulescu ME, Frahm J, Livesey FJ, and Stoykova A (2013a). Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 25, 256–269. [DOI] [PubMed] [Google Scholar]
- Tuoc TC, Narayanan R, and Stoykova A (2013b). BAF chromatin remodeling complex: cortical size regulation and beyond. Cell Cycle 12, 2953–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 1–33, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella MC, Choi EY, Lin SY, Reinert K, and Slack FJ (2004). The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3’UTR. Genes Dev. 18, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, and Crabtree GR (1996). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- Wang C, Pan Y, and Wang B (2010a). Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development 137, 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010b). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware JS, Samocha KE, Homsy J, and Daly MJ (2015). Interpreting de novo variation in human disease using denovolyzeR. Curr. Protoc. Hum. Genet 87, 1–15, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C, Eberhart DE, and Curran T (2000). The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer Res. 60, 2239–2246. [PubMed] [Google Scholar]
- Wetmore KM, Price MN, Waters RJ, Lamson JS, He J, Hoover CA, Blow MJ, Bristow J, Butland G, Arkin AP, and Deutschbauer A (2015). Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. MBio 6, e00306–e00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, and Yamanaka S (2014). The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 14, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Wang Z, Sharova L, Sharov AA, Ling C, Piao Y, Aiba K, Matoba R, Wang W, and Ko MS (2008). BAF250B-associated SWI/SNF chromatin-remodeling complex is required to maintain undifferentiated mouse embryonic stem cells. Stem Cells 26, 1155–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming GL, and Song H (2016). Epigenetic mechanisms in neurogenesis. Nat. Rev. Neurosci 17, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, and Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comp. Biol 11, 377–394. [DOI] [PubMed] [Google Scholar]
- Yu G, Yang Y, and Tian G (2010). Expressing and characterization of mLIN-41 in mouse early embryos and adult muscle tissues. J. Mol. Histol 41, 295–305. [DOI] [PubMed] [Google Scholar]
- Yung YC, Mutoh T, Lin M-E, Noguchi K, Rivera RR, Choi JW, Kingsbury MA, and Chun J (2011). Lysophosphatidic acid signaling may initiate fetal hydrocephalus. Sci. Transl. Med 3, 99ra87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zega K, Jovanovic VM, Vitic Z, Niedzielska M, Knaapi L, Jukic MM, Partanen J, Friedel RH, Lang R, and Brodski C (2017). Dusp16 deficiency causes congenital obstructive hydrocephalus and brain overgrowth by expansion of the neural progenitor pool. Front. Mol. Neurosci 10, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Williams MA, and Rigamonti D (2006). Genetics of human hydrocephalus. J. Neurol 253, 1255–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data for congenital hydrocephalus case-parent trios reported in this manuscript have been deposited in the NCBI database of Genotypes and Phenotypes (dbGaP). The accession number for the data reported in this paper is dbGaP: phs000744.