

Abstract
Background
Aberrant DNA methylation is significantly associated with breast cancer.
Methods
In this study, we aimed to determine novel methylation biomarkers using a bioinformatics analysis approach that could have clinical value for breast cancer diagnosis and prognosis. Firstly, differentially methylated DNA patterns were detected in breast cancer samples by comparing publicly available datasets (GSE72245 and GSE88883). Methylation levels in 7 selected methylation biomarkers were also estimated using the online tool UALCAN. Next, we evaluated the diagnostic value of these selected biomarkers in two independent cohorts, as well as in two mixed cohorts, through ROC curve analysis. Finally, prognostic value of the selected methylation biomarkers was evaluated breast cancer by the Kaplan-Meier plot analysis.
Results
In this study, a total of 23 significant differentially methylated sites, corresponding to 9 different genes, were identified in breast cancer datasets. Among the 9 identified genes, ADCY4, CPXM1, DNM3, GNG4, MAST1, mir129-2, PRDM14, and ZNF177 were hypermethylated. Importantly, individual value of each selected methylation gene was greater than 0.9, whereas predictive value for all genes combined was 0.9998. We also found the AUC for the combined signature of 7 genes (ADCY4, CPXM1, DNM3, GNG4, MAST1, PRDM14, ZNF177) was 0.9998 [95% CI 0.9994–1], and the AUC for the combined signature of 3 genes (MAST1, PRDM14, and ZNF177) was 0.9991 [95% CI 0.9976–1]. Results from additional validation analyses showed that MAST1, PRDM14, and ZNF177 had high sensitivity, specificity, and accuracy for breast cancer diagnosis. Lastly, patient survival analysis revealed that high expression of ADCY4, CPXM1, DNM3, PRDM14, PRKCB, and ZNF177 were significantly associated with better overall survival.
Conclusions
Methylation pattern of MAST1, PRDM14, and ZNF177 may represent new diagnostic biomarkers for breast cancer, while methylation of ADCY4, CPXM1, DNM3, PRDM14, PRKCB, and ZNF177 may hold prognostic potential for breast cancer.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12957-021-02124-6.
Keywords: Breast cancer, Methylation, Biomarkers, Diagnosis, Prognosis
Background
Breast cancer is the most commonly diagnosed cancer and the leading cause of cancer-associated death among women worldwide [1]. Early diagnosis and accurate prognostic assessment of breast cancer are crucial for timely targeted treatment [2]. Accumulating evidence suggests that DNA methylation may hold an important role for the development and progression of breast cancer [3–5].
DNA methylation consists in the addition of a methyl group to carbon 5-position of cytosine within a cytosine guanine (CpG) dinucleotide [6]. This molecular process is critical for several important cellular mechanisms, including embryonic development, regulation of gene expression, X-chromosome inactivation, and genomic imprinting and stability [7]. Aberrant hypo- and hypermethylation patterns of the DNA have been identified as critical players in tumorigenesis, promoting the expression or silencing of oncogenes and tumor suppressor genes, respectively [8–10]. Therefore, abnormal DNA methylation, acting as a cancer-related biomarker, could be helpful for cancer early detection and prognosis, as well as for predicting response to treatment of cancer.
DNA methylation markers are not currently in use in clinical settings for breast cancer assessment. This is mostly due to lack of evidence on their clinical value in large cohorts of breast cancer patients [4, 11–13]. Indeed, available data on the clinical potential of cancer-specific methylated markers rely on platforms with low genomic coverage, small sample datasets, or missing appropriate healthy counterparts for comparison [14].
In the present study, we aimed to evaluate methylation changes specific to breast cancer that could be used as tools in the clinical setting for diagnostic and prognostic assessment of patients. To achieve this goal, we used different bioinformatics approaches to analyze several publicly available methylation datasets of samples collected from cancer patients and healthy counterparts.
Methods
Description of breast cancer and control samples
Breast cancer and control samples publicly available at the Gene Expression Omnibus database (GEO, https://www.ncbi.nlm.nih.gov/geo/) were used for the different bioinformatics analyses. Cancer samples were obtained from GSE72308, which includes three sets (GSE72245, N = 118; GSE72251, N = 119; GSE72254, N = 58), as well as from GSE141338 (N = 42), GSE100850 (N = 34), and GSE117439 (N = 52). DNA methylation data from normal tissue samples was used as control and was obtained from GSE88883 (N = 100), GSE74214 (N = 18), GSE141338 (N = 6), GSE100850 (N = 5), and GSE101961 (N = 121) datasets. Data from GSE41169 (N = 95) were used as a blood control dataset. Information of all samples is compiled and available in supplementary information (Fig. 1; Table S1).
Fig. 1.

Workflow of the study. A multistep marker discovery analysis was performed to identify differentially methylated gene-based biomarkers of breast cancer
Differentially methylated analyses
Data from 118 breast cancer samples (GSE72245) and 50 normal samples (GSE88883) was analyzed by R package ChAMP, according to a previously described protocol [15]. Probe signal was removed when detected p value was above 0.05, or when more than 1% of the dataset contained no information. Briefly, differential methylation analysis was performed at probe (lmFit from limma; adjusted p ≤ 1 × 10−35; minimum delta beta value of 0.35) or region level (bumphunter from minfi; regions represented by at least two probes with L ≥ 2). In order to minimize the risk of false positive detection in blood tests, methylation in leukocytes was excluded (GSE41169; maximum beta value allowed = 0.1). Differentially methylated probes were limited to those overlapping differentially methylated regions, which was distant of a maximum 150 bp, not located in centromeres or telomeres. Lastly, methylation level of differentially methylated genes in control and breast cancer samples was plotted with the use of GraphPad Prism software.
UALCAN database analysis
UALCAN online tool (http://ualcan.path.uab.edu) is designed to provide easy access to publicly available cancer transcriptome data (TCGA and MET500 transcriptome sequencing), including 793 breast cancer samples and 97 normal samples. Therefore, it was used to perform a comprehensive analysis of promoter DNA methylation patterns in control and breast cancer samples [16]. In this study, the beta value indicated level of DNA methylation ranging from 0 (unmethylated) to 1 (fully methylated), and different beta value cutoff was considered as hypomethylation [beta value 0.3–0.25] or hypermethylation [beta value 0.7–0.5]. Additionally, mRNA expression of the identified genes in breast cancer was also analyzed using UALCAN.
Marker discovery analysis
Receiver operating characteristic (ROC) analyses were performed in GSE72251 and GSE88883 with the pROC package in R Bioconductor to establish thresholds, considering normal and adjacent mucosa as positive outcome and cancer as negative; only loci showing a threshold below 0.35 were kept. ROC curve was generated, and area under the curve (AUC) with the binomial exact confidence interval was calculated. For AUC values above 0.9, the differentially methylated gene was deemed able to distinguish between control and breast cancer with excellent specificity and sensitivity. AUC for the combined epigenetic signature was assessed using a logistic regression model [17]. Each threshold was used to stratify the two mixed cohorts, defining a positive predictive value and negative predictive value for discriminating normal adjacent from tumor tissue. The two mixed cohorts were as follows: mixed cohort 1 included breast cancer (GSE141338, GSE100850, and GSE117439) and control (GSE101961) samples, whereas mixed cohort 2 included breast cancer (GSE72254) and control (GSE74214, GSE141338, and GSE100850) samples.
Survival analysis
Prognostic value of the selected DNA methylation-driven genes was evaluated through the Kaplan-Meier plot assessment (http://kmplot.com/analysis/) with data from the mRNA breast cancer database [18]. Median value of all gene expression levels was used as threshold to identify and separate cases with high or low gene expression. p < 0.05 was considered significant.
Results
Identification of differentially methylated genes
To evaluate the DNA methylation pattern in breast cancer, we started by comparing 118 breast cancer and 50 control samples. We identified 105,143 differentially methylated positions and 8764 regions in breast cancer cases compared to controls (Fig. 2). Next, we filtered these differentially methylated sites as described in the “Methods” section, allowing us to refine our findings to a total of 23 differentially methylated sites. Importantly, these sites were directly linked to the transcription of 9 genes, namely adenylyl cyclase 4 (ADCY4), carboxypeptidase X (CPXM1), dynamin 3 (DNM3), guanine nucleotide binding-protein gamma subunit 4 (GNG4), microtubule associated serine-threonine kinase 1 (MAST1), microRNA 129-2 (mir129-2), PR domain zinc finger protein 14 (PRDM14), protein kinase C beta (PRKCB), and zinc finger protein 177 (ZNF177) (Table S2; Fig. 3; Table 1). All genes, with exception of PRKCB, had significantly higher levels of DNA methylation in breast cancer samples compared to controls.
Fig. 2.
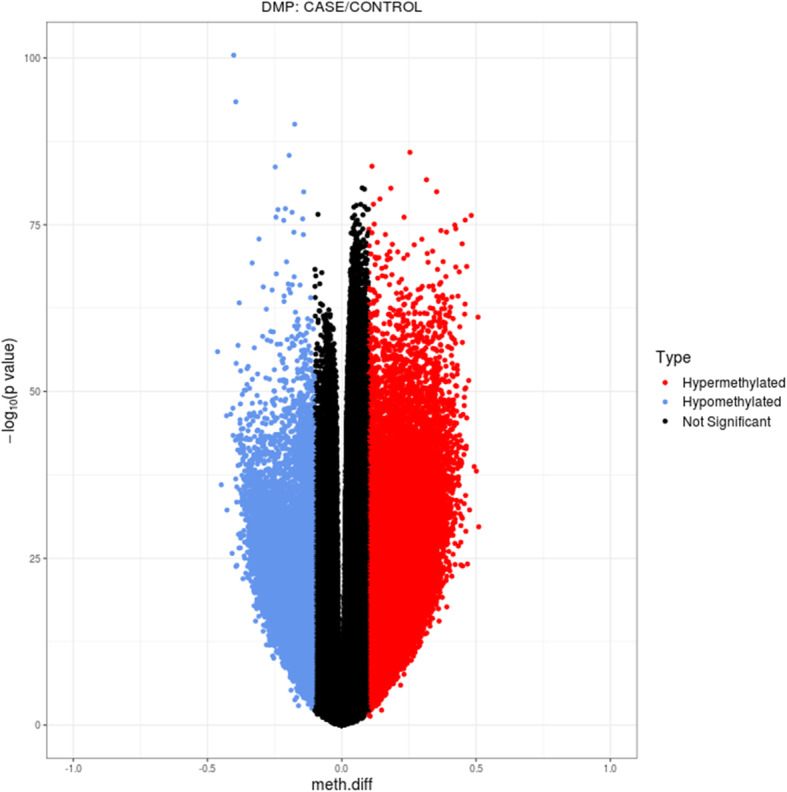
Volcano plot of DMPs. DMPs, differentially methylated positions
Fig. 3.

The selected methylation genes had significantly higher levels of DNA methylation in breast cancer as compared with controls. a ADCY4. b CPXM1. c DNM3. d GNG4. e MAST1. f mir129-2. g PRDM14. h ZNF177. ***p < 0.001
Table 1.
Twenty-three differentially methylated sites corresponding to 9 genes
| Site | CHR | MAPINFO | Gene | Relation to CpG island | McaM | McoM | p value | BH.adjust | DMR |
|---|---|---|---|---|---|---|---|---|---|
| cg27429080 | 1 | 171810300 | DNM3 | N_Shore | 0.5053 | 0.0615 | 9.76E−25 | 3.94E−24 | DMR_94 |
| cg17154724 | 1 | 171810322 | DNM3 | N_Shore | 0.5041 | 0.0359 | 7.13E−25 | 2.90E−24 | DMR_94 |
| cg04399751 | 1 | 171810376 | DNM3 | N_Shore | 0.4987 | 0.0487 | 1.43E−24 | 5.75E−24 | DMR_94 |
| cg04986015 | 1 | 171810433 | DNM3 | N_Shore | 0.4563 | 0.0678 | 3.25E−25 | 1.34E−24 | DMR_94 |
| cg17816394 | 1 | 235814009 | GNG4 | Island | 0.4519 | 0.0852 | 6.46E−29 | 3.12E−28 | DMR_179 |
| cg09649610 | 1 | 235814039 | GNG4 | Island | 0.5248 | 0.0813 | 1.26E−30 | 6.61E−30 | DMR_179 |
| cg00384539 | 8 | 70983567 | PRDM14 | Island | 0.4421 | 0.0848 | 1.01E−28 | 4.83E−28 | DMR_612 |
| cg13267264 | 8 | 70983600 | PRDM14 | Island | 0.4375 | 0.0826 | 3.64E−26 | 1.56E−25 | DMR_612 |
| cg14416371 | 11 | 43602847 | mir129-2 | Island | 0.4446 | 0.0395 | 6.48E−30 | 3.28E−29 | DMR_166 |
| cg01939477 | 11 | 43602879 | mir129-2 | Island | 0.4054 | 0.0260 | 1.98E−30 | 1.03E−29 | DMR_166 |
| cg16407471 | 11 | 43602914 | mir129-2 | Island | 0.4381 | 0.0688 | 5.40E−29 | 2.62E−28 | DMR_166 |
| cg05376374 | 11 | 43602920 | mir129-2 | Island | 0.4158 | 0.0452 | 2.27E−33 | 1.36E−32 | DMR_166 |
| cg23179456 | 14 | 24803873 | ADCY4 | Island | 0.4366 | 0.0623 | 7.06E−20 | 2.29E−19 | DMR_181 |
| cg12265829 | 14 | 24804022 | ADCY4 | Island | 0.4506 | 0.0883 | 4.94E−22 | 1.77E−21 | DMR_181 |
| cg03217795 | 16 | 23847556 | PRKCB | Island | 0.4211 | 0.0246 | 1.49E−32 | 8.54E−32 | DMR_224 |
| cg05436658 | 16 | 23847568 | PRKCB | Island | 0.3785 | 0.0173 | 1.11E−29 | 5.53E−29 | DMR_224 |
| cg08065231 | 19 | 9473684 | ZNF177 | Island | 0.4413 | 0.0755 | 1.61E−27 | 7.33E−27 | DMR_244 |
| cg09578475 | 19 | 9473688 | ZNF177 | Island | 0.5619 | 0.1547 | 1.38E−29 | 6.86E−29 | DMR_244 |
| cg08305551 | 19 | 12978611 | MAST1 | Island | 0.4241 | 0.0545 | 3.83E−30 | 1.96E−29 | DMR_340 |
| cg02776664 | 19 | 12978624 | MAST1 | Island | 0.4069 | 0.0455 | 4.54E−33 | 2.68E−32 | DMR_340 |
| cg06537894 | 19 | 12978706 | MAST1 | Island | 0.4117 | 0.0575 | 3.50E−24 | 1.38E−23 | DMR_340 |
| cg22304612 | 20 | 2781241 | CPXM1 | Island | 0.4544 | 0.0626 | 2.41E−32 | 1.37E−31 | DMR_97 |
| cg07113642 | 20 | 2781262 | CPXM1 | Island | 0.4436 | 0.0294 | 3.51E−29 | 1.72E−28 | DMR_97 |
CHR and MAPINFO represent chromosome and position information; McaM represents the mean methylation percentage of the cases, and the McoM represents the mean methylation percentage of the controls; DMR represents differentially methylated region; p value is calculated through the Wilcoxon rank-sum test followed by FDR (false discovery rate) adjustment for multiple correction
In order to validate the correlation between DNA methylation levels and mRNA expression of the identified genes in breast cancer, we used the online tool UALCAN. As expected, methylation levels of ADCY4, CPXM1, DNM3, GNG4, MAST1, PRDM14, and ZNF177 were found to be increased in breast cancer, and all with p values lower than 0.001 (Fig. 4). Note that information related to mir129-2 was not available in UALCAN, so we could not conduct this analysis. Then, we found the mRNA expression of ADCY4, CPXM1, GNG4, and ZNF177 were significantly decreased in breast cancer, the mRNA expression of MAST1 was significantly upregulated, but there was no difference of DNM3 mRNA expression between breast cancer samples and controls (Fig. 5).
Fig. 4.

Validation of DNA methylation level using the online tool UALCAN. The methylation levels of a ADCY4, b CPXM1, c DNM3, d GNG4, e MAST1, f PRDM14, and g ZNF177 were also significantly hypermethylated in breast cancer. ***p < 0.001
Fig. 5.

The mRNA expression of selected genes in breast cancer and controls. a ADCY4. b CPXM1. c DNM3. d GNG4. e MAST1. f ZNF177. *p < 0.05; ***p < 0.001
Diagnostic potential of differentially methylated genes
In this study, ROC analyses were performed in GSE72251 and GSE88883 with the pROC package in R Bioconductor to establish thresholds, and only loci showing a threshold below 0.35 were kept. Then, AUC values of ADCY4 (0.9471), CPXM1 (0.9856), DNM3 (0.9506), GNG4 (0.9589), MAST1 (0.9950), PRDM14 (0.9883), and ZNF177 (0.9786) were all above 0.9 (Fig. 6). Then, we validated the diagnostic value of the combined logistic regression model in these two cohorts, and found the AUC for the combined signature of 7 genes (ADCY4, CPXM1, DNM3, GNG4, MAST1, PRDM14, ZNF177) was 0.9998 [95% CI 0.9994–1] (Fig. 7a) and the AUC for the combined signature of 3 genes (MAST1, PRDM14, and ZNF177) was 0.9991 [95% CI 0.9976–1] (Fig. 7b).
Fig. 6.

ROC curves and AUC with 95% confidence intervals for the candidate markers. The AUC for individual markers: a ADCY4, b CPXM1, c DNM3, d GNG4, e MAST1, f PRDM14, and g ZNF177
Fig. 7.

Validating the diagnostic value of the combined logistic regression model in two cohorts. a The AUC for the combined signature of 7 genes (ADCY4, CPXM1, DNM3, GNG4, MAST1, PRDM14, ZNF177) using a logistic regression model. b The AUC for the combined signature of 3 genes (MAST1, PRDM14, and ZNF177) using a logistic regression model
Next, each threshold was used to stratify the two mixed cohorts. Our results showed that the breast cancer specificity of each gene ranged from 50.41 to 98.35%, while the sensitivity ranged from 84.25 to 97.64%, and accuracy from 67.82 to 91.13% in mixed cohort 1 (Table 2). Particularly, the specificity, sensitivity, and accuracy of MAST1 were 81.82%, 97.64%, and 89.92%; those of PRDM14 were 97.52%, 84.25%, and 90.73%; and those of ZNF177 were 80.17%, 89.76%, and 85.08%, respectively (Table 2). Results obtained in mixed cohort 2 also followed the same trend, with specificity, sensitivity, and accuracy of MAST1 being 75.86%, 100%, and 91.95%; of PRDM14 being 89.66%, 86.21%, and 87.36%; and of ZNF177 being 89.66%, 93.10%, and 91.95%, respectively (Table 3).
Table 2.
Validation of 7 differentially methylated genes through mixed cohort 1, including breast cancer (GSE141338, GSE100850, GSE117439) and normal (GSE101961)
| Gene | Case, N | Control, N | Specificity (%) | Sensitivity (%) | Accuracy (%) |
|---|---|---|---|---|---|
| ADCY4 | 81 | 121 | 50.41 | 93.83 | 67.82 |
| CPXM1 | 127 | 121 | 85.12 | 95.28 | 90.32 |
| DNM3 | 127 | 121 | 59.50 | 89.76 | 75.00 |
| GNG4 | 127 | 121 | 98.35 | 84.25 | 91.13 |
| MAST1 | 127 | 121 | 81.82 | 97.64 | 89.92 |
| PRDM14 | 127 | 121 | 97.52 | 84.25 | 90.73 |
| ZNF177 | 127 | 121 | 80.17 | 89.76 | 85.08 |
Table 3.
Validation of 7 differentially methylated genes through mixed cohort 2, including breast cancer (GSE72254) and normal tissues (GSE74214, GSE141338, GSE100850)
| Gene | Case, N | Control, N | Specificity (%) | Sensitivity (%) | Accuracy (%) |
|---|---|---|---|---|---|
| ADCY4 | 58 | 29 | 62.07 | 87.93 | 79.31 |
| CPXM1 | 58 | 29 | 72.41 | 79.31 | 77.01 |
| DNM3 | 58 | 29 | 58.62 | 62.07 | 60.92 |
| GNG4 | 58 | 29 | 93.10 | 63.79 | 73.56 |
| MAST1 | 58 | 29 | 75.86 | 100.00 | 91.95 |
| PRDM14 | 58 | 29 | 89.66 | 86.21 | 87.36 |
| ZNF177 | 58 | 29 | 89.66 | 93.10 | 91.95 |
Prognosis analyzed by K-M plotter
To further explore the clinical value of these biomarkers, we evaluated whether 6 of our differentially methylated genes—ADCY4, CPXM1, DNM3, PRDM14, PRKCB, and ZNF177—had any relation with overall survival of breast cancer patients. Hazard ratios of these 6 genes showed significant differences between the high-expression and the low-expression groups, with high expression of all genes being significantly associated with longer overall survival (Fig. 8).
Fig. 8.

Analysis of differentially methylated genes’ prognosis in breast cancer patients using the Kaplan-Meier plotter. logrank p < 0.05 was statistically significant. HR, hazard ratio; the greater the absolute value of (HR-1), the greater the difference between groups in overall survival
Discussion
Breast cancer has extremely high mortality worldwide, mostly due to late diagnosis. Cancer-specific DNA methylation patterns are correlated with gene silencing or activation in several types of cancers [19, 20]. Recent studies highlight that aberrant DNA methylation is significantly associated with breast cancer and demonstrated that DNA methylation analysis may help predict the outcome of patients with breast cancer [12, 21]. In this study, we identified differentially methylated genes and confirmed the diagnostic and prognostic value of 6 of these methylation-based biomarkers in breast cancer using a bioinformatics approach.
We first identified 23 differentially methylated CpG sites in breast cancer samples as compared to control counterparts. And the 23 differentially methylated CpG sites correspond to 9 genes, and then, 8 of these 9 genes were coding genes—ADCY4, CPXM1, DNM3, GNG4, MAST1, mir129-2, PRDM14, and ZNF177—that had significantly higher levels of DNA methylation in breast cancer. Similarly, methylation levels described in UALCAN analysis for all these genes were found to be significantly higher in patients with breast cancer, with exception for mir129-2 that was not possible to assess. Further analysis revealed the potential of these 8 differentially methylated genes to accurately predict the outcome of patients in training and validation datasets, suggesting that they could be used as biomarkers for breast cancer diagnosis. Additionally, combination of 7 of these methylation markers significantly improved our ability to predict the outcome of breast cancer patients. Overall, we found that MAST1, PRDM14, and ZNF177 had high sensitivity, specificity, and accuracy for the diagnosis of breast cancer.
Growing evidence shows a strong relationship between epigenetic and genetic aberrations of MAST1, PRDM14 [22], and ZNF177 [17] in tumorigenesis. Previous studies reported that abnormal MAST1 expression is significantly associated with worse cancer prognosis [23, 24]. Oishi et al. found that aberrant promoter demethylation of MAST1 could be responsible for overexpression of this gene in malignant pheochromocytoma and paraganglioma [25]. Other studies have shown that silencing of PRDM14 can suppress tumorigenicity and metastasis potential of breast cancer cells [26], while methylation-mediated gene silencing of PRDM14 leads to apoptosis evasion in human papillomavirus-positive cancer cells [27]. Several reports have also shown that methylation of ZNF177 is associated with different types of cancer including gastric and endometrial cancers [28], as well as non-small cell lung carcinoma. ZNF177 is methylation-silenced in gastric cancer cell lines, whereas methylation of its promotor is a frequent epigenetic event in endometrial cancer. Indeed, ROC analysis of ZNF177 has demonstrated that it can identify endometrial carcinomas cases with a sensitivity, specificity, and accuracy of 92.3%, 94.4%, and 95.1%, respectively. Furthermore, hypermethylated CpG islands within ZNF177 were selected as candidate biomarker for further validation in NSCLC. Nakakido et al. demonstrated that ZNF177 is overexpressed in breast cancer and plays a critical role in cancer cell proliferation [29]. However, the role of MAST1, PRDM14, and ZNF177 in diagnosis and prognosis of breast cancer remains unclear.
Our findings add a new layer of evidence to the epigenetic landscape of breast cancer, providing convincing clues that MAST1, PRDM14, and ZNF177 are differentially methylated in breast cancer, as well as that they may serve as potential drivers and biomarkers for breast cancer. Furthermore, our study demonstrates that high expression of ADCY4, CPXM1, DNM3, PRDM14, PRKCB, and ZNF177 are significantly associated with longer patient survival. This finding supports the hypothesis that methylation-driven genes are likely to be associated with clinical outcomes in cancer and can be used as potential biomarkers for predicting the outcome of breast cancer patients.
Conclusions
In summary, we have identified and independently validated abnormal DNA methylation patterns in MAST1, PRDM14, and ZNF177 as potential biomarkers for breast cancer diagnosis. Moreover, we showed that DNA methylation landscape of ADCY4, CPXM1, DNM3, PRDM14, PRKCB, and ZNF177 could be selected as accurate biomarkers for the prognosis of breast cancer. Overall, these findings provide a novel epigenetic predictive model that may help improve the diagnosis and prognosis of breast cancer.
Supplementary Information
Additional file 1: Table S1. The information of all samples. Table S2. A total of 23 differentially methylated sites were identified.
Acknowledgements
Not applicable.
Authors’ contributions
ZX.X, Q.Y, and P.H designed this study. GB.Z, JY.J, HY.Z, and YF.S performed the data collection and collation. All the authors were involved in the analysis and interpretation of data. ZQ.Y and XH.M wrote the paper. Q.Y, XH.M, and P.H reviewed and revised the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work is appropriately investigated and resolved.
Funding
This study was supported in part by the Project of Application on Public Welfare Technology in Zhejiang Province (No. LGF18H160022), the Major Project for Medical and Health Science and Technology of Zhejiang Province (No. WKJ-ZJ-1716), and the General Project Funds from the Health Department of Zhejiang Province (No. 2016KYB026).
Availability of data and materials
The datasets used during the present study are available from the corresponding author upon reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiao-hong Mao and Qiang Ye contributed equally to this work.
Contributor Information
Zi-xue Xuan, Email: xuanzixue0222@163.com.
Ping Huang, Email: huangping1841@zjcc.org.cn.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Goldhirsch A, Winer EP, Coates AS, Gelber RD, Piccart-Gebhart M, Thürlimann B, Senn HJ. Personalizing the treatment of women with early breast cancer: highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann Oncol. 2013;24:2206–2223. doi: 10.1093/annonc/mdt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang Q, Cheng J, Cao X, Surowy H, Burwinkel B. Blood-based DNA methylation as biomarker for breast cancer: a systematic review. Clin Epigenetics. 2016;8:115. doi: 10.1186/s13148-016-0282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen X, Zhang J, Dai X. DNA methylation profiles capturing breast cancer heterogeneity. BMC Genomics. 2019;20:823. doi: 10.1186/s12864-019-6142-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Downs BM, Mercado-Rodriguez C, Cimino-Mathews A, Chen C, Yuan JP, Van Den Berg E, Cope LM, Schmitt F, Tse GM, Ali SZ, et al. DNA methylation markers for breast cancer detection in the developing world. Clin Cancer Res. 2019;25:6357–6367. doi: 10.1158/1078-0432.CCR-18-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch A, Joosten SC, Feng Z, de Ruijter TC, Draht MX, Melotte V, Smits KM, Veeck J, Herman JG, Van Neste L, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15:459–466. doi: 10.1038/s41571-018-0004-4. [DOI] [PubMed] [Google Scholar]
- 7.Mahmood N, Rabbani SA. Targeting DNA hypomethylation in malignancy by epigenetic therapies. Adv Exp Med Biol. 2019;1164:179–196. doi: 10.1007/978-3-030-22254-3_14. [DOI] [PubMed] [Google Scholar]
- 8.Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- 9.Picardo F, Romanelli A, Muinelo-Romay L, Mazza T, Fusilli C, Parrella P, Barbazán J, Lopez-López R, Barbano R, De Robertis M, et al. Diagnostic and prognostic value of B4GALT1 hypermethylation and its clinical significance as a novel circulating cell-free DNA biomarker in colorectal cancer. Cancers (Basel) 2019;11:1598. doi: 10.3390/cancers11101598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong S, Li W, Wang L, Hu J, Song Y, Zhang B, Ren X, Ji S, Li J, Xu P, et al. Histone-related genes are hypermethylated in lung cancer and hypermethylated HIST1H4F could serve as a pan-cancer biomarker. Cancer Res. 2019;79:6101–6112. doi: 10.1158/0008-5472.CAN-19-1019. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S, Wang Y, Gu Y, Zhu J, Ci C, Guo Z, Chen C, Wei Y, Lv W, Liu H, et al. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Mol Oncol. 2018;12:1047–1060. doi: 10.1002/1878-0261.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Ruijter TC, van der Heide F, Smits KM, Aarts MJ, van Engeland M, Heijnen VCG. Prognostic DNA methylation markers for hormone receptor breast cancer: a systematic review. Breast Cancer Res. 2020;22:13. doi: 10.1186/s13058-020-1250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moss J, Zick A, Grinshpun A, Carmon E, Maoz M, Ochana BL, Abraham O, Arieli O, Germansky L, Meir K, et al. Circulating breast-derived DNA allows universal detection and monitoring of localized breast cancer. Ann Oncol. 2020;31:395–403. doi: 10.1016/j.annonc.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Sangtani A, Wang C, Weaver A, Hoppman NL, Kerr SE, Abyzov A, Shridhar V, Staub J, Kocher JA, Voss JS, et al. Combining copy number, methylation markers, and mutations as a panel for endometrial cancer detection via intravaginal tampon collection. Gynecol Oncol. 2020;156:387–392. doi: 10.1016/j.ygyno.2019.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barault L, Amatu A, Siravegna G, Ponzetti A, Moran S, Cassingena A, Mussolin B, Falcomata C, Binder AM, Cristiano C, et al. Discovery of methylated circulating DNA biomarkers for comprehensive non-invasive monitoring of treatment response in metastatic colorectal cancer. Gut. 2018;67:1995–2005. doi: 10.1136/gutjnl-2016-313372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diaz-Lagares A, Mendez-Gonzalez J, Hervas D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga LM, Zulueta J, et al. A novel epigenetic signature for early diagnosis in lung cancer. Clin Cancer Res. 2016;22:3361–3371. doi: 10.1158/1078-0432.CCR-15-2346. [DOI] [PubMed] [Google Scholar]
- 18.Lv L, Cao L, Hu G, Shen Q, Wu J. Methylation-driven genes identified as novel prognostic indicators for thyroid carcinoma. Front Genet. 2020;11:294. doi: 10.3389/fgene.2020.00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Almeida BP, Apolónio JD, Binnie A, Castelo-Branco P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer. 2019;19(1):219. [DOI] [PMC free article] [PubMed]
- 20.Sun X, Tian Y, Wang J, Sun Z, Zhu Y. Genome-wide analysis reveals the association between alternative splicing and DNA methylation across human solid tumors. BMC Med Genet. 2020;13:4. doi: 10.1186/s12920-019-0654-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Ruijter TC, Smits KM, Aarts MJ, van Hellemond IEG, Van Neste L, de Vries B, Peer PGM, Veeck J, van Engeland M, Tjan-Heijnen VCG. The trans-DATA study: aims and design of a translational breast cancer prognostic marker identification study. Diagn Progn Res. 2019;3:20. doi: 10.1186/s41512-019-0065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sybirna A, Tang WWC, Pierson Smela M, Dietmann S, Gruhn WH, Brosh R, Surani MA. A critical role of PRDM14 in human primordial germ cell fate revealed by inducible degrons. Nat Commun. 2020;11:1282. doi: 10.1038/s41467-020-15042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin L, Chun J, Pan C, Li D, Lin R, Alesi GN, Wang X, Kang HB, Song L, Wang D, et al. MAST1 drives cisplatin resistance in human cancers by rewiring cRaf-independent MEK activation. Cancer Cell. 2018;34:315–330. doi: 10.1016/j.ccell.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu X, Sheng P, Sun J, Zhao X, Zhang J, Li Y, Zhang Y, Zhang W, Wang J, Liu K, et al. The circular RNA circMAST1 promotes hepatocellular carcinoma cell proliferation and migration by sponging miR-1299 and regulating CTNND1 expression. Cell Death Dis. 2020;11:340. doi: 10.1038/s41419-020-2532-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oishi T, Iino K, Okawa Y, Kakizawa K, Matsunari S, Yamashita M, Taniguchi T, Maekawa M, Suda T, Oki Y. DNA methylation analysis in malignant pheochromocytoma and paraganglioma. J Clin Transl Endocrinol. 2017;7:12–20. doi: 10.1016/j.jcte.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taniguchi H, Imai K. Silencing PRDM14 via oligonucleotide therapeutics suppresses tumorigenicity and metastasis of breast cancer. Methods Mol Biol. 1974;2019:233–243. doi: 10.1007/978-1-4939-9220-1_18. [DOI] [PubMed] [Google Scholar]
- 27.Snellenberg S, Cillessen SA, Van Criekinge W, Bosch L, Meijer CJ, Snijders PJ, Steenbergen RD. Methylation-mediated repression of PRDM14 contributes to apoptosis evasion in HPV-positive cancers. Carcinogenesis. 2014;35:2611–2618. doi: 10.1093/carcin/bgu197. [DOI] [PubMed] [Google Scholar]
- 28.Chen YC, Tsao CM, Kuo CC, Yu MH, Lin YW, Yang CY, Li HJ, Yan MD, Wang TJ, Chou YC, Su HY. Quantitative DNA methylation analysis of selected genes in endometrial carcinogenesis. Taiwan J Obstet Gynecol. 2015;54:572–579. doi: 10.1016/j.tjog.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Nakakido M, Tamura K, Chung S, Ueda K, Fujii R, Kiyotani K, Nakamura Y. Phosphatidylinositol glycan anchor biosynthesis, class X containing complex promotes cancer cell proliferation through suppression of EHD2 and ZIC1, putative tumor suppressors. Int J Oncol. 2016;49:868–876. doi: 10.3892/ijo.2016.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. The information of all samples. Table S2. A total of 23 differentially methylated sites were identified.
Data Availability Statement
The datasets used during the present study are available from the corresponding author upon reasonable request.