

Abstract
Atherosclerotic cardiovascular disease (ASCVD) proceeds through a series of stages: initiation, progression (or regression), and complications. By integrating known biology regarding molecular signatures of each stage with recent advances in high-dimensional molecular data acquisition platforms (to assay the genome, epigenome, transcriptome, proteome, metabolome, and gut microbiome), “snapshots” of each phase of ASCVD development can be captured. In this review, we will summarize emerging approaches for assessment of ASCVD disease risk in humans using peripheral blood molecular signatures and molecular imaging approaches. We will then discuss the potential (and challenges) for these snapshots to be integrated into a personalized movie providing dynamic readouts of an individual’s ASCVD risk status throughout the life course.
Keywords: Cardiovascular disease risk prediction, prevention, genomics, proteomics, metabolomics, Biomarkers, Cardiovascular Disease, Primary Prevention, Atherosclerosis
INTRODUCTION
Atherosclerotic cardiovascular disease (ASCVD) remains the leading cause of mortality worldwide accounting for approximately 17.6 million deaths annually.1 At the population level, traditional clinical risk factors explain a large proportion of the attributable risk,2 but their ability to predict future CVD events in individuals is more limited.3 Combining recent advances in our understanding of the molecular biology of atherosclerosis development in experimental models4 with the emerging capability to ascertain corresponding molecular data in vivo in humans may herald the next frontier for ASCVD risk assessment.5
Atherosclerotic CVD can be conceptualized in three phases: initiation, progression, and complications.4 “Snapshots” of each phase can be ascertained by integrating known biology with information gained through high-throughput, high-dimensional molecular data acquisition platforms, genomics, and molecular imaging. The challenge (and opportunity) is to integrate these static snapshots into a dynamic movie that details the evolution of ASCVD over the life course. In this review, we will summarize the primary biological mechanisms underlying ASCVD, discuss state-of-the-art approaches for assessment of disease risk and activity in humans, and offer projections for how molecular insights may be integrated into clinical risk prediction approaches in the coming years.
ATHEROSCLEROSIS INITIATION
Molecular Biology
Initiation of atherosclerosis fundamentally involves three processes: atherogenic lipid deposition, pro-inflammatory conditions, and endothelial dysfunction (Figure 1). Atherogenic apolipoprotein B-containing lipoproteins (mainly low-density lipoprotein cholesterol, LDL-C) initiate atherosclerosis by depositing in the arterial intima.4,6 This deposition is directly related to circulating levels of atherogenic lipoproteins, with recent evidence suggesting that atherosclerosis would probably not occur with LDL-C levels not in excess of physiologic needs (10–20 mg/dL).7 Retained LDL-C particles contribute to atherogenesis via promotion of macrophage transition to atherogenic foam cells, by stimulating immunologic responses, and through the formation of reactive oxygen species and other inflammatory mediators.4,8,9 LDL-C in the arterial wall also directly binds to intimal proteoglycans and lipid-proteoglycan aggregates enter smooth muscle cells to further support growth of the developing atheroma (plaque).10,11 Other lipid particles are involved in atherogenesis. Triglycerides are causally linked with atherosclerosis12 and may act primarily through pro-inflammatory pathways.13 Despite strong inverse associations for high-density lipoprotein cholesterol (HDL-C) with atherosclerotic events in observational studies,14 its causal role in protecting against atherosclerosis has been questioned by some studies of genetically-mediated HDL-C levels, and by trials of HDL-C raising drugs that have not yet proven to be clinically beneficial.4 Lipoprotein(a), which has a similar structure to the LDL-C particle but with the addition of an apolipoprotein(a) molecule, has both pro-inflammatory and pro-atherogenic effects15 that explain its causal relationship with atherosclerosis.16
Figure 1. The molecular biology of ASCVD and its peripheral blood signature.
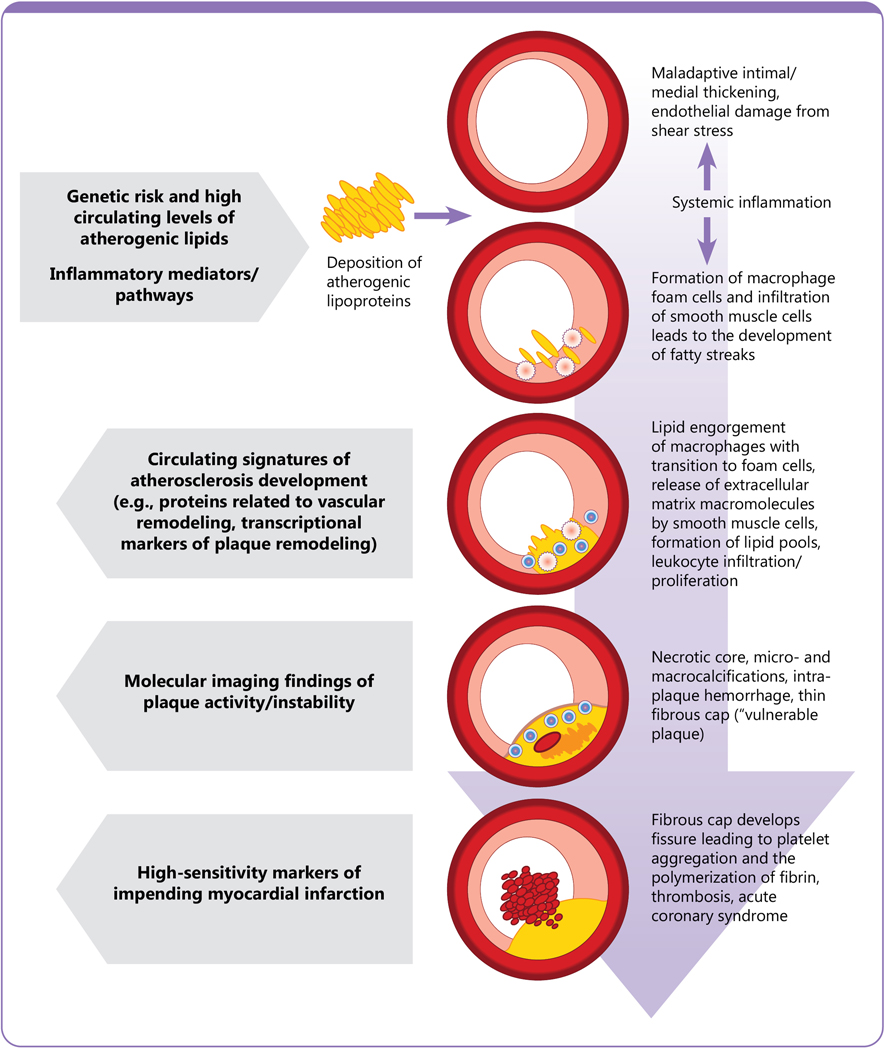
Initiation of atherosclerosis commences with a pro-atherogenic, lipid rich, inflammatory milieu leading to focal inflammation, maladaptive intimal and medial thickening, and endothelial damage. Markers of elevated blood lipid values and systemic inflammation (and their genetic and epigenetic determinants) can be detected in circulating blood. As atherosclerosis progresses, fatty streaks develop into atherosclerotic plaques through migration of smooth muscle cells and lipid accumulation. This process is heavily influenced by the functions of different leukocyte species including macrophage polarization and transition to foam cells and lymphocyte infiltration and proliferation within developing plaques. Programmed leukocyte cell death and micro-calcifications can promote a necrotic core, which, coupled with a thin fibrous cap, are characteristic of ‘vulnerable plaques’ more prone to rupture. Epigenetic, proteomic, and metabolomic fingerprints of developing plaque characteristics can be detected in circulating blood. Molecular imaging and cell-type specific profiling via cell sorting and single cell sequencing may also identify adverse plaque features. Complications of ASCVD primarily occur as a result of critical blockages and/or plaque rupture events (frequently preceded by subclinical intra-plaque hemorrhages) leading to thrombosis and acute organ ischemia. Identification of early signs of vessel occlusion prior to symptom development may be facilitated by assaying high-sensitivity transcriptomic, proteomic, and metabolomic markers in circulating blood.
Inflammation is integral to the initiation of atherogenesis. High levels of circulating LDL-C promote arterial wall inflammation and stimulate the conversion of the pro-inflammatory mediator interleukin-1ß to its bioactive cytokine.17 Indeed, the promotion of systemic inflammation represents an important mechanistic link between clinical risk factors and ASCVD. High blood pressure, smoking, and greater amounts of visceral adiposity – which correlates with insulin resistance and type 2 diabetes mellitus – all lead to upregulation of systemic inflammatory responses.4,18 This systemic inflammatory response can be measured by pro-inflammatory markers such as C-reactive protein (CRP), which is robustly – but perhaps not causally19 – linked to CVD risk.20 One end-result of a heightened inflammatory milieu is the activation of vascular endothelium to express chemoattractants and adhesion molecules, thereby leading to leukocyte migration and adhesion.21 Adaptive immunity also plays an important role in atherogenesis and both B and T lymphocytes can be identified within the microarchitecture of developing plaques.4,22 The observation that certain helper T-cell subsets promote atherosclerosis, while others counteract it,17 suggests that adaptive immunity may be a key modulator of the atherosclerotic process.
The vascular endothelium represents the interface between circulating blood and the arterial intima, where atherosclerosis formation occurs. Endothelial damage and dysfunction (due to exposure to risk factors) contribute directly to atherogenesis. Lower bioavailability of endothelial nitric oxide results from endothelial damage and impairs the maintenance of laminar blood flow and vascular hemostasis.23 Dysfunctional endothelium expresses vascular adhesion molecules that attract leukocytes and further contribute to the development and growth of plaques.23,24
Assessing atherosclerosis initiation in humans
Atherosclerosis initiation, therefore, relies on a systemic pro-atherogenic milieu that may be particularly amenable to interrogation via detailed profiling of circulating molecules and their genetic determinants. Evidence supporting the measurement of various “omics” levels to improve prediction and identification of atherosclerosis CVD development are discussed in the following section.
Genetics
Identification of genetic predispositions to ASCVD have in many cases validated decades of observational and experimental data. Principally, investigations of genetic determinants of lipid levels confirm the powerful role of elevated LDL-C in atherosclerosis development.25 Loss of function mutations in the LDL-C receptor lead to reduced clearance of LDL-C from the blood and resulting high circulating levels of LDL-C. Individuals inheriting LDL-C receptor mutations from one parent (heterozygous) often have LDL-C levels >200 mg/dL and can have premature clinical CVD events in their third and fourth decades of life, and individuals inheriting two copies of the defective LDL-C receptor genes (homozygous familial hypercholesterolemia) have LDL-C levels of 650–1000 mg/dL and can experience CVD events in their teens or early twenties.26,27 Conversely, loss of function mutations of the proprotein convertase subtilisin/kexin type 9 protein (PCSK9), which removes LDL-C receptors from the surface of hepatocytes, lead to ≈28% reductions in LDL-C cholesterol levels and accordingly are associated with an 88% percent relative risk reduction in coronary heart disease.28 The causal role for LDL-C in ASCVD pathogenesis is further supported by Mendelian randomization studies demonstrating that each genetically-determined 38.7 mg/dl lower LDL-C is associated with a 55% reduction in coronary heart disease.29
Common variant genetic association studies have also provided evidence supporting causal roles for other putative contributors to atherosclerosis initiation such as atherogenic lipid particles (triglycerides), pro-inflammatory pathways (insulin resistance and inflammation), and endothelial dysfunction (cell adhesion and proliferation, nitric oxide signaling, and vascular remodeling).30–32 For example, loss of function variants in the ANGPTL4 gene, which encodes the angiopoietin-like 4 protein, result in 35% lower triglyceride levels and reduced coronary artery disease (CAD) risk.33
In addition, loss of function mutations in the nitric oxide receptor component guanylate cyclase 1, soluble, alpha 3 (GUCY1A3) are associated with early CVD34 attesting to the central role of endothelial dysfunction in promoting atherosclerosis. Similarly, multiple lines of evidence including genome wide association,35 Mendelian randomization,36 and mechanistic studies37,38 implicate loss of function mutations in the CXCR4/CXCL12 pathway (which is integral to maintaining vascular integrity) as causal for ASCVD development.
On the other hand, the majority of CVD-associated variants are located in non-coding chromosomal regions and their mechanistic links to CVD pathogenesis can be challenging to elucidate.30 One example of this is the Chr9p21 risk locus, which is consistently observed to be the most highly significant ASCVD risk locus in genome-wide association studies.39 The Chr9p21 locus contains no protein-coding genes, but does include some exons of the antisense noncoding RNA in the INK4 locus (ANRIL).30 Recently, studies have shown that variants affecting ANRIL abundance and splicing may contribute to atherogenesis, but the exact mechanisms linking variants in the Chr9p21 region with ASCVD risk remain elusive.39
With the use of ever-larger discovery datasets, the current list of genetic variants associated with ASCVD now numbers >150.30 As the effect size for each of these variants is low, combining many (or even all) of the potential risk variants in polygenic risk scores may provide the ability to more comprehensively assess an individual’s CVD risk.40–43 In addition to including genetic variants reaching genome-wide statistical significance, several groups have recently expanded the concept of polygenic risk to include all risk-predicting variants across the human genome.44–46 With this approach, a genome-wide polygenic score comprising >6 million single nucleotide polymorphisms can identify 8% of the population to be at >3-fold higher risk of CAD, which rivals the risk for rare monogenic mutations.44
Whereas the discovery of genetic determinants of ASCVD have validated experimental observations of contributors to atherosclerosis initiation, and identified important druggable targets,28 their translation to clinical risk prediction is currently limited. For elevated lipid levels, understanding the genetic risk is scientifically important, but it is unlikely to provide incremental predictive information beyond lipid measurement itself. Polygenic CVD risk scores may be useful in identifying individuals who are most likely to benefit from lipid-lowering therapies,40 but the ability of polygenic risk scores to provide incremental information to clinical risk factors alone for clinical prediction and management of CVD events has not been fully established.47,48 Several other unanswered questions regarding the clinical translation of polygenic risk scores remain including optimal modeling strategies, most appropriate ages for assessment, and how to account for gene-gene, gene-environment, or gene-risk factor interactions.49 Moreover, polygenic risk scores have primarily been derived and validated in populations of European descent; calculation of polygenic risk scores in other racial and ethnic groups is necessary before they can be used widely for ASCVD risk assessment.30,50,51
Epigenetics
Epigenetic modifications both reflect and contribute to the development of ASCVD and, therefore, represent intriguing biomarkers for risk prediction.52 Variability in DNA methylation (binding of a methyl group to the 5’ carbon of cytosine in cytosine-guanine dinucleotide sequences) is heritable, relates to aging, and can be altered by environmental exposures and CVD risk factors.53 Differential DNA methylation also exerts important regulatory effects on gene expression, which can affect drivers of atherosclerosis such as lipid levels54 and pro-inflammatory cytokines.55 While analysis of DNA methylation requires collection and preparation of cells (frequently circulating white blood cells), non-coding RNAs serve as epigenetic biomarkers that are released into the bloodstream and are readily detectable in peripheral blood samples. MicroRNAs (miRNAs) are small, single-stranded, noncoding RNAs that modulate myriad biological processes including lipid metabolism,56,57 endothelial cell dysfunction, and inflammation,56 and may prove as useful biomarkers for CVD risk assessment.58–60 MiRNA profiling and methylation signatures can be combined to provide an even broader view of epigenomic regulation of gene expression in relation to ASCVD risk.61
Transcriptomics
Numerous studies have sought to identify gene expression signatures of atherosclerosis and CVD risk by measuring messenger RNA (mRNA) in peripheral blood, but few candidate transcripts have validated across studies.62 In one intriguing investigation, Yao and colleagues integrated genetic and transcriptomic data to identify 13 CVD-related gene expression modules that were associated with inter-individual phenotypic variation in clinical risk factors.63 While these observations require further validation, they demonstrate the potential to combine genetics and transcriptional profiling of circulating cells to more precisely characterize CVD risk phenotypes.
Transcriptional profiling of individual cell types has the potential to provide dynamic insights regarding atherosclerosis initiation and development. In experimental models, single-cell RNA sequencing of cell types within plaques, such as macrophages, reveal unique cellular populations with distinct gene expression profiles and different relations to stages of atherosclerosis.64,65 Methods are improving to harvest specific cell types directly from humans enabling more targeted transcriptional profiling or single-cell RNA sequencing.66 In one example of the information that can be gained from transcriptional profiling of specific types of cell, activated human platelet phenotypes in circulating blood can be detected in response to circulating inflammatory mediators,67,68 potentially identifying “higher risk” platelet phenotypes that may be amenable to targeted interventions.
Proteomics
Multiplexed, high-throughput proteomic profiling platforms are now capable of assaying 1,000 to 5,000 of the ≈20,000 proteins in the human proteome. Human plasma represents the ‘largest and deepest version’ of the human proteome (the secretome reflects 15% of the body’s proteins).69,70 Blood, therefore, is one of the best ‘reporter systems’ for detecting disease onset and progression. Blood tests are easy to obtain, inexpensive, and can be repeated even in frail, elderly people.71 Yet, measuring blood proteins is challenging. The concentration range of plasma proteins spans ten orders of magnitude resulting in the propensity for high-abundance proteins (range of g/ml) to mask low-abundance proteins (e.g., cytokines, with levels in μg/ml to pg/ml). The advent of microfluidics, multiplexing, and miniaturization have enabled ‘lab-on-chip’ tests compatible with point-of-care testing and population screening. Aptamer array platforms using affinity capture methods72,73 now facilitate automated multiplexed quantification of thousands of very low abundance proteins with small sample volumes, high throughput, and rapid scalability enabling profiling of large samples. These platforms have excellent analytic and clinical validity, with very high correlations with conventional laboratory-based assays for a variety of low abundance markers.74–76
These newer proteomic technologies have been applied to the discovery of novel protein biomarkers of ASCVD risk in the blood,74 with identification of proteins related to ASCVD risk factor burden,77 varied measures of cardiovascular health,78 and clinical CVD events.79 Indeed, multi-protein scores may predict CVD with better accuracy than risk scores based solely on traditional risk factors.79,80 Thus far, these broad, unbiased, proteomic profiling efforts have identified novel ASCVD risk proteins, some that have been associated with atheroma biology in experimental models and some for which the function is unknown and require further investigation. Initial efforts in proteomic discovery highlight the possibility that by assaying thousands of circulating proteins, we may obtain a more precise “snapshot” of an individual’s health status.
Metabolomics
Small molecule chemical intermediates (metabolites) representing different chemical categories such as amino acids, lipids, or by-products of drug or food metabolism can be detected in biological samples.81 Several of these metabolites have been demonstrated to relate to cardiometabolic risk in humans. Higher levels of circulating branched chain amino acids, for example, are correlated with insulin resistance,82,83 decrease with weight loss,84 are associated with future development of diabetes in community-dwelling individuals,85 and relate to future ASCVD risk.86 Metabolite profiling methods can also be used to assay a large number of bioactive lipids, thereby enabling more precise quantification of the atherogenic lipidome.87 Higher levels of circulating monoglycerides produced via hydrolysis of triglycerides, for instance, are associated with higher CVD risk, while higher levels of circulating lysophosphatidylcholines may inhibit macrophage biosynthesis and cellular cholesterol accumulation and therefore provide cardioprotection.88,89 Certain types of the bioactive sphingolipid ceramide and sphingomyelin species are also associated with higher CVD risk.90,91
The circulating metabolome may also contain exogenous substances reflecting relevant external exposures.92 For example, Breitner et al. demonstrated that metabolite concentrations are affected by ambient particulate matter and ozone concentration in hospitalized patients.93 Such assessments could enable precise quantification of individual exposures (exposome), and host-exposure interactions.
Hence, circulating metabolite levels reflect multiparametric host responses and external exposures, and can, therefore, integrate a large amount of potential information for ASCVD risk assessment. One important application of metabolomics in clinical practice may be its ability to explain some of the inter-individual variability in the links between clinical risk factors and future clinical ASCVD. In obese individuals, metabolite profiles can help distinguish between metabolically healthy and metabolically unhealthy obesity subphenotypes.94 Beyond improving the precision of traditional risk factor assessment, metabolomic profiling can also be used to define novel phenotypes. An example of this is the emerging endophenotype termed “metabolic flexibility,” which represents an organism’s adaptive capability to maintain homeostasis under a variety of metabolic conditions or differences in the availability of energy sources.95 An important challenge to the application of metabolomic data to clinical use is the relatively high intra-individual variation that can be observed in metabolite levels over time.96
Gut microbiome
Atherosclerotic plaques contain DNA from the same bacteria taxa as that found in the gut and oral cavity,97,98 indicating that microbial communities might contribute to and affect plaque formation. The composition of the gut microbiota can also impact host lipid levels, especially triglycerides.99–102 One product of microbial metabolism – trimethylamine N-oxide (TMAO), the hepatic oxidation product of the microbial metabolite trimethylamine (TMA) – has been shown to have a particularly strong association with atherosclerosis risk,103 however there are conflicting reports of inverse association of TMAO and ASCVD.104,105 TMA is rapidly oxidized to TMAO by flavin monooxygenase enzymes in the liver and released into the circulation.106 Circulating TMAO levels are associated with the presence of atherosclerotic plaques99 and CVD events.107 The causal role of TMAO in atherosclerosis is supported by a recent observation that a small molecule inhibitor of TMA lyase activity suppressed TMAO formation, macrophage foam cell formation, and atherosclerosis in mice.108 It remains to be seen whether profiling of gut microbiome features and/or TMAO levels may prove to be an important adjunct to current risk prediction methods.
Clonal hematopoiesis of indeterminate potential
Clonal hematopoiesis of indeterminant potential (CHIP) characterizes somatic mutations in bone marrow hematopoietic stem cells that confer a proliferative advantage and thereby promote clonal proliferation of myeloid lineages in the peripheral blood. CHIP clones can be identified via genetic sequencing of peripheral blood. They are present in 10% of individuals above age 70 years and are known to promote thrombosis and accelerate atherosclerosis.109,110 Although precise mechanisms are incompletely elucidated, CHIP is theorized to contribute to ASCVD risk via stimulation of inflammation and innate immunity. Given its association with ASCVD independent of clinical risk factors,109 CHIP represents a promising avenue for further study to assess its ability, if any, to improve clinical ASCVD risk prediction.
ATHEROSCLEROSIS PROGRESSION
Molecular Biology
Developing atherosclerotic plaques can be influenced by a number of diverse features serving to either promote or inhibit their growth and evolution (Figure 1). Atherosclerotic plaque progression traditionally proceeds through continued accumulation of lipid and formation of lipid-engorged cells.4 Migration of smooth muscle cells from the vascular media into the intima support the growing plaque and release extracellular matrix macromolecules,111 which bind lipoproteins causing extracellular lipid accumulation.4 The growing plaque remodels eccentrically initially driven partially by the release of proteinases (such as matrix metalloproteinase 3) by smooth muscle cells.112 Cysteine cathepsins are a family of proteins that perform varied functions in developing plaques including promoting extracellular matrix degradation, mediating LDL-C aggregation and accumulation and foam cell formation, and increasing immune cell recruitment.113 A growing number of resident leukocytes localize within the plaque through both infiltration and proliferation.114 Leukocytes are retained within growing plaques by locally secreted proteins such as semaphorins.115 Specific plaque characteristics are determined by diverse features such as programmed cell death of leukocytes, resulting in a growing necrotic core.116 Both micro- and macro-calcifications deposit in growing plaques as a result of impaired clearance and dysregulated deposition of extracellular matrix components.117 Within developing plaques, macrophages regulate inflammation, clear apoptotic cells, and determine plaque stability by secreting matrix metalloproteinases and processing and storing lipids.118 Depending on the local environment, these macrophages can ‘polarize’ to diverse cellular phenotypes, which serve either to promote or inhibit atherogenesis.118 Developing plaques therefore have specific characteristics and can be broadly categorized as fibrous plaques (fibroatheroma) or fibrocalcific plaques. Plaques displaying features that have been shown to predispose to rupture are termed ‘vulnerable plaques,’ which are characterized by large lipid cores with thin fibrous caps.4 Other high risk plaque features include expansive positive (outward) vessel remodeling and scattered (not dense) calcifications.119,120
Assessing atherosclerosis progression in humans
Within individuals, and even within the same blood vessel, many different plaques exist simultaneously, each with distinctive structural and molecular characteristics. Indeed, atherosclerosis proceeds heterogeneously across vascular beds and among different plaques within a given vascular territory. The atherosclerotic process is influenced both by the systemic factors reviewed previously and by the local microarchitecture of individual plaques. In the following section, we review methods to identify plaque characteristics through circulating markers and focused molecular imaging techniques, with a particular focus on identifying features of high-risk plaques.
Circulating molecular profiles
Detection of molecular signatures of unstable or higher risk plaques in the blood have been reported. By measuring >300 different circulating lipid molecules, Meikle et al. identified distinct lipid profiles of stable versus unstable CAD.121 In individuals with established ASCVD, Ganz et al. used broad proteomic profiling to identify 9 novel proteins that predicted CVD events over the following 4 years.79 Several of the proteins highlighted by this study, such as angiopoietin-2 and matrix metalloproteinase-12, are likely to reflect heightened metabolic activity among remodeling plaques.122 Moreover, features of the gut microbiome identified by metagenomic sequencing have been shown to differ in individuals with stable versus unstable plaques: persons with unstable plaques demonstrated lower fecal levels of the genus Roseburiam.123 Higher circulating TMAO levels have also been found to correlate with unstable plaque morphology.124
In addition to systemic profiles associated with adverse plaque morphology, methods are evolving to examine cell-specific molecular profiles that may provide insight into individual plaque biology. Single cell molecular profiling enables comprehensive study of distinct cell types. This approach has been used to interrogate different leukocyte species within atherosclerotic plaques, demonstrating that the higher proportion of memory T-cell lineages within carotid plaques is more predictive of future CVD events than macrophage number.65 Cell typing by flow cytometry has also been used to analyze and compare different leukocyte cell types in humans, with relevance for plaque biology.125 The next steps are to use these single cell profiling techniques to leverage the distinct proteomic,126 epigenetic,127,128 transcriptomic,129 and other molecular profiles of cellular constituents of high risk plaques to improve ASCVD risk prediction.
Molecular imaging
Cardiovascular imaging modalities presently used in clinical practice are focused primarily on the diagnosis and treatment of the symptoms and complications of ASCVD. One modality that has been shown to add complementary information to traditional risk factor assessment is coronary calcium scoring, which can be used to reclassify, and in some cases refine, risk prediction estimates.130 Coronary computed tomography angiography (CTA), and assessment of carotid intima-media thickness and ankle-brachial index are also used to assist with the diagnosis of subclinical ASCVD and can aid in ASCVD risk assessment.131 However, such techniques rely primarily on anatomical quantifications of plaque burden and, therefore, do not assess plaque ‘activity’ or high-risk plaque features.
New imaging approaches can harness molecular probes to assess plaque biology with real-time in vivo individualized assessments. Molecular tracers targeted to pathological processes of interest are labeled with an imaging reporter and injected into the body where they accumulate at sites of increased disease activity.132 Theoretically, any disease process can be imaged with this technique once suitable tracers and imaging platforms are identified, but regulatory approval and cost remain prohibitive in many cases.132 Here we review examples of molecular imaging probes that are being used to assess atherosclerosis progression with the goal of highlighting individual plaque characteristics that can be identified in real time. Detailed and comprehensive reviews are available elsewhere.132,133
18F-fluorodeoxyglucose (18F-FDG) is a PET tracer and glucose analog that is taken up by metabolically active cells.132 It is useful in identifying vascular inflammation as macrophages utilize glucose at a higher rate than surrounding cells,134 and FDG-PET activity has been found to correspond with macrophage content in excised carotid plaques.135 Endarterectomy specimens demonstrate correlations between areas of increased 18F-FDG uptake and regions of inflammation, microvascular permeability and microvascularization.136 In a retrospective study of 513 patients undergoing PET imaging for evaluation of cancer, 18F-FDG uptake in the aorta was associated with CVD events over a mean follow up of 4.2 years.137 Vascular 18F-FDG PET studies are time-efficient and relatively low cost.132 While aortic imaging is relatively simple, 18F-FDG uptake in the coronary arteries is more challenging to image. Glucose is the predominant energy source of the myocardium, which increases the “background” signal and renders coronary artery 18F-FDG uptake difficult to identify. Other PET tracers with more optimal characteristics for imaging coronary inflammation are in development.132
Ultrasmall superparamagnetic particles of iron oxide (USPIO) are removed from circulating blood by the reticuloendothelial system and accumulate in macrophages present in atherosclerotic plaques, thereby serving as a marker of plaque activity.132 Plaques can then be imaged using T2*-weighted magnetic resonance imaging (MRI) sequences.138,139 Intriguingly, the USPIO signal intensity has been shown to decrease with 3 months of statin therapy, suggesting that it may be a useful tool to assess plaque regression in response to treatment.140
As opposed to vascular macrocalcification, which may represent plaque chronicity and stability, microvascular calcification is associated with active inflammation and higher risk features.117 18F-fluoride binds to calcified deposits within atherosclerotic plaques with high selectivity and differentially binds to micro- as opposed to macrocalcification, thereby identifying calcium deposits too small to be recognized by CT imaging.141,142 In a study of 40 patients with recent MI, culprit plaque uptake of 18F-fluoride was elevated in 37 individuals.143 One challenge to applying this technique more broadly is that microcalcification of the vascular media (i.e., medial sclerosis) can occur, especially in the setting of chronic kidney disease or diabetes, but is not localized to atherosclerotic plaques and, therefore, may therefore distort ASCVD risk assessments based solely on vascular microcalcification.144
Other techniques such as near-infrared fluorescence (NIRF) imaging and MR spectroscopy hold promise for improving imaging of molecular characteristics of atherosclerotic plaques.133,145 MR spectroscopy, in particular, combines the spatial imaging of MRI with spectral analysis to detect chemical composition of cardiovascular tissue.145 MR spectroscopy of carotid arteries, for instance, can quantify cholesterol esters within atherosclerotic plaque.145
As molecular imaging techniques become more widely available, their use to further delineate not only the presence and burden of atherosclerotic plaques, but also their biological activity, and to combine this with other molecular ‘snapshots’ of an individual’s risk profile has the potential to potently improve the precision of individual-level risk prediction. For this vision to reach clinical care, there will need to be consensus on optimal imaging modalities and isotopes, standardized protocols, and better delineation of the associations of imaging findings with clinical outcomes.
ATHEROSCLEROSIS COMPLICATIONS
Molecular Biology
Atherosclerotic plaques lead to overt events when they limit myocardial blood flow via progressive stenosis or thrombus formation (Figure 1). Plaque growth may eventually impair coronary arterial perfusion, resulting in ischemia during rest or exercise (i.e., angina pectoris). The most common cause of myocardial infarction is atherosclerotic plaque rupture.146 High-risk (or “vulnerable”) plaque features that predispose to rupture include large lipid cores with a thin fibrous cap.4 Inflammation-mediated dysregulation of extracellular matrix homeostasis disrupts the normal balance between collagen synthesis and removal, with resultant degradation of the protective fibrous cap by interstitial collagenases.146,147 Intraplaque hemorrhage often precedes full plaque rupture and may derive from plaque neovascularization or from small ruptures of the plaque’s fibrous cap causing blood to infiltrate the plaque from the vessel lumen.148,149 When plaque rupture occurs, the thrombogenic material in the plaque core is exposed to the blood compartment leading to platelet aggregation and the polymerization of fibrin. An alternative thrombotic mechanism is plaque erosion, which typically occurs in plaques with few inflammatory leukocytes, lower lipid concentrations, and a rich extracellular matrix without a thin fibrous cap. Innate immune activation and the participation of polymorphonuclear leukocytes may be particularly important in fostering plaque erosion.4,150 Alternatively, plaques may undergo favorable remodeling leading reduced risk of rupture or erosion. Plaque stabilization or regression has mostly been demonstrated in response to lipid lowering therapy.151
Assessing atherosclerosis complications in humans
The primary complication of ASCVD is myocardial infarction (MI), which results from inadequate blood flow to the myocardium usually due to an occlusion or stenosis of a coronary artery. Early identification of impending MI is essential to mitigate the adverse effects and consequences. Here, we review evolving methods to improve the upstream diagnosis of MI using molecular profiling.
Genetics/Epigenetics
Genetic markers and epigenetic modifications related to platelet reactivity152 and thrombus formation may identify individuals who are more likely to have atherosclerotic CVD complications. These may include transcriptional profiling to recognize gene expression signatures of higher risk (or hyperreactive) platelets.153,154 Moreover, methylation signatures of pro-thrombotic hemostatic profiles can be identified in the blood representing markers of heightened thrombotic risk.155 Circulating MiRNAs may also prove useful as early indicators of myocardial damage.156 Studies have also investigated miRNA biomarkers of plaque instability (vulnerable plaques)157–159 and have shown that miRNAs may be important biomarkers for future CVD events in patients with established CVD.59,128,160
Proteomics
Profiling of lower concentration circulating proteins may enable upstream detection of MI prior to manifest heart damage. High-sensitivity troponin assays are now deployed clinically to facilitate early and accurate identification of myocardial infarction, often enabling treatment before clinically relevant myocardial damage has occurred.161 Efforts are ongoing to identify novel proteins that provide complementary information regarding early indicators of MI. Broad proteomic profiling approaches have been applied to detection of myocardial damage identifying a number of novel proteins that may improve the diagnostic accuracy, and perhaps the prevention, of myocardial infarction,80,162 and its complications.163 In one example, Jacob et al. measured nearly 5000 circulating proteins after MI and found 29 mostly intracellular proteins that were increased post-MI, which require further study regarding their clinical utility in MI diagnosis. In another study, plasma levels of biliverdin reductase B were shown to correlate with intraplaque hemorrhages that antedate clinical MI symptoms, and which may therefore enable even earlier detection of plaque rupture events prior to coronary occlusion.164
Metabolomics
Metabolite profiling is similarly being investigated to identify circulating signatures of impending/evolving MI prior to their detection by traditional methods. For example, peripheral blood metabolite profiles can predict the presence of CAD and associate with future events in individuals undergoing coronary angiography.165,166 Metabolomic profiling of circulating blood can also be used to identify evidence of myocardial injury,167,168 the extent of ischemia-reperfusion injury,169 and potentially to distinguish between stable and unstable CAD.121
CONSIDERATIONS FOR INCORPORATING MULTI-DIMENSIONAL DATA INTO RISK PREDICTION
This review has primarily focused on highlighting the tremendous progress in elucidating molecular signatures of ASCVD risk. However, there are a number of challenges and important considerations that must be addressed in the process of transitioning these discoveries to clinical use (Table 1 and Supplemental Table), several of which are discussed below.
Table 1.
Challenges to clinical translation of molecular biomarkers
| Challenges | Potential considerations/solutions |
|---|---|
| Threshold for abnormal values vary according to clinical context, background risk profile, medications, comorbidities | • Incorporation of biomarkers into multivariable equations using standard ASCVD risk markers and other relevant clinical information • Clinical utility studies, serial measures • Biomarker testing in variety of clinical contexts and patient samples |
| Differences in specificity of biomarkers in blood vs. tissue | • Blood assays require egress from tissue or overt injury for markers to appear in circulation • Novel methods (e.g., molecular imaging, cell sorting) to measure biomarkers within tissues |
| Differences in prognostic significance of biomarkers based on age, sex, race, geography | • Replication of the results in independent multi-ethnic samples • Careful study design with inclusion of appropriate controls, adequate sample size |
| Rigor and reproducibility | • Standardization of pre-analytical biosample processing • Analytical standardization, especially in studies with multiple batch runs and longitudinal samples • Analytical validation using orthogonal methods |
| Optimal design relative to role of biomarker investigated | • Clearly defined purpose of the study to address a specific clinical question • Distinct study designs are needed to assess screening role, cross-sectional disease correlates, prognostic significance, predictive capability, clinical utility, and potential to guide treatment • Time horizons over which utility of biomarkers are assessed |
Single points in time
Traditional observational methods for biomarker discovery rely on demonstrating an association between a biomarker of interest and the presence or development of the outcome. Accordingly, biomarker discovery often begins with measurements made at one timepoint. Longitudinal assessments to measure the cumulative burden, growth trajectory, or variability of biomarkers can add incremental value for risk assessment. In addition, the timing of the measurement within the patient’s overall risk profile and health trajectory is of paramount importance. A straightforward example that illustrates this point is the vastly different implications of an elevated high-sensitivity troponin level for a 30-year-old who has just run 5 miles versus a 65-year-old with chest pain or a 50-year old patient with end-stage renal disease and mild heart failure. This point holds true even for perhaps the most commonly assessed cardiovascular risk biomarker: LDL-C. Current guidelines support distinct recommendations for LDL-C lowering therapy depending on the individual’s global ASCVD risk assessment.170 The same LDL-C level will carry distinctive prognostic significance and, as a consequence, different treatment recommendations in individuals with very different ASCVD risk profiles.171 For LDL-C, this approach has largely evolved from an effort to balance the risk of adverse medication effects with the absolute risk reduction expected to be derived from lipid-lowering therapy. Similar calculations will have to be considered for incorporation of novel risk markers in clinical care.
Considerations of differences based on sex, race, and geography
Inherent in risk prediction efforts is the assessment of how molecular risk markers vary according to patient characteristics. Widespread efforts must be made to replicate and compare findings across diverse populations to define specific normative limits based on age, sex, racial/ethnic background, and geographic location. Not only are the distribution of risk factors across populations important, however, but the association with the outcome must be assessed iteratively. For example, sex differences in high-sensitive troponin levels have been described with important considerations for clinical care.172
Rigor and reproducibility of measurements
The accuracy and precision with which novel molecular markers are identified is of substantial importance in their translation to the clinic. Novel assays provide the opportunity to measure molecules at a scale never before seen but the assay accuracy and methods of quantification must be validated. Moreover, protocols need to be standardized for pre-analytic biosample processing and quantification methods across laboratories.
Modeling techniques
In most populations, traditional risk factors can explain ~80% of the variation in CVD development.2,173 This is acceptable from a population level but the extrapolation of this variance-explained to treatment implications for an individual is a major challenge.3 A clear illustration of this challenge is exemplified by the observation that despite the strong association of high blood pressure with CVD events, the majority of individuals experiencing CVD events in the US have a normal blood pressure.174 Personalized, precise, risk assessments that account for variability in the individual’s response to risk factor burden are necessary therefore to improve individual-level risk prediction.
INTEGRATION OF MULTI-DIMENSIONAL MOLECULAR DATA FOR LIFE-COURSE RISK PREDICTION
Major progress is being made, as summarized in this review, in identifying molecular signatures that can be used to improve CVD risk assessment. In coming years, we anticipate that these multi-dimensional assessments will be integrated into a life-course risk assessment framework that harnesses the growing knowledge of the molecular biology of atherosclerotic plaques with dynamic assessments of a patient’s molecular profile (Figure 2).
Figure 2. Life course integration of multi-dimensional molecular data for ASCVD risk prediction.
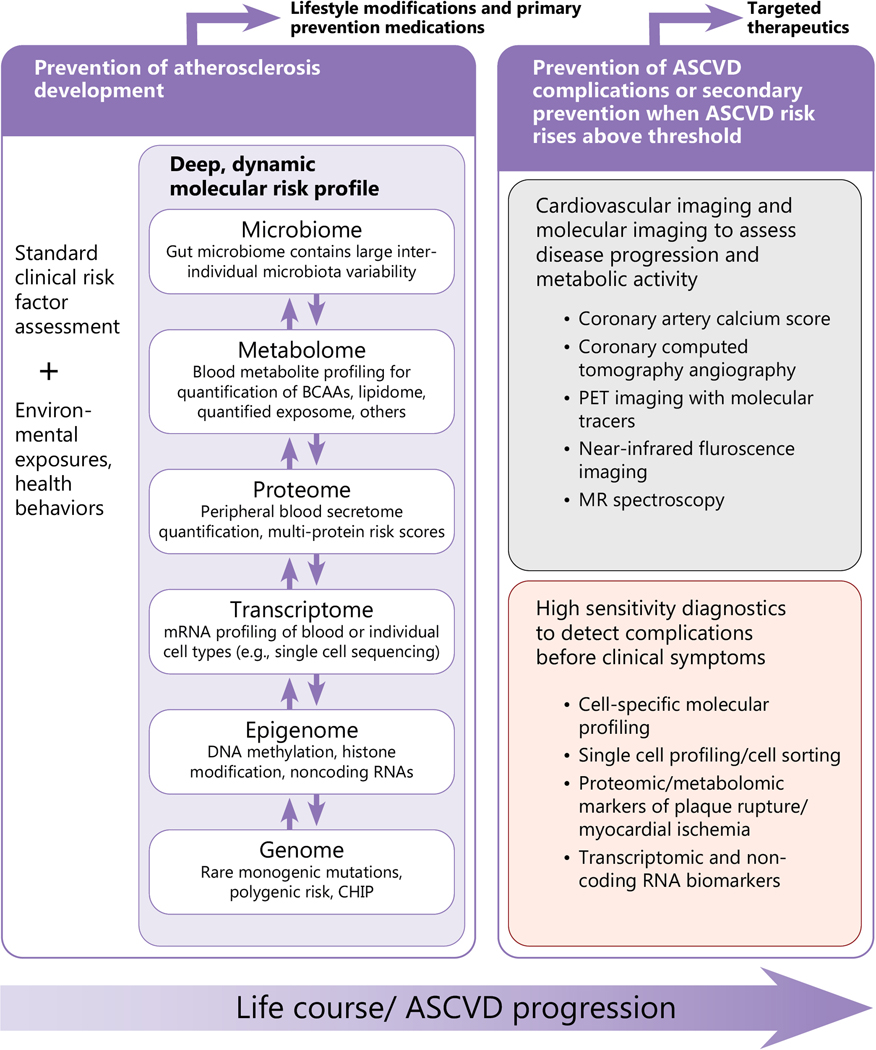
A conceptual view of how molecular diagnosis of ASCVD may proceed through the life course. Prevention of atherosclerosis development begins early in the life course with monitoring of standard clinical risk factors, environmental exposures, and deep, dynamic molecular risk profiles integrating information from the various “omics” layers. The connections between different “omic” layers displayed here is simplified for visualization purposes; the ‘post-genetic’ layers (epigenome, transcriptome, proteome, metabolome, microbiome) also influence each other bi-directionally. During early adulthood to mid-life, when atherosclerosis typically develops, personalized molecular risk profiles can inform risk status, leading to personalized treatment recommendations. In individuals with sufficient risk (typically in middle age and older adults), molecular imaging and cell-specific molecular profiling can be deployed to more precisely risk profile individual plaque features, with the goal of informing targeted therapeutic approaches. Lastly, high-sensitivity molecular diagnostics may reveal signs of impending ASCVD complications (e.g., plaque rupture) before clinical symptoms develop, enabling upstream treatment and prevention approaches.
Genetic risk is the obvious starting point for such assessments as it is the only “omic” domain that is predetermined and fixed. Genetic risk can be broadly divided into two categories. Modifiable genetic risk markers may affect known biological pathways, such as lipid levels, that can be measured and modulated with lifestyle and drug interventions. These genetic risk markers should not necessarily alter ASCVD risk prediction as they are not independent of known traditional risk factors. Other, non-modifiable genetic risk markers should be incorporated in global CVD risk prediction as they represent risk above what is routinely assessed.
Dynamic assessments of the epigenome, trancriptome, metabolome, proteome, and gut microbiome in circulating blood may be performed throughout the life course to gauge immediate health risks and identify targets for treatment (i.e., intermediate phenotypes). Molecular imaging and tissue-level molecular signatures could then serve as adjunct modalities in individuals identified as having sufficient risk for near-term events to warrant further evaluation of more precise markers of disease activity and potentially, to gauge therapeutic responses. Lastly, high-sensitivity, multi-dimensional markers of disease activity can facilitate identification of impending ASCVD events prior to symptom development or clinical relevance, allowing targeted preventative therapies. We foresee a new paradigm of multi-parametric, longitudinal, risk assessments that would adapt iteratively throughout an individual’s health course, with different sets of markers being more relevant at specific points in atherosclerosis progression.
FUTURE DIRECTIONS AND NEXT STEPS
Despite the tremendous progress to date in identifying and measuring molecular signals of atherosclerosis, much work is still required if we are to reach the aspirational vision for molecular targeting of CVD prevention depicted in the previous section.
The discovery phase of identifying novel risk markers and modalities relies primarily on studies of one or two “omics” domains at a time. Optimal risk assessment is likely to draw on trans-omics assessments integrating proteomic and metabolite responses with genetic and epigenetic information, for example, in a broad multi-omics multi-marker approach. This approach will be facilitated by advances in machine learning and big data analytics. An essential question that will arise is the optimal approach to ASCVD risk prediction for clinical care. Are “black box” machine learning approaches that arrive at the best prediction formula using obscure methodologies optimal, or will methods that are driven by empiric observations and known biology provide more actionable clinical insights?
Current risk prediction methods draw primarily on assessments made at a single time point in the resting state. Future models will incorporate lifetime burden, variability, and trajectories of risk features (which is already known to be predictive for a number of CVD risk factors including BMI, smoking and LDL-C175,176). In addition to the growing precision with which molecular markers of essential biological processes can be identified, more precise quantification of the exposome will be available in the future, facilitated by smartphone technologies, chemo-, mechano- and biosensors, and identification of the imprint of extrinsic factors in circulating blood.
Finally, it is our opinion that focusing our prevention efforts on specific organ-based disease systems may be a narrow perspective. Atherosclerosis, for example, leads not only to coronary artery disease, but also culminates in cerebrovascular disease and stroke, carotid disease, aortic disease, and peripheral vascular disease. Unifying the approaches to detection and monitoring of atherosclerosis development across multiple vascular beds and related organ systems is an important next step in comprehensive promotion of vascular health. In addition, many risk factors and circulating risk markers are not unique to CVD but are shared among other diseases of aging such as dementia and cancer. As such, a broader framework of chronic disease prevention as opposed to CVD prevention alone is likely to provide the most widespread health benefits for individuals and populations.
Summary
The molecular diagnosis of ASCVD is advancing at a rapid pace benefiting from tremendous progress in elucidating the pathobiology of atherosclerosis development and in high-throughput profiling of high-dimensional molecular data. Integration of multi-parametric molecular assessments has the potential to provide a longitudinal “snapshot” of an individual’s current status and risk profile and may enable more precise risk prediction and treatment approaches.
Supplementary Material
Acknowledgments
Funding sources: Dr. Nayor is supported by grant K23 HL138260 from the National Heart, Lung, and Blood Institute. Dr. Brown is supported by the John S. LaDue Memorial Fellowship at Harvard Medical School. Dr. Vasan is supported by an Evans Scholar award from the Department of Medicine, Boston University School of Medicine and the following: Contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute, and grants R01HL143227, R01HL131015, RF1AG063507, R01HL131029, R01HL132320, R01HL143818 and R01146860, and U01HL146382.
ABBREVIATIONS
- ASCVD
atherosclerotic cardiovascular disease
- CVD
cardiovascular disease
- LDL-C
low-density lipoprotein cholesterol
- HDL-C
high-density lipoprotein cholesterol
- CRP
C-reactive protein
- CAD
coronary artery disease
- miRNA
micro RNA
- mRNA
messenger RNA
- CHIP
clonal hematopoiesis of indeterminant potential
- CTA
computed tomography angiography
- 18F-FDG
18F-fluorodeoxyglucose
- USPIO
ultrasmall superparamagnetic particles of iron oxide
- MRI
magnetic resonance imaging
- NIRF
near-infrared fluorescence
- MI
myocardial infarction
Footnotes
Disclosures: None
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation. 2020;141:e139–e596. [DOI] [PubMed] [Google Scholar]
- 2.Cheng S, Claggett B, Correia AW, Shah AM, Gupta DK, Skali H, Ni H, Rosamond WD, Heiss G, Folsom AR, et al. Temporal trends in the population attributable risk for cardiovascular disease: the Atherosclerosis Risk in Communities Study. Circulation. 2014;130:820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hingorani AD and Psaty BM. Primary prevention of cardiovascular disease: time to get more or less personal? Jama. 2009;302:2144–5. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgozoglu L and Lewis EF. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56. [DOI] [PubMed] [Google Scholar]
- 5.Leopold JA and Loscalzo J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ Res. 2018;122:1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabas I, Williams KJ and Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–44. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein JL and Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller YI, Choi SH, Wiesner P, Fang L, Harkewicz R, Hartvigsen K, Boullier A, Gonen A, Diehl CJ, Que X, et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res. 2011;108:235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour N, et al. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. [DOI] [PubMed] [Google Scholar]
- 10.Llorente-Cortes V, Martinez-Gonzalez J and Badimon L. LDL receptor-related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2000;20:1572–9. [DOI] [PubMed] [Google Scholar]
- 11.Boren J and Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Current opinion in lipidology. 2016;27:473–83. [DOI] [PubMed] [Google Scholar]
- 12.Sarwar N, Sandhu MS, Ricketts SL, Butterworth AS, Di Angelantonio E, Boekholdt SM, Ouwehand W, Watkins H, Samani NJ, Saleheen D, et al. Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res. 2016;118:547–63. [DOI] [PubMed] [Google Scholar]
- 14.Di Angelantonio E, Gao P, Pennells L, Kaptoge S, Caslake M, Thompson A, Butterworth AS, Sarwar N, Wormser D, Saleheen D, et al. Lipid-related markers and cardiovascular disease prediction. Jama. 2012;307:2499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orso E and Schmitz G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin Res Cardiol Suppl. 2017;12:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Libby P, Lichtman AH and Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Despres JP. Body fat distribution and risk of cardiovascular disease: an update. Circulation. 2012;126:1301–13. [DOI] [PubMed] [Google Scholar]
- 19.Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, Erdmann J, Braund P, Engert JC, Bennett D, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. Jama. 2009;302:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridker PM. A Test in Context: High-Sensitivity C-Reactive Protein. J Am Coll Cardiol. 2016;67:712–723. [DOI] [PubMed] [Google Scholar]
- 21.Libby P, Nahrendorf M and Swirski FK. Leukocytes Link Local and Systemic Inflammation in Ischemic Cardiovascular Disease: An Expanded “Cardiovascular Continuum”. J Am Coll Cardiol. 2016;67:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ketelhuth DF and Hansson GK. Adaptive Response of T and B Cells in Atherosclerosis. Circ Res. 2016;118:668–78. [DOI] [PubMed] [Google Scholar]
- 23.Ignarro LJ and Napoli C. Novel features of nitric oxide, endothelial nitric oxide synthase, and atherosclerosis. Curr Diab Rep. 2005;5:17–23. [DOI] [PubMed] [Google Scholar]
- 24.Cybulsky MI and Gimbrone MA, Jr. Endothelial expression of a mononuclear leukocyte adhesion molecule during atherogenesis. Science. 1991;251:788–91. [DOI] [PubMed] [Google Scholar]
- 25.Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridker PM. LDL cholesterol: controversies and future therapeutic directions. Lancet. 2014;384:607–17. [DOI] [PubMed] [Google Scholar]
- 27.Singh S and Bittner V. Familial hypercholesterolemia--epidemiology, diagnosis, and screening. Curr Atheroscler Rep. 2015;17:482. [DOI] [PubMed] [Google Scholar]
- 28.Cohen JC, Boerwinkle E, Mosley TH, Jr. and Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. [DOI] [PubMed] [Google Scholar]
- 29.Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA Sr. and Flack JM Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease: a Mendelian randomization analysis. J Am Coll Cardiol. 2012;60:2631–9. [DOI] [PubMed] [Google Scholar]
- 30.Musunuru K and Kathiresan S. Genetics of Common, Complex Coronary Artery Disease. Cell. 2019;177:132–145. [DOI] [PubMed] [Google Scholar]
- 31.Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, Leed A, Weale ME, Spencer CCA, Aguet F, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khera AV and Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18:331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myocardial Infarction G, Investigators CAEC, Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med. 2016;374:1134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, de Wit C, Kaiser FJ, Braunholz D, Medack A, Fischer M, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504:432–6. [DOI] [PubMed] [Google Scholar]
- 35.Consortium CAD, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, Ingelsson E, Saleheen D, Erdmann J, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sjaarda J, Gerstein H, Chong M, Yusuf S, Meyre D, Anand SS, Hess S and Pare G. Blood CSF1 and CXCL12 as Causal Mediators of Coronary Artery Disease. J Am Coll Cardiol. 2018;72:300–310. [DOI] [PubMed] [Google Scholar]
- 37.Doring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, Atzler D, Santovito D, Rader DJ, Saleheen D, et al. CXCL12 Derived From Endothelial Cells Promotes Atherosclerosis to Drive Coronary Artery Disease. Circulation. 2019;139:1338–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doring Y, Noels H, van der Vorst EPC, Neideck C, Egea V, Drechsler M, Mandl M, Pawig L, Jansen Y, Schroder K, et al. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence From Mouse and Human Studies. Circulation. 2017;136:388–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holdt LM and Teupser D. Long Noncoding RNA ANRIL: Lnc-ing Genetic Variation at the Chromosome 9p21 Locus to Molecular Mechanisms of Atherosclerosis. Front Cardiovasc Med. 2018;5:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, Sartori S, Fuster V, Reilly DF, Butterworth A, et al. Polygenic Risk Score Identifies Subgroup With Higher Burden of Atherosclerosis and Greater Relative Benefit From Statin Therapy in the Primary Prevention Setting. Circulation. 2017;135:2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, Guiducci C, Perola M, Jula A, Sinisalo J, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. The Lancet. 2010;376:1393–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L, Tikkanen E, Perola M, Schunkert H, Sijbrands EJ, et al. Genomic prediction of coronary heart disease. Eur Heart J. 2016;37:3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Theriault S, Lali R, Chong M, Velianou JL, Natarajan MK and Pare G. Polygenic Contribution in Individuals With Early-Onset Coronary Artery Disease. Circ Genom Precis Med. 2018;11:e001849. [DOI] [PubMed] [Google Scholar]
- 44.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, Lai FY, Kaptoge S, Brozynska M, Wang T, et al. Genomic risk prediction of coronary artery disease in nearly 500,000 adults: implications for early screening and primary prevention. bioRxiv. 2018:250712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levin MG, Kember RL, Judy R, Birtwell D, Williams H, Arany Z, Giri J, Guerraty M, Cappola T, Regeneron Genetics C, et al. Genomic Risk Stratification Predicts All-Cause Mortality After Cardiac Catheterization. Circ Genom Precis Med. 2018;11:e002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mosley JD, Gupta DK, Tan J, Yao J, Wells QS, Shaffer CM, Kundu S, Robinson-Cohen C, Psaty BM, Rich SS, et al. Predictive Accuracy of a Polygenic Risk Score Compared With a Clinical Risk Score for Incident Coronary Heart Disease. JAMA. 2020;323:627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janssens A Validity of polygenic risk scores: are we measuring what we think we are? Hum Mol Genet. 2019;28:R143–R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lieb W and Vasan RS. An update on genetic risk scores for coronary artery disease: are they useful for predicting disease risk and guiding clinical decisions? Expert Rev Cardiovasc Ther. 2020:1–5. [DOI] [PubMed] [Google Scholar]
- 50.De La Vega FM and Bustamante CD. Polygenic risk scores: a biased prediction? Genome Med. 2018;10:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim MS, Patel KP, Teng AK, Berens AJ and Lachance J. Genetic disease risks can be misestimated across global populations. Genome Biol. 2018;19:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khyzha N, Alizada A, Wilson MD and Fish JE. Epigenetics of Atherosclerosis: Emerging Mechanisms and Methods. Trends Mol Med. 2017;23:332–347. [DOI] [PubMed] [Google Scholar]
- 53.Huan T, Joehanes R, Song C, Peng F, Guo Y, Mendelson M, Yao C, Liu C, Ma J, Richard M, et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat Commun. 2019;10:4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hedman AK, Mendelson MM, Marioni RE, Gustafsson S, Joehanes R, Irvin MR, Zhi D, Sandling JK, Yao C, Liu C, et al. Epigenetic Patterns in Blood Associated With Lipid Traits Predict Incident Coronary Heart Disease Events and Are Enriched for Results From Genome-Wide Association Studies. Circ Cardiovasc Genet. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aslibekyan S, Agha G, Colicino E, Do AN, Lahti J, Ligthart S, Marioni RE, Marzi C, Mendelson MM, Tanaka T, et al. Association of Methylation Signals With Incident Coronary Heart Disease in an Epigenome-Wide Assessment of Circulating Tumor Necrosis Factor alpha. JAMA Cardiol. 2018;3:463–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feinberg MW and Moore KJ. MicroRNA Regulation of Atherosclerosis. Circ Res. 2016;118:703–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. [DOI] [PubMed] [Google Scholar]
- 58.Zampetaki A, Willeit P, Tilling L, Drozdov I, Prokopi M, Renard JM, Mayr A, Weger S, Schett G, Shah A, et al. Prospective study on circulating MicroRNAs and risk of myocardial infarction. J Am Coll Cardiol. 2012;60:290–9. [DOI] [PubMed] [Google Scholar]
- 59.Karakas M, Schulte C, Appelbaum S, Ojeda F, Lackner KJ, Munzel T, Schnabel RB, Blankenberg S and Zeller T. Circulating microRNAs strongly predict cardiovascular death in patients with coronary artery disease-results from the large AtheroGene study. Eur Heart J. 2017;38:516–523. [DOI] [PubMed] [Google Scholar]
- 60.Peters LJF, Biessen EAL, Hohl M, Weber C, van der Vorst EPC and Santovito D. Small Things Matter: Relevance of MicroRNAs in Cardiovascular Disease. Front Physiol. 2020;11:793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huan T, Mendelson M, Joehanes R, Yao C, Liu C, Song C, Bhattacharya A, Rong J, Tanriverdi K, Keefe J, et al. Epigenome-wide association study of DNA methylation and microRNA expression highlights novel pathways for human complex traits. Epigenetics. 2019:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen HH and Stewart AF. Transcriptomic Signature of Atherosclerosis in the Peripheral Blood: Fact or Fiction? Curr Atheroscler Rep. 2016;18:77. [DOI] [PubMed] [Google Scholar]
- 63.Yao C, Chen BH, Joehanes R, Otlu B, Zhang X, Liu C, Huan T, Tastan O, Cupples LA, Meigs JB, et al. Integromic analysis of genetic variation and gene expression identifies networks for cardiovascular disease phenotypes. Circulation. 2015;131:536–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE and Zernecke A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res. 2018;122:1661–1674. [DOI] [PubMed] [Google Scholar]
- 65.Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ Res. 2018;122:1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beckman JA, Doherty SP, Feldman ZB, Banks ES, Moslehi J, Jaffe IZ, Hamburg NM, Sheng Q and Brown JD. Comparative Transcriptomics of Ex Vivo, Patient-Derived Endothelial Cells Reveals Novel Pathways Associated With Type 2 Diabetes Mellitus. JACC Basic Transl Sci. 2019;4:567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, Genco CA, Tanriverdi K, Larson MG, Benjamin EJ, et al. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. 2014;34:552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McManus DD, Beaulieu LM, Mick E, Tanriverdi K, Larson MG, Keaney JF, Jr., Benjamin EJ and Freedman JE. Relationship among circulating inflammatory proteins, platelet gene expression, and cardiovascular risk. Arterioscler Thromb Vasc Biol. 2013;33:2666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 70.Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG and Fagan R. The human plasma proteome. Mol Cell Proteomics. 2004;3:311–326. [DOI] [PubMed] [Google Scholar]
- 71.Thambisetty M and Lovestone S. Blood-based biomarkers of Alzheimer’s disease: challenging but feasible. Biomark Med. 2010;4:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Davies DR, Gelinas AD, Zhang C, Rohloff JC, Carter JD, O’Connell D, Waugh SM, Wolk SK, Mayfield WS, Burgin AB, et al. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc Natl Acad Sci U S A. 109:19971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smith JG and Gerszten RE. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation. 2017;135:1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lollo B, Steele F and Gold L. Beyond antibodies: new affinity reagents to unlock the proteome. Proteomics. 2014;14:638–44. [DOI] [PubMed] [Google Scholar]
- 76.Mehan MR, Ostroff R, Wilcox SK, Steele F, Schneider D, Jarvis TC, Baird GS, Gold L and Janjic N. Highly multiplexed proteomic platform for biomarker discovery, diagnostics, and therapeutics. Adv Exp Med Biol. 2013;735:283–300. [DOI] [PubMed] [Google Scholar]
- 77.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O’Sullivan JF, Keshishian H, Farrell LA, et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Williams SA, Kivimaki M, Langenberg C, Hingorani AD, Casas JP, Bouchard C, Jonasson C, Sarzynski MA, Shipley MJ, Alexander L, et al. Plasma protein patterns as comprehensive indicators of health. Nature medicine. 2019;25:1851–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG and Williams SA. Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients With Stable Coronary Heart Disease. JAMA. 2016;315:2532–41. [DOI] [PubMed] [Google Scholar]
- 80.Ngo D, Sinha S, Shen D, Kuhn EW, Keyes MJ, Shi X, Benson MD, O’Sullivan JF, Keshishian H, Farrell LA, et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation. 2016;134:270–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McGarrah RW, Crown SB, Zhang GF, Shah SH and Newgard CB. Cardiovascular Metabolomics. Circ Res. 2018;122:1238–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9:311–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheng S, Rhee EP, Larson MG and Lewis GD. Metabolite profiling identifies pathways associated with metabolic risk in humans. Circulation. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah SH, Crosslin DR, Haynes CS, Nelson S, Turer CB, Stevens RD, Muehlbauer MJ, Wenner BR, Bain JR, Laferrere B, et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia. 2012;55:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. Metabolite profiles and the risk of developing diabetes. Nature medicine. 2011;17:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tobias DK, Lawler PR, Harada PH, Demler OV, Ridker PM, Manson JE, Cheng S and Mora S. Circulating Branched-Chain Amino Acids and Incident Cardiovascular Disease in a Prospective Cohort of US Women. Circ Genom Precis Med. 2018;11:e002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lagerborg KA, Watrous JD, Cheng S and Jain M. High-Throughput Measure of Bioactive Lipids Using Non-targeted Mass Spectrometry. Methods Mol Biol. 2019;1862:17–35. [DOI] [PubMed] [Google Scholar]
- 88.Ganna A, Salihovic S, Sundstrom J, Broeckling CD, Hedman AK, Magnusson PK, Pedersen NL, Larsson A, Siegbahn A, Zilmer M, et al. Large-scale metabolomic profiling identifies novel biomarkers for incident coronary heart disease. PLoS Genet. 2014;10:e1004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cavus E, Karakas M, Ojeda FM, Kontto J, Veronesi G, Ferrario MM, Linneberg A, Jørgensen T, Meisinger C, Thorand B, et al. Association of Circulating Metabolites With Risk of Coronary Heart Disease in a European Population. JAMA Cardiology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Peterson LR, Xanthakis V, Duncan MS, Gross S, Friedrich N, Volzke H, Felix SB, Jiang H, Sidhu R, Nauck M, et al. Ceramide Remodeling and Risk of Cardiovascular Events and Mortality. Journal of the American Heart Association. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, Hunt SC, Holland WL, Summers SA and Playdon MC. Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest. 2020;130:1363–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Athersuch TJ and Keun HC. Metabolic profiling in human exposome studies. Mutagenesis. 2015;30:755–62. [DOI] [PubMed] [Google Scholar]
- 93.Breitner S, Schneider A, Devlin RB, Ward-Caviness CK, Diaz-Sanchez D, Neas LM, Cascio WE, Peters A, Hauser ER, Shah SH, et al. Associations among plasma metabolite levels and short-term exposure to PM2.5 and ozone in a cardiac catheterization cohort. Environ Int. 2016;97:76–84. [DOI] [PubMed] [Google Scholar]
- 94.Cirulli ET, Guo L, Leon Swisher C, Shah N, Huang L, Napier LA, Kirkness EF, Spector TD, Caskey CT, Thorens B, et al. Profound Perturbation of the Metabolome in Obesity Is Associated with Health Risk. Cell Metab. 2019;29:488–500 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goodpaster BH and Sparks LM. Metabolic Flexibility in Health and Disease. Cell Metab. 2017;25:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sampson JN, Boca SM, Shu XO, Stolzenberg-Solomon RZ, Matthews CE, Hsing AW, Tan YT, Ji BT, Chow WH, Cai Q, et al. Metabolomics in epidemiology: sources of variability in metabolite measurements and implications. Cancer Epidemiol Biomarkers Prev. 2013;22:631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koren O, Spor A, Felin J, Fak F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4592–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ott SJ, El Mokhtari NE, Musfeldt M, Hellmig S, Freitag S, Rehman A, Kuhbacher T, Nikolaus S, Namsolleck P, Blaut M, et al. Detection of diverse bacterial signatures in atherosclerotic lesions of patients with coronary heart disease. Circulation. 2006;113:929–37. [DOI] [PubMed] [Google Scholar]
- 99.Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nature medicine. 2013;19:576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fu J, Bonder MJ, Cenit MC, Tigchelaar EF, Maatman A, Dekens JAM, Brandsma E, Marczynska J, Imhann F, Weersma RK, et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circulation Research. 2015;117:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015;10:326–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tang WH, Kitai T and Hazen SL. Gut Microbiota in Cardiovascular Health and Disease. Circ Res. 2017;120:1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yin J, Liao SX, He Y, Wang S, Xia GH, Liu FT, Zhu JJ, You C, Chen Q, Zhou L, et al. Dysbiosis of Gut Microbiota With Reduced Trimethylamine-N-Oxide Level in Patients With Large-Artery Atherosclerotic Stroke or Transient Ischemic Attack. Journal of the American Heart Association. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Landfald B, Valeur J, Berstad A and Raa J. Microbial trimethylamine-N-oxide as a disease marker: something fishy? Microb Ecol Health Dis. 2017;28:1327309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bennett Brian J, Vallim Thomas Qde A, Wang Z, Shih Diana M, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metabolism. 2013;17:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y and Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell. 2015;163:1585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N Engl J Med. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bennett MR, Sinha S and Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Unemori EN, Lark MW, Amento E and Libby P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–9. [DOI] [PubMed] [Google Scholar]
- 113.Liu CL, Guo J, Zhang X, Sukhova GK, Libby P and Shi GP. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nature reviews Cardiology. 2018;15:351–370. [DOI] [PubMed] [Google Scholar]
- 114.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nature medicine. 2013;19:1166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wanschel A, Seibert T, Hewing B, Ramkhelawon B, Ray TD, van Gils JM, Rayner KJ, Feig JE, O’Brien ER, Fisher EA, et al. Neuroimmune guidance cue Semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler Thromb Vasc Biol. 2013;33:886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tabas I, Garcia-Cardena G and Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ruiz JL, Hutcheson JD and Aikawa E. Cardiovascular calcification: current controversies and novel concepts. Cardiovasc Pathol. 2015;24:207–12. [DOI] [PubMed] [Google Scholar]
- 118.Baidzajevas K, Hadadi E, Lee B, Lum J, Shihui F, Sudbery I, Kiss-Toth E, Wong SC and Wilson HL. Macrophage polarisation associated with atherosclerosis differentially affects their capacity to handle lipids. Atherosclerosis. 2020;305:10–18. [DOI] [PubMed] [Google Scholar]
- 119.Ferraro RA, van Rosendael AR, Lu Y, Andreini D, Al-Mallah MH, Cademartiri F, Chinnaiyan K, Chow BJW, Conte E, Cury RC, et al. Non-obstructive high-risk plaques increase the risk of future culprit lesions comparable to obstructive plaques without high-risk features: the ICONIC study. Eur Heart J Cardiovasc Imaging. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee SE, Sung JM, Andreini D, Al-Mallah MH, Budoff MJ, Cademartiri F, Chinnaiyan K, Choi JH, Chun EJ, Conte E, et al. Differences in Progression to Obstructive Lesions per High-Risk Plaque Features and Plaque Volumes With CCTA. JACC Cardiovasc Imaging. 2020;13:1409–1417. [DOI] [PubMed] [Google Scholar]
- 121.Meikle PJ, Wong G, Tsorotes D, Barlow CK, Weir JM, Christopher MJ, MacIntosh GL, Goudey B, Stern L, Kowalczyk A, et al. Plasma lipidomic analysis of stable and unstable coronary artery disease. Arterioscler Thromb Vasc Biol. 2011;31:2723–32. [DOI] [PubMed] [Google Scholar]
- 122.Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG and Williams SA. Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients With Stable Coronary Heart Disease. JAMA. 2016;315:2532–2541. [DOI] [PubMed] [Google Scholar]
- 123.Karlsson FH, Fak F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Backhed F and Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fu Q, Zhao M, Wang D, Hu H, Guo C, Chen W, Li Q, Zheng L and Chen B. Coronary Plaque Characterization Assessed by Optical Coherence Tomography and Plasma Trimethylamine-N-oxide Levels in Patients With Coronary Artery Disease. Am J Cardiol. 2016;118:1311–1315. [DOI] [PubMed] [Google Scholar]
- 125.Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, Bobik A and Toh BH. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. 2011;109:830–40. [DOI] [PubMed] [Google Scholar]
- 126.Herrington DM, Mao C, Parker SJ, Fu Z, Yu G, Chen L, Venkatraman V, Fu Y, Wang Y, Howard TD, et al. Proteomic Architecture of Human Coronary and Aortic Atherosclerosis. Circulation. 2018;137:2741–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Valencia-Morales Mdel P, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Moran S, et al. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genomics. 2015;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zeller T, Keller T, Ojeda F, Reichlin T, Twerenbold R, Tzikas S, Wild PS, Reiter M, Czyz E, Lackner KJ, et al. Assessment of microRNAs in patients with unstable angina pectoris. Eur Heart J. 2014;35:2106–14. [DOI] [PubMed] [Google Scholar]
- 129.Papaspyridonos M, Smith A, Burnand KG, Taylor P, Padayachee S, Suckling KE, James CH, Greaves DR and Patel L. Novel candidate genes in unstable areas of human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2006;26:1837–44. [DOI] [PubMed] [Google Scholar]
- 130.Greenland P, Blaha MJ, Budoff MJ, Erbel R and Watson KE. Coronary Calcium Score and Cardiovascular Risk. Journal of the American College of Cardiology. 2018;72:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–45. [DOI] [PubMed] [Google Scholar]
- 132.Dweck MR, Aikawa E, Newby DE, Tarkin JM, Rudd JH, Narula J and Fayad ZA. Noninvasive Molecular Imaging of Disease Activity in Atherosclerosis. Circ Res. 2016;119:330–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chowdhury MM, Tawakol A and Jaffer FA. Molecular Imaging of Atherosclerosis: A Clinical Focus. Curr Cardiovasc Imaging Rep. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, Di Carli MF and Libby P. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. J Am Coll Cardiol. 2011;58:603–14. [DOI] [PubMed] [Google Scholar]
- 135.Tawakol A, Migrino RQ, Bashian GG, Bedri S, Vermylen D, Cury RC, Yates D, LaMuraglia GM, Furie K, Houser S, et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. 2006;48:1818–24. [DOI] [PubMed] [Google Scholar]
- 136.Taqueti VR, Di Carli MF, Jerosch-Herold M, Sukhova GK, Murthy VL, Folco EJ, Kwong RY, Ozaki CK, Belkin M, Nahrendorf M, et al. Increased microvascularization and vessel permeability associate with active inflammation in human atheromata. Circulation Cardiovascular imaging. 2014;7:920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rudd JH, Myers KS, Bansilal S, Machac J, Rafique A, Farkouh M, Fuster V and Fayad ZA. (18)Fluorodeoxyglucose positron emission tomography imaging of atherosclerotic plaque inflammation is highly reproducible: implications for atherosclerosis therapy trials. J Am Coll Cardiol. 2007;50:892–6. [DOI] [PubMed] [Google Scholar]
- 138.Ruehm SG, Corot C, Vogt P, Kolb S and Debatin JF. Magnetic resonance imaging of atherosclerotic plaque with ultrasmall superparamagnetic particles of iron oxide in hyperlipidemic rabbits. Circulation. 2001;103:415–22. [DOI] [PubMed] [Google Scholar]
- 139.Alam SR, Shah AS, Richards J, Lang NN, Barnes G, Joshi N, MacGillivray T, McKillop G, Mirsadraee S, Payne J, et al. Ultrasmall superparamagnetic particles of iron oxide in patients with acute myocardial infarction: early clinical experience. Circulation Cardiovascular imaging. 2012;5:559–65. [DOI] [PubMed] [Google Scholar]
- 140.Tang TY, Howarth SP, Miller SR, Graves MJ, Patterson AJ, JM UK-I, Li ZY, Walsh SR, Brown AP, Kirkpatrick PJ, et al. The ATHEROMA (Atorvastatin Therapy: Effects on Reduction of Macrophage Activity) Study. Evaluation using ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging in carotid disease. J Am Coll Cardiol. 2009;53:2039–50. [DOI] [PubMed] [Google Scholar]
- 141.Irkle A, Vesey AT, Lewis DY, Skepper JN, Bird JL, Dweck MR, Joshi FR, Gallagher FA, Warburton EA, Bennett MR, et al. Identifying active vascular microcalcification by (18)F-sodium fluoride positron emission tomography. Nat Commun. 2015;6:7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Youn T, Al’Aref SJ, Narula N, Salvatore S, Pisapia D, Dweck MR, Narula J, Lin FY, Lu Y, Kumar A, et al. (18)F-Sodium Fluoride Positron Emission Tomography/Computed Tomography in Ex Vivo Human Coronary Arteries With Histological Correlation. Arterioscler Thromb Vasc Biol. 2020;40:404–411. [DOI] [PubMed] [Google Scholar]
- 143.Joshi NV, Vesey AT, Williams MC, Shah ASV, Calvert PA, Craighead FHM, Yeoh SE, Wallace W, Salter D, Fletcher AM, et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: a prospective clinical trial. The Lancet. 2014;383:705–713. [DOI] [PubMed] [Google Scholar]
- 144.Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, St Hilaire C and Shanahan C. Medial vascular calcification revisited: review and perspectives. Eur Heart J. 2014;35:1515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Syed MB, Fletcher AJ, Forsythe RO, Kaczynski J, Newby DE, Dweck MR and van Beek EJ. Emerging techniques in atherosclerosis imaging. Br J Radiol. 2019;92:20180309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bentzon JF, Otsuka F, Virmani R and Falk E. Mechanisms of Plaque Formation and Rupture. Circulation Research. 2014;114:1852–1866. [DOI] [PubMed] [Google Scholar]
- 147.Galis ZS, Sukhova GK, Lark MW and Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. [DOI] [PubMed] [Google Scholar]
- 149.Falk E, Nakano M, Bentzon JF, Finn AV and Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. 2013;34:719–28. [DOI] [PubMed] [Google Scholar]
- 150.Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G and Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015;36:1394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, Davignon J, Erbel R, Fruchart JC, Tardif JC, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. Jama. 2006;295:1556–65. [DOI] [PubMed] [Google Scholar]
- 152.Eicher JD, Chami N, Kacprowski T, Nomura A, Chen MH, Yanek LR, Tajuddin SM, Schick UM, Slater AJ, Pankratz N, et al. Platelet-Related Variants Identified by Exomechip Meta-analysis in 157,293 Individuals. Am J Hum Genet. 2016;99:40–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schubert S, Weyrich AS and Rowley JW. A tour through the transcriptional landscape of platelets. Blood. 2014;124:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bongiovanni D, Santamaria G, Klug M, Santovito D, Felicetta A, Hristov M, von Scheidt M, Aslani M, Cibella J, Weber C, et al. Transcriptome Analysis of Reticulated Platelets Reveals a Prothrombotic Profile. Thromb Haemost. 2019;119:1795–1806. [DOI] [PubMed] [Google Scholar]
- 155.Ward-Caviness CK, Huffman JE, Everett K, Germain M, van Dongen J, Hill WD, Jhun MA, Brody JA, Ghanbari M, Du L, et al. DNA methylation age is associated with an altered hemostatic profile in a multiethnic meta-analysis. Blood. 2018;132:1842–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zhu GF, Yang LX, Guo RW, Liu H, Shi YK, Ye JS and Yang ZH. microRNA-155 is inversely associated with severity of coronary stenotic lesions calculated by the Gensini score. Coron Artery Dis. 2014;25:304–10. [DOI] [PubMed] [Google Scholar]
- 157.Eken SM, Jin H, Chernogubova E, Li Y, Simon N, Sun C, Korzunowicz G, Busch A, Backlund A, Osterholm C, et al. MicroRNA-210 Enhances Fibrous Cap Stability in Advanced Atherosclerotic Lesions. Circ Res. 2017;120:633–644. [DOI] [PubMed] [Google Scholar]
- 158.Cipollone F, Felicioni L, Sarzani R, Ucchino S, Spigonardo F, Mandolini C, Malatesta S, Bucci M, Mammarella C, Santovito D, et al. A unique microRNA signature associated with plaque instability in humans. Stroke. 2011;42:2556–63. [DOI] [PubMed] [Google Scholar]
- 159.Maitrias P, Metzinger-Le Meuth V, Massy ZA, M’Baya-Moutoula E, Reix T, Caus T and Metzinger L. MicroRNA deregulation in symptomatic carotid plaque. J Vasc Surg. 2015;62:1245–50 e1. [DOI] [PubMed] [Google Scholar]
- 160.Schulte C, Molz S, Appelbaum S, Karakas M, Ojeda F, Lau DM, Hartmann T, Lackner KJ, Westermann D, Schnabel RB, et al. miRNA-197 and miRNA-223 Predict Cardiovascular Death in a Cohort of Patients with Symptomatic Coronary Artery Disease. PLoS One. 2015;10:e0145930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Januzzi JL Jr., Mahler SA, Christenson RH, Rymer J, Newby LK, Body R, Morrow DA and Jaffe AS. Recommendations for Institutions Transitioning to High-Sensitivity Troponin Testing: JACC Scientific Expert Panel. J Am Coll Cardiol. 2019;73:1059–1077. [DOI] [PubMed] [Google Scholar]
- 162.Jacob J, Ngo D, Finkel N, Pitts R, Gleim S, Benson MD, Keyes MJ, Farrell LA, Morgan T, Jennings LL, et al. Application of Large-Scale Aptamer-Based Proteomic Profiling to Planned Myocardial Infarctions. Circulation. 2018;137:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Deleon-Pennell KY, Ero OK, Ma Y, Padmanabhan Iyer R, Flynn ER, Espinoza I, Musani SK, Vasan RS, Hall ME, Fox ER, et al. Glycoproteomic Profiling Provides Candidate Myocardial Infarction Predictors of Later Progression to Heart Failure. ACS Omega. 2019;4:1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Matic LP, Jesus Iglesias M, Vesterlund M, Lengquist M, Hong MG, Saieed S, Sanchez-Rivera L, Berg M, Razuvaev A, Kronqvist M, et al. Novel Multiomics Profiling of Human Carotid Atherosclerotic Plaques and Plasma Reveals Biliverdin Reductase B as a Marker of Intraplaque Hemorrhage. JACC Basic Transl Sci. 2018;3:464–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shah SH, Bain JR, Muehlbauer MJ, Stevens RD, Crosslin DR, Haynes C, Dungan J, Newby LK, Hauser ER, Ginsburg GS, et al. Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ Cardiovasc Genet. 2010;3:207–14. [DOI] [PubMed] [Google Scholar]
- 166.Shah SH, Sun JL, Stevens RD, Bain JR, Muehlbauer MJ, Pieper KS, Haynes C, Hauser ER, Kraus WE, Granger CB, et al. Baseline metabolomic profiles predict cardiovascular events in patients at risk for coronary artery disease. Am Heart J. 2012;163:844–850 e1. [DOI] [PubMed] [Google Scholar]
- 167.Lewis GD, Wei R, Liu E, Yang E, Shi X, Martinovic M, Farrell L, Asnani A, Cyrille M, Ramanathan A, et al. Metabolite profiling of blood from individuals undergoing planned myocardial infarction reveals early markers of myocardial injury. Journal of Clinical Investigation. 2008;118:3503–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bodi V, Sanchis J, Morales JM, Marrachelli VG, Nunez J, Forteza MJ, Chaustre F, Gomez C, Mainar L, Minana G, et al. Metabolomic profile of human myocardial ischemia by nuclear magnetic resonance spectroscopy of peripheral blood serum: a translational study based on transient coronary occlusion models. J Am Coll Cardiol. 2012;59:1629–41. [DOI] [PubMed] [Google Scholar]
- 169.Surendran A, Aliani M and Ravandi A. Metabolomic characterization of myocardial ischemia-reperfusion injury in ST-segment elevation myocardial infarction patients undergoing percutaneous coronary intervention. Sci Rep. 2019;9:11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella-Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nayor M and Vasan RS. Recent Update to the US Cholesterol Treatment Guidelines: A Comparison With International Guidelines. Circulation. 2016;133:1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lee KK, Ferry AV, Anand A, Strachan FE, Chapman AR, Kimenai DM, Meex SJR, Berry C, Findlay I, Reid A, et al. Sex-Specific Thresholds of High-Sensitivity Troponin in Patients With Suspected Acute Coronary Syndrome. J Am Coll Cardiol. 2019;74:2032–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, Ellis SG, Lincoff AM and Topol EJ. Prevalence of conventional risk factors in patients with coronary heart disease. Jama. 2003;290:898–904. [DOI] [PubMed] [Google Scholar]
- 174.Tajeu GS, Booth JN 3rd, Colantonio LD, Gottesman RF, Howard G, Lackland DT, O’Brien EC, Oparil S, Ravenell J, Safford MM, et al. Incident Cardiovascular Disease Among Adults With Blood Pressure <140/90 mm Hg. Circulation. 2017;136:798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Song M, Hu FB, Wu K, Must A, Chan AT, Willett WC and Giovannucci EL. Trajectory of body shape in early and middle life and all cause and cause specific mortality: results from two prospective US cohort studies. BMJ. 2016:i2195–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee EY, Yang Y, Kim HS, Cho JH, Yoon KH, Chung WS, Lee SH and Chang K. Effect of visit-to-visit LDL-, HDL-, and non-HDL-cholesterol variability on mortality and cardiovascular outcomes after percutaneous coronary intervention. Atherosclerosis. 2018;279:1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.