

Abstract
Brain pericytes regulate diverse aspects of neurovascular development and function, including blood-brain barrier (BBB) induction and maintenance. Primary brain pericytes have been widely employed in coculture-based in vitro models of the BBB, and a method to generate brain pericytes from human pluripotent stem cells (hPSCs) could provide a renewable, genetically tractable source of cells for BBB modeling and studying pericyte roles in development and disease. Here we describe a protocol to differentiate hPSCs to NG2+PDGFRβ+⍺SMAlow brain pericyte-like cells in 22–25 days through a p75-NGFR+HNK-1+ neural crest intermediate, which mimics the developmental origin of forebrain pericytes. The resulting brain pericyte-like cells have molecular and functional attributes of brain pericytes. We also provide protocols for maintenance, cryopreservation, and recovery of the neural crest intermediate, and for molecular and functional characterization of the resulting cells.
Keywords: brain pericytes, neural crest, human pluripotent stem cells, neurovascular unit, blood-brain barrier
INTRODUCTION
Pericytes are mural cells that line the outer surface of microvessels and regulate diverse aspects of vascular development and function (reviewed in Armulik et al., 2011). In the brain, pericytes are a constituent of the neurovascular unit (NVU) and among several specialized functions, regulate blood-brain barrier (BBB) development and maintenance (Armulik et al., 2010; Daneman et al., 2010; Bell et al., 2010). Primary rodent, bovine, and human pericytes have been widely employed in in vitro models of the NVU, where they generally improve brain microvascular endothelial cell (BMEC) barrier properties (see Commentary). As primary human brain pericytes have limited availability and rapidly undergo dedifferentiation in culture (Boado 1994; Ramsauer 1998), acute differentiation of human pluripotent stem cells (hPSCs) to brain pericytes is a promising alternative for in vitro modeling applications. hPSCs have been used to generate other NVU cell types (reviewed in Gastfriend et al., 2018) and are a tractable model system for isogenic modeling of the NVU and genetic manipulation.
Quail-chick chimeras and lineage tracing studies revealed a neural crest origin of forebrain pericytes (Etchevers et al., 2001; Korn et al., 2002; Yamanishi et al., 2012; Ando et al., 2016), which contrasts with the mesodermal developmental origin of mural cells in other organs. This motivates the development of methods to differentiate pericyte-like cells from hPSC-derived neural crest. Recently, we and others showed that pericyte-like cells could be differentiated from hPSC-derived neural crest (Stebbins et al., 2019; Faal et al., 2019; Griffin and Bajpai, 2019). We adapted existing protocols that rely on inhibition of TGF-β superfamily signaling and activation of Wnt signaling to differentiate hPSCs to p75-NGFR+HNK-1+ neural crest cells (Lee et al., 2007, 2010; Menendez et al., 2012, 2013; Chambers et al., 2013). We found that E6 medium (Chen et al., 2011) supplemented with 10% FBS efficiently directed these hPSC-derived neural crest cells to NG2+PDGFRβ+ mural cells that also expressed brain mural cell-enriched genes (ZIC1 and FOXF2) and expressed low levels of the vascular smooth muscle cell (vSMC) marker ⍺-smooth muscle actin (⍺SMA) at the protein and transcript level (Stebbins et al., 2019), but did express other vSMC-related contractile proteins such as calponin and SM22⍺. Knowledge of markers that distinguish pericytes, vSMCs, and other stromal cells in the brain is incomplete (Vanlandewijck et al., 2018; He et al., 2018; Alarcon-Martinez et al., 2018; Chasseigneaux et al., 2018), but given the low level of ⍺SMA expression, we termed the resulting cells brain pericyte-like. Importantly, the hPSC-derived brain pericyte-like cells have functional attributes of brain pericytes, self-assemble with endothelial cell cords, and induce BBB properties in hPSC-derived BMEC-like cells and primary rat BMECs (Stebbins et al., 2019; Canfield et al., 2019).
Here, we describe methods to differentiate hPSCs to brain pericyte-like cells via a neural crest intermediate (Figure 1A). We first outline methods to differentiate hPSCs to neural crest over a 15-day period and isolate p75-NGFR+ neural crest cells via magnetic activated cell sorting (MACS) (Basic Protocol 1). We next describe methods to differentiate neural crest cells to brain pericyte-like cells over a 6–9 day period (Basic Protocol 2), after which they may be used for downstream applications such as in coculture NVU models. We also provide protocols for maintenance, cryopreservation, and recovery of neural crest cells for subsequent differentiation to brain pericyte-like cells (Support Protocol 2), methods for characterization of neural crest cells and brain pericyte-like cells by flow cytometry, immunocytochemistry, and quantitative PCR (qPCR) (Support Protocol 1 and Support Protocol 3), and a method to evaluate the ability of brain pericyte-like cells to self-assemble with endothelial cords (Support Protocol 4).
Figure 1. Protocol overview.
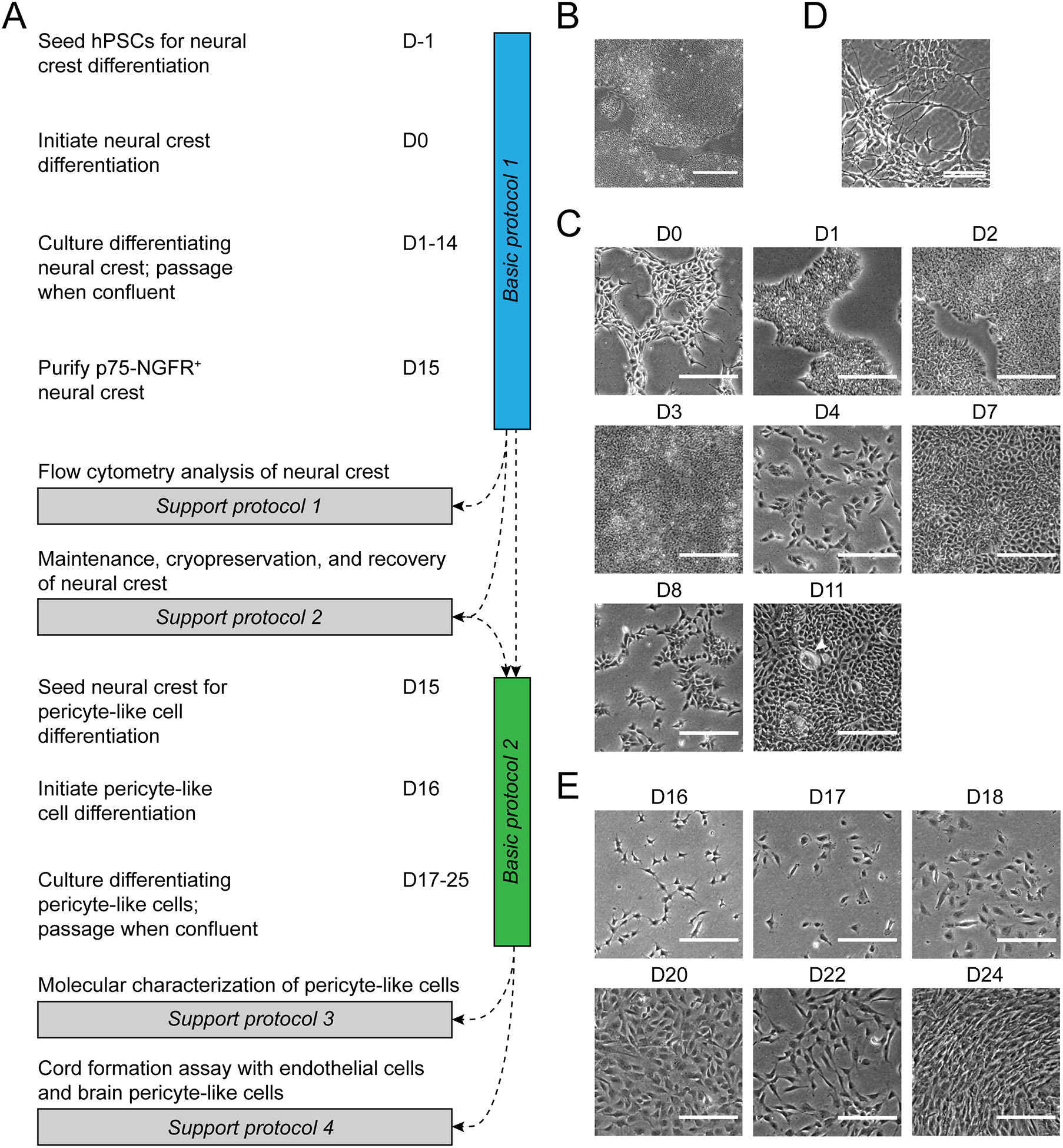
(A) Schematic of protocol steps and timing. (B) Brightfield image of hPSC colony morphology at a density appropriate for seeding. Scale bar: 500 μm. (C) Brightfield images of typical cell morphology during the differentiation of hPSCs to neural crest. Arrowhead indicates a non-neural crest colony. Scale bars: 200 μm. (D) Example of spontaneous differentiation in a maintained neural crest culture. Scale bar: 100 μm. (E) Brightfield images of typical cell morphology during the differentiation of neural crest to brain pericyte-like cells. Scale bars: 200 μm.
BASIC PROTOCOL 1: Differentiation of hPSCs to neural crest
This protocol describes a method to generate p75-NGFR+HNK-1+ neural crest cells from hPSCs. First, hPSCs are singularized and seeded on Matrigel-coated plates using the ROCK inhibitor Y-27632 to permit attachment and survival of singularized hPSCs. Next, hPSCs are treated with a neural crest-inductive medium termed E6-CSFD, composed of E6 medium (Chen et al., 2011) supplemented with CHIR 99021 to activate canonical Wnt signaling, SB 431542 to inhibit TGF-β/Activin/Nodal signaling, FGF2, dorsomorphin to inhibit BMP signaling, and heparin to stabilize FGF2. Differentiating neural crest cells are expanded in E6-CSFD medium for 15 days, passaging 1:6 when confluent. On D15, MACS is used to isolate p75-NGFR+ neural crest cells. We previously demonstrated that the resulting cells express known neural crest markers including SOX9, SOX10, TFAP2A, and ETS1, have multipotency characteristic of cranial neural crest as they are able to differentiate to peripheral neurons and mesenchymal derivatives, and can be derived from multiple induced pluripotent stem cell (iPSC) and human embryonic stem cell (hESC) lines (Stebbins et al., 2019).
Materials
hPSCs (e.g. H9 hESCs or IMR90–4 iPSCs, WiCell; or CS03iCTR-n2 iPSCs, Cedars-Sinai iPSC Core Repository)
Accutase (Innovative Cell Technologies, AT104)
DMEM/F12 medium (Gibco, 11330032)
E8 medium (TeSR-E8, STEMCELL Technologies, 05990; or Essential 8 Medium, Gibco, A1517001; or manufactured in-house according to Chen et al., 2011)
Y-27632 (10 mM stock solution; see recipe)
Matrigel-coated 6-well plates (see recipe)
E6-CSFD medium (see recipe)
MACS buffer (see recipe)
Neural Crest Stem Cell Microbeads, human (Miltenyi, 130-097-127)
FcR Blocking Reagent, human (Miltenyi, 130-059-901)
40 μm cell strainers (Falcon, 352340)
Hemocytometer (e.g. Hausser Scientific, 3110)
Automated cell counter (optional; e.g. Countess II, Invitrogen, AMQAX1000)
MidiMACS Separator (Miltenyi, 130-042-302)
MACS MultiStand (Miltenyi, 130-042-303)
LS Columns (Miltenyi, 130-042-401)
All operations should be performed using sterile technique in a biological safety cabinet.
Seeding hPSCs for neural crest differentiation
-
1
When hPSCs are at an appropriate density for passaging (approximately 70% confluence or when colonies begin to touch; Figure 1B), aspirate medium from three wells of a 6-well plate and add 1 mL Accutase to each well. Incubate 5–7 min at 37°C.
Three wells of a 6-well plate of hPSCs will be sufficient to seed at least three wells of a 6-well plate for neural crest differentiation.
-
2
Using a p1000 pipette, dissociate and singularize cells, and add the 3 mL of single cell suspension to 12 mL DMEM/F12 in a 15 mL tube. Mix well by gentle pipetting.
If cells do not easily dissociate, incubate for another 1–2 minutes rather than harshly triturating.
-
3
Transfer 10 μL of the cell suspension to a hemocytometer and count cells. Centrifuge the remaining cell suspension 5 min at 200×g, room temperature.
Alternatively, if the cell suspension is dilute (fewer than ~30 cells per 1 mm × 1 mm hemocytometer square), cells can be counted after centrifugation and resuspension in 1 mL of E8 medium. An automated cell counter can also be used instead of a hemocytometer.
-
4
Aspirate supernatant and resuspend the cell pellet in 1 mL of E8 medium.
-
5
Prepare 6.5 mL of E8 medium + 10 μM Y-27632 (add 6.5 μL 10 mM Y-27632 to 6.5 mL E8 in a 15 mL tube). Add a volume of cell suspension from step 4 containing 2.84 × 106 cells to the prepared medium. This achieves a final concentration of 4.38 × 105 cells/mL, equivalent to 8.75 × 105 cells/well.
-
6
Aspirate Matrigel coating solution from 3 wells of a 6-well plate and add 2 mL of the prepared cell suspension in E8 medium + 10 μM Y-27632 to each well.
-
7
Place the plate in a 37°C, 5% CO2 incubator. To evenly distribute cells, shake the plate back and forth, then side to side, repeating three times. Do not disturb the plate for 24 h.
Neural crest differentiation
-
8
The following day (D0), replace medium with 2 mL E6-CSFD per well.
-
9
Continue to feed cells daily with 2 mL E6-CSFD medium per well until cells reach 100% confluence (typically by D1–3, Figure 1C).
-
10
When cells reach 100% confluence, aspirate medium from one well and add 1 mL Accutase. Replace medium in remaining wells with E6-CSFD to retain as backup. Incubate 5–6 min at 37°C.
A backup well can be used the following day if poor reattachment is observed (see step 16).
-
11
Using a p1000 pipette, dissociate and singularize cells, and add the single cell suspension to 4 mL DMEM/F12 in a 15 mL tube.
If cells do not easily dissociate, incubate for another 1–2 minutes rather than harshly triturating.
-
12
Centrifuge the cell suspension 5 min at 200×g, room temperature.
-
13
Aspirate supernatant and resuspend the cell pellet in 12 mL E6-CSFD medium (1:6 split ratio).
-
14
Aspirate Matrigel coating solution from all wells of a 6-well plate and add 2 mL of the cell suspension to each well.
-
15
Place the plate in a 37°C, 5% CO2 incubator. To evenly distribute cells, shake the plate back and forth, then side to side, repeating three times. Do not disturb the plate for 24 h.
-
16
The following day, observe cells under a microscope.
Some cell death after the first passaging step is expected. However, if reattachment is poor (see example of expected cell density one day after passaging in Figure 1C, D4), discard the newly-passaged cells and repeat steps 10–15 using one of the wells retained as backup in step 10. Supplementation of E6-CSFD with 10 μM Y-27632 may improve attachment of cells from some hPSC lines during the first passage.
-
17
Continue to feed cells daily with 2 mL E6-CSFD medium per well.
-
18
When cells reach 100% confluence, repeat steps 10–16 to passage the differentiating neural crest cells 1:6. To ensure an adequate number of cells are available for sorting on D15, if cells reach 100% confluence on D12 or D13, passage 1:2 or 1:3 (i.e., dissociate cells from 3 wells or 2 wells and add to a new 6-well plate), and if cells reach 100% confluence on D14, passage all wells 1:2 to two new 6-well plates.
Differentiating neural crest cells typically do not require more than 5 min of Accutase treatment after the first passage. Differentiating neural crest cells typically require 3–5 passages prior to D15.
Purification of p75-NGFR+ neural crest cells
-
8
On D15, aspirate medium from all wells and add 1 mL Accutase to each well. Incubate 5 min at 37°C.
-
9
Using a p1000 pipette, dissociate and singularize cells, and pass the single cell suspension (6 mL total) through a 40 μm cell strainer into 24 mL DMEM/F12 in a 50 mL tube.
Gentle trituration and filtration through a 40 μm cell strainer is critical to achieve a single cell suspension, as clumps of cells may clog the MACS LS column and/or contribute to poor post-sort neural crest purity.
-
10
Transfer 10 μL of the cell suspension to a hemocytometer and count cells. Centrifuge the remaining cell suspension 5 min at 180×g, 4°C.
-
11
Aspirate supernatant and resuspend the cell pellet in 60 μL MACS buffer per 107 cells. If there are less than 107 cells, resuspend in 60 μL MACS buffer.
-
12
Add 20 μL FcR blocking reagent per 107 cells, then add 20 μL neural crest stem cell microbeads per 107 cells. Mix gently and incubate 15 min at 4°C.
-
13
Add 2 mL MACS buffer per 107 cells and mix gently. At this time, aliquots of cells can be removed for flow cytometry analysis (see Support Protocol 1). Centrifuge the cell suspension 5 min at 180×g, 4°C.
-
14
During centrifugation, prepare the MACS apparatus: place a LS column in the Midi MACS Separator (magnet) on the stand. Position a 15 mL tube beneath the column to collect flow through. To prime the column, add 3 mL MACS buffer.
-
15
Aspirate supernatant and resuspend the cell pellet in 500 μL MACS buffer per 2×107 cells. If there are less than 2×107 cells, resuspend in 500 μL MACS buffer. Add 500 μL cell suspension (i.e. ≤ 2×107 cells) to the column. Allow the cell suspension to flow through the column until no liquid remains in the reservoir.
2×107 cells is the recommended maximum capacity of the LS column. If desired, the remaining cells may be sorted in parallel if multiple MACS separators are available, or placed on ice and sorted after completing sorting of the first aliquot.
-
16
Add 3 mL MACS buffer to the column. Allow the buffer to flow through the column until no liquid remains in the reservoir. Repeat this step for a total of three washes. Discard flow through.
-
17
Remove the LS column from the MACS separator (magnet) and place in an empty 15 mL tube. Add 5 mL MACS buffer and use the plunger provided with the LS column to elute cells. Discard the column.
-
18
Centrifuge the eluate 5 min at 180×g, 4°C.
-
19
During centrifugation, prepare another LS column for the second sorting step as described in step 25.
-
20
Aspirate supernatant and resuspend the cell pellet (the eluate from the first sorting step) in 500 μL MACS buffer. Add this cell suspension to the column and complete the second sorting step as described in steps 26–28.
If desired, an aliquot of cells can be removed prior to the second sorting step for subsequent flow cytometry analysis (see Support Protocol 1). Place this aliquot on ice until sorting is complete and analyze concurrently with the pre-sort and post-sort samples. If one MACS step routinely enriches neural crest cells to ≥ 95% p75-NGFR+HNK-1+, the second MACS step can be eliminated.
-
21
Transfer 10 μL of the cell suspension to a hemocytometer and count cells. At this time, an aliquot of cells can be removed for flow cytometry analysis (see Support Protocol 1). Centrifuge the cell suspension 5 min at 180×g, 4°C.
-
22
Aspirate supernatant. At this time, cells may be seeded for differentiation of brain pericyte-like cells (see Basic Protocol 2), or expanded or cryopreserved (see Support Protocol 2).
Brightfield images of neural crest cells throughout the differentiation protocol are shown in Figure 1C.
BASIC PROTOCOL 2: Differentiation of neural crest to brain pericyte-like cells
This protocol describes a method to differentiate brain pericyte-like cells, beginning with hPSC-derived neural crest cells that have been either freshly sorted (Basic Protocol 1), dissociated from a maintained neural crest culture, or thawed from a cryopreserved stock (Support Protocol 2). The neural crest cells are seeded on uncoated, tissue culture-treated well plates, with the ROCK inhibitor Y-27632 included to promote attachment. The cells are subsequently treated with E6 + 10% FBS medium for 6–9 days, depending on downstream application (see Commentary), and passaged 1:2 when confluent. Treatment of neural crest cells with E6 + 10% FBS medium yields NG2+ PDGFRβ+ mural cells in the absence of exogenous growth factors. We previously demonstrated that the resulting cells express low levels of the smooth muscle cell marker ACTA2 (encoding ⍺SMA) on both the transcript and protein levels, and therefore classified these mural cells as pericyte-like (Stebbins et al., 2019). The resulting brain pericyte-like cells can be used for molecular profiling or functional analyses, such as coculture with endothelial cells in a Transwell system for BBB modeling (see Commentary) or in a Matrigel-based cord formation assay (Support Protocol 4).
Materials
Neural crest cells differentiated from hPSCs according to Basic Protocol 1 or cultured according to Support Protocol 2
E6-CSFD medium (see recipe)
Y-27632 (10 mM stock solution; see recipe)
E6 + 10% FBS medium (see recipe)
Accutase (Innovative Cell Technologies, AT104)
DMEM/F12 medium (Gibco, 11330032)
Tissue culture-treated well plates (6-well, 12-well, 24-well, 48-well, or 96-well)
Resuspend the neural crest cell pellet in 1 mL E6-CSFD medium.
-
Prepare an appropriate volume of E6-CSFD medium + 10 μM Y-27632 for your desired application. For example, to seed one 6-well plate, prepare 12.5 mL medium (add 12.5 μL 10 mM Y-27632 to 12.5 mL E6-CSFD in a 15 mL tube).
Medium volumes and seeding densities for other well plate types are listed in Table 1. 6-well plates are appropriate for flow cytometry and PCR analyses, 12-well and 24-well plates are appropriate for Transwell-based cocultures, and 48-well and 96-well plates are appropriate for immunocytochemistry. Pericyte-like cells typically require one passage prior to D22–25, and therefore may be seeded initially in 6-well plates and subsequently passaged to a smaller-sized well plate depending on the intended downstream application.
Add a volume of the cell suspension to the prepared E6-CSFD + 10 μM Y-27632 to achieve the appropriate seeding density as shown in Table 1.
Add an appropriate volume of cell suspension to each well of an uncoated, tissue-culture treated well plate.
Place the plate in a 37°C, 5% CO2 incubator. To evenly distribute cells, shake the plate back and forth, then side to side, repeating three times. Do not disturb the plate for 24 h.
The following day (D16), replace medium with E6 + 10% FBS. Continue to feed cells daily with E6 + 10% FBS.
-
When cells reach 90–100% confluence (typically after 3–4 days in E6 + 10% FBS), aspirate medium add a half medium volume of Accutase. Replace medium in at least 1 well with E6 + 10% FBS to retain as backup. Incubate 5–15 min at 37°C, observing cells under a microscope every 5 min until > 90% of cells appear detached from the plate.
If cultures of differentiating pericyte-like cells do not singularize after ~15 min of Accutase treatment, consider passing the cell suspension through a 40 μm cell strainer, or passaging one day earlier. Insufficient singularization of cells will lead to uneven distribution of cells after replating and difficulty counting cells, if applicable.
Using a p1000 pipette, dissociate and singularize cells, and add the single cell suspension to 4× volume of DMEM/F12 in a 15 mL tube.
Centrifuge the cell suspension 5 min at 200×g, room temperature.
-
Aspirate supernatant and resuspend the cell pellet in a volume of E6 + 10% FBS appropriate to achieve a 1:2 split ratio.
For example, if the cell pellet was derived from 3 confluent wells of a 6-well plate, resuspend in a volume of E6 + 10% FBS sufficient to seed 6 wells of a 6- well plate, i.e. 12 mL. Alternatively, pericyte-like cells can be counted and replated at a specific cell density, which may be beneficial for some applications (e.g. Transwell-based cocultures comparing barrier-inductive capacity of multiple cell types).
Place the plate in a 37°C, 5% CO2 incubator. To evenly distribute cells, shake the plate back and forth, then side to side, repeating three times. Do not disturb the plate for 24 h.
Continue to feed cells daily with 2 mL E6 + 10% FBS until D22–25. At this time, the cells may be used for molecular and functional assays (Support Protocols 3 and 4).
Table 1.
Well plate surface areas, medium volumes, and seeding densities.
| Well plate type | Surface area per well (cm2) | Medium volume per well | Neural crest cell seeding density (cells per well) |
|---|---|---|---|
| 6-well | 9.5 | 2 mL | 1×105 |
| 12-well | 3.8 | 800 μL | 4×104 |
| 24-well | 1.9 | 400 μL | 2×104 |
| 48-well | 0.95 | 200 μL | 1×104 |
| 96-well | 0.32 | 67 μL | 3.4×103 |
Brightfield images of pericyte-like cells during the differentiation are shown in Figure 1E.
SUPPORT PROTOCOL 1: Flow cytometry analysis of neural crest cells
This protocol describes a method for two-color flow cytometry analysis for the neural crest markers p75-NGFR and HNK-1. This protocol can be used to validate the presence of neural crest cells in D15 cultures differentiated according to Basic Protocol 1, and to validate the efficiency of p75-NGFR MACS used to isolate neural crest cells. Prior to MACS, neural crest purity (% p75-NGFR+HNK-1+) varies by hPSC line and differentiation from approximately 40–90%, and MACS reproducibly enriches neural crest purity to ≥ 95% (Figure 2).
Figure 2. Expected results of flow cytometry analysis of hPSC-derived neural crest.
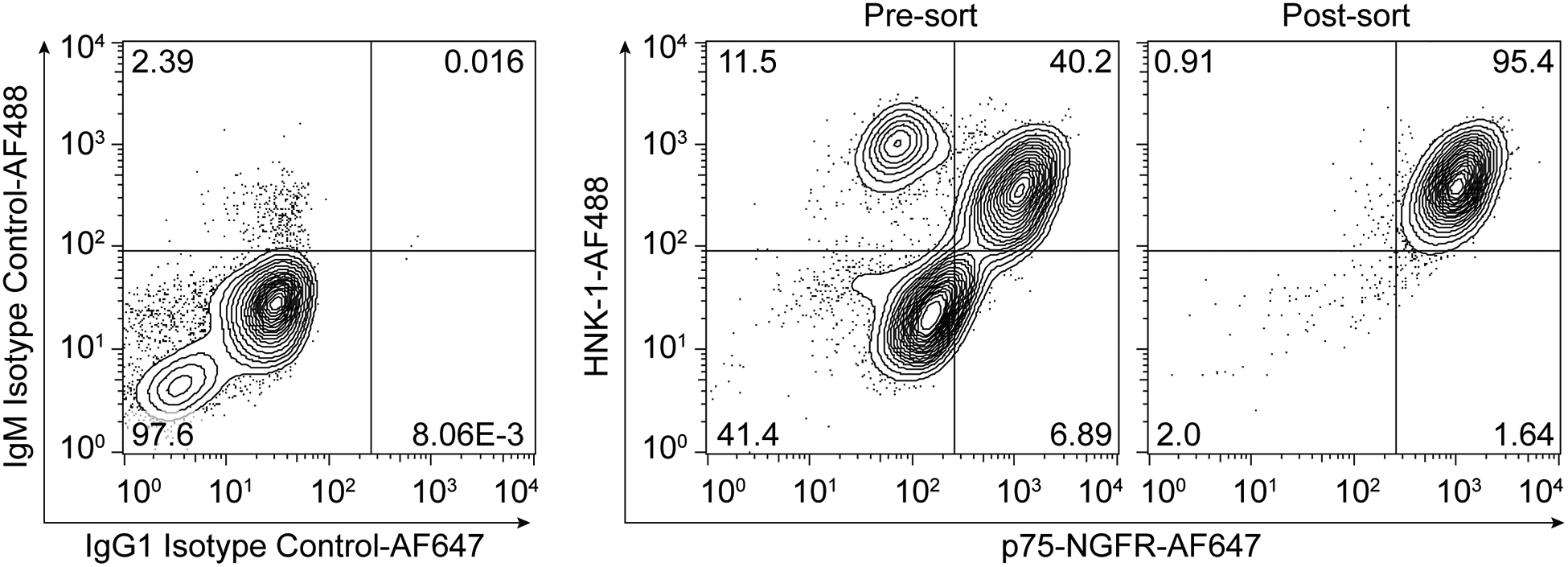
Flow plots show isotype control staining and p75-NGFR/HNK-1 staining of hPSC-derived neural crest cells before MACS (pre-sort) and after two MACS steps (post-sort).
Materials
Neural crest cells differentiated from hPSCs according to Basic Protocol 1
Dulbecco’s phosphate buffered saline, no Ca, no Mg (DPBS, Gibco, 14190144)
Antibodies (see Table 2)
4% PFA solution (see recipe)
Round bottom flow cytometry tubes (Falcon, 352054)
- Flow cytometer
- During purification of neural crest cells (Basic Protocol 1, steps 19–32), collect 106 cells prior to MACS for isotype control staining, 106 cells prior to MACS for p75-NGFR/HNK-1 staining (“pre-sort” sample), and 106 cells after MACS for p75-NGFR/HNK-1 staining (“post-sort” sample). Keep cells on ice in 15 mL tubes while completing the purification protocol.
- Centrifuge all samples 5 min at 180×g, 4°C.
- Prepare primary antibody solutions in cold (4°C) DPBS:
- 200 μL DPBS + 0.4 μL (0.4 μg) p75-NGFR antibody + 0.4 μL (~0.08 μg) HNK-1 antibody
-
100 μL DPBS + mouse IgG1 isotype control antibody + mouse IgM isotype control antibodyIsotype control antibodies should be used at the same concentrations as corresponding primary antibodies.
- Aspirate supernatant from each sample and resuspend the pre-sort and post-sort samples in 100 μL each of the p75-NGFR/HNK-1 antibody solution and resuspend the isotype control sample in the 100 μL isotype control antibody solution. Incubate 30 min on ice.
- Add 2 mL cold DPBS to each tube and mix gently. Centrifuge 5 min at 180×g, 4°C.
- Prepare secondary antibody solution: 300 μL DPBS + 0.6 μL goat anti-Mouse IgM-Alexa Fluor 488 antibody + 0.6 μL goat anti-Mouse IgG1-Alexa Fluor 647 antibody.
- Aspirate supernatant from each sample and resuspend each sample in 100 μL of the prepared secondary antibody solution. Incubate 30 min on ice, protected from light.
- Add 2 mL cold DPBS to each tube and mix gently. Centrifuge 5 min at 180×g, 4°C.
- Aspirate supernatant from each sample and resuspend each sample in 500 μL of 4% PFA solution. Incubate 15 min at room temperature, protected from light.
- Centrifuge samples 5 min at 180×g, room temperature.
- Remove PFA supernatant from each sample and dispose according to institutional protocols. Resuspend each sample in 300 μL DPBS. Transfer samples to round bottom flow cytometry tubes and analyze on a flow cytometer.
Table 2.
Antibodies.
| Target | Species/isotype | Manufacturer, clone (product number) | Fluor.a | App.b | Dilution | Blocking bufferd | Incubation bufferd |
|---|---|---|---|---|---|---|---|
| Primary antibodies | |||||||
| p75-NGFR | Mouse IgG1 | Advanced Targeting Systems, ME20.4 (AB-N07) | U | FC | 0.2 μL / 106 cells | N/A | N/A |
| HNK-1 | Mouse IgM | Sigma, VC1.1 (C6680) | U | FC | 0.2 μL / 106 cells | N/A | N/A |
| PDGFRβ | Mouse IgG2a | BD Biosciences, 28D4 (558820) | U | FC | 1.25 μL / 106 cells | N/A | N/A |
| PDGFRβ | Rabbit IgG | Cell Signaling Technology, 28E1 (3169) | U | ICC | 1:100 | 5% GS + 0.4% TX-100 | 0.4% TX-100 |
| NG2 | Mouse IgG2a | Millipore, 9.2.27 (MAB2029) | U | FC | 2 μL / 106 cells | N/A | N/A |
| ICC | 1:100 | 5% GS + 0.4% TX-100 | 0.4% TX-100 | ||||
| SM22⍺ | Rabbit polyclonal | Abcam, (ab14106) | U | ICC | 1:1000 | 3% BSA + 0.1% TX-100 | 3% BSA |
| Calponin | Mouse IgG1 | Sigma, hCP (C2678) | U | ICC | 1:15000 | 3% BSA + 0.1% TX-100 | 3% BSA |
| ⍺SMA | Mouse IgG2a | Lab Vision, 1A4 (MS-113-P) | U | ICC | 1:100 | 5% milk + 0.1% TX-100 | 5% milk |
| CD31 | Rabbit polyclonal | Lab Vision (RB-10333-P) | U | ICC | 1:25 | 5% GS + 0.4% TX-100 | 0.4% TX-100 |
| Isotype control | Mouse IgG1 | BD Biosciences, MOPC-21 (554121) | U | FC | Note (c) | N/A | N/A |
| Isotype control | Mouse IgM | BD Biosciences, G155–228 (553472) | U | FC | Note (c) | N/A | N/A |
| Isotype control | Mouse IgG2a | BD Biosciences, G155–178 | U | FC | Note (c) | N/A | N/A |
| Secondary antibodies | |||||||
| Mouse IgG1 | Goat polyclonal | Invitrogen, (A-21240) | AF647 | FC | 1:500 | N/A | N/A |
| Mouse IgM | Goat polyclonal | Invitrogen, (A-21042) | AF488 | FC | 1:500 | N/A | N/A |
| Mouse IgG | Goat polyclonal | Invitrogen, (A-11002) | AF488 | FC | 1:500 | N/A | N/A |
| ICC | 1:200 | N/A | Note (e) | ||||
| Rabbit IgG | Goat polyclonal | Invitrogen, (A-21428) | AF555 | ICC | 1:200 | N/A | Note (e) |
| Rabbit IgG | Goat polyclonal | Invitrogen, (A-21245) | AF647 | ICC | 1:200 | N/A | Note (e) |
Fluorophore: U, unconjugated; AF, Alexa Fluor
Application: FC, Flow cytometry; ICC, immunocytochemistry
Use isotype control antibodies at the same concentration as the corresponding primary antibodies
See recipe; N/A, not applicable (for FC or secondary antibodies for ICC)
Use incubation buffer for the corresponding primary antibody
We acquire data using a BD FACSCalibur flow cytometer (excitation at 488 nm and 635 nm, Alexa Fluor 488 emission detected using FL1 with a 530/30 filter and Alexa Fluor 647 emission detected using FL4 with a 661/16 filter). We use FlowJo software to gate cellular events using side scatter (SSC-H) vs. forward scatter (FSC-H) plots and to determine the % p75-NGFR/HNK-1+ cells.
Example flow cytometry data are shown in Figure 2.
SUPPORT PROTOCOL 2: Maintenance, cryopreservation, and recovery of neural crest cells
This protocol describes methods to maintain hPSC-derived neural crest cells in culture, cryopreserve neural crest cells, and recover cryopreserved cells. This allows a large batch of neural crest cells to be differentiated, purified via MACS on D15, and expanded or cryopreserved for subsequent differentiation to brain pericyte-like cells. These strategies reduce the total duration of the brain pericyte-like cell differentiation and may reduce differentiation-to-differentiation variability. However, we recommend performing initial validation of the brain pericyte-like cell differentiation procedures using freshly-sorted D15 neural crest cells.
Materials
Neural crest cells differentiated from hPSCs according to Basic Protocol 1
E6-CSFD medium (see recipe)
Matrigel-coated 6-well plates (see recipe)
Accutase (Innovative Cell Technologies, AT104)
DMEM/F12 medium (Gibco, 11330032)
Neural Crest Cell Cryopreservation medium (see recipe)
Cryovials (e.g. Thermo Scientific, 5000–0020)
Controlled rate freezing device (e.g. Mr. Frosty, Thermo Scientific, 5100–0001)
37°C water bath
Maintenance of neural crest cells
-
1a.
After completing MACS (Basic Protocol 1) or thawing a vial of neural crest cells (step 3c), resuspend the neural crest cell pellet in 1 mL E6-CSFD medium.
-
2a.
Add 12.5 mL E6-CSFD medium to a 15 mL tube and add an appropriate volume of the cell suspension to achieve a final concentration of 5×104–105 cells/mL.
-
3a.
Aspirate Matrigel coating solution from all wells of a Matrigel-coated 6-well plate and add 2 mL of the cell suspension to each well.
-
4a.
Place the plate in a 37°C, 5% CO2 incubator. To evenly distribute cells, shake the plate back and forth, then side to side, repeating three times. Do not disturb the plate for 24 h.
-
5a.Feed cells daily with 2 mL E6-CSFD medium per well. When cells reach 100% confluence, they can be passaged and/or seeded for the brain pericyte-like cell differentiation:
- To passage neural crest cells for continued culture, repeat steps 10–16 of Basic Protocol 1. Continue to feed cells daily with 2 mL E6-CSFD, passaging when confluent.
- To seed neural crest cells for the brain pericyte-like cell differentiation, aspirate medium and add 1 mL Accutase to each well. Incubate 5 min at 37°C. Using a p1000 pipette, dissociate and singularize cells, and add the single cell suspension to 4× volume of DMEM/F12. Transfer 10 μL of the cell suspension to a hemocytometer and count cells. Centrifuge the remaining cell suspension 5 min at 180×g, 4°C. Aspirate the supernatant and proceed to Basic Protocol 2.
Neural crest cells undergo phenotype changes during extended culture (Lee et al., 2007). Thus, although neural crest cells will continue to expand for at least 6 months under these conditions, we recommend using early passage cells (1–3 passages post-MACS) for the pericyte-like cell differentiation. Discard neural crest cultures that exhibit spontaneous differentiation (often appearing as isolated regions of putative neuronal differentiation, Figure 1D) or develop compact non-neural crest colonies similar to those observed prior to MACS (Figure 1C, D11, arrowhead).
Cryopreservation of neural crest cells
-
1b.
Resuspend the neural crest cell pellet in Neural Crest Cell Cryopreservation medium to achieve a concentration of 106 cells per mL, or another concentration appropriate for your downstream application.
-
2b.
Transfer 1 mL cell suspension to each cryovial. Transfer cryovials to a controlled rate freezing device and place in a −80°C freezer overnight.
-
3b.
The following day, transfer cryovials to liquid nitrogen.
We recommend using freshly sorted or early passage (1–3 passages post-MACS) neural crest cells for cryopreservation.
Recovery of cryopreserved neural crest cells
-
1c.
Retrieve a vial of cryopreserved neural crest cells from liquid nitrogen and thaw rapidly by swirling in a 37°C water bath until only a small ice crystal remains.
-
2c.
Using a p1000 pipette, transfer the thawed cell suspension to a 15 mL tube. Add 4 mL DMEM/F12 dropwise. Transfer 10 μL of the cell suspension to a hemocytometer and count cells.
-
3c.
Centrifuge the remaining cell suspension 5 min at 180×g, 4°C. Aspirate supernatant.
-
4c.Thawed neural crest cells can be seeded directly onto uncoated plates for brain pericyte-like cell differentiation, or seeded onto a Matrigel-coated plate to expand neural crest cells.
- To seed thawed neural crest cells for the pericyte-like cell differentiation and differentiate pericyte-like cells, follow steps 1–12 in Basic Protocol 2.
- To seed thawed neural crest cells for expansion, follow steps 1a–5a above.
SUPPORT PROTOCOL 3: Molecular characterization of brain pericyte-like cells
This protocol describes methods for molecular characterization of brain pericyte-like cells, including flow cytometry analysis for NG2 and PDGFRβ expression, immunocytochemistry analysis of NG2, PDGFRβ, calponin, SM22⍺, and ⍺SMA expression, and qPCR analysis of several neural crest-, mural cell-, and pericyte-associated transcripts.
Materials
Pericyte-like cells differentiated from hPSCs according to Basic Protocol 2
Accutase (Innovative Cell Technologies, AT104)
DMEM/F12 medium (Gibco, 11330032)
Dulbecco’s phosphate buffered saline, no Ca, no Mg (DPBS, Gibco, 14190144)
Antibodies (see Table 2)
MACS buffer (see recipe)
4% PFA solution (see recipe)
Hoechst solution (see recipe)
ELIMINase (Decon Labs, 1102)
RNA extraction kit (e.g. RNeasy Mini Kit, Qiagen, 74104; QIAshredder homogenization columns, Qiagen, 79654; RNase-free DNase Set, Qiagen, 79254)
β-mercaptoethanol (Sigma Aldrich, M6250)
Nuclease-free water (e.g. Qiagen, 129115)
70% ethanol
Reverse transcriptase kit (e.g. Omniscript RT Kit, Qiagen, 205111)
Oligo(dT)12–18 primers, diluted to 20 μM (e.g. Invitrogen, 18418012)
RNaseOUT Recombinant Ribonuclease Inhibitor, diluted to 10 U/μL (Invitrogen, 10777019)
SYBR Green qPCR master mix (e.g. Applied Biosystems, A25742)
40 μm cell strainers (Falcon, 352340)
Hemocytometer (e.g. Hausser Scientific, 3110)
Automated cell counter (optional; e.g. Countess II, Invitrogen, AMQAX1000)
Round bottom flow cytometry tubes (Falcon, 352054)
Flow cytometer
Fluorescence microscope
NanoDrop spectrophotometer
8-Strip PCR tubes, 0.2 mL (e.g. Axygen PCR0208CPC)
qPCR plates and covers
qPCR thermal cycler
Table 3.
Primer sequences.
| Gene | Forward primer sequence | Reverse primer sequence | Ampa |
|---|---|---|---|
| NANOG | CGA AGA ATA GCA ATG GTG TGA CG | TTC CAA AGC AGC CTC CAA GTC | 329 |
| NGFR | GTG GGA CAG AGT CTG GGT GT | AAG GAG GGG AGG TGA TAG GA | 201 |
| ZIC1 | TGG CCC GGA GCA GAG TAA T | CCC TGT GTG CGT CCT TTT G | 199 |
| ETS1 | CCT GCA GAA AGA GGA TGT GAA | TGG GCA TGC TCA ATA CCA TAG | 111 |
| PDGFRB | GCT CAC CAT CAT CTC CCT TAT C | CTC ACA GAC TCA ATC ACC TTC C | 89 |
| CSPG4 | Integrated DNA Technologies Predesigned Probe, Hs.PT.58.39417158 | 115 | |
| TBX18 | CCC AGG ACT CCC TCC TAT GT | TAG GAA CCC TGA TGG GTC TG | 200 |
| FOXF2b | ACC AGA GCG TCT GTC AGG ATA TT | GTG ACT TGA ATC CGT CCC AGT TTC | 124 |
| RGS5 | GGA GGC TCC TAA AGA GGT GAA TA | CCA TCA GGG CAT GGA TTC TTT | 119 |
| KCNJ8 | GTG ATT GCC GTC CGA AAT GG | AGT TGG TGA ATA GGA ACC ACC T | 146 |
| ABCC9 | GGG ATT CGT TGC CAC TAC AA | CCA ATA CAG GAA CAG GGC TAA A | 94 |
| ACTA2 | TGT TCC AGC CAT CCT TCA TC | GCA ATG CCA GGG TAC ATA GT | 202 |
| GAPDH | GAA GGT GAA GGT CGG AGT CAA CG | TCC TGG AAG ATG GTG ATG GGA T | 231 |
Amplicon size expected
From (Everson et al., 2017)
Flow cytometry analysis of brain pericyte-like cells
-
1a.
At D22–25 of the pericyte-like cell differentiation, aspirate medium from wells of pericyte-like cells and add a half medium volume of Accutase. Incubate 5–6 min at 37°C.
We typically perform flow cytometry analysis on pericyte-like cells differentiated in 6-well plates.
-
2a.
Using a p1000 pipette, dissociate and singularize cells, and pass the single cell suspension through a 40 μm cell strainer into a 15 mL tube containing 4× volume of DMEM/F12.
If cells do not easily dissociate, incubate for another 1–2 minutes rather than harshly triturating.
-
3a.
Transfer 10 μL of the cell suspension to a hemocytometer and count cells. Transfer volumes containing 106 cells each to three 15 mL tubes, one tube for isotype control antibody staining, one tube for NG2 staining, and one tube for PDGFRβ staining.
Alternatively, if the cell suspension is dilute (fewer than ~30 cells per 1 mm × 1 mm hemocytometer square), cells can be centrifuged, resuspended in 1 mL of DMEM/F12, counted, and then split into tubes each containing 106 cells. An automated cell counter can also be used instead of a hemocytometer.
-
4a.
Centrifuge all samples 5 min at 180×g, 4°C.
-
5a.Prepare primary antibody solutions in cold (4°C) MACS buffer:
- 100 μL DPBS + 2 μL NG2 antibody
- 100 μL DPBS + 1.25 μL PDGFRβ antibody
- 100 μL DPBS + mouse IgG2a isotype control antibody
-
6a.
Aspirate supernatant from each sample and resuspend the cell pellets in 100 μL of prepared primary antibody solutions. Incubate 30 min on ice.
-
7a.
Add 2 mL cold MACS buffer to each tube and mix gently. Centrifuge 5 min at 180×g, 4°C.
-
8a.
Prepare secondary antibody solution: 300 μL MACS buffer + 0.6 μL goat anti-Mouse IgG-Alexa Fluor 488 antibody.
-
9a.
Aspirate supernatant from each sample and resuspend each sample in 100 μL of the prepared secondary antibody solution. Incubate 30 min on ice, protected from light.
-
10a.
Add 2 mL cold MACS buffer to each tube and mix gently. Centrifuge 5 min at 180×g, 4°C.
-
11a.
Aspirate supernatant from each sample and resuspend each sample in 500 μL of 4% PFA solution. Incubate 15 min at room temperature, protected from light.
-
12a.
Centrifuge samples 5 min at 180×g, room temperature.
-
13a.
Remove PFA supernatant from each sample and dispose according to institutional protocols. Resuspend each sample in 300 μL DPBS. Transfer samples to round bottom flow cytometry tubes and analyze on a flow cytometer.
We acquire data using a BD FACSCalibur flow cytometer (excitation at 488 nm, Alexa Fluor 488 emission detected using FL1 with a 530/30 filter). We use FlowJo software to gate cellular events using SSC-H vs. FSC-H plots and to determine the % NG2+ and % PDGFRβ+ cells.
Example flow cytometry data are shown in Figure 3.
Figure 3. Expected results of molecular analysis of hPSC-derived brain pericyte-like cells.
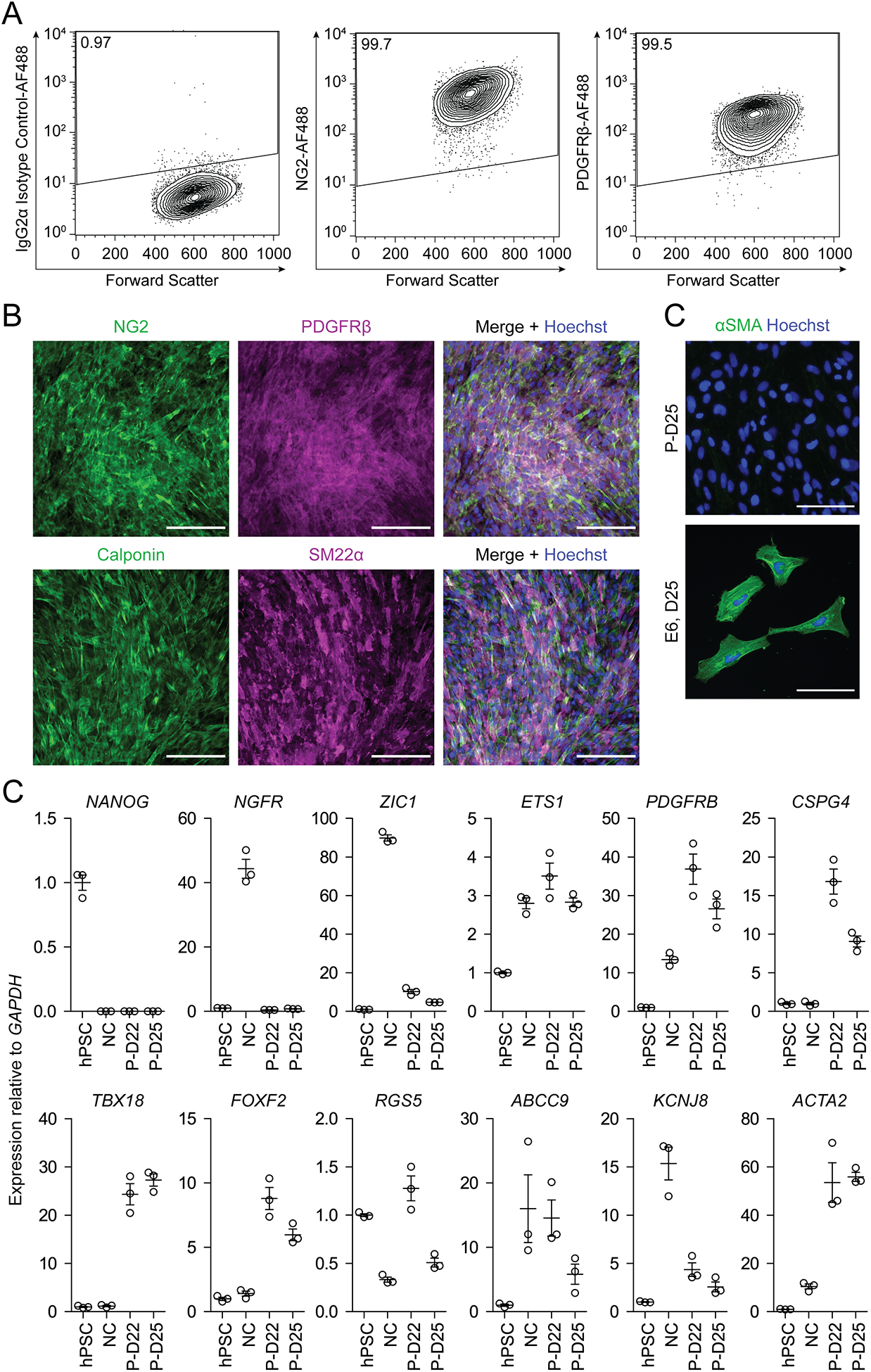
(A) Flow cytometry analysis of D25 brain pericyte-like cells differentiated from hPSCs. Flow plots show isotype control staining, NG2 staining, and PDGFRβ staining. (B) Immunocytochemistry analysis of D25 brain pericyte-like cells stained for NG2/PDGFRβ and calponin/SM22⍺. Hoechst nuclear counterstain is overlaid in the merged images. Scale bars: 200 μm. (C) Immunocytochemistry analysis of D25 brain pericyte-like cells (P-D25) and neural crest-derived cells maintained in E6 medium from D16–D25 (E6, D25) stained for ⍺SMA. Cells in both conditions were passaged once, 1:2, upon reaching confluence. Hoechst nuclear counterstain is also shown. Scale bars: 100 μm. (D) qPCR analysis of hPSCs, neural crest (NC), brain pericyte-like cells at D22 (P-D22), and brain pericyte-like cells at D25 (P-D25). Expression of each gene of interest is shown relative to GAPDH expression and normalized to hPSC expression. Error bars represent the standard deviation of three replicate wells.
Immunocytochemistry analysis of brain pericyte-like cells
-
1b.
At D22–25 of the pericyte-like cell differentiation, aspirate medium from wells of pericyte-like cells and add a half medium volume of 4% PFA solution. Incubate 15 min at room temperature.
We typically perform immunocytochemistry on pericyte-like cells in 48-well plates (one medium volume is 200 μL). See Table 1 for volumes for other well plate types.
-
2b.
Remove PFA supernatant from each well and dispose according to institutional protocols.
-
3b.
Add one medium volume of DPBS to each well. Aspirate DPBS and repeat this step for a total of 3 washes.
-
4b.
Add a half medium volume of blocking buffer to each well. Incubate 1 h at room temperature with gentle rocking.
Optimal blocking buffer is antibody-dependent. See Table 2 for a list of antibodies and suggested blocking buffers.
-
5b.
Prepare one medium volume of each primary antibody solution by adding primary antibodies (see Table 2 for appropriate dilution ratios) to the corresponding incubation buffer (Table 2).
-
6b.
Aspirate blocking buffer from each well and add the prepared primary antibody solutions. Incubate overnight at 4°C with gentle rocking.
-
7b.
Aspirate primary antibody solutions from each well and wash each well 3× with one medium volume of DPBS.
-
8b.
Prepare a half medium volume of each secondary antibody solution by adding secondary antibodies (see Table 2 for appropriate dilution ratios) to the corresponding incubation buffer (Table 2). Add prepared secondary antibody solutions to each well. Incubate 1 h at room temperature with gentle rocking, protected from light.
-
9b.
Aspirate secondary antibody solutions from each well and wash each well 3× with one medium volume DPBS.
-
10b.
Add a half medium volume of Hoechst solution to each well. Incubate 10 min at room temperature, protected from light.
-
11b.
Image samples on a fluorescence microscope.
Example immunocytochemistry images are shown in Figure 3. For a positive control for ⍺SMA, cells differentiated according to Basic Protocol 2, substituting E6 medium for E6 + 10% FBS medium, can be employed. Primary human brain vascular pericytes (ScienCell, 1200) can also serve as a positive control for ⍺SMA immunocytochemistry (Stebbins et al., 2019).
qPCR analysis of neural crest and brain pericyte-like cells
-
1c.
At desired timepoints during the neural crest and/or pericyte-like cell differentiations, aspirate medium and wash each well intended for analysis with a half medium volume of DPBS.
We typically harvest triplicate wells from 6-well plates for PCR analysis of neural crest and pericyte-like cells. Below, we provide a protocol for RNA extraction using the Qiagen RNeasy Mini Kit and reverse transcription using the Qiagen Omniscript RT Kit, but any validated in-house protocols could be employed.
-
2c.
Aspirate DPBS and add 350 μL of Buffer RLT (lysis buffer) supplemented with 1% β-mercaptoethanol to each well. Use a p1000 pipette to collect and transfer each lysate to a QIAshredder homogenization column. Centrifuge 30 s at maximum speed (≥ 8000×g), room temperature.
-
3c.
Add 350 μL of 70% ethanol to each lysate. Mix well and transfer each lysate to an RNeasy spin column. Centrifuge 30 s at maximum speed, room temperature. Discard flow through.
-
4c.
Add 350 μL of Buffer RW1 to each column. Centrifuge 30 s at maximum speed, room temperature. Discard flow through.
-
5c.
Prepare 80 μL of DNase I solution per sample by mixing 1 part DNase I stock solution and 7 parts Buffer RDD. Add 80 μL of this solution to the membrane of each column and incubate 15 min at room temperature.
-
6c.
Add 350 μL of Buffer RW1 to each column. Centrifuge 30 s at maximum speed, room temperature. Discard flow through.
-
7c.
Add 500 μL of Buffer RPE to each column. Centrifuge 30 s at maximum speed, room temperature. Discard flow through. Repeat this step a second time.
-
8c.
Transfer each spin column to a new 2 mL collection tube. Centrifuge 1 min at maximum speed, room temperature.
-
9c.
Transfer each spin column to a 1.5 mL collection tube. Add 40 μL nuclease-free water to the membrane of each column. Centrifuge 1 min at maximum speed, room temperature to elute RNA.
-
10c.
Quantify RNA concentration in each sample using a NanoDrop spectrophotometer. For each sample, add a volume containing 1000 ng RNA to one tube of an 8-strip PCR tube, and a volume of nuclease-free water sufficient to achieve a total volume of 13 μL.
-
11c.
Prepare 7 μL reverse transcriptase (RT) master mix per sample by mixing 2 parts Buffer RT, 2 parts dNTP Mix, 1 part Oligo(dT)12–18 primers, 1 part RNaseOUT, and 1 part RT. Add 7 μL of this mix to each sample in the 8-strip PCR tube. Incubate 1 h at 37°C.
-
12c.
Add 80 μL of nuclease-free water to each sample (for a final volume of 100 μL and a cDNA concentration of approximately 10 ng/μL). Store at −20°C until ready to proceed with qPCR.
-
13c.
Prepare solutions containing SYBR Green qPCR master mix and forward/reverse primer mix for each gene of interest according to master mix manufacturer instructions. Prepare enough solution for 2 reactions per sample plus 10% excess.
For example, for each 20 μL reaction, combine 11 μL of PowerUp SYBR Green Master Mix, 2.2 μL of forward/reverse primer mix (final concentration of 500 nM), and 7.7 μL of nuclease-free water.
-
14c.
Add 1 μL (10 ng) of cDNA from each sample to duplicate wells on a qPCR plate. Repeat for each gene of interest. Centrifuge or spin the plate briefly to settle droplets to the bottom of wells.
-
15c.
Add prepared solutions of SYBR Green qPCR master mix and forward/reverse primer mix to each well on the qPCR plate according to master mix manufacturer instructions.
For example, add 19 μL of the mixes described in step 13c to each well.
-
16c.
Cover the qPCR plate and centrifuge or spin briefly to settle solutions to the bottom of wells.
-
17c.
Run the qPCR plate in a qPCR thermal cycler using conditions specified by the master mix and an annealing temperature of 60°C.
Example qPCR results are shown in Figure 3.
SUPPORT PROTOCOL 4: Cord formation assay with endothelial cells and brain pericyte-like cells
This protocol describes a method to evaluate the ability of brain pericyte-like cells to associate with and stabilize endothelial cell cords, a general property of both brain and non-brain pericytes. When endothelial cells, such as the human umbilical vein endothelial cells (HUVECs) used here, are plated on concentrated Matrigel in VEGF-containing medium, they organize into cord structures reminiscent of blood vessel networks. When pericytes are cocultured with HUVECs under these conditions, pericytes attach to the outside of endothelial cords and permit longer cords to form (“stabilization”).
Materials
Pericyte-like cells at D22 differentiated according to Basic Protocol 2
Human umbilical vein endothelial cells cultured according to supplier protocols (HUVECs, ATCC, PCS-100–013)
Matrigel aliquots (see recipe)
Trypsin-EDTA, 0.25% (Gibco, 25200056)
Accutase (Innovative Cell Technologies, AT104)
DMEM + 10% FBS medium (see recipe)
DMEM/F12 medium (Gibco, 11330032)
EGM-2 medium (Lonza, CC-3162)
4% PFA solution (see recipe)
Dulbecco’s phosphate buffered saline, no Ca, no Mg (DPBS, Gibco, 14190144)
Antibodies (see Table 2)
Hoechst solution (see recipe)
ProLong Gold Antifade Mountant (Invitrogen, P36930)
Hemocytometer (e.g. Hausser Scientific, 3110)
Automated cell counter (optional; e.g. Countess II, Invitrogen, AMQAX1000)
Inverted brightfield or phase contrast microscope with 4× objective and camera
8-well chamber slides (Nunc Lab-Tek II, Thermo Scientific, 154534PK)
Xacto knife or scalpel
Coverslips, 22 × 50 mm or 22 × 40 mm, no. 1 thickness
Clear nail polish
Confocal microscope
Initiating cord formation
-
1
Thaw Matrigel aliquots at 4°C for 2 h, and chill p1000 pipette tips at −20°C for 2 h. Once thawed, transfer Matrigel aliquots to ice in a biological safety cabinet and place an 8-well chamber slide on ice.
-
2
Using a pre-chilled p1000 pipette tip, transfer ~250 μL of Matrigel to each chamber on the 8-well chamber slide. Use the pipette tip to spread the Matrigel into the corners and evenly coat the entire chamber surface. Avoid introducing bubbles.
To compare cord formation of HUVECs alone to HUVECs cocultured with pericyte-like cells, we recommend preparing 6 chambers (3 per condition).
-
3
Transfer the Matrigel-coated chamber slide to a 37°C incubator for 1 h to allow the Matrigel to solidify.
-
4
Aspirate medium from a plate or flask containing HUVECs and add a half medium volume of trypsin-EDTA. Aspirate medium from a plate containing brain pericyte-like cells at D22 and add a half medium volume of Accutase. Incubate cells 5–15 min at 37°C, observing cells under a microscope every 5 min until > 90% of cells appear detached.
For a coculture negative control, a non-cord stabilizing cell type such as hPSC-derived neural crest cells can be employed (Figure 4). Dissociate neural crest cells with Accutase and parallel the steps below for pericyte-like cells. Human embryonic kidney (HEK) cells can also be employed as a coculture negative control (Stebbins et al., 2019).
-
5
Using a p1000 pipette, dissociate and singularize HUVECs, and add the single cell suspension to 4× volume of DMEM + 10% FBS medium in a 15 mL tube. Dissociate and singularize pericyte-like cells and add the single cell suspension to 4× volume of DMEM/F12 medium in a 15 mL tube.
-
6
Transfer 10 μL of each cell suspension to a hemocytometer and count cells.
Alternatively, if the cell suspension is dilute (fewer than ~30 cells per 1 mm × 1 mm hemocytometer square), cells can be counted after centrifugation and resuspension in 1 mL of EGM-2 medium. An automated cell counter can also be used instead of a hemocytometer.
-
7
Centrifuge the cell suspensions 5 min at 200×g, room temperature.
-
8
Resuspend each cell pellet in 1 mL of EGM-2 medium.
-
9Prepare cell suspensions in 1.5 mL tubes:
- For the HUVEC-only control, add a volume of HUVEC suspension containing 2.2×104 cells per chamber. For example, for triplicate chambers, add a volume of HUVEC suspension containing 6.6×104 cells. Add a volume of EGM-2 medium to bring the total volume to 500 μL per chamber (1.5 mL for triplicate chambers).
- For the HUVEC + pericyte-like cell condition, add a volume of HUVEC suspension containing 2.2×104 cells, and a volume of pericyte-like cell suspension containing 6.6×104 cells, per chamber (i.e. a 1:3 ratio of HUVECs to pericyte-like cells). For example, for triplicate chambers, add a volume of HUVEC suspension containing 6.6×104 cells, and a volume of pericyte-like cell suspension containing 1.98×105 cells. Add a volume of EGM-2 medium to bring the total volume to 500 μL per chamber (1.5 mL for triplicate chambers).
-
10
Transfer 500 μL of the prepared cell suspensions to each chamber of the prepared 8-well chamber slide. Place the chamber slide in a 37°C, 5% CO2 incubator for 24 h.
Figure 4. Expected results of cord formation assay.
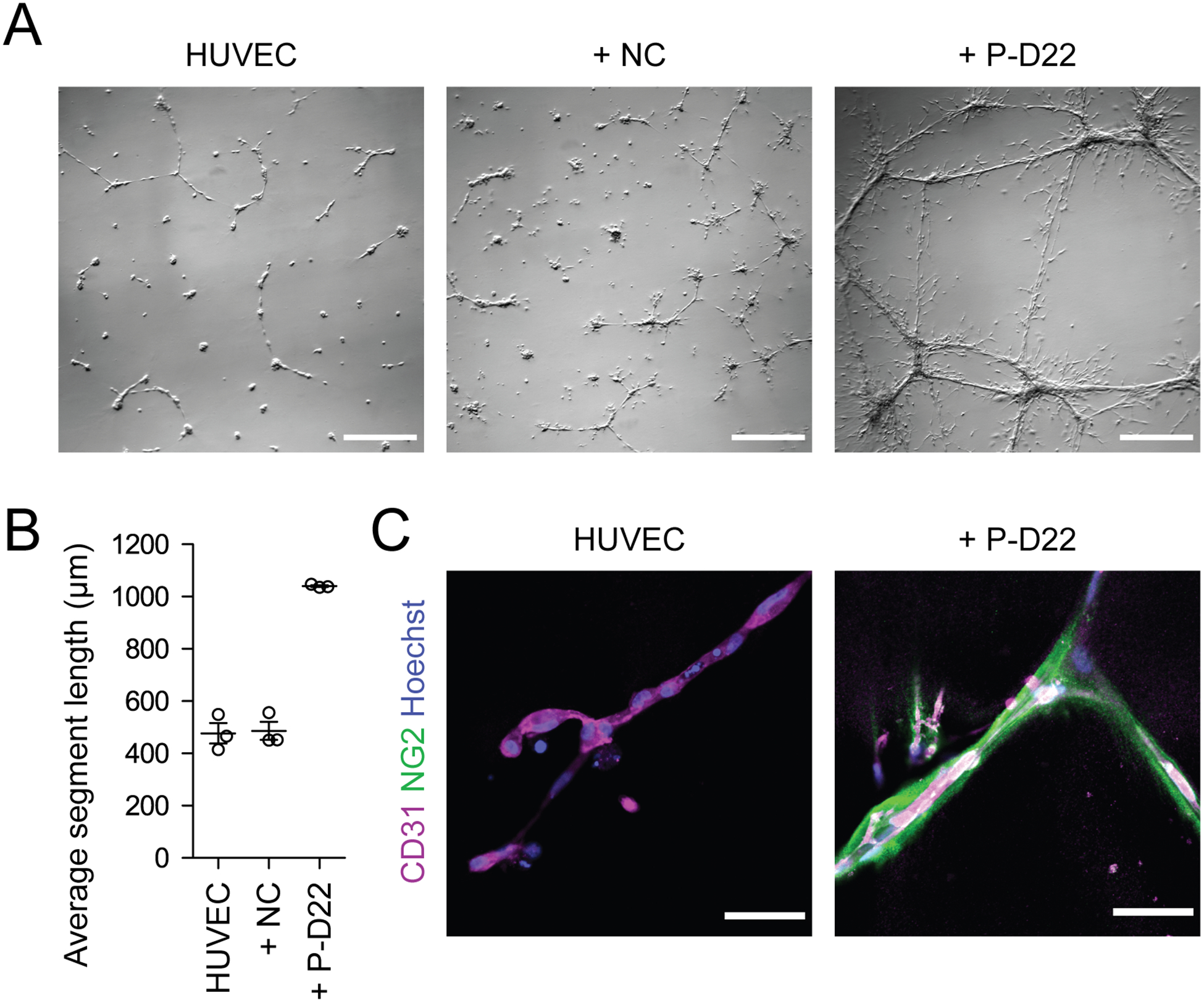
(A) Phase contrast images of cells 24 h after initiating cord formation. Images of HUVECs alone, HUVECs cocultured with neural crest cells (+ NC), and HUVECs cocultured with brain pericyte like cells at D22 (+ P-D22) are shown. Scale bars: 1 mm. (B) Quantification of average segment length for the cord formation assay conditions described in (A). Error bars represent the standard deviation of three replicate chambers. (C) Confocal immunocytochemistry analysis of cords from HUVEC and + P-D22 conditions stained for CD31 and NG2. Hoechst nuclear counterstain is also shown. Scale bars: 50 μm.
Analyzing cord formation
-
11
24 h after seeding and prior to fixation, use an inverted microscope to acquire phase contrast or brightfield images of cells in each chamber.
Cords are best visualized using phase contrast microscopy. For quantification, we recommend acquiring two 4× fields for each chamber and using software such as FIJI/ImageJ to measure the length of cord segments (Figure 4B).
-
12
Carefully aspirate medium from each chamber and add 250 μL of 4% PFA solution to fix cells. Incubate 15 min at room temperature.
After fixation, cords are extremely fragile and loosely adhered to the Matrigel. Exercise extreme care when adding and removing solutions to prevent sample loss.
-
13Perform immunocytochemistry for NG2 and CD31 following steps 2b–10b of Support Protocol 3(Molecular characterization of brain pericyte-like cells: Immunocytochemistry analysis of brain pericyte-like cells) except:
- Use a volume of 250 μL per chamber for all wash, blocking, and antibody solutions.
- Conduct all incubation steps without rocking to limit cord detachment.
-
14
Carefully aspirate the Hoechst solution from each chamber.
-
15
Use an Xacto knife or scalpel to carefully cut around the perimeter of each chamber to detach the Matrigel from the plastic chamber walls. Use the tool provided with the chamber slides to carefully remove the plastic chambers from the glass slide.
Proceed slowly to ensure that the pieces of Matrigel remain affixed to the slide and do not lift away with the plastic chambers.
-
16
Place a drop of ProLong Gold Antifade mountant on top of each piece of Matrigel and use mountant to fill in the spaces between pieces of Matrigel.
Use a liberal amount of mountant as an insufficient volume will cause bubbles to form under the coverslip.
-
17
Place a coverslip on the slide. Use a Kimwipe to blot excess mountant from the edges of the slide.
-
18
Set the slide aside, protected from light, until the Matrigel is dehydrated and the coverslip contacts the slide. Typically, this process takes 24–48 hours.
-
19
Seal the edges of the slide with clear nail polish and allow to cure for ~1 h at room temperature. Store the slide at 4°C, protected from light, until ready to image.
-
20
Image cords using a confocal microscope.
Example results of the cord formation assay are shown in Figure 4.
REAGENTS AND SOLUTIONS
Materials
Dulbecco’s phosphate buffered saline, no Ca, no Mg (DPBS, Gibco, 14190144)
Triton X-100 (IBI Scientific, IB07100)
Bovine serum albumin (BSA, Sigma Aldrich, A3858)
Paraformaldehyde, 16% solution (Electron Microscopy Sciences, 15710-S)
Goat serum (e.g., Gibco, 16210072)
Nonfat dry milk (e.g., Lab Scientific, M0841)
Dimethyl sulfoxide (DMSO), sterile (Sigma Aldrich, D2650)
CHIR 99021, 10 mg (Tocris, 4423)
Dorsomorphin dihydrochloride, 10 mg (Tocris, 3093)
DMEM (Gibco, 11965092)
Fetal bovine serum (FBS, e.g. Peak Serum, PS-FB1)
E6 medium (TeSR-E6, STEMCELL Technologies, 05946; or Essential 6 Medium, Gibco, A1516401; or manufactured in-house according to Lippmann et al., 2014ba)
Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA, Sigma, E5134)
Water, sterile (e.g. Sigma Aldrich, W3500)
Sodium hydroxide (NaOH), 12 M solution
FGF2, 100 μg (Peprotech, 100–18B)
Heparin sodium salt from porcine intestinal mucosa (Sigma, H3393)
Hoechst 33342 solution, 20 mM (Thermo Scientific, 62249)
Matrigel, Growth Factor Reduced (GFR) Basement Membrane Matrix, LDEV-free (Corning, 354230)
DMEM/F12 medium (Gibco, 11330032)
SB 431542, 10 mg (Tocris, 1614)
Y-27632 dihydrochloride, 10 mg (Tocris, 1254)
0.2 μm sterile filters (e.g. Nalgene Rapid-Flow 250 mL, 5680020; and Millipore SteriFlip 50 mL, SCGP00525)
Recipes
Prepare media and reagent stock solutions intended for cell culture under sterile conditions.
0.4% TX-100 solution
In a 15 mL tube, combine 10 mL DPBS and 40 μL Triton-X 100. Vortex to dissolve Triton X-100. Store at 4°C for up to 6 months.
3% BSA and 3% BSA + 0.1% TX-100 solutions
In a 15 mL tube, combine 0.3 g BSA and 10 mL DPBS. Vortex to dissolve. Transfer 5 mL of the BSA solution to a new 15 mL tube and add 5 μL Triton-X 100. Vortex to dissolve. Store at 4°C for up to one week.
4% PFA solution
In a 15 mL tube, combine 1 part 16% paraformaldehyde (PFA) solution and 3 parts DPBS. Use the same day.
5% GS + 0.4% TX-100 solution
In a 15 mL tube, combine 0.5 mL goat serum (GS), 9.5 mL DPBS, and 40 μL Triton X-100. Vortex to dissolve Triton-X 100. Store at 4°C for up to one week.
5% milk and 5% milk + 0.1% TX-100 solutions
In a 15 mL tube, combine 0.5 g nonfat dry milk and 10 mL DPBS. Vortex to dissolve. Transfer 5 mL of the milk solution to a new 15 mL tube and add 5 μL Triton-X 100. Vortex to dissolve. Store at 4°C for up to one week.
CHIR 99021 stock solution
Dissolve CHIR 99021 in sterile DMSO to a concentration of 10 mM to make a 10000× stock. Prepare 50 μL aliquots and store at −20°C for up to one year.
Dorsomorphin stock solution
Dissolve dorsomorphin dihydrochloride in sterile DMSO to a concentration of 10 mM to make a 10000× stock. Prepare 50 μL aliquots and store at −20°C for up to one year.
DMEM + 10% FBS medium
In a 250 mL filter, combined 225 mL DMEM and 25 mL FBS. Filter sterilize and store at 4°C for up to one month.
E6 + 10% FBS medium
In a 250 mL filter, combine 225 mL E6 medium and 25 mL FBS. Filter sterilize and store at 4°C for up to two weeks.
E6-CSFD medium
In a 250 mL filter, combine 250 mL E6 medium, 25 μL of CHIR 99021 stock solution, 250 μL of SB 431542 stock solution, 25 μL of FGF2 stock solution, 25 μL of dorsomorphin stock solution, and 250 μL of heparin stock solution. Filter sterilize and store at 4°C for up to one week.
EDTA stock solution
Add 9.3 g EDTA to a graduated cylinder and add water to a final volume of 50 mL to make a 500 mM stock. Titrate to pH 8.0 by dropwise addition of 12 M NaOH. Filter sterilize and store at 4°C for up to 6 months.
FGF2 stock solution
Dissolve FGF2 in sterile water to a concentration of 100 μg/mL to make a 10000× stock. Prepare 50 μL aliquots and store at −80°C for up to one year.
Forward/reverse primer mix
Resuspend primers to 100 μM in nuclease-free water. For each gene of interest, make a 5 μM forward/reverse primer mix by combining 10 μL forward primer, 10 μL reverse primer, and 180 μL nuclease-free water. Store primers and forward/reverse primer mixes at −20°C for up to 2 years.
Heparin stock solution
Dissolve 1.125 g heparin sodium salt in 50 mL of sterile water to make a 22.5 mg/mL (1000×) stock. Filter sterilize and store at 4°C for up to 6 months.
Hoechst solution
In a 50 mL tube, combine 50 mL DPBS and 10 μL of 20 mM Hoechst 33342 solution. Store at 4°C, protected from light, for up to one month.
MACS buffer
In a 50 mL tube, dissolve 1.25 g BSA in 48 mL DPBS by gentle inversion. In a 250 mL filter, combine 48 mL BSA solution with an additional 200 mL DPBS. Add 2 mL EDTA stock solution. Filter sterilize and store at 4°C for up to six months.
Matrigel and Matrigel-coated plates
Thaw Matrigel overnight at 4°C. Chill p1000 pipette tips and sterile 1.5 mL tubes overnight at −20°C. The next day, place thawed Matrigel and 1.5 mL tubes on ice in a biological safety cabinet. Using chilled pipette tips, make aliquots of a volume containing 2.5 mg Matrigel (Matrigel concentration varies). Transfer aliquots to −80°C for up to one year. To make Matrigel-coated plates, transfer 30 mL cold DMEM/F12 medium to a 50 mL tube. Use 1 mL of this DMEM/F12 medium to rapidly thaw and dissolve a 2.5 mg aliquot of Matrigel by pipetting up and down. Transfer to the tube containing DMEM/F12, then use a serological pipet to mix the solution and transfer 1 mL of the diluted Matrigel-DMEM/F12 mixture to each well of five 6-well plates. Incubate at least 1 h, 37°C. Plates may be stored at 37°C for up to one week. Add additional DMEM/F12 to prevent drying.
Neural Crest Cell Cryopreservation medium
In a 50 mL tube, combine 6 parts E6-CSFD medium, 3 parts FBS, and 1 part DMSO. Filter sterilize and store at 4°C for up to one week. Make 1 mL cryopreservation medium per 106 cells intended for cryopreservation.
SB 431542 stock solution
Dissolve SB 431542 in sterile DMSO to a concentration of 10 mM to make a 1000× stock. Prepare 500 μL aliquots and store at −20°C for up to one year.
Y-27632 stock solution
Dissolve Y-27632 dihydrochloride in sterile water to a concentration of 10 mM to make a 1000× stock. Prepare 50 μL aliquots and store at −20°C for up to one year.
COMMENTARY
Background Information
Pericytes regulate vascular development and function; brain pericytes also regulate BBB induction and maintenance. Pericyte-deficient mice demonstrate high BBB permeability to small and large molecule tracers, abnormal endothelial tight junctions, increased endothelial cell vesicular density, and increased endothelial expression of leukocyte adhesion molecules (Daneman et al., 2010; Armulik et al., 2010; Bell et al., 2010). Brain pericytes also regulate neuronal and glial phenotype (Montagne et al., 2018; Nikolakopoulou et al., 2019), inflammation (Duan et al., 2018; Rustenhoven et al., 2017), and potentially neurovascular coupling (Peppiatt et al., 2006; Fernandez-Klett et al., 2010; Hall et al., 2014; Hill et al., 2015; Mishra et al., 2016; Kisler et al., 2017, 2020; Hartmann et al., 2020; Grubb et al., 2020). Furthermore, emerging evidence suggests that pericyte dysfunction contributes to neurological disorders, including Alzheimer’s disease (Sagare et al., 2013; Sengillo et al., 2013; Montagne et al., 2018; Nortley et al., 2019; Halliday et al., 2015).
Because of their role in BBB function, pericytes are a common component of in vitro coculture models of the BBB/NVU. In Transwell-based coculture systems, pericytes increase transendothelial electrical resistance and decrease small molecule permeability (Dohgu et al., 2005; Nakagawa et al., 2007, 2009; Lippmann et al., 2014a; Cecchelli et al., 2014; Thomsen et al., 2015; Stebbins et al., 2019; Canfield et al., 2019; Jamieson et al., 2019), improve endothelial cell tight junction protein expression or localization (Hori et al., 2004; Stebbins et al., 2019), decrease the rate of non-specific endothelial transcytosis (Stebbins et al., 2019), and modulate efflux transporter expression and function (Berezowski et al., 2004; Thomsen et al., 2015; Praça et al., 2019).
While several protocols exist to differentiate pericyte-like cells from hPSC-derived mesodermal progenitors (Kusuma et al., 2013; Orlova et al., 2014; Kumar et al., 2017) and efforts have been made to model brain pericyte function and dysfunction using mesoderm-derived pericytes (Jamieson et al., 2019; Blanchard et al., 2020), pertinent differences in molecular characteristics between brain and non-brain pericytes exist (Vanlandewijck et al., 2018; He et al., 2018). Here, we present a method to differentiate brain pericyte-like cells from hPSCs through a developmentally-relevant neural crest intermediate. Our protocol is complementary to existing methods to differentiate smooth muscle cells from hPSC-derived neural crest via treatment with TGF-β1 and/or PDGF-BB (Cheung et al., 2014; Wang et al., 2012). We found that E6 + 10% FBS medium was sufficient to produce NG2+PDGFRβ+⍺SMAlow pericyte-like cells, and TGF-β1 and PDGF-BB treatment produced cells with markedly lower NG2 expression (Stebbins et al., 2019). While other recent methods to derive similar pericyte-like cells from hPSC-derived neural crest employ commercial “pericyte” medium (Faal et al., 2019; Griffin and Bajpai, 2019), the E6-CSFD and E6 + 10% FBS media used in this protocol are readily prepared in-house.
hPSC-derived brain pericyte-like cells differentiated using this protocol can be included in coculture models of the BBB/NVU to improve endothelial cell phenotype and better understand molecular mechanisms by which pericytes regulate the BBB. Similarly, emerging in vitro models may facilitate improved understanding of pericyte roles in other neurovascular functions, neuronal and glial regulation, and neurological disease.
Critical Parameters and Troubleshooting
Prior to initiating differentiation, hPSCs should be maintained on Matrigel- or vitronectin-coated plates in E8 medium and passaged using non-enzymatic methods (e.g. Versene treatment) when colonies begin to touch (WiCell, 2018). hPSCs should form round, compact colonies with sharp borders and exhibit little to no spontaneous differentiation as assessed by morphology or flow cytometry for pluripotency markers (Chen et al., 2011). hPSC cultures should have ≥ 95% viable cells as assessed by trypan blue exclusion (Strober, 2015).
The hPSC seeding density on D-1 is a critical parameter; low seeding densities result in poor cell survival, and high seeding densities can yield a confluent monolayer requiring passaging prior to cells undergoing sufficient differentiation to permit reattachment after singularization. We have found a seeding density of 8.75 × 105 hPSCs per well of a 6-well plate to work well for multiple hPSC lines, but evaluation of other seeding densities may be pursued for cell lines that exhibit suboptimal reattachment or neural crest yield. The MACS step on D15 is critical to isolate p75-NGFR+ neural crest cells prior to further differentiation to pericyte-like cells.
The neural crest and pericyte-like cell differentiation protocols are robust across multiple hPSC lines, including iPSCs and hESCs (Stebbins et al., 2019). We have, however, observed some line-to-line variability, primarily in reattachment of differentiating neural crest after the first passaging step and neural crest differentiation efficiency as quantified by p75-NGFR/HNK-1 flow cytometry. Suggestions for troubleshooting these and other potential problems are provided in Table 4.
Table 4.
Troubleshooting
| Step | Problem | Possible reason | Potential solution |
|---|---|---|---|
| Basic Protocol 1, step 16 | Little to no cell reattachment after first passage of differentiating neural crest | Cells were passaged prior to 100% confluence. | Passage one of the back-up wells retained in step 10. |
| Some hPSC lines show poor reattachment at first passage. | Include 10 μM Y-27632 during first passage. | ||
| Support protocol 1, step 11 | Low % p75-NGFR+ HNK-1+ cells in pre-sort sample. | Differentiation efficiency is hPSC line-dependent. | Optimize hPSC seeding density and/or molecule concentrations in E6-CSFD medium (see recipe), especially CHIR and/or FGF2 concentration (Basic Protocol 1, step 6). |
| Support protocol 1, step 11 | < 95% p75-NGFR+HNK-1+ cells in post-sort sample | Low % p75-NGFR+HNK-1+ cells in pre-sort sample. | See above. |
| Cells were incompletely singularized prior to MACS. | Extend Accutase treatment time; filter cell suspension through 40 μm cell strainer prior to MACS. (Basic Protocol 1, steps 19 and 20). | ||
| MACS column was overloaded or insufficiently washed. | Load ≤ 2×107 cells per column. Allow MACS buffer to stop flowing before adding next 3 mL. Perform at least 3 washes (Basic Protocol 1, steps 26 and 27). | ||
| Support protocol 3, step 13a | Low % NG2+ cells | Timing of increase in NG2 expression is variable and may be hPSC line-dependent. | Continue to culture differentiating pericyte-like cells in E6 + 10% FBS for an additional ~3 days. |
Understanding Results
Differentiation of neural crest (Basic Protocol 1) begins by seeding hPSCs at moderate density on Matrigel-coated plates in E8 medium supplemented with ROCK inhibitor. The ROCK inhibitor prevents apoptosis of dissociated hPSCs (Watanabe et al., 2007) and causes the cells to adopt a larger and less compact morphology than typical hPSC colonies (Figure 1C, D0). After changing medium to E6-CSFD, cells should regain typical hPSC morphology with sharp colony borders (Figure 1C, D1) and reach confluence by D1–D3. After passaging cells for the first time, the cells should adopt a larger, polygonal morphology (Figure 1C, D4). These large, polygonal cells are putative neural crest progenitors and should continue to expand over the course of the 15 day differentiation. At later timepoints, we often observe colonies of more compact cells among the larger putative neural crest cells (Figure 1C, D11, arrowhead). We previously observed that these colonies are p75-NGFR– (Stebbins et al., 2019), and are thus eliminated by MACS. Differentiating neural crest cells typically require passaging 2–4 times prior to D15 and yield approximately 4×106 cells per confluent 6-well on D15 (i.e. > 2×107 cells per 6-well plate), corresponding to a ~100-fold to ~1000-fold expansion relative to the number of seeded hPSCs. MACS is the yield-limiting step of the procedure as LS columns have a capacity of 2×107 input cells. Depending on pre-sort purity, after two rounds of MACS, we typically recover 10–40% of input cells (i.e. 2×106–8×106 cells) and achieve a purity of 95–99% p75-NGFR+HNK-1+ based on flow cytometry (Support Protocol 1). Example flow cytometry results are shown in Figure 2.
Differentiation of brain pericyte-like cells (Basic Protocol 2) begins by seeding sorted neural crest either immediately after MACS, from a maintained neural crest culture, or from a thawed stock (Support Protocol 2) on uncoated plates in E6-CSFD medium supplemented with ROCK inhibitor. The following day, the cells should appear sparsely distributed and have polygonal morphology with small processes (Figure 1E, D16). After changing the medium to E6 + 10% FBS, the cells should lose these processes (Figure 1E, D17) and gradually adopt a more elongated morphology over the course of the next several days. The differentiating pericyte-like cells typically expand ~10-fold by D22, at which time 60–80% of cells should be NG2+ based on flow cytometry analysis (Support Protocol 3). The pericyte-like cells typically require passaging one time prior to D25, at which time 90–100% of cells should be NG2+. Example flow cytometry results are shown in Figure 3. PDGFRβ is expressed by nearly 100% of neural crest cells and pericyte-like cells throughout the differentiation as quantified by flow cytometry; however, the expression of PDGFRB transcript should increase in pericyte-like cells compared to neural crest (Figure 3C). Similarly, we observe CSPG4, TBX18, FOXF2, and ACTA2 transcript upregulation in pericyte-like cells compared to neural crest (Figure 3C). We also observe KCNJ8 expression in pericyte-like cells that is elevated compared to hPSCs, but lower than in neural crest, and observe only transient upregulation of the pericyte marker RGS5, potentially reflecting immaturity (Figure 3C). Expected expression patterns of neural crest, mural cell, and pericyte markers as assessed by immunocytochemistry and qPCR are shown in Figure 3. In a cord formation assay (Support Protocol 4), coculturing pericyte-like cells with HUVECs should lead to increased average segment length compared to HUVECs alone and HUVECs cocultured with neural crest cells (Figure 4A,B). Pericytes should localize to the outside of HUVEC cords and extend processes along the cords, as visualized by immunocytochemistry and confocal imaging (Figure 4C).
Time considerations
Below, we describe the estimated time required to complete each protocol if working with a single neural crest or pericyte-like cell culture. Additional time should be allotted if working with multiple cultures in parallel (e.g. cultures from multiple hPSC lines).
Basic Protocol 1
On D-1, seeding hPSCs for neural crest differentiation takes ~30 min. On D0–D14, medium changes take < 5 min, except when differentiating neural crest is confluent and requires passaging. These passaging steps take ~15 min, and passaging is typically required 2–3 times prior to D14. On D15, purification of p75-NGFR+ neural crest cells by MACS takes ~1.5 h.
Basic Protocol 2
On D15, seeding neural crest for brain pericyte-like cell differentiation takes ~15 min. On D16–D25, medium changes take < 5 min, except when differentiating pericyte-like cells are confluent and require passaging. This passaging step takes ~15 min and is typically required once prior to D25.
Support Protocol 1
Preparing neural crest samples for flow cytometry takes ~2 h, including incubation steps. Running samples on a flow cytometer and analyzing data takes an additional ~1 h.
Support Protocol 2
Cryopreservation of neural crest takes ~30 min. An overnight freezing step is required prior to transferring vials to liquid nitrogen. Recovery of cryopreserved neural crest and seeding these cells for brain pericyte-like cell differentiation takes ~30 min.
Support Protocol 3
Preparing brain pericyte-like cell samples for flow cytometry takes ~2 h, including incubation steps. Running samples on a flow cytometer and analyzing data takes an additional ~1 h. Preparing pericyte-like cells for immunocytochemistry analysis takes ~3.5 h: On the first day, ~2 h is required, including incubation steps for fixation and blocking, but excluding the overnight incubation with primary antibodies. The following day, an additional ~1.5 h is required, including incubation with secondary antibodies. Time required for imaging varies based on the number of images desired. Preparing cell samples for qPCR analysis takes ~3 h, excluding a 1 h incubation step for reverse-transcription and a 1–2 h qPCR run.
Support Protocol 4
Thawing Matrigel takes ~2 h, and preparing Matrigel-coated chamber slides takes ~30 min followed by a 1 h incubation to allow the Matrigel to solidify. Dissociating and seeding cells for the cord formation assay takes ~30 min. The following day, acquiring phase contrast or brightfield images takes ~30 min. For performing immunocytochemistry, on the first day, ~2 h is required, including incubation steps for fixation and blocking, but excluding the overnight incubation with primary antibodies. The following day, an additional ~2 h is required, including incubation with secondary antibodies. After removing chambers and adding the coverslip, 24–48 is required for the Matrigel to dehydrate. Time required for confocal imaging varies based on the number of images desired.
ACKNOWLEDGEMENTS
The authors thank Scott G. Canfield, Ming-Song Lee, Drew Richards, Madeline G. Faubion, Wan-Ju Li, Richard Daneman, Allison M. Bosworth, and Ethan S. Lippmann for their assistance in development and validation of the protocols presented here. We thank the University of Wisconsin-Madison Biochemistry Optical Core for use of a confocal microscope. This work was supported by NIH grants NS083688, NS109486, and NS103844 (to EVS and SPP) and DTRA grant HDTRA-15-1-0012 (to EVS). MJS and BDG were supported by NIH Biotechnology Training Program grant T32 GM008349. BDG was supported by the National Science Foundation Graduate Research Fellowship Program under grant no. 1747503.
Footnotes
CONFLICT OF INTEREST
M.J.S, E.V.S, and S.P.P. are inventors on a patent application related to this work filed by the Wisconsin Alumni Research Foundation (US20200017827A1). The authors declare no other conflicts of interest.
E6 medium is E8 medium (Chen et al., 2011) lacking FGF2 and TGF-β1.
LITERATURE CITED
- Alarcon-Martinez L, Yilmaz-Ozcan S, Yemisci M, Schallek J, Kılıç K, Can A, Di Polo A, and Dalkara T 2018. Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. eLife 7:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando K, Fukuhara S, Izumi N, Nakajima H, Fukui H, Kelsh RN, and Mochizuki N 2016. Clarification of mural cell coverage of vascular endothelial cells by live imaging of zebrafish. Development (Cambridge) 143:1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genové G, and Betsholtz C 2011. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Developmental Cell 21:193–215. Available at: https://linkinghub.elsevier.com/retrieve/pii/S1534580711002693. [DOI] [PubMed] [Google Scholar]
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, et al. 2010. Pericytes regulate the blood-brain barrier. Nature 468:557–561. [DOI] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, and Zlokovic BV 2010. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 68:409–427. Available at: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezowski V, Landry C, Dehouck MP, Cecchelli R, and Fenart L 2004. Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Research 1018:1–9. [DOI] [PubMed] [Google Scholar]
- Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, Victor MB, Bonner JM, Mathys H, Lin Y, et al. 2020. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nature Medicine 26:952–963. Available at: 10.1038/s41591-020-0886-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield SG, Stebbins MJ, Faubion MG, Gastfriend BD, Palecek SP, and Shusta EV 2019. An isogenic neurovascular unit model comprised of human induced pluripotent stem cell-derived brain microvascular endothelial cells, pericytes, astrocytes, and neurons. Fluids and Barriers of the CNS 16:1–12. Available at: 10.1186/s12987-019-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchelli R, Aday S, Sevin E, Almeida C, Culot M, Dehouck L, Coisne C, Engelhardt B, Dehouck MP, and Ferreira L 2014. A stable and reproducible human blood-brain barrier model derived from hematopoietic stem cells. PLoS ONE 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Mica Y, Lee G, Studer L, and Tomishima MJ 2013. Dual-SMAD Inhibition/WNT Activation-Based Methods to Induce Neural Crest and Derivatives from Human Pluripotent Stem Cells. In Methods in Molecular Biology pp. 329–343. Available at: http://link.springer.com/10.1007/7651_2013_59. [DOI] [PubMed] [Google Scholar]
- Chasseigneaux S, Moraca Y, Cochois-Guégan V, Boulay A, Gilbert A, Le Crom S, Blugeon C, Firmo C, Cisternino S, Laplanche J-L, et al. 2018. Isolation and differential transcriptome of vascular smooth muscle cells and mid-capillary pericytes from the rat brain. Scientific Reports 8:12272 Available at: http://www.nature.com/articles/s41598-018-30739-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Gulbranson DR, Hou Z, Bolin JM, Ruotti V, Probasco MD, Smuga-Otto K, Howden SE, Diol NR, Propson NE, et al. 2011. Chemically defined conditions for human iPSC derivation and culture. Nature Methods 8:424–429. Available at: http://www.nature.com/doifinder/10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Goh YYT, Zhang J, Wu C, and Guccione E 2014. Modeling cerebrovascular pathophysiology in amyloid-β metabolism using neural-crest-derived smooth muscle cells. Cell Reports 9:391–401. Available at: 10.1016/j.celrep.2014.08.065. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, and Barres BA 2010. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 468:562–566. Available at: http://www.nature.com/doifinder/10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohgu S, Takata F, Yamauchi A, Nakagawa S, Egawa T, Naito M, Tsuruo T, Sawada Y, Niwa M, and Kataoka Y 2005. Brain pericytes contribute to the induction and up-regulation of blood–brain barrier functions through transforming growth factor-β production. Brain Research 1038:208–215. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0006899305000764. [DOI] [PubMed] [Google Scholar]
- Duan L, Zhang X-D, Miao W-Y, Sun Y-J, Xiong G, Wu Q, Li G, Yang P, Yu H, Li H, et al. 2018. PDGFRβ Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 100:183–200.e8. Available at: 10.1016/j.neuron.2018.08.030. [DOI] [PubMed] [Google Scholar]
- Etchevers HC, Vincent C, Le Douarin NM, and Couly GF 2001. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development (Cambridge, England) 128:1059–1068. Available at: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=11245571&retmode=ref&cmd=prlinks%5Cnpapers3://publication/uuid/B1B2C213-3F0E-4C85-9737-2C87C6B0AC8A. [DOI] [PubMed] [Google Scholar]
- Everson JL, Fink DM, Yoon JW, Leslie EJ, Kietzman HW, Ansen-Wilson LJ, Chung HM, Walterhouse DO, Marazita ML, and Lipinski RJ 2017. Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development 144:2082–2091. Available at: http://dev.biologists.org/lookup/doi/10.1242/dev.149930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faal T, Phan DTTT, Davtyan H, Scarfone VM, Varady E, Hughes CCWW, Inlay MA, Blurton-Jones M, Hughes CCWW, and Inlay MA 2019. Induction of Mesoderm and Neural Crest-Derived Pericytes from Human Pluripotent Stem Cells to Study Blood-Brain Barrier Interactions. Stem Cell Reports 12:451–460. Available at: 10.1016/j.stemcr.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Klett F, Offenhauser N, Dirnagl U, Priller J, and Lindauer U 2010. Pericytes in capillaries are contractile in vivo, but arterioles mediate functional hyperemia in the mouse brain. Proceedings of the National Academy of Sciences 107:22290–22295. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.1011321108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastfriend BD, Palecek SP, and Shusta EV 2018. Modeling the blood–brain barrier: Beyond the endothelial cells. Current Opinion in Biomedical Engineering 5:6–12. Available at: 10.1016/j.cobme.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin C, and Bajpai R 2019. Neural crest-derived human cranial pericytes model primary forebrain pericytes and predict disease-specific cranial vasculature defects. SSRN. Available at: 10.2139/ssrn.3189103. [DOI] [Google Scholar]
- Grubb S, Cai C, Hald BO, Khennouf L, Murmu RP, Jensen AGK, Fordsmann J, Zambach S, and Lauritzen M 2020. Precapillary sphincters maintain perfusion in the cerebral cortex. Nature Communications 11:395 Available at: http://www.nature.com/articles/s41467-020-14330-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, Oâ Farrell FM, Buchan AM, Lauritzen M, and Attwell D 2014. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508:55–60. Available at: 10.1038/nature13165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, and Zlokovic BV 2015. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 36:1–9. Available at: http://www.ncbi.nlm.nih.gov/pubmed/25757756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann DA, Berthiaume A-A, Grant RI, Harill SA, Noonan T, Costello J, Tieu T, McDowell K, Faino A, Kelly A, et al. 2020. Brain capillary pericytes exert a substantial but slow influence on blood flow. bioRxiv:2020.03.26.008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Vanlandewijck M, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, et al. 2018. Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Scientific data 5:180160 Available at: http://www.nature.com/protocolexchange/protocols/6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, and Grutzendler J 2015. Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 87:95–110. Available at: 10.1016/j.neuron.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Ohtsuki S, Hosoya KI, Nakashima E, and Terasaki T 2004. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. Journal of Neurochemistry 89:503–513. [DOI] [PubMed] [Google Scholar]
- Jamieson JJ, Linville RM, Ding YY, Gerecht S, and Searson PC 2019. Role of iPSC ‑ derived pericytes on barrier function of iPSC ‑ derived brain microvascular endothelial cells in 2D and 3D. Fluids and Barriers of the CNS:1–16. Available at: 10.1186/s12987-019-0136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, et al. 2017. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler K, Nikolakopoulou AM, Sweeney MD, Lazic D, Zhao Z, and Zlokovic BV 2020. Acute Ablation of Cortical Pericytes Leads to Rapid Neurovascular Uncoupling. Frontiers in Cellular Neuroscience 14:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn J, Christ B, Kurz H, Kurz R, Kurz H, Kurz R, and Kurz H 2002. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. Journal of Comparative Neurology 442:78–88. [DOI] [PubMed] [Google Scholar]
- Kumar A, D’Souza SS, Moskvin OV, Toh H, Wang B, Zhang J, Swanson S, Guo LW, Thomson JA, and Slukvin II 2017. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Reports 19:1902–1916. Available at: 10.1016/j.celrep.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusuma S, Shen Y-I, Hanjaya-Putra D, Mali P, Cheng L, and Gerecht S 2013. Self-organized vascular networks from human pluripotent stem cells in a synthetic matrix. Proceedings of the National Academy of Sciences of the United States of America 110:12601–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Chambers SM, Tomishima MJ, and Studer L 2010. Derivation of neural crest cells from human pluripotent stem cells. Nature Protocols 5:688–701. Available at: http://www.nature.com/doifinder/10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, and Studer L 2007. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nature Biotechnology 25 Available at: http://www.nature.com/doifinder/10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, and Shusta EV 2014a. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Scientific reports 4:4160 Available at: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3932448&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Estevez-Silva MC, and Ashton RS 2014b. Defined human pluripotent stem cell culture enables highly efficient neuroepithelium derivation without small molecule inhibitors. Stem Cells 32:1032–1042. [DOI] [PubMed] [Google Scholar]
- Menendez L, Kulik MJ, Page AT, Park SS, Lauderdale JD, Cunningham ML, and Dalton S 2013. Directed differentiation of human pluripotent cells to neural crest stem cells. Nature Protocols 8:203–212. Available at: http://www.nature.com/doifinder/10.1038/nprot.2012.156. [DOI] [PubMed] [Google Scholar]
- Menendez L, Yatskievych T. a., Antin PB, Dalton S, Menendez L, Yatskievych T. a., Antin PB, and Dalton S 2012. Wnt signaling and a Smad pathway blockade direct the differentiation of human pluripotent stem cells to multipotent neural crest cells. Proceedings of the National Academy of Sciences 109:9220–9220. Available at: http://www.pnas.org/cgi/doi/10.1073/pnas.1207810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, and Attwell D 2016. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A, Nikolakopoulou AM, Zhao Z, Sagare AP, Si G, Lazic D, Barnes SR, Daianu M, Ramanathan A, Go A, et al. 2018. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nature Medicine 24:326–337. Available at: 10.1038/nm.4482. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel Á, Tanaka K, Niwa M, Kittel A, Tanaka K, et al. 2009. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochemistry International 54:253–263. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, and Niwa M 2007. Pericytes from brain microvessels strengthen the barrier integrity in primary cultures of rat brain endothelial cells. Cellular and Molecular Neurobiology 27:687–694. Available at: http://link.springer.com/10.1007/s10571-007-9195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, Sagare AP, Lazic D, Sweeney MD, Kong P, et al. 2019. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nature Neuroscience 22:1089–1098. Available at: 10.1038/s41593-019-0434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Gong H, Richard-loendt A, Huang W, Saito T, and Saido TC 2019. Amyloid β oligomers constrict human capillaries in Alzheimer ‘ s disease via signaling to pericytes. 9518:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova VV, van den Hil FE, Petrus-Reurer S, Drabsch Y, ten Dijke P, and Mummery CL 2014. Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nature Protocols 9:1514–1531. Available at: http://www.nature.com/doifinder/10.1038/nprot.2014.102. [DOI] [PubMed] [Google Scholar]
- Peppiatt CM, Howarth C, Mobbs P, and Attwell D 2006. Bidirectional control of CNS capillary diameter by pericytes. Nature 443:700–704. Available at: http://www.nature.com/doifinder/10.1038/nature05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praça C, Rosa SC, Sevin E, Cecchelli R, Dehouck M-PP, and Ferreira LS 2019. Derivation of Brain Capillary-like Endothelial Cells from Human Pluripotent Stem Cell-Derived Endothelial Progenitor Cells. Stem Cell Reports 13:599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustenhoven J, Jansson D, Smyth LC, and Dragunow M 2017. Brain Pericytes As Mediators of Neuroinflammation. Trends in Pharmacological Sciences 38:291–304. [DOI] [PubMed] [Google Scholar]
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, and Zlokovic BV 2013. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nature Communications 4:1–14. Available at: http://www.nature.com/doifinder/10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, and Zlokovic BV 2013. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in alzheimer’s disease. Brain Pathology 23:303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins MJ, Gastfriend BD, Canfield SG, Lee M-SS, Richards D, Faubion MG, Li W-JJ, Daneman R, Palecek SP, and Shusta EV 2019. Human pluripotent stem cell–derived brain pericyte–like cells induce blood-brain barrier properties. Science Advances 5:eaau7375 Available at: http://advances.sciencemag.org/lookup/doi/10.1126/sciadv.aau7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W 2015. Trypan Blue Exclusion Test of Cell Viability. Current protocols in immunology 111:A3.B.1–A3.B.3. Available at: http://www.ncbi.nlm.nih.gov/pubmed/26529666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen LB, Burkhart A, and Moos T 2015. A triple culture model of the blood-brain barrier using porcine brain endothelial cells, astrocytes and pericytes. PLoS ONE 10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, et al. 2018. A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480. [DOI] [PubMed] [Google Scholar]
- Wang A, Tang Z, Li X, Jiang Y, Tsou DA, and Li S 2012. Derivation of Smooth Muscle Cells with Neural Crest Origin from Human Induced Pluripotent Stem Cells. Cells Tissues Organs 195:5–14. Available at: http://www.karger.com/doi/10.1159/000331412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, et al. 2007. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. 25:681–686. [DOI] [PubMed] [Google Scholar]
- WiCell 2018. Feeder-Independent Pluripotent Stem Cell Protocols, E8 Medium. Madison, WI: Available at: https://www.wicell.org/media.acux/394cbfb1-021e-44e8-9805-512c30a66979. [Google Scholar]
- Yamanishi E, Takahashi M, Saga Y, and Osumi N 2012. Penetration and differentiation of cephalic neural crest-derived cells in the developing mouse telencephalon. Development, Growth & Differentiation 54:785–800. Available at: http://doi.wiley.com/10.1111/dgd.12007. [DOI] [PubMed] [Google Scholar]