

Abstract
Drug-induced cardiotoxicity is a potentially lethal and yet one of the most common side effects with the drugs in clinical use. Most of the drug-induced cardiotoxicity is associated with an off-target pharmacological blockade of K+ currents carried out by the cardiac Human-Ether-a-go-go-Related (hERG1) potassium channel. There is a compulsory pre-clinical stage safety assessment for the hERG1 blockade for all classes of drugs, which adds substantially to the cost of drug development. The availability of a high-resolution Cryogenic Electron Microscopy (cryo-EM) structure for the channel in its open/depolarized state solved in 2017 enabled the application of molecular modeling for rapid assessment of drug blockade by molecular docking and simulation techniques. More importantly, if successful, in-silico methods may allow a path to lead-compound salvaging by mapping out key block determinants. Here, we report the blind application of the Site-Identification by Ligand Competitive Saturation (SILCS) protocol to map out druggable/regulatory hotspots in the hERG1 channel available for blockers and activators. The SILCS simulations use small solutes representative of common functional groups to sample the chemical space for the entire protein and its environment using all-atom simulations. The resulting chemical maps, FragMaps, explicitly account for receptor flexibility, protein-fragment interactions, and fragment desolvation penalty allowing for rapid ranking of potential ligands as blockers or non-blockers of hERG1. To illustrate the power of the approach SILCS was applied to a test set of 55 blockers with diverse chemical scaffolds and pIC50 values measured under uniform conditions. The original SILCS model was based on the all-atom modelling of the hERG1 channel in an explicit lipid bilayer and was further augmented with a Bayesian-Optimization/Machine-Learning (BML) stage employing an independent literature-derived training set of 163 molecules. BML approach was used to determine weighting factors for the FragMaps contributions to the scoring function. pIC50 predictions from the combined SILCS/BML approach to the 55 blockers showed a Pearson Correlation (P.C.) coefficient of > 0.535 relative to the experimental data. SILCS/BML model was shown to yield substantially improved performance as compared to commonly used rigid and flexible molecular docking methods for a well-established cohort of hERG1 blockers where no correlation to experimental data was recorded. SILCS/BML results also suggest that a proper weighting of protonation states of common blockers present at physiological pH is essential for accurate predictions of blocker potency. The pre-calculated and optimized SILCS FragMaps can now be used for the rapid screening of small molecules for their cardiotoxic potential as well as for exploring alternative binding pockets in the hERG1 channel with applications to the rational design of activators.
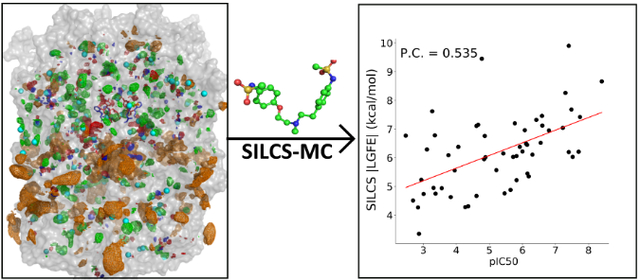
Graphical Abstract
Introduction
Drug-induced cardiotoxicity is by far one of the most common and dangerous side effects associated with the use of drugs. The majority of the drug-induced cardiotoxicity is associated with an off-target pharmacological block of the cardiac Human-Ether-a-go-go-Related (hERG1) potassium channel 1–3. The channel is found throughout the body including the gall bladder 4, the digestive tract 5, the mid-brain6, and the abnormal expression of the channel is associated with cancer 7. However, the function and the role of the channel is most studied and understood in the heart 8, where the channel carries the majority of the IKr current corresponding to phase 3 of the Action Potential (AP) in cardiomyocytes 9–10. One of the major problems in drug development is the cardiovascular side effects associated with off-target drug binding particularly, to the hERG1. Binding of drugs to channels, alters cardiac IKr current, potentially causing a long-QT syndrome (LQTS) which may lead to irregular heartbeats, fainting, and sudden cardiac death 1, 11–12. The sudden cardiac death of patients and prolongation of the QT interval in the heart due to the drug-induced block of hERG1 is the main cause of the withdrawal of drugs from the market and mandatory drug screening for hERG1 interactions was implemented by Food and Drug Administration (FDA)13–14 and European Medicine Agency (EMEA). The drug-induced long-QT syndrome and drug arrhythmogenic activity associated with the hERG1 block made the channel infamous in the pharmacological field, making it one of the most studied ion channels. A massive effort by the scientific alliance using high-throughput screening of several drugs provided additional evidence for the central role of the hERG1 blockade in drug safety assessments 15. Several methods have been developed and tested in attempts to find and predict drug binding pockets by screening large datasets of drug libraries, including assays based on binding, flux, electrophysiology, and fluorescence 1, 16. However, hERG1-mediated toxicity detection remains a challenging task due to the inherent variability of the methodologies used at laboratories throughout the world. Moreover, in vitro studies for the determination of drug binding pockets in hERG1 are costly, labor-intensive, and technically demanding. Accordingly, affinity prediction using in silico screening is considered a potential cost-effective screening tool 17–18.
Despite years of experimental and theoretical studies aimed at studying and determining interactions of drugs with the hERG1 channel, a-priori prediction of the cardiotoxic potential for novel compounds without expensive testing remains an elusive problem in drug development. Therefore, many efforts were pushed towards developing predictive models of different complexity ranging from traditional Quantitative Structure-Activity Relationship (QSAR) and pharmacophore-based models to Deep-Learning (DL) techniques for the screening of a large cohort compound. While very useful, DL- or QSAR- based approaches are focused on ligand-based inference driven by biological assay data and therefore provide little information about the molecular underpinnings of the drug-receptor interactions 19–20. The DL/QSAR-based approaches are often focused on the binary classification e.g. blocker vs. non-blocker providing very little guidance on potential modifications of the blocker chemical structure needed for salvaging of the promising drug lead. In addition, these ligand- or knowledge-based methods face serious challenges in predicting drug blockade by a compound without strong similarity to the chemical scaffolds used to train the model 17–18.
Therefore, mapping atomistic details of the hERG1 high-affinity site for blockers potentially opens up the potential of using target structure-based approaches for the prediction of the hERG1 blockers as well as a potential strategy for “de-toxifying” a drug during development. In 2017, the cryo-EM structure of the hERG1 was solved at 3.8 Å resolution 21. The structure of the transmembrane region of the channel consists of four repeating chains comprised of the six transmembrane segments, S1-S6. The segments S1-S4 make up a Voltage Sensing Domain (VSD) that responds to the voltage changes across the membrane and the S5-S6 segments make up the Pore Domain (PD) which open and close in response to the VSD, allowing selective passage of the potassium ions (Figure 1). Experimental and theoretical studies have firmly established a water-filled intra-cellular central cavity (IC) located in the PD as the main site for blockers with diverse scaffolds 22–23. It has also been postulated that the large size of the IC binding site for blockers was one of the culprits resulting in the promiscuity in drug binding to the hERG1 28. The cryo-EM resolved structure revealed key features that were not predicted experimentally or observed in the structural models developed based on the 3D templates from bacterial or mammalian K+ channels with known structures 24. In contrast to the previously established wide IC which was proposed to accommodate drugs of various chemical structures, the cryo-EM structure features a long and narrow IC in its presumably depolarized (open) state 21. Several recent efforts to extend molecular modelling of the hERG1 blockade using the new cryo-EM structure have been made to develop structure-based models to predict hERG1 blockade by drug-like molecules 25–29. However, the narrow and elongated cavity presented a challenge to traditional applications of molecular docking due to the steric constraints in the cavity with the apparent need to re-pack aromatic residues such as F656 upon ligand binding (Figure 1) 27, 29–30. Our studies utilizing multi-microsecond MD simulations have shown that the IC site remains narrow but hydrated allowing both K+ permeation and binding of several canonical blockers of hERG1 (dofetilide, d-sotalol) 25, which is also in agreement with known binding pockets for channel activators 25, 31. In addition, the availability of the structure enabled the mapping of alternative mechanisms for drug access to the main blockade site located in the IC. For example, a heart-pace lowering agent, ivabradine, was proposed to block the hERG1 via a lipophilic route 26, while moxifloxacin was found to bind in a “shallow” portion of the IC 9. Thus, the new structure presents a natural challenge to direct applications of traditional all-atom simulation techniques due to its apparent flexibility, need to sample slow conformational dynamics of residues comprising the binding pocket and located at the protein-membrane interface while correctly accounting for conformational dynamics of the bound drug. For example, to reduce the complexity of the problem at hand, Dickson et al. 25 extracted specific frames from a large-scale MD simulation trajectory based on the principal component analysis (PCA) analysis to assess contributions of different conformational states of key coordinating residues on hERG1-drug interactions. The authors noted significant challenges in determining optimal binding pockets in an integral membrane protein such as hERG1, issues arising from the choices of drug protonation, and inherent flexibility of the receptor. A very poor correlation between molecular docking results with the receptor structure based on the cryo-EM conformation and measured binding affinities were also cited as major challenges in several recent modeling studies 28–29, 32–33.
Figure 1.

The general topology of the hERG1 channel is shown in a sagittal view and features a Voltage-Sensing-Domain (VSD) labeled as S1-S4, pore domain (PD), and the cytoplasmic region (C-linker). Only one monomer is shown for clarity as a red cartoon while the rest of the channel is shown as a transparent surface. The pore domain is comprised of S5-Pore helix-S6 helix and features a large intra-cellular central cavity (IC) which is shown as a cyan surface generated by HOLLOW program42. The key chemical moieties in the IC are shown in the inset. VSD domain is formed by S1-S4 helices and corresponding linkers. For the intracellular region (C-linker), only a fully-resolved CNBD domain is shown.
An alternative to traditional molecular docking approaches that utilize a rigid structure or ensemble of structures for the receptor is the use of co-solvent drug design approaches that produce functional group affinity maps that account for protein flexibility among other terms, including the Site Identification by Ligand Competitive Saturation (SILCS) method 34–37. In SILCS simulations, the combination of solute size and chemical diversity allows for direct mapping and ranking of putative binding pockets available in the system for drug binding. Notably, the SILCS FragMaps may also be used for the docking of drug-like molecules in which the presence of several rotamer and protonation states along with complexities of chiral centers contribute to the significant sampling challenges in computational modeling of drug binding to the proteins. Here, we used the SILCS approach to identify hotspots in the hERG1 channel taking advantage of the oscillating chemical potential Grand Canonical Monte Carlo/Molecular Dynamics approach to calculate the receptor FragMaps 38. The computed FragMaps were then used to explore binding affinities of fragment-like molecules for the identification of hotspots throughout the entire channel including the lipid exposed surface of the channel 39. The hotspot analysis shows the presence of two distinct binding regions in the intracellular cavity of the hERG1 channel lending direct support to the postulated presence of “shallow” and “deep” binding pockets in the cavity/distal S6 helix in hERG1 channel 40. Subsequently, the SILCS FragMaps were used to produce a predictive model of the affinity of drug-like molecules for these binding sites. To improve the determination capabilities of SILCS to predict the affinities of blockers we combined the FragMaps protocol with a Bayesian Machine Learning (BML) FragMap reweighting method 34. The BML optimization was based on a training set of 163 hERG1 blockers that were used to train the SILCS affinity model. The power of the SILCS/BML approach is illustrated with a set of 55 blockers/non-blockers where uniformly measured IC50 is available 41 and compared to data from the Glide molecular docking approach.
Methods
Site Identification by Ligand Competitive Saturation (SILCS) Simulations:
SILCS methodology generates a functional group affinity pattern of a receptor, termed FragMaps, through simulations of a macromolecule in the presence of probe or solute molecules and explicit water molecules. SILCS simulations were initialized based on the cryo-EM derived structure of the hERG1 (PDB: 5VA1) channel. The building of the unresolved region of the hERG1 structure and additional refinement has been described in our previous publication 43. The channel was embedded in 2-oleoyl-1-pamlitoyl-sn-glyecro-3-phosphocholine (POPC) lipids along with cholesterol at a 9:1 ratio, respectively, with explicit water molecules, eight types of solutes each at a concentration of 250 mM. The system setup was performed with the MolCal package 36, 44 (SilcsBio LLC) in conjunction with GROMACS tools 45. The eight fragments used in our study are benzene (benz), propane (prpa), methanol, formamide, acetaldehyde, imidazole, methyl-ammonium (mamm), and acetate (acet). The simulation box is selected to be larger than the hERG1 dimensions by 20 Å. The REDUCE software 46 was used to place missing hydrogens and to optimally rearrange ring orientations of the following side chains: Asn, His, and Gln. Ten independent systems with the same settings were set with different distributions of water and solutes (Figure 2).
Figure 2.

The SILCS simulation setup for the hERG1 channel is embedded in an explicit POPC membrane, eight solutes, water, and ions. The channel is shown as the colorful cartoon, the membrane is a red transparent surface, cholesterol embedded in the membrane are shown as red sticks, water molecules are shown as blue transparent surface, and solutes are shown as sticks and lines.
The SILCS simulations involve iterative oscillating chemical potential, μex, Grand-Canonical Monte Carlo (GCMC) 38, and MD simulations using GROMACS. GCMC is used to sample the distribution of water and the eight small solutes in and around the protein while the MD simulations explore the conformational changes within the protein along with additional sampling of the solvent environment as previously described 34. Simulations used the CHARMM36m protein forcefield for proteins and lipids 47, and the CHARMM TIP3P model for explicit water molecules 48–49. The solutes were modelled using the CHARMM general force field (CGenFF) 50–51. Timestep for MD runs was set to 2 fs and the Nose−Hoover method was used to keep the temperature at 298 K 52. Using the Parrinello−Rahman barostat the pressure was kept at 1 bar while the time constant for pressure and temperature coupling was 1ps 53, and the LINCS algorithm was used to maintain bond geometries 54. The rotation and potential denaturation of the protein due to the solutes in the system 55 were circumvented by applying harmonic restraints to the protein. A force constant of 2.4 kcal/mol*Å2 was used as a harmonic positional restraint on all non-hydrogen atoms of the protein during the minimization and equilibration steps. During the production run, the restraint was applied only on Cα carbon backbone atoms of the protein with a force constant of 0.12 kcal/mol*A2. To prevent aggregation between the solutes a repulsive intermolecular wall was applied to the following fragment pairs: benz:benz, benz:prpa, prpa:prpa, mamm:acet, mamm:mamm, and acet:acet. Following the minimization and equilibration procedure, the system was subjected to 25×200,000 cycles of the GCMC simulations with the oscillating μex to re-equilibrate the solutes and water around the protein. Then, the production run of 100 cycles of oscillating μex GCMC/MD involving 200,000 GCMC steps followed by 1ns of MD simulation producing 100 ns data for each of the 10 simulation systems is performed (1μs in total).
The 1μs of MD trajectories were used to calculate the 3D-occupancy maps for the selected solute atoms that encompass the entire system by binning each atom into 1 Å* 1 Å* 1 Å cubic volumes elements (voxels) every 10 ps. The voxel occupancy for each selected atom from each solute was calculated for each voxel. To normalize the occupancy, the distribution of solutes in solution was calculated without the protein. This 3D probability distribution (FragMaps) was constructed for different atoms: benzene carbons, propane carbons, methanol polar hydrogens, methanol oxygens, formamide polar hydrogens, formamide oxygen, acetaldehyde oxygens, methylammonium polar hydrogens, and acetate oxygens. In addition to these specific FragMaps, generic FragMaps were generated including apolar (benzene and propane carbons), generic hydrogen bond donor (formamide and imidazole polar hydrogens, and generic hydrogen bond acceptors (formamide and acetaldehyde oxygens and imidazole acceptors nitrogen). The methylammonium nitrogen comprises the positive FragMaps and the acetate carbonyl carbon comprises the negative FragMaps 34. The Boltzmann transformation was used on the normalized probability distributions to calculate the Grid Free Energy (GFE) for each type of FragMap (T) yielding GFE FragMaps using the following equation 1:
| (1) |
With GFE value capped at 3 kcal/mol. To assess the convergence in the calculated GFE FragMaps, the ten independent simulations were split into two equally-sized sets and then used independently to calculate FragMaps. This comparative analysis showed that the Overlap Coefficient (OC) between two maps was greater than 0.6 indicating the convergence in the SILCS simulations 44. In addition to the FragMaps, Exclusion Maps were defined as regions of the grids in which no sampling by water or the solutes occurred during the SILCS simulations. All visualizations shown in this work were generated using PYMOL and VMD 56,57.
Ligand Grid Free Energy Calculation and SILCS-MC Sampling:
The SILCS FragMaps allow for rapid fragment and ligand scoring using the SILCS Monte Carlo (SILCS-MC) algorithm on the pre-generated FragMap grid. In this approach, each classified atom in the ligand is associated with a FragMap type and a GFE score assigned to each atom based on the value of the FragMap at that position combined with a scale factor as previously presented 34. The assignments are based on the translation of CGenFF atom types into FragMaps classes via an Atom-Classification Scheme (ACS) (see ref. 34, 36 for more information). A ligand GFE (LGFE) score is then calculated based on a summation over the atomic GFE scores as shown in Equation 2:
| (2) |
where iT corresponds to FragMaps types and GFET are the Grid-Free Energies for various FragMaps types. The FragMaps types include the Exclusion Map to which a GFE value of 1000 kcal/mol is assigned, disallowing sampling of the interior region of the protein. During SILCS-MC ligand sampling the ligands are sampled with respect to translational, rotational, and dihedral degrees of freedom where the CGenFF parametrization is used for intramolecular energies including dihedral (dihe), vdW, and electrostatic (elec) terms in addition to the LGFE score, as previously described 34, 36. The LGFE scores are an approximation of the binding affinity as they do not account for the covalent connectivity of the atoms of the ligands. The intramolecular energies are not considered in the final ranking scores as we assume those terms do not contribute significantly in the binding process where the ligand is transferred from the bulk phase to a binding pocket. As LGFE scores are representative of binding affinities they may be used to rank ligands and perform correlation analysis as performed below.
SILCS-MC was performed on all accessible protonation states of the studied ligands. The pKa values from the DrugBank dataset were used in analyzing possible protonation states for the compound and in assessing protonation equilibrium. The protonation sites of drugs were determined using the Chemicalize ChemAxon pKa distribution calculator under biological constraints (pH=7.4) 58. Then, the 3D-protonated structures were generated using Avogadro software where the optimization plugin was used to produce optimized intramolecular conformations 59. These optimized structures were used in the SILCS-MC calculations.
Bayesian Machine-Learning Re-Scoring of FragMaps:
To improve model performance of the SILCS method for predicting relative affinities, we adopted the optimization of the FragMap weighting factors, used to calculate the LGFE scores, using a Bayesian Machine Learning (BML) FragMap reweighting method34. This method allows for optimization of the GFE contribution of each class of atoms present in the fragments or ligands. To perform the BML optimization, a dataset of 163 compounds with diverse scaffolds and functional groups from our previous study was used 17. A flat penalty potential of 1000 kcal/mol was used to cap upper- and lower- boundaries of the FragMap weighting factors. The error function is the Pearson R between the predicted LGFE scores and the experimental pIC50 values. Overfitting was avoided using upper and lower boundaries set as double and half of the initial FragMap weight. These compounds were subjected to SILCS-MC and the most energetically favorable poses and LGFE scores were used as an input for the BML optimization 34. The optimized weighting parameters based on FragMap weights were used to verify that no overfitting by performing SILCS-MC on the 163 training ligands using the new optimized 2018 ACS and redetermining the Pearson R. When overfitting does occur the resulting poses from SILCS-MC degraded significantly as does the metric PC value 34. Following the optimization of weighting factors, the re-weighted FragMaps were used with SILCS-MC to predict LGFE scores for the 55 blockers assayed in the MICE study (listed in Table S1) 41.
SILCS-Hotspots:
SILCS-Hotspots is based on comprehensive fragment screening using the SILCS-MC docking approach to generate an ensemble of fragments poses with associated LGFE scores for the entire hERG1 channel 39. Approximately 100 monocyclic and bicyclic rings found in drug-like molecules were used as fragments in this study 39, 60–61. In SILCS-Hotspots, the full hERG1 channel is partitioned into subspaces that encompass the full protein to identify all possible fragment binding sites. Specifically, the simulation box was partitioned to sampling boxes with a size of 14.14 Å ×14.14 Å ×14.14 Å, covering the hERG1 channel and surrounding area. Using SILCS-MC, the fragment is randomly positioned in a sphere of 10 Å centered in each sampling box where the random variation of one rotatable bond of the fragment is generated. Subsequently, the fragment is subjected to 10,000 MC steps (at 300 K) followed by 40,000 MC annealing steps from 0 to 300 °K. This procedure is applied 1000 times on each fragment in each subspace. The collection of posed orientations is subjected to two rounds of Center-of-Mass (COM) clustering to identify orientations with the highest neighbor population thereby identifying putative fragment binding sites in round one followed by clustering of the different fragment types to identify those fragments that occupy each site. The radius of COM clustering used values of 3 Å and 4 Å for the first and second round, respectively.
Molecular Docking:
The cryo-EM solved structure of the tetrameric hERG1 was taken from the PDB, specifically biological assembly 1 (PDB: 5VA2). The receptor structure was prepared using Schrödinger’s restrained minimization Protein Preparation Wizard 62. The hydrogen atoms and missing parts of the residues were added, and the positions of hydrogen atoms were re-optimized. The Ligprep Wizard, available in the Schrödinger suite (Schrodinger 2012), was utilized to prepare 55 ligands which include restraint of bond lengths, bond angles, conversion of the 2D representation into the 3D format, adding hydrogens, sampling ring conformations, and minimizing the structure based on the OPLS force field 63–64. Charged states of the drugs were generated using EPIK at a pH of 7±2 for drugs and using PROPKA at pH 7. We utilized an automated Glide cross-docking script available in Maestro to automate the docking of 55 ligands to the receptor. The Glide-XP (extra-precision) 65 cross-docking modules of the Maestro suit in Schrödinger were used for all docking calculations with a ligand vdW scale factor of 0.80 and an RMSD cut-off of 2.0 Å. We restricted the outcome to a maximum of 3 docking poses for each drug. In order to generate a receptor grid for docking, we used an inner box edge length of 10 Å which represents the space explored by Glide as acceptable positions for the center of the drugs and the outer box of 30 Å, the space that all atoms of the drug must occupy. The coordinates used for the generation of the receptor grids for the docking was defined as a centroid of F656 and Y652 from each four subunits as these residues are key determinants of the drug-induced block of the hERG1. The main purpose of the Glide-XP docking studies was to evaluate the usefulness of the receptor state captured in the cryo-EM for high-throughput screening of large ligand libraries. Several recent studies utilizing large-scale MD simulations pointed out that the conformational arrangement of the aromatic cassette (F557, F656, and Y652) captured in the cryo-EM structure may inhibit high-affinity binding to the intra-cavitary site 9, 25, 29, 66. GLIDE outputs GScore, which is calculated in kcal/mol and accounts for the hydrophobic interactions, π-π stacking interactions between aromatic rings, root mean square deviation (RMSD), desolvation, protein-ligand interaction, and hydrogen bond formation 62. For the purposes of our study, we only show results of the best docking score of all ligands binding to the high-affinity site present the central cavity of the hERG1. To distinguish between outcomes of docking from GLIDE and SILCS approaches we will use GLIDE|Gscore| and SILCS|LGFE| for predicted binding scores, respectively.
Results and Discussion
SILCS is advantageous over the majority of molecular docking approaches in that it directly considers the flexibility of the protein along with protein and ligand desolvation and functional group-protein interactions in the form of the FragMaps allowing for comprehensive mapping of both orthosteric and possible allosteric binding sites of the hERG1 channel. The calculated FragMaps were then used in the SILCS-MC method to estimate the binding affinities of drugs in the pockets of interest.
FragMaps from Site Identification by Ligand Competitive Saturation Simulations:
SILCS uses Grand Canonical Monte Carlo and Molecular Dynamics (GCMC/MD) simulations for solute and protein conformational sampling which enables the determination of regions explored by the solute and water molecules during the simulation. In addition to orthosteric sites, hidden pockets of allosteric sites may be identified by the SILCS method as it comprehensively maps the 3D space of the protein 44. SILCS FragMaps, which represents the predicted binding affinity of functional groups around the hERG1 channel, are shown in Figure 3 and the detailed description of the maps is in Table 1. SILCS identifies and characterizes all possible binding sites via the FragMaps. The regions in the vicinity of the most important residues involved in drug binding, determined from a plethora of experimental studies, were excellently mapped by our SILCS simulations. The central cavity mapped residues by our simulations include F557, S624, Y652, and F656, all of which are crucial for drug binding (Figure 3b) 29, 31, 40, 67. The FragMaps of the central cavity show the preferential binding of apolar fragments including aromatic and aliphatic moieties (Figure 4a). The positively charged fragments also favorably bind to the central cavity of the hERG1 in agreement with the reported significant negative electrostatic potential inside the cavity 21.
Figure 3.

(a) A subset of SILCS FragMaps presented as isocontour meshes are shown for the hERG1 channel from the side perspective. The channel is shown as a transparent surface in grey, colored areas are the proposed binding spots for various chemical groups. (b) SILCS FragMaps distribution of the central cavity of the channel, specifically in the vicinity of Y652 and F656 that are the key residues involved in drug binding to the hERG1. (energy is shown as the GFE contour level of FragMaps according to Table 1). The FragMaps also occupied a pocket located around M651 in the lipid facing portion of the S6 helix, important for lipophilic drugs such as ivabradine 33. The lipid facing sides of the hERG1 revealed the presence of Apolar, Benzene, and Propane FragMaps with, notably, those maps penetrating through the biological membrane, consistent with the tendency of lipids to specifically bind to the cardiac channel 68–70.
Table 1.
FragMaps identifiers, their representative colors and the energy cutoffs used for visualization
| FragMaps | Label | Color | Level (kcal/mol) |
|---|---|---|---|
| apolar | Generic Apolar Map | Green | −1.2 |
| hbdon | Generic Donor Map | Blue | −1.2 |
| hbacc | Generic Acceptor Map | Red | −1.2 |
| excl | Exclusion Map | Sand | 0.6 |
| benc | Benzene Carbon Map | Purple | −1.2 |
| prpc | Propane Carbon Map | Lime | −1.2 |
| meoo | Methanol Oxygen Map | Red | −1.2 |
| forn | Formamide Nitrogen Map | Blue | −1.2 |
| foro | Formamide Oxygen Map | Red | −1.2 |
| mamn | Methylammonium Nitrogen Map | Cyan | −1.2 |
| aceo | Acetate Oxygen Map | Orange | −1.2 |
| aalo | Red | −1.2 | |
| imin | Imidazole Acceptor Nitrogen | Red | −1.2 |
| tipo | Water Oxygen Map | Red | −0.5 |
Figure 4.

(a) Top-down perspective view of the pore cavity of the hERG1 channel. The most well studied binding pocket is shown around the crucial residues Y652 and F656. Adjacent to both residues are generic apolar, benzene carbon, propane carbon, methylammonium nitrogen (above Y652), imidazole donor nitrogen, and imidazole acceptor nitrogen, and acetaldehyde oxygen FragMap types (b) The region below the central cavity (Y652 and F656) is occupied by negative acetate oxygen FragMaps. (c,d) SILCS Exclusion Map shows the regions of the hERG1 channel which are not accessible to the water and solute molecules. The SILCS Exclusion Map (red) for hERG1 shows how the dynamics of the protein allow for regions beyond those defined by the solvent-accessible surface of the initial hERG1 structure (transparent white) to open thereby allowing permeation of small molecules to the hidden pockets of hERG1 channel that are not evident in the rigid hERG1 structure. The FragMaps color codes are provided in Table 1.
The pocket around L529 vicinity has also been mapped. The residue is in the VSD and has been proposed as an important binding determinant of the NS-1643 activator 71. Our FragMaps of this pocket reveal the presence of the apolar, hydrogen-bond donor, and acceptor maps. The FragMaps for anionic (acetate) and cationic functional groups (methylammonium) indicate favorable binding to the region between the intracellular gate in the pore-domain and the cytoplasmic region (Figure 4b). This corresponds to the presence of a highly polar intra-cellular surface with multiple salt-bridges and interactions between the transmembrane (TM) domain and the cytoplasmic domains.
SILCS Hotspots Binding Site Identification in the hERG1 channel:
SILCS-Hotspots employs the SILCS-MC method to sample all possible binding sites for fragments on the hERG1 channel including low affinity and/or occluded sites in the interior of the protein 39. Such sites are not immediately evident from the examination of the experimental structure and unlikely to be detected from blind docking studies. Several recent publications have commented on the apparent difficulties in docking or simulations of binding of diverse ligands known to bind to the very constricted and hydrophobic environment of the IC cavity found in the cryo-EM structure 28–29, 33. The receptor flexibility plays an apparent role in the high-affinity binding to the intra-cavitary site and SILCS-MC has the potential to map the site directly in the presence of explicit lipid bilayer and solvent molecules. SILCS-Hotspots for the hERG1 identified favorable binding spots for the ring based fragments, which are termed as “hotspots”. 145 hotspots were identified with average LGFE scores for the fragments occupying each Hotspot ranging between –2.3 kcal/mol to –4 kcal/mol in the PD, VSD, and the intra-cellular terminal of the hERG1 channel (Figure 5). These hotspots were further organized into 32 binding pockets. The presence of multiple binding sites with relatively small differences in binding affinities for both activators and blockers have also been shown by previous modeling and electrophysiology studies 40, 72–73.
Figure 5. Hotspots analysis of the hERG1 channel.

TMD/Cytoplasmic domains considered for the analysis are shown as the transparent grey surface with identified Hotspots shown as cyan spheres. Two major binding areas with a preference for different functional groups were found in the distal S6/IC region. The classical drug binding region around F656 and Y652 is labeled as S1. The second detected binding region (S2) is located near the intra-cellular gate located in the distal S6 helix in the vicinity of T675 and R665. The Y652, F656, R665, and T675 are depicted as red, blue, orange, and magenta sticks, respectively. FragMaps are presented are described in Table 1. The top 10 hotspots scores are shown in Figure S1.
Experimentally, the IC cavity is the high-affinity drug binding location and our results show several hotspots in the cavity. These hotspots can be grouped as two distinct binding pockets labeled as S1 and S2. Similar finding of two distinct pockets is recently published for dofetilide 33. The S1 corresponds to the deeper classic drug binding pocket in the immediate vicinity of F656 or Y652. These Hotspots are mapping the primary binding pocket in the IC reported in many previous studies 22. On the other hand, the S2 is in the distal S6 (R665) and closer to the CNBD domain (T675, L678, and R679), represents a shallow drug binding region.
Hotspots #1 and #2, based on the average LGFE score ranking, represent nearly identical binding pockets determined by the location of R665 in the distal S6 and T675/R679 in the CNDB domain. These represent the “shallow” easily accessible site S2 (Figure 5). R665 is one of the key residues establishing a direct link between VSD of the channel and pore gate located in the S6 helix 76. However, the cryo-EM structure used in the calculations of the SILCS maps was lacking the PAS domain, which may impact its accuracy in assessing accessibility to the intracellular surface of channel 49. Hotspot #3 is defined by Y569 and F431 on the S5 and S4 helices, respectively. Both residues are known to be essential for the channel’s activation and were proposed to facilitate binding to channel agonists 74–75. Interestingly, this inter-digitated cluster of aromatic residues was also proposed to be essential for conformational coupling to the pore-opening during repolarization dynamics and drug access to the main sites. Hotspot #4 is located in the “deep”/high-affinity part of the IC and can be classified as part of the site S1 binding pocket. The site is formed by polar, aromatic, and amphipathic residues with well-established roles in the interactions with the channel blockers 22. It is important to emphasize that FragMaps featured in Figure 5 may be impacted by the truncated PAS domain in the cryo-EM, thus missing in our simulations. Overall, it can be concluded that fragment hotspot mapping of the accessible binding pockets of the cryo-EM structure of hERG1 is in general agreement with known sites for blockers and/or for activators.
Binding Scores for Blockers from SILCS and High-Precision Docking:
As a proof-of-principle assessment of the ability of SILCS to predict binding scores using maps generated with the hERG1 structure solved by cryo-EM, we selected 55 blockers with IC50 values measured in the same cell-line and under controlled experimental conditions (temperature, N of experiments, voltage-ramp protocols, etc) as part of the MICE study 41. The ligands were selected to contain diverse chemical groups, topologies, and binding affinities, with the list including very high μM to low nM blockers. This set was selected to reduce cell-type or temperature-dependent variations of IC50 values across a large cohort of blockers/non-blockers compounds which is a well-known issue in safety pharmacology 76–77. We considered both protonated and neutral forms of all the compounds in Table S1 including stable zwitterionic forms. An example of neutral and protonated structures for the well-known hERG1 blocker, dofetilide, is shown in Figure S1. The neutral, protonated, and zwitterionic states of the drugs were considered in the SILCS-MC and GLIDE docking. The resulting LGFE and Gscore values were weighted using the Henderson–Hasselbach equation using pH=7.4 (Equation 3) 78.
| (3) |
The initial SILCS|LGFE| scores were calculated using SILCS-MC on the 55 blockers dataset with the default FragMap weighting in the 2018 ACS. As is evident in Figure 6 weak correlations are present with experimentally observed pIC50 values. This motivated scaling of the default FragMap ACS weighting factors using the published Bayesian Machine Learning (BML) approach. Target data for the BML approach was the dataset of 163 hERG1 blockers with diverse chemical scaffolds and standardized IC50 values selected from our previous study 17. The compounds from this dataset were chosen for SILCS-MC affinity determination in the intra-cellular cavity of the hERG1 channel using the default 2018 ACS. The resulting optimal binding conformations for compounds from the dataset were used as input for training the FragMap weighting factors. The default ACS and BML optimized ACS weighting factors are provided in Table S2. The optimization of ACS weighting factors for this 163 molecules dataset, improved the Pearson correlation coefficient between calculated |LGFE| and experimental pIC50 from 0.091 to 0.467 for the 163 compounds (Figure S3).
Figure 6.

The correlation analysis of binding scores obtained with SILCS and Glide Docking for neutral, charged, and HH-weighted dugs blocking S1 or S2 sites in hERG1 using default ACS 2018 and BML optimized ACS 2018. Pearson correlation coefficients are shown relative to the experimentally measured pIC50 values from the MICE study.
Subsequently, the BML optimized weighting factors were used to re-pose and score these 55 drugs from the MICE study. Using this optimized ACS, these blockers (Table S1) were subjected to SILCS-MC sampling within the sphere of radius 10 Å in two binding regions located in IC (S1 and S2) as shown in Figure 5. The sphere centers were determined by the location of Cα atoms from Y652 and F656 for S1 while for the S2 the Cα atoms selected are from R665 and T675 residues. Spheres of radius 10 Å and 15 Å were used to test the effects of sampling space on the LGFE scores. The spherical boundaries for GLIDE docking were selected according to the hotspots and binding pockets that are shown in Figure 5 to explore blocker predicted binding affinity through the whole IC. The Glide molecular docking using the cryo-EM structure of the hERG1 channel shows drugs only bind to one location in the cavity, potentially due to the lack of dynamic movements within the protein (Figure S4) 21. However, the dynamic structure from MD simulations shows a wider cavity with deep and shallow binding regions (S1 and S2, Figure S4.). Table S1 shows the drug pIC50 values from the experimental studies for the wildtype hERG1 channel and the |LGFE| scores based on the optimized ACS obtained for the hERG1 channel.
The resulting binding scores for the various methods and protonation states are compared to experimental pIC50 values in Figure 6. The BML optimization yields a significant improvement in the Pearson correlation coefficient and show our predictive model to be capable of discriminating between hERG1 blockers and non-blockers. The BML optimized SILCS |LGFE| has a correlation coefficient of 0.535 with the experimental pIC50 whereas the GLIDE |Gscore| shows no correlation (Pearson correlation =0.016) for the high-affinity site S1 (Figure 5). We note that the poor correlation of GLIDE is consistent with previous docking studies on hERG1. Several groups have reported a complete lack of correlation in direct application of docking to the pore-domain of hERG1 captured by the cryo-EM structure. The incorporation of different rotamers for -OH and -SH has no impact on the lack of correlation in predicted scores (data not shown). Several issues may contribute to the lack of correlations. One is the low resolution of the structure (3.8 Å), albeit the pore-domain architecture is highly conserved across the family of K+-selective channels. Helliwell et al.29 noted that they were unable to observe binding to experimentally-characterized high-affinity pocket referred in the present study as site S1 due to steric clashes with the aromatic cassette (F557, F656, and Y652). Helliwell et al. constructed a different conformation of F656 and obtained a better agreement with experiment for a small set of compounds 29. Several recent studies employing multi-microsecond long MD simulations also corroborated the importance of S1 plasticity and conformational dynamics of F656 in high-affinity drug binding33. Studies based on homology models of the hERG1 pore domain reported reasonable performance for different docking algorithms 24, 72, 79. The key differences between the models and the experimentally derived structure were in the orientation of the aromatic cassette residues F557, F656, and Y65231. Therefore, docking for hERG1 blockade prediction has to account for receptor flexibility as is illustrated by the present lack of correlation in GLIDE |Gscore| for the studied cohort of compounds.
One approach to address this challenge is to perform long atomistic simulations followed by identification of ensembles of conformational states for docking from those simulations25, 33, 66 using clustering algorithms or experimentally-inspired structural insights. However, assigning statistical weights for the selected rotameric states of specific residues such as those in the aromatic cassette is challenging and their contributions to protein-ligand affinity therefore is somewhat arbitrary. An alternative is to apply free-energy simulations, ensuring sampling sufficient for affinity estimation 9. While attractive, approaches based on free-energy simulations are hard to extend to high-throuhput screening of thousands, if not millions, of drug candidates due to their high CPU or GPU requirements.
The SILCS/FragMaps explicitly accounts for the flexibility of the binding pockets allowing direct evaluation of binding of various substrates to the constricted IC cavity of the hERG1 resulting in better correlation with the experimental pIC50 than docking. It should be noted that IC50 is not a representation of the experimental ΔGbind, and LGFE is not directly analogous to binding affinity due to the lack of parameters such as the configurational entropy loss coming from differences between solute based functional groups and covalent connections of a full ligand 34. Therefore, correlation analysis suggests that the direct incorporation of conformational plasticity in the binding pocket with statistical-weighting (FragMaps) led to better performance than a blind docking study considering single receptor state with the experimentally determined structure.
Considering only neutral states of drugs show a better correlation of SILCS |LGFE| scores to the experimental pIC50 values than strictly considering cationic states, in agreement with recent studies that employed Replica-Exchange/Umbrella-Sampling simulations for dofetilide and moxifloxacin9. It is evident that the weighting of binding scores to account for various charge states of the compounds improves correlation to both SILCS and GLIDE scores to pIC50 values. The SILCS |LGFE| scores in both S1 and S2 (Figure 5) have a higher correlation coefficient than Glide |Gscore|. The high-affinity hydrophobic pocket S1, defined by Y652 and F656 results in the highest correlation with experimental pIC50 and a higher average of |LGFE| than the shallower pocket S2, around R665/T675 in agreement with previous studies3, 23. The SILCS-MC allows individual atom contributions to SILCS|LGFE| scores to be determined based on atomic GFE scores allowing for the determination of key functional groups of the ligand responsible for binding to be identified. Atoms that don’t make a significant contribution to the |LGFE| may still play a role in scaffolding between moieties on the ligand as required to properly orient favorable functional groups in the molecule 80–81. Information on these atomic contributions could be used for further ligand optimization to decrease affinity for hERG1, while increasing the ligand predicted binding affinity for the specific biological targets as illustrated by the atomic contribution analysis for the – Saquinavir (Figure S6). As an example of the role of analysis of the atomic GFE contributions, we show our results of neutral and protonated states of an antihistamine drug, astemizole, which was withdrawn from the market due to off-target hERG1 block (pIC50 = 8.40, HH weighted SILCS|LGFE|= 8.66 kcal/mol)41, 82. Astemizole has an estimated pKa value of 8.75 as calculated from Chemicalize ChemAxon, thus the drug is assumed to mostly exist in the protonated state at pH=7.4. The protonated drug (SILCS|LGFE|=8.74 kcal/mol) has a higher affinity to the hERG1 cavity by ~2kcal/mol compared to the neutral state (SILCS|LGFE|=6.84 kcal/mol). The favorable energetic contribution of the SILCS|LGFE| scores of the cation is shown to mostly arise due to the basic nitrogen of the drug (NLGFE score = −2.17 kcal/mol), which is consistent with the experimental predictions that speculate protonated basic nitrogen may play a crucial role in the hERG1 block 83–84 as shown in Figure 7. The secondary amine and pyridine amine play a role in increasing the |LGFE| due to the stabilization of the cationic form of astemizole. These two groups also overlap with methylammonium nitrogen map in the protonated state and with generic apolar, benzene carbon, and propane carbon maps in the neutral state. The para-methoxyphenyl is interacting with generic apolar, benzene carbon, and propane carbon in both states while in the neutral state and protonated state there is an overlap with methanol oxygen maps and acetaldehyde oxygen maps, respectively. In practice, during a medicinal chemistry campaign, atomic GFE information for the hERG1 could be combined with analogous information for a drug candidate based on SILCS FragMaps for the desired protein target thereby simultaneously maximizing binding to the target protein and minimizing that to hERG1.
Figure 7.

The overlap of the neutral vs protonated states of astemizole with FragMaps is shown in panels (a) and (c) and the atomic GFE contribution to overall LGFE are shown in (b) and (d), respectively. The best pose conformation of neutral vs protonated astemizole shows a change of conformation due to the protonation on pyridine amine. The astemizole is shown as sticks and FragMaps are shown as iso-surfaces according to Table 1.
Analysis of the atomic GFE contribution was also applied to an outlier case for ligand binding to S1 and S2 to identify the reason for the discrepancy with the experimental data. Saquinavir was predicted by SILCS to be a high-affinity ligand for site S2. Saquinavir is an anti-retroviral compound featuring a large number of aromatic groups and aliphatic linkages. Analysis of the GFE contributions to the predicted binding score from SILCS |LGFE| indicates the overestimation to be due to favourable contribution arising from non-polar and aromatic moieties, indicating an area for future improvements of the method. Overall, the data clearly shows that SILCS-MC allows a quantitative evaluation of binding affinities with direct account for complex membrane environment on par with cutting edge methodologies such as all-atom MD or Free Energy simulations29, 32, 85–86. SILCS-MC affinity ranking clearly supersedes the ranking obtained with GLIDE docking in agreement with the numerous docking reports to the cryo-EM structure. The report of Helliwell et al. shows the manual manipulation of the F656 was necessary to obtain reasonable results from molecular dockings to the intra-cavitary site in hERG1 channel29. While SILCS FragMap generation is a time-consuming step in the protocol, once the FragMaps are developed SILCS-MC allows rapid assessment of the drug cohorts. The advantage of the hERG1 flexibility consideration in the generation of FragMaps is critical as studies have been proposed the dynamic gating mechanism of the central cavity residues involving intricate interplay network of F557, M651, and Y652, and F65626, 72, 87.
One of the natural limitations of this study is its focus on only one functional state of the channel – open state captured in the cryo-EM structure 21. The state-dependence of hERG1 blockade has been shown to be an important determinant of drug cardiotoxicity. Blocker binding to an inactivated state was proposed to be associated with drug propensity to cause Torsades de Pointes 88. There are relatively minor structural differences (RMSD ~ 2.7 Å) in the topology/organization of the site S1 in the homologous channel EAG1 with PD in the closed state and the pore domain of hERG1 in the open state 43. At the same time, there is substantial experimental evidence for the conformational re-packing of the aromatic cassette in S6 (Y652 and F656) and slow inactivation itself being key determinants of the high-affinity blockade87. The only available cryo-EM structure of hERG1 captures the pore-domain in its open state. Most of the blockers assessed in the MICE study are targeting the high-affinity intra-cavitary site, which requires either opening of the intra-cellular gate (open-state blockade) or lipophilic access. There is also substantial experimental evidence for a pivotal role of drug access to the open-state in the process of hERG1 block 89–90. Therefore, we believe that directly accounting for receptor flexibility in the FragMaps provides a platform for assessment of the molecular determinants for high-affinity block, thus providing the community with an essential tool for use in “safety pharmacology”9.
Conclusions
The availability of the high-resolution cryo-EM provides a unique opportunity for us to apply rapidly evolving fragment-based approaches to map potentially druggable sites in the hERG1 channel. The direct application of popular molecular docking techniques leads to low correlations in predictions of the hERG1 blockade by small molecules compared to SILCS. Molecular docking approaches when applied to integral membrane proteins suffer from challenges arising from the inherent flexibility of the receptor, apparent need in accurate account for protein-lipid interface and drug lipophilicity or dynamic ionization and the potential role of the lipid-exposed areas of the channel involved in drug binding91–92. The chemical fragments used in SILCS allow for in-depth sampling of hard-to-reach binding pockets and/or mapping general preferences of flexible and challenging to map binding regions such as the intra-cavitary site in the hERG1 channel. Unlike the traditional methods for ligand identification and optimization, SILCS as a functional group mapping approach that provides rigorous functional group affinity free energy patterns for the entire space around and inside the biological target. The resulting FragMaps include contributions from protein flexibility, functional group and protein desolvation, and functional group-protein interactions. Accordingly, the calculated FragMaps may be used in database screening, fragment-based drug design, lead optimization, and mapping potentially relevant binding sites. The intrinsic flexibility of the Y652-F656 binding cassette in the deep binding pocket S1 site is an important determinant for drug binding; thus the conformational dynamics of these residues must explicitly be accounted. This is indeed accounted for in the SILCS approach and once FragMaps are calculated the approach can easily be scaled up to a large number of new compounds without additional all-atom MD simulations. The power of the approach was further illustrated by direct consideration of various states for the drug with pre-computed FragMaps. We showed that a BML optimization with a literature-derived training set of diverse chemical compounds allows for improvements in the pIC50 prediction. Together, the present results yield a predictive model of the relative affinity of the hERG1 blockers that has the potential to be used in drug design efforts to minimize the inadvertent off-target hERG1 block. This predictive tool could combine with Deep Learning methods using large drug datasets to classify fragments that may cause hERG1 blockade at different binding pockets.
Supplementary Material
Acknowledgments
Authors gratefully appreciate the valuable comments, help with the database of hERG1 blockers, and general feedback from Mr. Williams Miranda. The work in SYN’s group was partially supported by the National Institutes of Health (NIH USA) (Grant R01HL128537-03) and by the Canadian Institutes for Health Research (Project Program FRN-CIHR: 156236). MK was supported by the Queen Elizabeth II graduate scholarship, Jake Duerksen Memorial scholarship. ADM was supported by NIH USA grant R35GM131710. MM is supported partially by Graeme Bell and Norma Kay Sullivan-Bell Graduate Scholarship in Biology.
Footnotes
Conflict of Interest Disclosure: A.D.M. Jr. is co-founder and Chief Scientific Officer of SilcsBio, LLC.; S.Y.N. is a co-founder of Achlys Inc.
Supporting Information Available:
A list of 55 blockers assayed in the MICE study associated with their predicted binding affinity from SILCS-MC (LGFE) and Glide (Gscore) docking methods are provided. The top 10 Hotspots and their relative position to the hERG1 channel are depicted. A sample of protonated and neutral states of hERG1 blockers 2D structure and the protonation site are shown. The power of BML optimization to improve the correlation coefficient for 163 literature-derived blockers is provided. The 2D electrostatic potential map of hERG1 from cryo-EM and MD-refined structures is provided. The weighting factors for the default 2018 ACS and BML optimized ACS is provided. The conformational space sampled by the 55 blockers from the MICE protocol is shown. The atomic contributions to the SILCS |LGFE| for Saquinavir as an outlier of the predictive model is provided.
References.
- 1.Vandenberg JI; Perry MD; Perrin MJ; Mann SA; Ke Y; Hill AP, hERG K(+) channels: structure, function, and clinical significance. Physiol Rev 2012, 92, 1393–1478. [DOI] [PubMed] [Google Scholar]
- 2.Arcangeli A; Becchetti A, hERG Channels: From Antitargets to Novel Targets for Cancer Therapy. Clin Cancer Res 2017, 23, 3–5. [DOI] [PubMed] [Google Scholar]
- 3.Sanguinetti MC; Chen J; Fernandez D; Kamiya K; Mitcheson J; Sanchez-Chapula JA, Physicochemical basis for binding and voltage-dependent block of hERG channels by structurally diverse drugs. Novartis Found Symp 2005, 266, 159–166. [PubMed] [Google Scholar]
- 4.Parr E; Pozo MJ; Horowitz B; Nelson MT; Mawe GM, ERG K+ channels modulate the electrical and contractile activities of gallbladder smooth muscle. Am J Physiol Gastrointest Liver Physiol 2003, 284, G392–G398. [DOI] [PubMed] [Google Scholar]
- 5.Farrelly AM; Ro S; Callaghan BP; Khoyi MA; Fleming N; Horowitz B; Sanders KM; Keef KD, Expression and function of KCNH2 (HERG) in the human jejunum. Am J Physiol Gastrointest Liver Physiol 2003, 284, G883–G895. [DOI] [PubMed] [Google Scholar]
- 6.Huffaker SJ; Chen J; Nicodemus KK; Sambataro F; Yang F; Mattay V; Lipska BK; Hyde TM; Song J; Rujescu D; Giegling I; Mayilyan K; Proust MJ; Soghoyan A; Caforio G; Callicott JH; Bertolino A; Meyer-Lindenberg A; Chang J; Ji Y; Egan MF; Goldberg TE; Kleinman JE; Lu B; Weinberger DR, A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med 2009, 15, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arcangeli A, Expression and role of hERG channels in cancer cells. Novartis Found Symp 2005, 266, 225–232. [PubMed] [Google Scholar]
- 8.Wymore RS; Gintant GA; Wymore RT; Dixon JE; McKinnon D; Cohen IS, Tissue and species distribution of mRNA for the IKr-like K+ channel, erg. Circ Res 1997, 80, 261–268. [DOI] [PubMed] [Google Scholar]
- 9.Yang PC; DeMarco KR; Aghasafari P; Jeng MT; Dawson JRD; Bekker S; Noskov SY; Yarov-Yarovoy V; Vorobyov I; Clancy CE, A Computational Pipeline to Predict Cardiotoxicity: From the Atom to the Rhythm. Circ Res 2020, 126, 947–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trudeau MC; Warmke JW; Ganetzky B; Robertson GA, HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [DOI] [PubMed] [Google Scholar]
- 11.Zhang S; Zhou Z; Gong Q; Makielski JC; January CT, Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res 1999, 84, 989–998. [DOI] [PubMed] [Google Scholar]
- 12.Zimmermann M; Duruz H; Guinand O; Broccard O; Levy P; Lacatis D; Bloch A, Torsades de Pointes after treatment with terfenadine and ketoconazole. Eur Heart J 1992, 13, 1002–1003. [DOI] [PubMed] [Google Scholar]
- 13.Strauss DG; Gintant G; Li Z; Wu W; Blinova K; Vicente J; Turner JR; Sager PT, Comprehensive In Vitro Proarrhythmia Assay (CiPA) Update from a Cardiac Safety Research Consortium / Health and Environmental Sciences Institute / FDA Meeting. Ther Innov Regul Sci 2019, 53, 519–525. [DOI] [PubMed] [Google Scholar]
- 14.Vicente J; Zusterzeel R; Johannesen L; Mason J; Sager P; Patel V; Matta MK; Li Z; Liu J; Garnett C; Stockbridge N; Zineh I; Strauss DG, Mechanistic Model-Informed Proarrhythmic Risk Assessment of Drugs: Review of the “CiPA” Initiative and Design of a Prospective Clinical Validation Study. Clin Pharmacol Ther 2018, 103, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Veroli GY; Davies MR; Zhang HG; Abi-Gerges N; Boyett MR, High-throughput screening of drug-binding dynamics to HERG improves early drug safety assessment. Am J Physiol-Heart C 2013, 304, H104–H117. [DOI] [PubMed] [Google Scholar]
- 16.Wallis R; Benson C; Darpo B; Gintant G; Kanda Y; Prasad K; Strauss DG; Valentin JP, CiPA challenges and opportunities from a non-clinical, clinical and regulatory perspectives. An overview of the safety pharmacology scientific discussion. J Pharmacol Toxicol Methods 2018, 93, 15–25. [DOI] [PubMed] [Google Scholar]
- 17.Wacker S; Noskov SY, Performance of Machine Learning Algorithms for Qualitative and Quantitative Prediction Drug Blockade of hERG1 channel. Comput Toxicol 2018, 6, 55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hemmerich J; Ecker GF, In silico toxicology: From structure-activity relationships towards deep learning and adverse outcome pathways. Wires Comput Mol Sci 2020, e1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q; Jorgensen FS; Oprea T; Brunak S; Taboureau O, hERG classification model based on a combination of support vector machine method and GRIND descriptors. Mol Pharm 2008, 5, 117–127. [DOI] [PubMed] [Google Scholar]
- 20.Wang S; Sun H; Liu H; Li D; Li Y; Hou T, ADMET Evaluation in Drug Discovery. 16. Predicting hERG Blockers by Combining Multiple Pharmacophores and Machine Learning Approaches. Mol Pharm 2016, 13, 2855–2866. [DOI] [PubMed] [Google Scholar]
- 21.Wang W; MacKinnon R, Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 2017, 169, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitcheson J; Perry M; Stansfeld P; Sanguinetti MC; Witchel H; Hancox J, Structural determinants for high-affinity block of hERG potassium channels. Novartis Found Symp 2005, 266, 136–150. [PubMed] [Google Scholar]
- 23.Sanguinetti MC; Tristani-Firouzi M, hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [DOI] [PubMed] [Google Scholar]
- 24.Stary A; Wacker SJ; Boukharta L; Zachariae U; Karimi-Nejad Y; Aqvist J; Vriend G; de Groot BL, Toward a consensus model of the HERG potassium channel. ChemMedChem 2010, 5, 455–467. [DOI] [PubMed] [Google Scholar]
- 25.Dickson CJ; Velez-Vega C; Duca JS, Revealing Molecular Determinants of hERG Blocker and Activator Binding. J Chem Inf Model 2020, 60, 192–203. [DOI] [PubMed] [Google Scholar]
- 26.Perissinotti L; Guo J; Kudaibergenova M; Lees-Miller J; Ol’khovich M; Sharapova A; Perlovich GL; Muruve DA; Gerull B; Noskov SY; Duff HJ, The Pore-Lipid Interface: Role of Amino-Acid Determinants of Lipophilic Access by Ivabradine to the hERG1 Pore Domain. Mol Pharm 2019, 96, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munawar S; Vandenberg JI; Jabeen I, Molecular Docking Guided Grid-Independent Descriptor Analysis to Probe the Impact of Water Molecules on Conformational Changes of hERG Inhibitors in Drug Trapping Phenomenon. Int J Mol Sci 2019, 20, 3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Negami T; Araki M; Okuno Y; Terada T, Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci Rep 2019, 9, 16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helliwell MV; Zhang Y; El Harchi A; Du C; Hancox JC; Dempsey CE, Structural implications of hERG K(+) channel block by a high-affinity minimally structured blocker. J Biol Chem 2018, 293, 7040–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cernuda B; Fernandes CT; Allam SM; Orzillo M; Suppa G; Chia Chang Z; Athanasopoulos D; Buraei Z, The molecular determinants of R-roscovitine block of hERG channels. PLoS One 2019, 14, e0217733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zangerl-Plessl EM; Berger M; Drescher M; Chen Y; Wu W; Maulide N; Sanguinetti M; Stary-Weinzinger A, Toward a Structural View of hERG Activation by the Small-Molecule Activator ICA-105574. J Chem Inf Model 2020, 60, 360–371. [DOI] [PubMed] [Google Scholar]
- 32.Cavalluzzi MM; Imbrici P; Gualdani R; Stefanachi A; Mangiatordi GF; Lentini G; Nicolotti O, Human ether-a-go-go-related potassium channel: exploring SAR to improve drug design. Drug Discov Today 2020, 25, 344–366. [DOI] [PubMed] [Google Scholar]
- 33.Kudaibergenova M; Guo J; Khan HM; Zahid F; Lees-Miller JP; Noskov SY; Duff HJ, Allosteric Coupling Between Drug Binding and the Aromatic Cassette in the Pore Domain of the hERG1 Channel: Implications for a State-Dependent Blockade. Front Pharmacol 2020, 11, 914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ustach VD; Lakkaraju SK; Jo S; Yu W; Jiang W; MacKerell AD Jr., Optimization and Evaluation of Site-Identification by Ligand Competitive Saturation (SILCS) as a Tool for Target-Based Ligand Optimization. J Chem Inf Model 2019, 59, 3018–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faller CE; Raman EP; MacKerell AD; Guvench O, Site Identification by Ligand Competitive Saturation (SILCS) simulations for fragment-based drug design In Fragment-Based Methods in Drug Discovery, Springer: 2015; pp 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raman EP; Yu W; Lakkaraju SK; MacKerell AD Jr., Inclusion of multiple fragment types in the site identification by ligand competitive saturation (SILCS) approach. J Chem Inf Model 2013, 53, 3384–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raman EP; Yu W; Guvench O; MacKerell AD Jr, Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations. J Chem Inf Model 2011, 51, 877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakkaraju SK; Raman EP; Yu W; MacKerell AD Jr., Sampling of Organic Solutes in Aqueous and Heterogeneous Environments Using Oscillating Excess Chemical Potentials in Grand Canonical-like Monte Carlo-Molecular Dynamics Simulations. J Chem Theory Comput 2014, 10, 2281–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacKerell AD Jr.; Jo S; Lakkaraju SK; Lind C; Yu W, Identification and characterization of fragment binding sites for allosteric ligand design using the site identification by ligand competitive saturation hotspots approach (SILCS-Hotspots). Biochim Biophys Acta Gen Subj 2020, 1864, 129519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duff HJ; Feng ZP; Sheldon RS, High- and low-affinity sites for [3H]dofetilide binding to guinea pig myocytes. Circ Res 1995, 77, 718–725. [DOI] [PubMed] [Google Scholar]
- 41.Kramer J; Obejero-Paz CA; Myatt G; Kuryshev YA; Bruening-Wright A; Verducci JS; Brown AM, MICE Models: Superior to the HERG Model in Predicting Torsade de Pointes. Sci Rep-Uk 2013, 3, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ho BK; Gruswitz F, HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct Biol 2008, 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perissinotti LL; De Biase PM; Guo J; Yang PC; Lee MC; Clancy CE; Duff HJ; Noskov SY, Determinants of Isoform-Specific Gating Kinetics of hERG1 Channel: Combined Experimental and Simulation Study. Front Physiol 2018, 9, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lakkaraju SK; Yu W; Raman EP; Hershfeld AV; Fang L; Deshpande DA; MacKerell AD Jr., Mapping functional group free energy patterns at protein occluded sites: nuclear receptors and G-protein coupled receptors. J Chem Inf Model 2015, 55, 700–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hess B; Kutzner C; van der Spoel D; Lindahl E, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- 46.Word JM; Lovell SC; Richardson JS; Richardson DC, Asparagine and glutamine: using hydrogen atom contacts in the choice of side-chain amide orientation. J Mol Biol 1999, 285, 1735–1747. [DOI] [PubMed] [Google Scholar]
- 47.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein M, Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983, 79, 926–935. [Google Scholar]
- 49.Reiher WH III Theoretical studies of hydrogen bonding. Harvard University, Boston, MA, 1985. [Google Scholar]
- 50.Vanommeslaeghe K; Raman EP; MacKerell AD Jr., Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 2012, 52, 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr., CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Feller SE; Zhang Y; Pastor RW; Brooks BR, Constant pressure molecular dynamics simulation: The Langevin piston method. J Chem Phys 1995, 103, 4613–4621. [Google Scholar]
- 53.Berendsen HJ; Postma J. v.; van Gunsteren WF; DiNola A; Haak JR, Molecular dynamics with coupling to an external bath. J Chem Phys 1984, 81, 3684–3690. [Google Scholar]
- 54.Berk Hess HB, Berendsen Herman JC, Fraaije Johannes GEM, LINCS: a linear constraint solver for molecular simulations. J Comp Chem 1997, 18, 1463–1472. [Google Scholar]
- 55.Foster TJ; MacKerell AD Jr.; Guvench O, Balancing target flexibility and target denaturation in computational fragment-based inhibitor discovery. J Comput Chem 2012, 33, 1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrodinger, LLC, The PyMOL Molecular Graphics System, Version 1.8. 2015.
- 57.Humphrey W; Dalke A; Schulten K, VMD: visual molecular dynamics. J Mol Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 58.Chemicalize https://chemicalize.com/ developed by ChemAxon (http://www.chemaxon.com). (accessed 03-26-2020).
- 59.Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012, 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG, ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor RD; MacCoss M; Lawson AD, Rings in drugs. J Med Chem 2014, 57, 5845–5859. [DOI] [PubMed] [Google Scholar]
- 62.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 63.Jorgensen WL; Maxwell DS; TiradoRives J, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996, 118, 11225–11236. [Google Scholar]
- 64.Jorgensen WL; Tirado-Rives J, The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J Am Chem Soc 1988, 110, 1657–1666. [DOI] [PubMed] [Google Scholar]
- 65.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 66.Kalyaanamoorthy S; Lamothe SM; Hou X; Moon TC; Kurata HT; Houghton M; Barakat KH, A structure-based computational workflow to predict liability and binding modes of small molecules to hERG. Sci Rep-Uk 2020, 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Perry M; Degroot M; Helliwell R; Leishman D; Mitcheson J, Structural determinants of HERG potassium channel block by ibutilide and clofilium. J Physiol-London 2002, 544, 26p–26p. [Google Scholar]
- 68.Stegemann C; Pechlaner R; Willeit P; Langley SR; Mangino M; Mayr U; Menni C; Moayyeri A; Santer P; Rungger G; Spector TD; Willeit J; Kiechl S; Mayr M, Lipidomics profiling and risk of cardiovascular disease in the prospective population-based Bruneck study. Circulation 2014, 129, 1821–1831. [DOI] [PubMed] [Google Scholar]
- 69.Bohannon BM; Perez ME; Liin SI; Larsson HP, omega-6 and omega-9 polyunsaturated fatty acids with double bonds near the carboxyl head have the highest affinity and largest effects on the cardiac IK s potassium channel. Acta Physiol (Oxf) 2019, 225, e13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bohannon BM; de la Cruz A; Wu X; Jowais JJ; Perez ME; Dykxhoorn DM; Liin SI; Larsson HP, Polyunsaturated fatty acid analogues differentially affect cardiac NaV, CaV, and KV channels through unique mechanisms. Elife 2020, 9, e51453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo J; Cheng YM; Lees-Miller JP; Perissinotti LL; Claydon TW; Hull CM; Thouta S; Roach DE; Durdagi S; Noskov SY; Duff HJ, NS1643 interacts around L529 of hERG to alter voltage sensor movement on the path to activation. Biophys J 2015, 108, 1400–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saxena P; Zangerl-Plessl EM; Linder T; Windisch A; Hohaus A; Timin E; Hering S; Stary-Weinzinger A, New potential binding determinant for hERG channel inhibitors. Sci Rep-Uk 2016, 6, 24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perry M; Sachse FB; Sanguinetti MC, Structural basis of action for a human ether-a-go-go-related gene 1 potassium channel activator. P Natl Acad Sci U S A 2007, 104, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perry M; Sachse FB; Abbruzzese J; Sanguinetti MC, PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. P Natl Acad Sci USA 2009, 106, 20075–20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Butler A; Zhang YH; Stuart AG; Dempsey CE; Hancox JC, Functional and pharmacological characterization of an S5 domain hERG mutation associated with short QT syndrome. Heliyon 2019, 5, e01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kernik DC; Morotti S; Wu HD; Garg P; Duff HJ; Kurokawa J; Jalife J; Wu JC; Grandi E; Clancy CE, A computational model of induced pluripotent stem-cell derived cardiomyocytes incorporating experimental variability from multiple data sources. J Physiol-London 2019, 597, 4533–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Muszkiewicz A; Britton OJ; Gemmell P; Passini E; Sanchez C; Zhou X; Carusi A; Quinn TA; Burrage K; Bueno-Orovio A; Rodriguez B, Variability in cardiac electrophysiology: Using experimentally-calibrated populations of models to move beyond the single virtual physiological human paradigm. Prog Biophys Mol Biol 2016, 120, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hills AG, pH and the Henderson-Hasselbalch equation. Am J Med 1973, 55, 131–133. [DOI] [PubMed] [Google Scholar]
- 79.Durdagi S; Deshpande S; Duff HJ; Noskov SY, Modeling of open, closed, and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J Chem Inf Model 2012, 52, 2760–2774. [DOI] [PubMed] [Google Scholar]
- 80.Heinzl GA; Huang W; Yu W; Giardina BJ; Zhou Y; MacKerell AD Jr.; Wilks A; Xue F, Iminoguanidines as Allosteric Inhibitors of the Iron-Regulated Heme Oxygenase (HemO) of Pseudomonas aeruginosa. J Med Chem 2016, 59, 6929–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lanning ME; Yu W; Yap JL; Chauhan J; Chen L; Whiting E; Pidugu LS; Atkinson T; Bailey H; Li W; Roth BM; Hynicka L; Chesko K; Toth EA; Shapiro P; MacKerell AD Jr.; Wilder PT; Fletcher S, Structure-based design of N-substituted 1-hydroxy-4-sulfamoyl-2-naphthoates as selective inhibitors of the Mcl-1 oncoprotein. Eur J Med Chem 2016, 113, 273–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leurs R; Church MK; Taglialatela M, H1-antihistamines: inverse agonism, anti-inflammatory actions and cardiac effects. Clin Exp Allergy 2002, 32, 489–498. [DOI] [PubMed] [Google Scholar]
- 83.Durdagi S; Subbotina J; Lees-Miller J; Guo J; J Duff H; Y Noskov S, Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr Med Chem 2010, 17, 3514–3532. [DOI] [PubMed] [Google Scholar]
- 84.Choe H; Nah KH; Lee SN; Lee HS; Lee HS; Jo SH; Leem CH; Jang YJ, A novel hypothesis for the binding mode of HERG channel blockers. Biochem Biophys Res Comm 2006, 344, 72–78. [DOI] [PubMed] [Google Scholar]
- 85.Anwar-Mohamed A; Barakat KH; Bhat R; Noskov SY; Tyrrell DL; Tuszynski JA; Houghton M, A human ether-a-go-go-related (hERG) ion channel atomistic model generated by long supercomputer molecular dynamics simulations and its use in predicting drug cardiotoxicity. Toxicol Lett 2014, 230, 382–392. [DOI] [PubMed] [Google Scholar]
- 86.DeMarco KR; Dawson JRD; Yang P-C; Bekker S; Ngo VA; Noskov SY; Yarov-Yarovoy V; Clancy CE; Vorobyov I, Atomistic modeling towards predictive cardiotoxicity. bioRxiv 2019, 635441. [Google Scholar]
- 87.Chen J; Seebohm G; Sanguinetti MC, Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. P Natl Acad Sci U S A 2002, 99, 12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hancox JC; McPate MJ; El Harchi A; Zhang YH, The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther 2008, 119, 118–132. [DOI] [PubMed] [Google Scholar]
- 89.Perrin MJ; Kuchel PW; Campbell TJ; Vandenberg JI, Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-a-go-go-related gene channels. Mol Pharmacol 2008, 74, 1443–1452. [DOI] [PubMed] [Google Scholar]
- 90.Li Z; Dutta S; Sheng J; Tran PN; Wu W; Chang K; Mdluli T; Strauss DG; Colatsky T, Improving the In Silico Assessment of Proarrhythmia Risk by Combining hERG (Human Ether-a-go-go-Related Gene) Channel-Drug Binding Kinetics and Multichannel Pharmacology. Circ Arrhythm Electrophysiol 2017, 10, e004628. [DOI] [PubMed] [Google Scholar]
- 91.De Vivo M; Cavalli A, Recent advances in dynamic docking for drug discovery. WIREs Comp Mol Sci 2017, 7, e1320. [Google Scholar]
- 92.Abel R; Manas ES; Friesner RA; Farid RS; Wang L, Modeling the value of predictive affinity scoring in preclinical drug discovery. Curr Opin Struct Biol 2018, 52, 103–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.