

Key Points
Neutrophils from patients with GPS have reduced SG content and show defective NETosis.
NBEAL2 contributes to SG retention in neutrophils.
Abstract
Gray platelet syndrome (GPS) is an autosomal recessive bleeding disorder characterized by a lack of α-granules in platelets and progressive myelofibrosis. Rare loss-of-function variants in neurobeachin-like 2 (NBEAL2), a member of the family of beige and Chédiak-Higashi (BEACH) genes, are causal of GPS. It is suggested that BEACH domain containing proteins are involved in fusion, fission, and trafficking of vesicles and granules. Studies in knockout mice suggest that NBEAL2 may control the formation and retention of granules in neutrophils. We found that neutrophils obtained from the peripheral blood from 13 patients with GPS have a normal distribution of azurophilic granules but show a deficiency of specific granules (SGs), as confirmed by immunoelectron microscopy and mass spectrometry proteomics analyses. CD34+ hematopoietic stem cells (HSCs) from patients with GPS differentiated into mature neutrophils also lacked NBEAL2 expression but showed similar SG protein expression as control cells. This is indicative of normal granulopoiesis in GPS and identifies NBEAL2 as a potentially important regulator of granule release. Patient neutrophil functions, including production of reactive oxygen species, chemotaxis, and killing of bacteria and fungi, were intact. NETosis was absent in circulating GPS neutrophils. Lack of NETosis is suggested to be independent of NBEAL2 expression but associated with SG defects instead, as indicated by comparison with HSC-derived neutrophils. Since patients with GPS do not excessively suffer from infections, the consequence of the reduced SG content and lack of NETosis for innate immunity remains to be explored.
Visual Abstract

Introduction
Rare variants in NBEAL2 (neurobeachin-like 2) are causal of gray platelet syndrome (GPS), an autosomal recessive bleeding disorder characterized by formation of large platelets devoid of α-granules, splenomegaly, and bone marrow fibrosis.1,2 NBEAL2 encodes a protein of 2754 amino acids and is, among others, expressed in hematopoietic cells and particularly abundant in platelets, monocytes, and neutrophils. The NBEAL2 gene belongs to a family of genes containing a beige and Chédiak-Higashi syndrome (BEACH) domain.3 Most of these genes encode large multidomain scaffolding proteins, with a predicted function in fusion, fission, and trafficking of vesicles and granules.2,3 The function of NBEAL2 is in recent years predominantly investigated in platelets and megakaryocytes, reporting the possible role of NBEAL2 in granule cargo loading and/or retention.4-6 In a recent study, NBEAL2 was found to simultaneously bind P-selectin and membrane-resident trafficking protein SEC22B, where these interactions facilitate the suggested essential role of NBEAL2 in granule development and cargo stability.7 Furthermore, in a study using NBEAL2-Tag protein constructs in HEK cells or CHRFs (a megakaryoblastic cell line), NBEAL2 was found to be primarily localized in the cytoplasm.8 In addition, proteins like Dock7, Sec16a, and Vac14, which are suggested to function in signaling, were found to be interactors of NBEAL2.8 Studies in knockout mice also suggest that NBEAL2 may control the formation and retention of granules in neutrophils as well.3-6 In fact, the protein coding transcript of NBEAL2 is expressed in higher levels in granulocyte-monocyte progenitor cells compared with megakaryocyte-erythroid progenitor cells.9 These findings prompted us to perform a detailed assessment of the function of human GPS neutrophils and their granules.
We studied neutrophils obtained from the peripheral blood from 13 patients with GPS to examine their granule composition and functionality in detail. In addition, in an optimized culture system, in which peripheral blood CD34+ hematopoietic stem cells (HSCs) can be grown into fully differentiated mature human neutrophils, we explored the relationship of NBEAL2 and the levels of granule proteins. Although Nbeal2-knockout mouse studies have suggested a defect in neutrophil reactivity,10 previous results of neutrophil studies in patients with GPS have remained inconclusive.11-14
Neutrophils act against bacteria and fungi and use reactive oxygen species (ROS) generation by the NADPH oxidase system, the mobilization of antimicrobial proteases and proteins from granules to maintain immune fitness.15 In addition, neutrophils also possess a cytotoxic mechanism via the formation of neutrophil extracellular traps (NETs), which are web-like chromatin structures decorated with histones, granule-derived proteases, and antimicrobial peptides.16 Neutrophils can deploy this mechanism as an alternative to phagocytosis, to constrain the spread of fungi, large bacteria, and several other micro-organisms.17,18 Here, we show that patients with GPS have a strongly reduced amount of specific granule (SG) content and impaired NET formation. We conclude that NBEAL2 is critically important for neutrophils as regulator of SG release, where the lack of NET formation is caused by the absence of SG proteins and not by the NBEAL2 protein itself. Hence, we identify the SG compartment as another relevant factor in the process of NET formation, subsequent to ROS formation and the proteolytic activity of elastase. The consequences of the reduced SG content and lack of NET formation for innate immunity in GPS patients require further study.
Methods
Patients with GPS, control subjects, and cell isolation
Heparinized blood was obtained from patients with GPS and healthy adults after informed consent and according to the Declaration of Helsinki 2008 (Table 1). This study was approved by the Medical Ethical Committee of the Radboud University Medical Center (Nijmegen, The Netherlands). Blood leukocytes were separated based on density by centrifugation over isotonic Percoll (Pharmacia, Uppsala, Sweden) with a specific density of 1.076 g/mL. The interphase fraction, containing peripheral blood mononuclear cells (PBMCs), was harvested for isolation of CD34+ cells by magnetic-activated cell sorting with a human CD34 MicroBead Kit (Miltenyi-Biotec, Bergisch Gladbach, Germany) according to the manufacturer’s instructions. Neutrophils were obtained from the pellet fraction after erythrocyte lysis with hypotonic ammonium chloride solution at 4°C as previously described.19
Table 1.
Pathogenic variants in patients with GPS from unrelated pedigrees
| Patient | Sex | Age band (y) | Pathogenic and likely pathogenic NBEAL2 variants | Identifier |
|---|---|---|---|---|
| A | Female | 30-34 | NM_015175.2:c.1928A>T NP_055990.1:p.(Glu643Val) | C.II.31 |
| NM_015175.2:c.6299C>T NP_055990.1:p.(Pro2100Leu) | 20,126 | |||
| B | Female | 30-34 | NM_015175.2:c.1928A>T NP_055990.1:p.(Glu643Val) | C.II.4b1 |
| NM_015175.2:c.6299C>T NP_055990.1:p.(Pro2100Leu) | 20,226 | |||
| C | Male | 40-44 | NM_015175.2:c.6806C>T NP_055990.1:p.(Ser2269Leu) | D.II.31 |
| NM_015175.2:c.256A>G NP_055990.1:p.(Ile86Val) | 2426 | |||
| Splice acceptor variant NM_015175.2:c.6920-1 G>C | ||||
| Splice acceptor variant NP_055990.1:p.(Ser2269Ter) | ||||
| D | Male | 40-44 | Splice acceptor variant NM_015175.2:c.4485-1G>T NP_055990.1:p.(Phe646del) | 2626 |
| E | Female | 45-49 | NM_015175.2:c.6568del NP_055990.1:p.(Cys2190AlafsTer23) | 2526 |
| NM_015175.2:c.7937T>C | ||||
| NP_055990.1:p.(Leu2646Pro) | ||||
| F | Male | 45-50 | NM_015175.2:c.3058T>C NP_055990.1:p.(Tyr1020His) | 3126 |
| NM_015175.2:c.7134G>A NP_055990.1:p.(Glu2378 =) | ||||
| Splice acceptor variant NM_015175.2:c.2650-1G>A | ||||
| G | Female | 60-65 | NM_015175.2:c.4890del | 22,126 |
| NP_055990.1:p.(Arg1631Glyfs*3) | ||||
| H | Male | 19-24 | NM_015175.2:c.4501_4503del | |
| NP_055990.1:p.(Leu1501del) | ||||
| I | Male | 15-20 | NM_015175.2:c.5299C>T | P150 |
| NP_055990.1:p.(Gln1767) | ||||
| J | Male | 13-18 | NM_015175.2:c.5299C>T | P350 |
| NP_055990.1:p.(Gln1767) | ||||
| K | Male | 9-14 | NM_015175.2:c.5299C>T | P250 |
| NP_055990.1:p.(Gln1767) | ||||
| L | Male | 62 | NM_015175.2:c.6432delT | Dutch |
| NP_055990.1:p.(Phe2144Leufs*23) | ||||
| NM_015175.2:c.5497G>A | ||||
| NP_055990.1:p.(Glu1833Lys) | ||||
| M | Male | 62 | NM_015175.2:c.6432delT | Dutch |
| NP_055990.1:p.(Phe2144Leufs*23) | ||||
| NM_015175.2:c.5497G>A | ||||
| NP_055990.1:p.(Glu1833Lys) |
Neutrophil granules
The identification of myeloperoxidase (MPO)-positive azurophilic and lactoferrin-positive SGs was performed with a CM10 electron microscope (Philips, Eindhoven, The Netherlands).20 To assess degranulation, neutrophils (2 × 106/mL) were primed for 5 minutes with platelet-activating factor (PAF; Sigma-Aldrich, Steinheim, Germany) or cytochalasin B (Sigma-Aldrich) at 37°C and stimulated for 15 minutes with N-formyl-methionyl-leucyl-phenylalanine, (fMLF; Sigma-Aldrich). The cells were stained with the granule markers CD63-phycoerythrin (immunoglobulin G1 [IgG1], clone 435; US Biologicals, Suffolk, United Kingdom) and CD66b-fluorescein isothiocyanate (IgG1, CLBB13.9; Sanquin, The Netherlands) and analyzed by flow cytometry (LSRII; BD Pharmingen, San Diego, CA). Elastase and lactoferrin release were measured by enzyme-linked immunosorbent assay (HyCult-Biotech, Uden, The Netherlands).
Proteomic analysis
Mass spectrometry (MS) analysis was performed as described previously.21 All data were acquired using Xcalibur software and analyzed with the MaxQuant (1.5.3.30) computational platform.22 Proteins and peptides were identified using the Andromeda search engine by querying the human UniProt database (release 3-2017). Protein quantification was based on unique peptides with the “match between runs” option enabled. Results were loaded into R 3.6.2. Contaminants and reverse values were filtered out. Proteins were considered quantified based on the presence of 2 out of 3 valid values in ≥1 condition. Missing values were replaced with random values from a down shifted normal distribution (width = 0.3 standard deviations and down shift = 1.8 standard deviations of the nonmissing data23). Statistical analysis was performed using the limma package,24 and a P < .05 and an absolute log2 fold change >1 were considered significant and relevant. Enrichment for granule proteins was tested with Fisher exact tests followed by Benjamini-Hochberg multiple testing correction using a granule protein database.25
CD34+ HSCs culture
CD34+ HSCs were isolated from granulocyte colony-stimulating factor (G-CSF) mobilized peripheral blood (MPB) of healthy stem cell donors or from PBMCs of patients with GPS or healthy controls (HCs). CD34+ HSCs were cultured in Stemline II (Sigma-Aldrich) with 1% penicillin/streptomycin, 10 ng/mL interleukin-3 (StemCell Technologies, Vancouver, BC, Canada), 10 ng/mL granulocyte-macrophage colony-stimulating factor (Peprotech EC, London, United Kingdom), 30 ng/mL G-CSF (Neupogen, clinical grade), 50 ng/mL Flt-3 (StemCell Technologies), and 50 ng/mL human stem cell factor (Sanquin) for the first 3 days in a 24-well plate. After 3 days, the media was changed and the cells were cultured in Stemline II with 1% penicillin/streptomycin and 30 ng/mL G-CSF for the next 13 to 15 days until the end of differentiation. The cells were replated and received fresh media every 3 days.
Plasma proteomics
Plasma was obtained from EDTA anticoagulated blood of 11 patients with GPS and 13 age-matched controls and analyzed using protein MS (see supplemental Methods).
Microbial killing
The neutrophil-mediated killing of Candida albicans (strain:SC5314), Escherichia coli (strain:ML-35), and Staphylococcus aureus (strain:502A) was determined by a colony-forming unit assay on Luria broth agar plates as previously described.19 To assess the Aspergillus fumigatus hyphae killing, neutrophils (5.0 × 106 cells per mL) were incubated for 1 hour on a preformed monolayer at 37°C. Cells were lysed in water/NaOH, pH 11.0, and the monolayer was incubated with MTT (Sigma-Aldrich). The absorbance of acidic-isopropanol diluted samples was then measured on a plate reader (Tecan, Mannedorf, Switzerland).
NET formation
Neutrophils (2 × 106/mL) were stimulated with phorbol myristate acetate (PMA) (100 ng/mL; Sigma-Aldrich), monosodium urate (MSU) crystals (200 μg/mL; Invivogen, San Diego, CA), or opsonized C albicans (strain SC5314; multiplicity of infection of 10) for 180 or 240 minutes, respectively, at 37°C. C albicans was grown at 30°C in the presence of 30 μg/mL kanamycin, set to optical density 625 = 1.0, and subsequently opsonized with 10% (v/v) human pool serum for 10 minutes at 37°C. After stimulation, the neutrophils were fixed with 3.7% (wt/vol) paraformaldehyde and permeabilized with a kit (Beckman-Coulter, Brea, CA). DNA was stained with Hoechst (Sigma-Aldrich), anti-elastase (clone:AT15; Sanquin), and anti-MPO (clone:2C7; Abnova, Taipei, Taiwan). Primary antibodies were detected with anti-rabbit-Ig 488 and anti-mouse-Ig 633 antibodies (Life, Bleiswijk, The Netherlands) by confocal laser scanning microscopy (Zeiss, Sliedrecht, The Netherlands). NET formation was quantified as the mean number of NETs per field of 100 neutrophils for at least 5 fields per sample.
DNA release was determined with Quant-iT PicoGreen dsDNA Kit (Life) or via Sytox Green Nucleic Acid Stain (Thermo Fisher Scientific) by SPECTRA-Fluor-Plus plate reader (Tecan).
For live-cell imaging, neutrophils were stained with the Quant-iT PicoGreen dsDNA Kit (Life), stimulated with PMA (100 ng/mL; Sigma-Aldrich), and imaged for 4 hours with a Leica SP8 confocal laser microscope.
Statistics
Statistical analysis was performed with GraphPad Prism version 8.00 for Windows (GraphPad Software, San Diego, CA). Data were evaluated by unpaired, 2-tailed Student’s t test and analysis of variance (ANOVA) Benjamini-Hochberg test. The results are presented as mean ± standard error of the mean (SEM) and considered significant when P < .05.
For more information on materials and methods, see supplemental Data.
Results
Granule defects in neutrophils of patients with GPS
From the study cohort of 47 patients with GPS,26 we received and analyzed blood cells from 7 of these patients with GPS and 6 additional patients with GPS with causal variants in NBEAL2 from 9 genetically independent pedigrees in further detail (Table 1). In this study, we show by flow cytometric analysis a reduced side-scatter signal for neutrophils, suggesting a diminished level of granularity (supplemental Figure 1). To verify the presence and form of the granules, we performed immunoelectron microscopy (IEM) of neutrophils from patients with GPS to obtain maximal resolution about the formation and morphology of their azurophilic granules (AGs) and SGs, as we have performed previously in similar studies on the biogenesis of lysosomal organelles.27 We assumed that AGs are the more likely candidate organelle in neutrophils reminiscent of platelet α-granules, because both types of granules can only be released by strong activation, in contrast to SGs or dense granules in neutrophils and platelets, respectively. However, our electron microscopy study showed the distribution of AGs to be normal in neutrophils from patients with GPS. Instead, the lactoferrin-positive SGs were reduced by 60% (Figure 1A-C).
Figure 1.
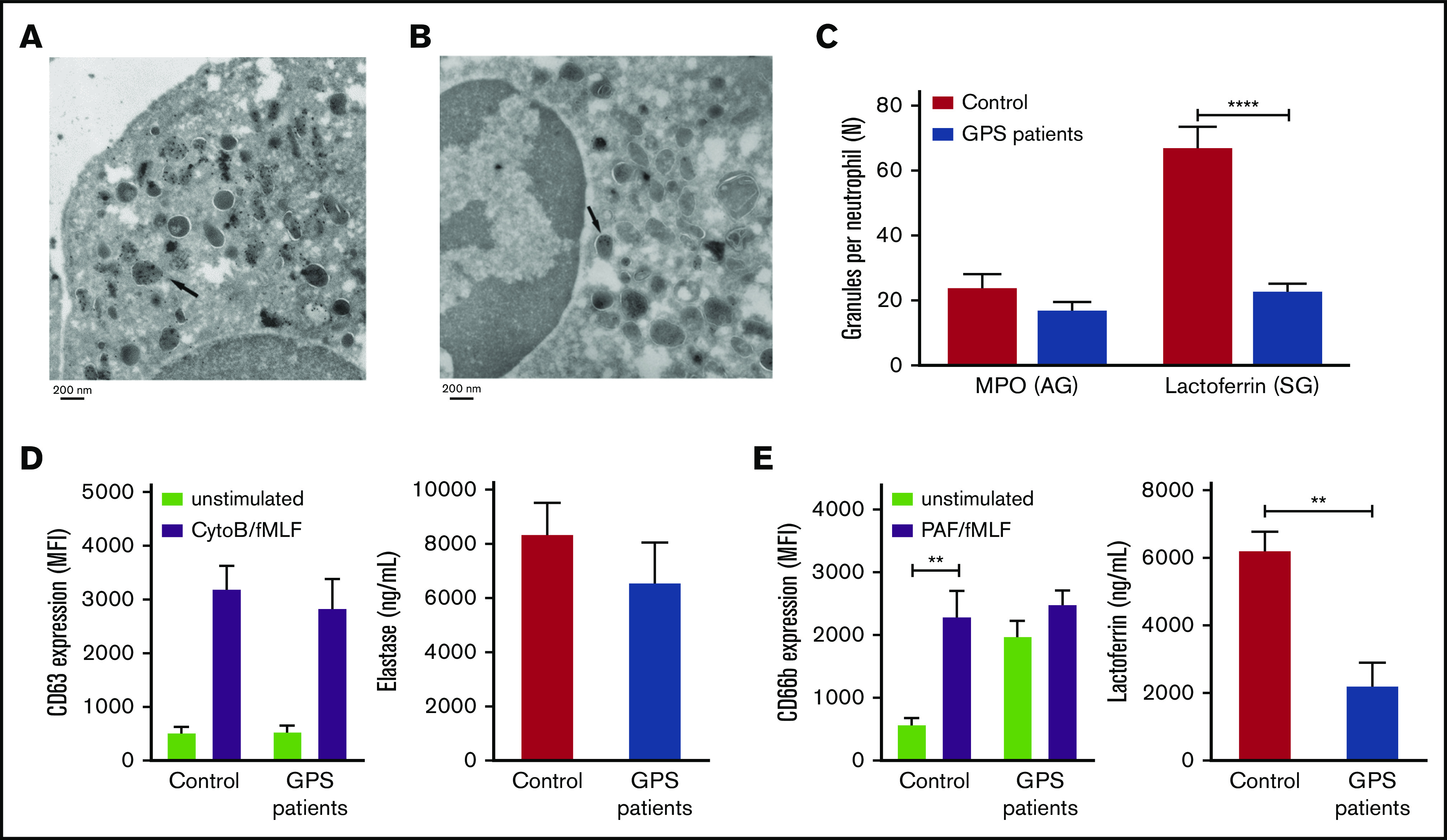
SG deficiency in GPS neutrophils. (A-B) IEM identification of MPO-positive AGs (A) and lactoferrin-positive SGs (B) in GPS neutrophils. The arrow indicates an example of positive granule labeling. (C) The numbers of lactoferrin- and MPO-positive granules per cell were determined by counting the number of positive granules in control and GPS neutrophils. (D-E) Neutrophils from controls and patients with GPS were stimulated with cytochalasin B (5 μg/mL)/fMLF (1 μM) or PAF (1 μM)/fMLF (1 μM), and plasma membrane expression of CD63 (azurophilic; D, left graph) and CD66b (specific; E, left graph) was measured by flow cytometry. The extracellular concentrations of elastase (azurophilic; D, right graph) and lactoferrin (specific; E, right graph) were measured by enzyme-linked immunosorbent assay. Results are means ± SEM, n = 3-5. An unpaired, 2-tailed t test was used. **P < .01, ****P < .0001. MFI, mean fluorescence intensity.
The mobilization of granules was evaluated after stimulation of neutrophils with cytochalasin B/fMLF or PAF/fMLF for AGs and SGs, respectively.28 Elastase secretion and expression of CD63, both markers for the former granules (AGs), did not differ between patients and controls (Figure 1D), reinforcing the normal formation and retention of this granule type. However, lactoferrin secretion, a marker for SGs, was significantly decreased in patients with GPS compared with controls (Figure 1E), corroborating our electron microscopy findings. The expression of the α-granule membrane protein P-selectin (CD62P) is abundantly present on resting platelets from patients with GPS compared with platelets from healthy controls (supplemental Figure 2). We show a similar signature on resting neutrophils from patients, which stain strongly positive for the SG membrane protein CD66b. In addition, its expression did not alter significantly upon activation (Figure 1E), which was relatively similar for platelets, where GPS platelets showed a near maximum of P-selectin surface expression which only slightly increased upon activation (supplemental Figure 2). From this we conclude that resting neutrophils from patients with GPS have prematurely released their SGs.
Proteome analysis of neutrophils from patients with GPS
To investigate which proteins were reduced in their abundance, we compared neutrophils isolated from the blood of patients with GPS with those obtained from healthy controls using label-free quantitative MS. In addition to the strongly reduced abundance of NBEAL2, the proteomes of GPS neutrophils showed differential expression of 72 proteins (Figure 2A-B; Table 2). Most interestingly, 3 patients with GPS (patients C, L, and M in Table 1) showed presence of NBEAL2 peptides. The peptide identifications were based on both MS/MS data and the match-between-run feature of the MaxQuant software package. These peptide identifications and intensities were used by MaxQuant to calculate label-free protein quantification values. This algorithm integrates data from at least 2 unique peptides to calculate protein intensities. For 1 patient (patient C in Table 1), multiple peptides were identified by MS/MS and matching, giving high confidence to the quantified NBEAL2 protein level (see patient 1 in supplemental Figure 3). Surprisingly, 2 other patients with GPS (patients L and M in Table 1) were suggested to also have some detectable NBEAL2 protein levels. These quantification values were based on matching instead of real MS/MS data, consisting of 2 matched peptide sequences (see patients 7 and 8 in supplemental Figure 3). Upon further inspection of these peptides, a protein blast of “LSAEEAAHR” resulted in 100% alignment with hCG15426, KLAA0540, and SQFE253 sequences. These sequences are not present in the UniProt-reviewed database used for our MaxQuant calculations and represent a case of misidentification by the database as a peptide unique to NBEAL2 (thus representing a limitation of the used sequence database). Therefore, the protein quantification intensity of NBEAL2 in these 2 patients with GPS should be interpreted with extreme caution.
Figure 2.

Neutrophils from patients with GPS show lower SG protein abundance compared with healthy control neutrophils (PMNs). (A) Volcano plot comparing PMN proteomes of patients with GPS vs healthy controls (HC). (B) Label-free intensity values of PMN of patients with GPS vs HCs. The white and red circles indicate patients 1, 7, and 8 in supplemental Figure 3, respectively. (C) GO term enrichment analysis of proteins with decreased abundancies in PMNs of patients with GPS. (D) Distribution of granule-annotated proteins with decreased abundancies in PMNs of patients with and corresponding granule protein enrichment analysis. BP, biological process; CC, cellular component.; CM, cell membrane; FG, ficolin-1-rich granule; LFQ, label free quantification; SV, secretory vesicle.
Table 2.
Differently abundant proteins between neutrophils from patients with GPS and healthy controls
| Gene names | logFC | Adjusted P | Direction | Granule |
|---|---|---|---|---|
| ADAM8 | −1.323657312 | 1.25871E-07 | Down | SG |
| AK1 | 1.404751471 | 0.006640861 | Up | |
| ALDH3B1 | −1.064129038 | 9,03216E-06 | Down | |
| ANTXR2 | −1.420443758 | 0.001203625 | Down | SV |
| AZU1 | −1.258930678 | 0.015537439 | Down | AG |
| C14orf142 | 1.944269282 | 0.038452512 | Up | |
| CD200R1 | −1.706012645 | 0.002598112 | Down | |
| CEACAM1 | −1.00298783 | 1.27051E-05 | Down | SG/GG |
| CD93 | −1.201001783 | 1.63897E-05 | Down | FG |
| CHIT1* | −2.580734627 | 0.004690497 | Down | SG |
| CIB1 | −1.553545064 | 2.0362E-10 | Down | SV |
| CLEC4D | −1.683458526 | 1.05203E-06 | Down | SG |
| CLEC5A | −1.438176815 | 0.001199003 | Down | SG |
| CRISP3 | −1,053756172 | 6.28887E-06 | Down | SG |
| CRISPLD2 | −3.028483512 | 1.30474E-06 | Down | FG |
| CTCF | 1.093635528 | 0.010377533 | Up | |
| DDI2 | 1.235789759 | 0.004754332 | Up | |
| FBXO7 | 1.86528072 | 0.018502334 | Up | |
| FCAR | −1.157873873 | 5.29839E-07 | Down | SG |
| FCN1 | −1.358795862 | 0.008014707 | Down | FG |
| FGL2 | −3.152449135 | 2.23273E-08 | Down | FG |
| FLNB | 1.591666804 | 0.028032967 | Up | |
| FPR2 | −3.215391757 | 2.87143E-07 | Down | FG |
| GP1BB | 2.489535838 | 0.002505912 | Up | |
| HBA1 | 1.425726403 | 0.036719775 | Up | |
| HBB | 1.499401526 | 0.024715387 | Up | † |
| HBD | 2.18089191 | 0.00805624 | Up | |
| IGHM | 4.564716704 | 0.001517623 | Up | |
| ISG15 | 1.50580967 | 0.013120869 | Up | |
| ITGA2B | 2.17313411 | 0.034495612 | Up | |
| ITGB3 | 3.256606076 | 0.000590977 | Up | |
| ITM2B | −1,776889478 | 0.005035806 | Down | SV |
| LCN2* | −1.067419243 | 9.36095E-07 | Down | SG |
| LILRA3 | −1.191339116 | 0.03617178 | Down | SG/GG |
| LONRF2 | −1.615776321 | 0.040658862 | Down | |
| LPCAT1 | −1.166004931 | 0.008161495 | Down | AG |
| LRG1 | −1.656966487 | 3.116E-08 | Down | SG |
| LST1 | −1.335978979 | 0.038055338 | Down | CM |
| LTB4R* | −1.144669658 | 0.001994964 | Down | SG |
| MCEMP1 | −1.726928642 | 1.71313E-09 | Down | SG |
| MGAM | −1.311041164 | 3.77199E-08 | Down | FG |
| MMP8* | −1.524649658 | 8.97816E-08 | Down | SG |
| MMP9* | −2.520262691 | 3.71309E-10 | Down | GG |
| MMP25 | −1.166679848 | 0.034288334 | Down | SV |
| NBEAL2 | −5.458711875 | 1.10983E-16 | Down | GG |
| NUDT3 | 1.047414287 | 0.018146866 | Up | |
| OLR1* | −1.275030481 | 9.03216E-06 | Down | SG |
| ORM2* | −1.92503296 | 0.003408997 | Down | SG |
| OSCAR* | −1.663676016 | 0.000146155 | Down | SG |
| PF4 | −3.245437395 | 3.87295E-05 | Down | † |
| PGLYRP1 | −1.730698451 | 1.1023E-10 | Down | SG |
| PPBP | −1.735672277 | 0.016495775 | Down | GG |
| PTPRJ | −1.009341488 | 5.32917E-09 | Down | SG |
| PTX3 | −1.237195701 | 8.33899E-07 | Down | SG |
| QPCT | −1.871870602 | 5.32917E-09 | Down | GG |
| QSOX1 | −1.319014529 | 1.3305E-08 | Down | SG |
| RNF123 | 2.052297543 | 0.000170734 | Up | |
| RPL37A | 1.619666522 | 7.9634E-07 | Up | |
| SDPR | 1.423250247 | 0.028032967 | Up | |
| SIRPB1 | −1,04135498 | 0.010205733 | Down | CM |
| SLC44A1 | −1,298388194 | 0.015698875 | Down | CM |
| SPAG9 | 1.612871903 | 0.01723901 | Up | |
| SPTB | 2.478165209 | 0.014456575 | Up | CM |
| SRGN* | −2.369263809 | 7.17865E-08 | Down | Preferential SG |
| TBC1D24 | 1.064763056 | 0.016157178 | Up | |
| THEMIS | 2.939359331 | 0.02286843 | Up | |
| TPM4 | 1.033728537 | 0.001199003 | Up | |
| TSPAN14* | −1.199040302 | 0.000158716 | Down | SG |
| TTN | 2.775421702 | 0.034495612 | Up | |
| TUBB1 | 2.386733997 | 0.020660207 | Up | |
| TXNDC17 | 1.464401309 | 0.022068896 | Up | |
| YOD1 | 1.670178267 | 0.002453601 | Up |
Results of the comparison between GPS (n = 11) and control (n = 15) neutrophils, as represented in the volcano plot of Figure 2A. Shown are the proteins more (up direction) or less (down direction) abundant in GPS neutrophils compared to healthy control neutrophils, with a restriction of a twofold increase or decrease in abundance. Granule annotation was defined according to Rorvig et al25 and adjudicated using cluster enrichment from Hoogendijk et al.21
CM, cell membrane; FG, ficolin-1-rich granule; logFC, log fold change; SV, secretory vesicle.
Deficient in SGD neutrophil proteomics according to Serwas et al.40
Possible contamination.
Analysis for proteins with reduced levels in neutrophils of patients with GPS showed an enrichment for Gene Ontology (GO) terms related to granule release (Figure 2C). This association with granule biology was supported by a striking enrichment of proteins we found to be reduced in levels to be present in sets of proteins assumed to be localized in SGs (n = 17) and gelatinase granules (GGs; n = 5) (Figure 2D).25 Although other SG proteins, such as lactoferrin, did not meet the strict requirements of significant fold change (ie, a twofold increase or decrease in abundance) as shown in Table 2, they did show a significant lower abundance in GPS neutrophils (supplemental Table 1). Furthermore many of the proteins with reduced levels in GPS neutrophils are known by IEM to be localized to SG, adding an additional level of evidence that SG and GG proteins are specifically depleted from GPS neutrophils.21
The MS analysis results were accompanied by the sequencing of the RNA of the same neutrophil population. The analysis showed that the transcript levels for the proteins that are reduced in GPS neutrophils are not significantly different from those observed in matched controls (Figure 3).
Figure 3.
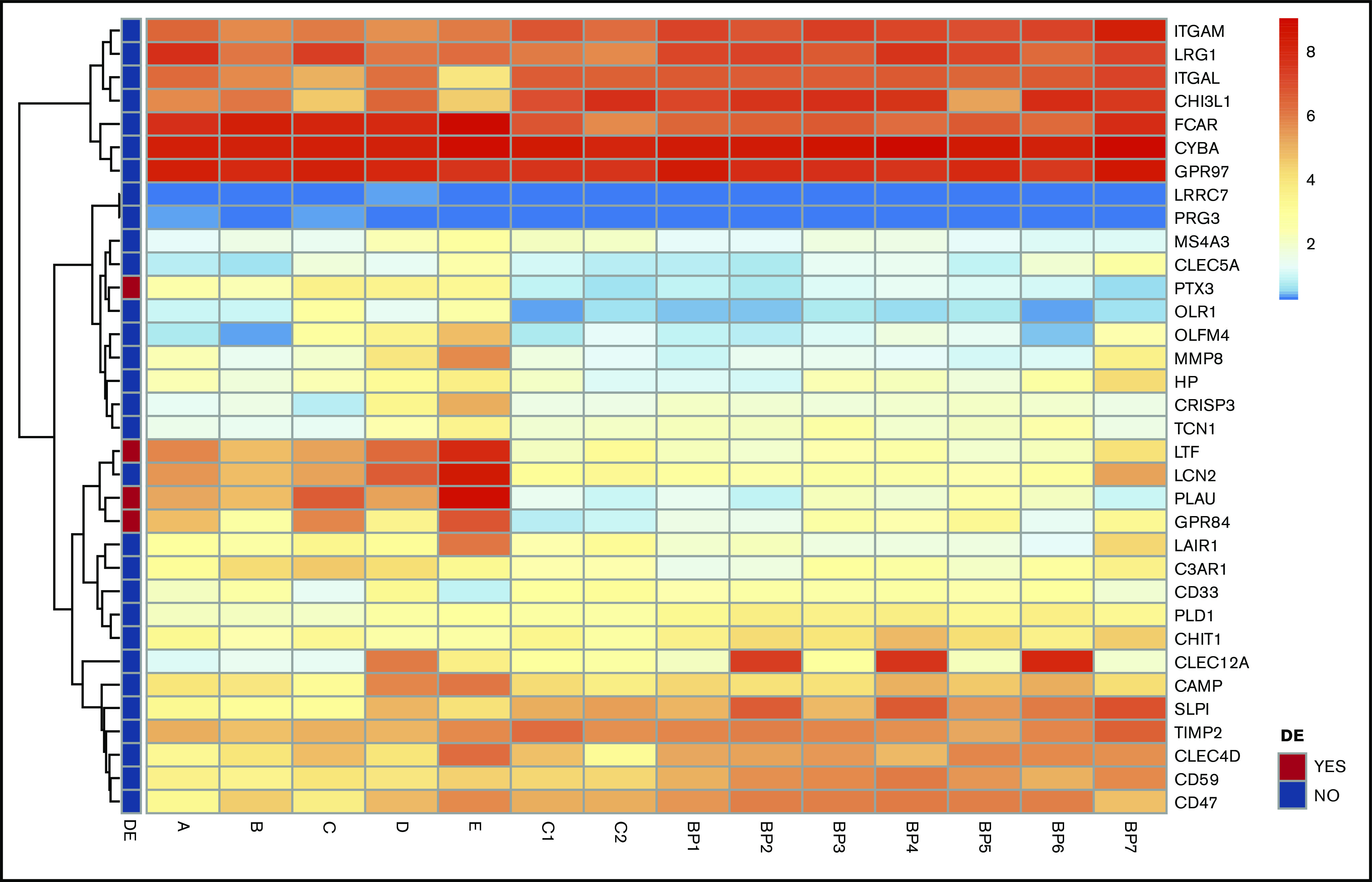
Transcript analysis of mRNA for SG proteins revealed no distinct differences in neutrophils of patients with GPS. Heatmaps of mRNA gene expression in granulocytes of GPS patients (A-E), controls (C1, C2) and BluePrint neutrophil reference transcriptomes (BP1-BP7). Of the 34 SG transcripts expressed, 4 were differentially expressed in GPS neutrophils (red boxes in the left panel indicate differentially expressed genes). See also the study by Sims et al.26 SG proteins according to Rorvig et al.25 DE, differentially expressed.
Transcriptome and proteome analysis of neutrophils differentiating from HSCs
We postulated that the discrepancy between reduced SG protein abundance with normal RNA transcript levels for these same proteins may be explained by the premature release of SGs from neutrophils lacking functional NBEAL2 protein, possibly prior to or during egress from the bone marrow. Since direct studies on separated myeloid cell fractions from bone marrow aspirates of patients with GPS were ethically not permissible because of the risk of bleeding, we developed an in vitro culture system for neutrophil development from peripheral blood–derived CD34+ HSCs from patients with GPS and healthy controls. To study the formation of SGs and their content, we initiated our cultures with CD34+ HSCs isolated from G-CSF MPB of healthy stem cell donors. In the next paragraph, we describe the results of the CD34+ HSC culture isolated from patients with GPS.
The CD34+ HSCs derived from healthy stem cell donors were expanded with a cocktail of different growth factors (interleukin-3, granulocyte-macrophage colony-stimulating factor, G-CSF, Flt-3, and human stem cell factor) for the first 3 days, followed by culturing with G-CSF until days 16 to 18. By the end of the culture, the majority of the cell population included cells with a fully differentiated neutrophil morphology (band-shaped and segmented nucleus) (supplemental Figure 4A). The entire cell population was positive for the myeloid marker CD15, of which 60% to 70% was observed to be positive for the neutrophil maturation markers CD11b and CD16 (Figure 4A). Functional tests showed the neutrophils derived from HSCs to produce normal levels of ROS production and contain normal protease activity when compared with control neutrophils freshly purified from peripheral blood (supplemental Figure 4B-C). HSC-derived neutrophils were also effective in the killing of bacterial and fungal micro-organisms (supplemental Figure 4D-F). Imaging by IEM showed normal distribution and abundance of MPO-positive azurophilic and lactoferrin-positive SG in the HSC-derived neutrophils (supplemental Figure 4G-H). In contrast to their distribution and abundance, the shape of the SGs was different, being round structures instead of the typical dumbbell-shaped vesicles in circulating neutrophils purified from blood. This was accompanied by reduced degranulation of SG, when assessed by CD66b upregulation (supplemental Figure 4I), while the degranulation of AG was normal compared with mature neutrophils derived from peripheral blood (supplemental Figure 4J).
Figure 4.
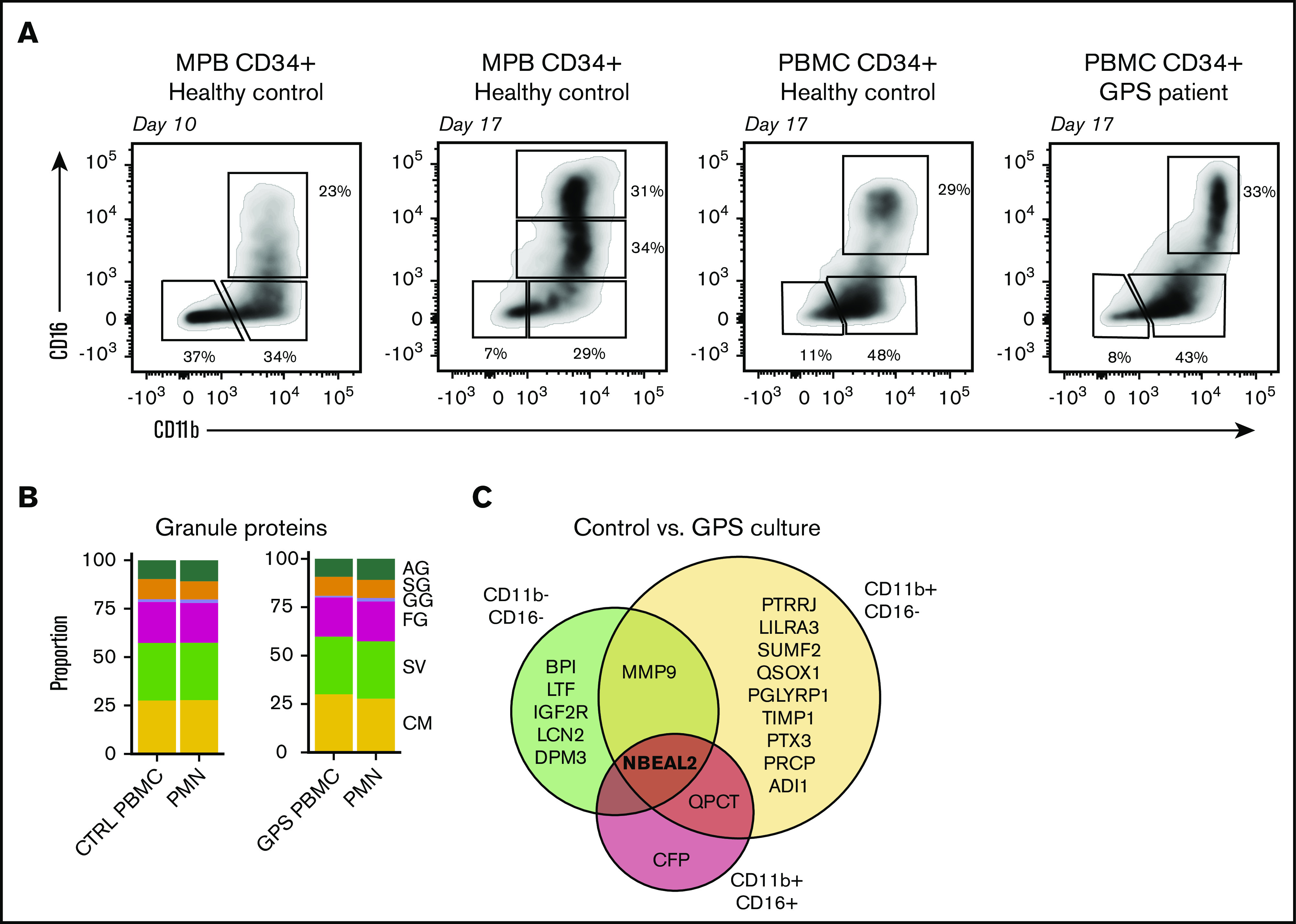
Proteomic comparison of control and GPS CD34+HSC-derived neutrophil culture. (A) Representative plots of the 3 isolated cell populations obtained by fluorescence-activated cell sorting based on cell surface markers CD11b and CD16 from control or GPS CD34+ HSC-derived cells at day 10 or 17 of the cell culture. (B) Stacked bar plots showing proportions of measured granule protein content between control (CTRL) PBMC CD34+ HSCs cultured CD16POS vs blood-derived PMNs (left panel) and GPS PBMC CD34+ HSCs cultured CD16POS vs blood-derived PMNs (right panel). (C) Comparisons among proteomes of the 3 different development stages (CD11bNEGCD16NEG, CD11bPOSCD16NEG, and CD11bPOSCD16POS) at the end of differentiation from the CD34+ HSC cultures derived from healthy controls or patients with GPS.
To further investigate the granule content in HSC-derived neutrophils, we performed proteomics analyses based on expression of the cell surface markers CD15, CD11b, and CD16, isolating 3 different cell fractions at days 10 and 17 of differentiation by fluorescence-activated cell sorting (Figure 4A). Upon comparing the proteomics profiles of the 3 fractions of myeloid cultured cells collected at day 17 (ie, end of differentiation) with that of primary differentiating neutrophils from bone marrow,21 we observed that the CD16− cells still resembled the (pro)myelocyte stage and CD16+ cells were most similar to segmented neutrophils (supplemental Figure 5). This analysis indicates that we can investigate the different neutrophil developmental stages from 1 time point of the cell culture. Furthermore, throughout the culturing protocol (CD11b+ CD16+), HSC-derived neutrophils resembled blood-derived neutrophils on both the transcriptomic and proteomic levels (supplemental Figures 6A-B and 7A-B).
We subsequently explored whether these MPB HSC-derived neutrophils had a normal level of the NBEAL2 protein. The analysis of the proteomics showed increasing levels of NBEAL2 with maturation, where maturation is based on the expression of cell-surface markers CD11b and CD16. At the most mature stage (CD16dim/CD16+ at day 17), the levels resembled those measured in blood circulating neutrophils (supplemental Figure 6C). The distribution of the granule proteins quantified in the most mature (CD11b+ CD16+) HSC-derived neutrophils showed no significant differences with neutrophils obtained from blood (supplemental Figure 6D). Pearson correlations between the levels of the granule proteins and those of NBEAL2 showed the highest correlation coefficients for SG/GG localized granule proteins (supplemental Figure 6E-F). The SG transcript levels seemed unaltered during the cell culture, whereas the NBEAL2 transcript levels increased during maturation (supplemental Figure 7C-D). These data suggest that transcription of SG proteins is independent of NBEAL2 and indicate normal SG content of HSC-derived neutrophils during their formation and differentiation in these cultures.
Transcriptome and proteome analysis of GPS neutrophils from patient-derived HSCs
Using the above-mentioned method, we initiated neutrophil cultures from CD34+ HSCs obtained from the peripheral blood of healthy controls. Even though the yield of mature neutrophils from these cultures was ∼15-fold less compared with G-CSF mobilized HSCs (supplemental Figure 8), the proteomic results for these neutrophils were remarkably similar at the end of differentiation at day 17 (Figure 4B). We then performed a proteomics analysis on HSC-derived neutrophils obtained after 16 to 18 days of culture (ie, end of differentiation) of GPS HSCs from peripheral blood for comparison. We found that in sharp contrast to the abnormal proteome signature of neutrophils obtained from the peripheral blood of GPS patients, the signatures obtained with neutrophils derived from GPS CD34+ HSCs were nearly identical to those from healthy controls, with only 18 statistically significant differences, with NBEAL2 showing the most outspoken difference in all 3 fractions (Figure 4B-C). Only 2 additional proteins were different in terms of expression level at the most mature neutrophil stage (ie, QPCT and CFP) and are considered to be of little relevance here. When taken together, these data indicate that the striking reduction in the levels of proteins localizing to SG in blood-derived circulating GPS neutrophils was not observed when studying GPS HSC-derived neutrophils. This indicates the normal abundance of SG content during neutrophil development from GPS HSC cells.
Having sorted the myeloid cells into different cell fractions at the end of the culture at day 17, again, we did not observe any major difference between the RNA-sequencing data from these HSC-derived neutrophil fractions from cultures of patients with GPS compared with cultures from control CD34+ HSCs (supplemental Figure 9). This added further evidence that the observed deficiencies of GPS neutrophil SG content were unlikely to be due to a developmental block as already indicated by the normal transcriptome analysis of SG protein in GPS neutrophils from peripheral blood.
Patient plasma and increased SG-derived protein concentrations
Our proteomics analyses indicated that the observed partial SG protein deficiency found in neutrophils obtained from the blood of patients with GPS is not due to a developmental block. The presence of the already high expression of the SG membrane marker CD66b on resting GPS neutrophils could further support the assumption of premature release from the SGs prior to or during egress from the bone marrow. The results of a proteomics analysis of the plasmas of 11 patients with GPS and 13 age-matched controls identified 51 discriminatory proteins, with 11 and 40 proteins having higher and lower concentrations, respectively.26 As indicated in Table 3, 4 of these plasma proteins (LCN2, CAMP, LYZ, and CRISP3) in patients with GPS are proteins known to localize to SGs and have a noticeable lower abundance by proteomics in GPS neutrophils (see Table 2). These data collectively suggest that the NBEAL2 protein may indeed be involved in the retention of SG, leading to early release of these granules from GPS neutrophils.
Table 3.
Differently abundant proteins between plasma from patients with GPS and healthy controls
| Description | Symbol | Direction | SG/GG |
|---|---|---|---|
| α-Actinin-4 | ACTN4 | Up | 0 |
| CD5 antigen-like | CD5L | Up | 0 |
| Carbonic anhydrase 1 | CA1 | Down | 0 |
| Apolipoprotein C-I | APOC1 | Down | 0 |
| Apolipoprotein C-III | APOC3 | Down | 0 |
| C-reactive protein | CRP | Up | 0 |
| Apolipoprotein D | APOD | Down | 0 |
| Coagulation factor XIII B chain | F13B | Down | 0 |
| Tetranectin | CLEC3B | Down | 0 |
| Gelsolin | GSN | Down | 0 |
| Vitamin K–dependent protein S | PROS1 | Down | 0 |
| Complement C8 β chain | C8B | Down | 0 |
| Complement C8 γ chain | C8G | Down | 0 |
| 60-kDa HSP, mitochondrial | HSPD1 | Up | 0 |
| Histidine-tRNA ligase, cytoplasmic | HARS | Down | 0 |
| Xaa-Pro dipeptidase | PEPD | Down | 0 |
| Bone marrow proteoglycan | PRG2 | Up | 1 |
| Lipopolysaccharide-binding protein | LBP | Up | 0 |
| Glutathione peroxidase 3 | GPX3 | Down | 0 |
| Serum paraoxonase/arylesterase 1 | PON1 | Down | 0 |
| Peroxiredoxin-2 | PRDX2 | Down | 0 |
| Utrophin | UTRN | Down | 0 |
| Cathelicidin antimicrobial peptide | CAMP | Up | 1 |
| Cysteine-rich secretory protein 3 | CRISP3 | Up | 1 |
| Phospholipid transfer protein | PLTP | Down | 0 |
| Laminin subunit β-2 | LAMB2 | Down | 0 |
| Ras-related protein Rab-14 | RAB14 | Down | 0 |
| Lysozyme C | LYZ | Up | 1 |
| Hemoglobin subunit β | HBB | Down | 0 |
| Hemoglobin subunit α | HBA1/HBA2 | Down | 0 |
| Peptidyl-prolyl cis-trans isomerase FKBP5 | FKBP5 | Down | 0 |
| Apolipoprotein F | APOF | Down | 0 |
| Dihydropyrimidinase-related protein 2 | DPYSL2 | Down | 0 |
| Peptidyl-prolyl cis-trans isomerase CWC27 homolog | CWC27 | Down | 0 |
| Proprotein convertase subtilisin/kexin type 9 | PCSK9 | Down | 0 |
| Mediator of RNA polymerase II transcription subunit 30 | MED30 | Down | 0 |
| β-Ala-His dipeptidase | CNDP1 | Down | 0 |
| RuvB-like 2 | RUVBL2 | Down | 0 |
| Adseverin | SCIN | Down | 0 |
| Carbonic anhydrase 2 | CA2 | Down | 0 |
| Hemoglobin subunit δ | HBD | Down | 0 |
| Protein disulfide-isomerase A3 | PDI | Down | 0 |
| 40S ribosomal protein S19 | RPS19 | Down | 0 |
| Cadherin-13 | CDH13 | Down | 0 |
| Neutrophil gelatinase-associated lipocalin | NGAL/LCN2 | Up | 1 |
| Exportin-1 | XPO | Up | 0 |
| Platelet basic protein | PBP | Down | 0 |
| Thrombospondin-1 | THSB1 | Down | 0 |
| Flavin reductase (NADPH) | BLVRB | Down | 0 |
| T-complex protein 1 subunit γ | CCT3 | Down | 0 |
| Catalase | CAT | Down | 0 |
Results of the plasma proteomic analysis using SWATH-MS. Shown are the results of the comparison between plasma of patients with GPS (n = 11) and age-matched controls. A total of 51 discriminatory proteins were identified, with 11 proteins being more abundant (up direction) and 40 proteins being less abundant in plasma of patients with GPS (down direction) compared with plasma of healthy controls, as reported by Sims et al.26 Presence of SG proteins (5 proteins in total, indicated in bold and in the SG/GG column) in the plasma of GPS patients was found among other significantly increased plasma proteins.
HSP, heat shock protein.
Functional activity of neutrophils from patients with GPS
The apparent reduced number of SGs in human neutrophils lacking functional NBEAL2 protein made us wonder what the consequences were for the antimicrobial response. We found that the ROS production was unaltered (supplemental Figure 10) between patients and controls, as was the level of released MPO (Figure 1) and protease activity of elastase and cathepsin G (supplemental Figure 10). Similarly, neutrophil-mediated killing of gram-positive (S aureus) and gram-negative bacteria (E coli) as well as fungal pathogens (C albicans and A fumigatus) was largely unaffected (Figure 5). As part of the antimicrobial potential of neutrophils,16,29 we also investigated NET formation in GPS neutrophils by confocal imaging and DNA release. We started our NETosis experiments by stimulating the neutrophils with PMA. PMA is a potent activator and a robust NET inducer, where calcium, NADPH oxidase–induced ROS, MPO, and neutrophil elastase play an important role, causing chromatin decondensation resulting in chromatin swelling, rupture of the nuclear envelope, and ultimately the rupture of the cell membrane.30-33 In the presence of normal ROS production and MPO and elastase reactivity in GPS neutrophils, NET formation was almost absent upon PMA stimulation of patient neutrophils (Figure 6 A-C). Both control and GPS neutrophils showed chromatin swelling and cell rounding, but the cell membrane of the GPS neutrophils did not rupture in the final stage of NET release (supplemental Videos 1-2; supplemental Figure 11).
Figure 5.

Normal microbial killing by GPS neutrophils. Killing of S aureus (A), E coli (B), C albicans (C), and A fumigatus (D) was assessed with neutrophils from healthy controls and patients with GPS. The microbial viability was quantified as colony-forming units and expressed as a percentage relative to the colony-forming units at the start of the assay. Results are means ± SEM, n = 3-10. An unpaired, 2-tailed t test was used. **P < .01.
Figure 6.
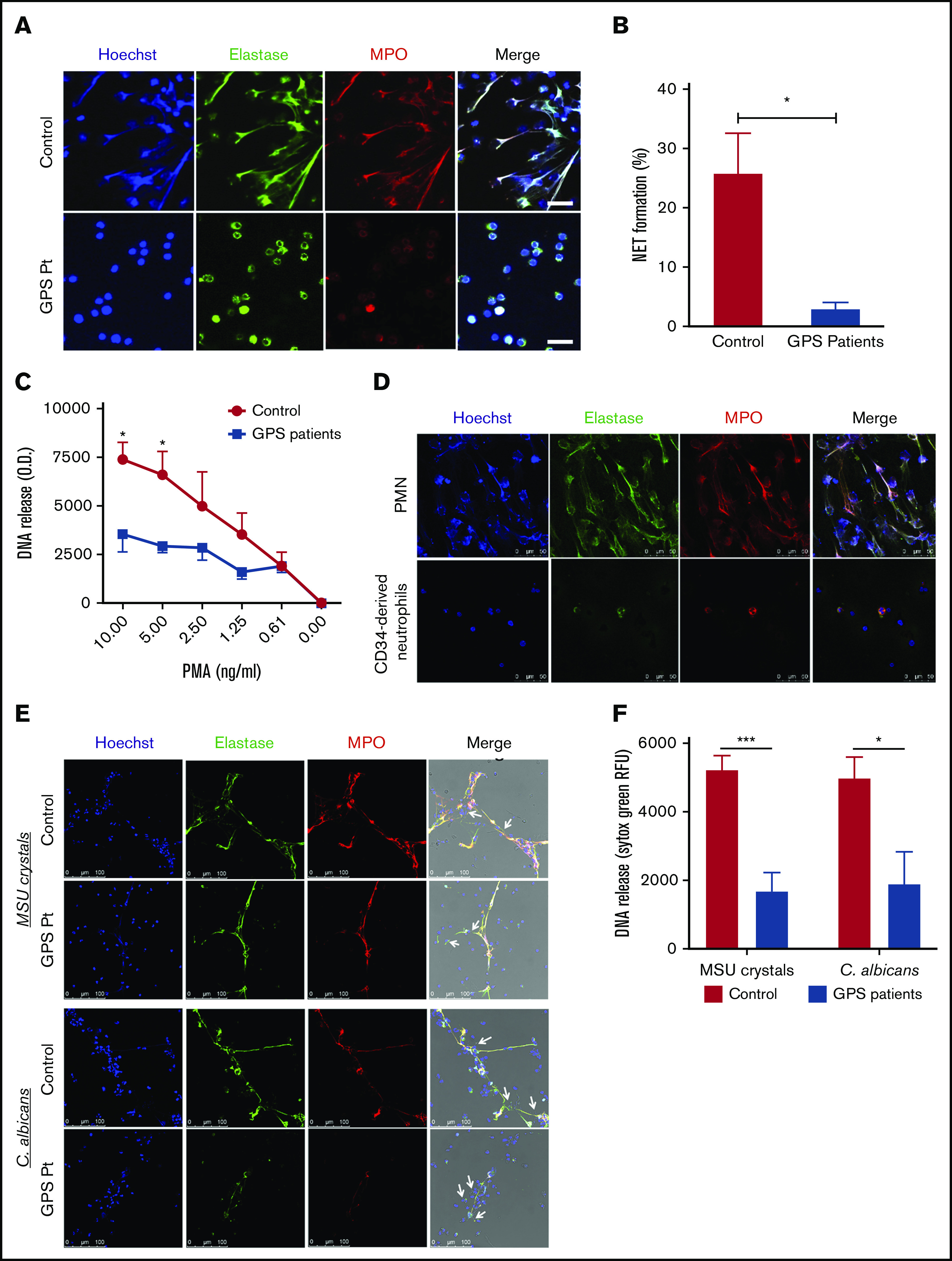
Impaired NETosis by GPS neutrophils and MPB CD34+HSC-derived neutrophils. (A) Neutrophils derived from blood from controls (upper panel) and patients with GPS (lower panel) were stimulated with PMA (100 ng/mL) for 3 hours, and NET formation was visualized by staining for DNA (blue), elastase (green), and MPO (red). Scale bars, 20 µm. (B) NET formation was determined by counting the number of NETs per field of 100 cells in control and GPS neutrophils, stimulated with PMA (100 ng/mL) for 3 hours, in ≥5 fields. (C) DNA release was measured in the supernatant from control and GPS neutrophils stimulated with increasing concentrations of PMA (n = 3-5). (D) Control neutrophils from blood (PMN, upper panel) or MPB CD34+ HSC-derived neutrophils (lower panel) were stimulated with PMA (100 ng/mL) for 3 hours, and NET formation was visualized by staining for DNA (Hoechst, blue), elastase (green), and MPO (red). Shown are representative images of n = 3. Scale bars, 50 µm. (E) Neutrophils derived from blood from controls and patients with GPS were stimulated with MSU crystals (200 μg/mL, upper panel) or opsonized C albicans (lower panel) for 4 hours. NET formation was visualized by staining for DNA (blue), elastase (green), and MPO (red). White arrows in the merge images indicate the MSU crystals (upper panel) or the C albicans hyphae (lower panel). Scale bars, 100 µm. Shown are representative images of n = 3-9. Pt, patient. (F) DNA release was measured in the supernatant from control (n = 7-9) and GPS (n = 3-4) neutrophils stimulated with MSU crystals (200 μg/mL) or opsonized C albicans for 4 hours. RFU, relative fluorescence units. Results are means ± SEM. An unpaired, 2-tailed t test was used. *P < .05, ***P < .001 compared with control neutrophils.
Because of the similarity with GPS neutrophils in a defective SG release in our cultured cells, we also investigated NET formation by CD34+ HSC-derived neutrophils. These cells also showed defective NETosis upon PMA stimulation (Figure 6D), while ROS production and activity of elastase (both important for PMA-induced NETosis30,32) were both intact in CD34+ HSC-derived neutrophils (supplemental Figure 4B-C). Since NBEAL2 is expressed in these control CD34+ HSC-derived neutrophils (supplemental Figure 6), it is most likely that NBEAL2 itself is not the culprit frustrating the process of PMA-induced NETosis, suggesting the unexpected role for SG components in the process of NET formation instead of NBEAL2. These data also imply that transfection of NBEAL2 into CD34+ HSCs will not yield additional insight in the process of either SG formation or NETosis as such.
NET formation by GPS neutrophils is broadly impaired
The phorbol ester PMA is a very potent NET inducer, but not a physiological stimulus, bypassing the plasma membrane and directly activating protein kinase C (PKC). It can therefore differ from NET release induced by microbial or endogenous stimuli. For instance, PMA-induced NETosis via direct PKC activation relies on NADPH oxidase–derived ROS, where C albicans–induced NETosis seems to be largely independent of ROS.30 Also, MSU crystals, present in joints of gout patients, can induce NET release independent of ROS and acts via the SYK-phosphatidylinositol 3-kinase-mTORC2 pathway instead of PKC.31,34 In contrast to PMA-induced NET release, GPS neutrophils were able to form a small number of NETs in the presence of MSU crystals or opsonized C albicans (Figure 6E), indicating that NETosis is not completely absent in GPS neutrophils but strongly reduced. Since the neutrophils were localized tightly around the MSU crystals and C albicans hyphae, we could not quantify NET formation properly. For this, we performed a DNA release assay to quantify our NET data. Although a small amount of NET formation was detected in our confocal images, MSU- and C albicans–induced NETosis was impaired in GPS neutrophils compared with control neutrophils (Figure 6F).
Thus, the GPS neutrophils did not undergo NETosis in response to PMA and were also significantly impaired in NET production in the presence of MSU crystals or hyphenating C albicans. These results indicate that even though the 3 NET-inducing stimuli tested engage in different pathways leading up to NET release, SG proteins may play an important role in neutrophil NETosis. Since the proteome data showed high overlap in GPS and control CD34+ HSC-derived neutrophils (Figure 4), we tried to narrow the SG protein candidates involved in NET formation. We compared differentially abundant proteins between blood-derived neutrophils of control vs patients with GPS and control mature (CD11b+CD16+) CD34+ HSC-derived neutrophils vs blood-derived neutrophils of controls. Between these 2 data sets, 22 proteins were overlapping (Figure 7A). Upon inspection of the dynamics of these proteins, we found that 13 were similarly downregulated (Figure 7B), where most of these form an interaction network, as is apparent from the STRING-DB analysis (Figure 7C). Their expression levels were similar between HSC-derived and blood-derived neutrophil data sets (Figure 7D), suggesting that one or more of these proteins, a combination of these, or as-yet-unidentified SG-dependent factors or proteins may play a role in neutrophil NETosis, as illustrated by the GPS studies presented here.
Figure 7.
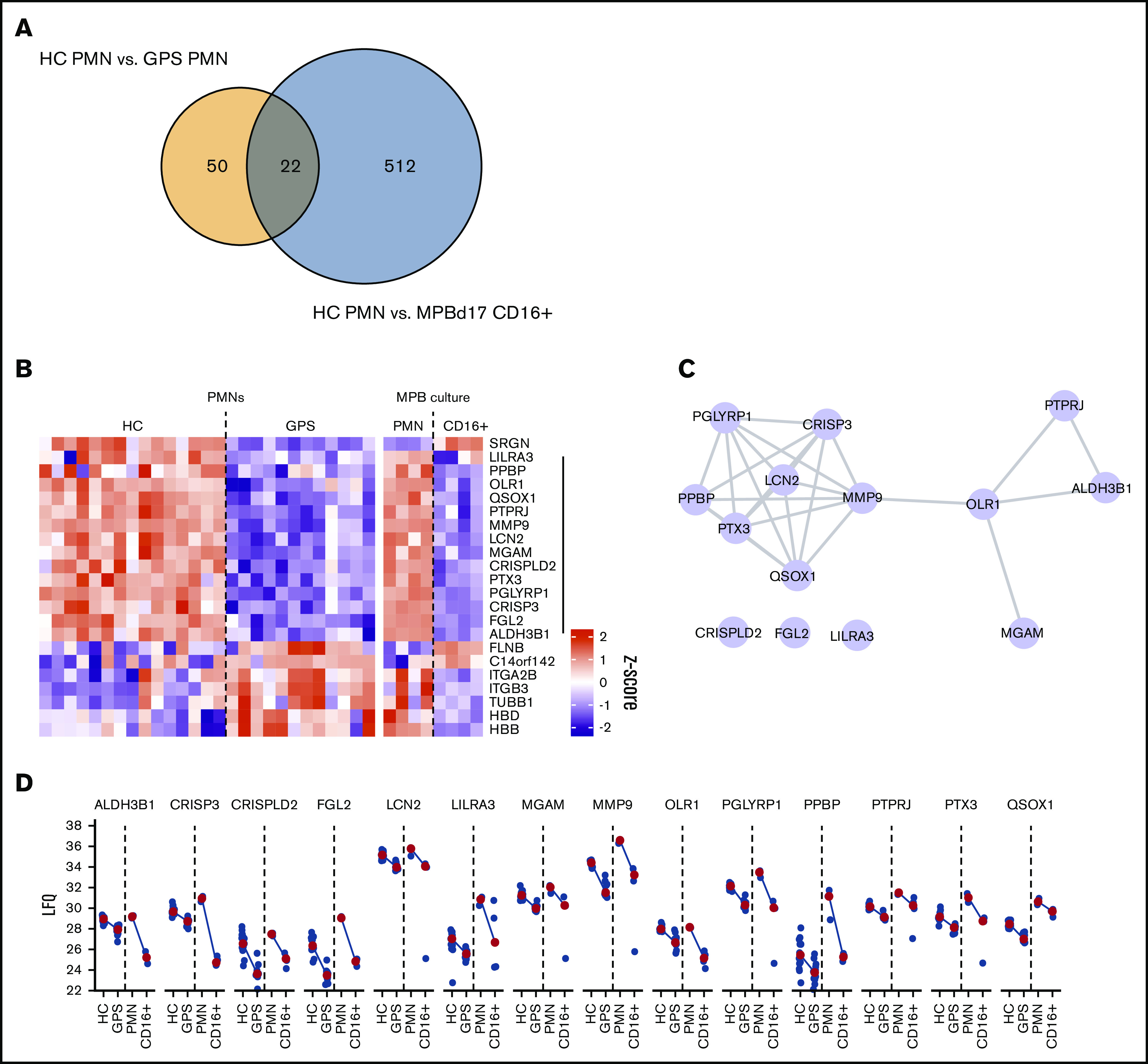
Proteomic basis of failed NETosis capacity. (A) Overlap in protein differences between circulating GPS and control blood neutrophils (HC PMN) vs MPB CD34+ HSC-derived neutrophils and circulating blood-derived neutrophils (HC PMN). (B) Heatmap of 22 overlapping proteins between the same cell groups as shown in panel A. (C) STRING-DB interaction network of 13 proteins with shared reduced abundancy. (D) Dot and line plots of 13 proteins with shared reduced abundancy. Blue dots present LFQ values, and red dots represent median LFQ values.
Discussion
Our study on GPS indicates that NBEAL2 deficiency is a defect that not only affects platelets but also manifests itself as a granular defect in human neutrophils. In NBEAL2 deficiency, we identify a defect that seems to be limited to a strongly reduced presence of specific lactoferrin-positive and gelatinase-positive granules. Both types of granules are formed during the final stages of myeloid progenitor differentiation into mature polymorphonuclear cells (PMNs).21 These granules are subject to the same transcriptional programming. The amount of protein generated and stored in these granules during neutrophil differentiation is a mere reflection rather than a biologically relevant difference in compartmentalization between SGs and GGs.21,35 SGs and GGs are highly distinct from AGs and secretory vesicles. AGs contain the bulk of proteolytic enzymes and are formed prior to the promyelocyte stage at the myeloblast-promyelocyte transitional stage. Secretory vesicles are believed to contain both neutrophil constituents such as the phosphatidyl inositol (PI)-linked IgG receptor FcγRIIIB (CD16), as well as endocytosed material from plasma (eg, albumin).36,37
To explain the difference between a partial deficiency of SGs, we investigated in detail if the reduced number of SGs was due to a failure in granule formation or the result of premature degranulation, knowing that early granule release has previously been suggested to occur in NBEAL2-deficient platelets.4 Fresh bone marrow samples from patients with GPS were not readily available, and such samples could already be altered due to manipulations of the isolation procedure itself or subsequent purification of the cell fractions of interest. Using an optimized CD34+ HSC culture system to grow neutrophils, we could now exclude that the formation of SGs in GPS is significantly different from control HSC-derived neutrophils. Normal mRNA and protein expression levels in neutrophils cultured from GPS HSCs argue against a failure of granule synthesis. The clear difference in granular protein content in circulating GPS neutrophils purified from blood suggests a process of early release due to NBEAL2 deficiency rather than a developmental failure. This was also shown for platelet development, where the in vivo megakaryocytes and pro-platelets from bone marrow of Nbeal2 knockout mice contained a normal number of α-granules (the same as wild-type mice), whereas these granules of platelets in the circulation were reduced.4
The hypothesis of early degranulation by GPS neutrophils, instead of a failure in the development and formation of SGs, is further supported by the fact that some of the significantly increased plasma proteins are of SG origin, including LCN2, CAMP, LYZ, and CRISP3. These data on the presence of SG proteins leaves us with an intriguing consideration about the timing of their release from NBEAL2-deficient neutrophils. Although there is no solid evidence on when during myeloid development release from the SG compartment may occur, the identical proteomic profiles of neutrophils cultured from peripheral blood CD34+ HSCs from either healthy or NBEAL2-deficient individuals suggests that release most likely occurs during or after egress from the bone marrow. The shear force to which neutrophils are subverted to once entering the bloodstream may constitute the trigger to premature SG release from GPS neutrophils. This trigger may take place during or directly after egress from the bone marrow into the bloodstream or perhaps in the circulation in the vascular bed of lung, liver, or spleen, for example. We have extensively studied neutrophil migration in the past and found SG displacement (and not AG) in neutrophils from healthy donors during movement across filters or endothelial cells.38 Whether this could explain a more definite release of SG content from neutrophils from patients with GPS because of the lack of NBEAL2 compared with control neutrophils remains unresolved. Exposure to gradients of neutrophil-activating substances in plasma and absent in the bone marrow niche may form an alternative explanation, although we did not observe any activation of patient neutrophils by classical surface markers such as a low CD62L or CD16 or the upregulation of CD64 when compared with neutrophils from healthy controls shipped simultaneously (supplemental Figure 12). Interestingly, the same phenomenon was found in resting platelets of patients with GPS. P-selectin, normally stored in the α-granules, is already strongly expressed on the cell surface compared with controls, while these circulating GPS platelets were not preactivated, as indicated by the absence of fibrinogen binding unless activated in vitro (supplemental Figure 2).
Apart from the bleeding tendency and autoimmune phenomena, these patients with GPS are not known to suffer from invasive or severe infections.26 Upper respiratory infections were most commonly reported in a fraction of the patients. In humans, the lack of severe and recurrent infectious disease in GPS contrasts with a rare but well-described neutrophil disorder known as specific granule deficiency (SGD). In SGD, mutations in the gene CEBPE, which encodes the essential transcription factor CCAAT/enhancer binding protein ε, have been identified, resulting in a complete lack of secondary and tertiary granules.39,40 Apart from a defect in SG formation, neutrophils from SGD patients also have an abnormal bilobar nuclear morphology,40-42 which clearly distinguishes SGD neutrophils from the normal nuclear morphology of GPS neutrophils. The SG deficiency in circulating neutrophils from patients with GPS is not as absolute as in SGD, which may contribute to the fact that patients with GPS do not suffer from recurrent or severe bacterial infections. In addition, the lack of clinical susceptibility to infection in the presence of a partial SG defect in human GPS neutrophils also differs from the observation in Nbeal2-deficient mice, which have a different and more severe phenotype in experimental infection models.10
Although a clinical neutrophil defect in patients with GPS has not been reported thus far, NBEAL2-deficient neutrophils showed impaired NETosis, a process thought to be key for innate immunity as entrapment of pathogens to enhance the concentration of antimicrobial proteases.30,32,43 Interestingly, carriers of the NBEAL2 mutation showed normal NBEAL2 and SG protein levels, where the SG marker CD66b was normally expressed on the cell surface of resting neutrophils. These neutrophils were able to form NETs upon stimulation with either PMA or physiological relevant stimuli (data not shown). Hence, heterozygous NBEAL2 mutations do not induce the GPS phenotype.
Several mechanisms contribute to NETosis, where it has been shown that several stimuli engage in different pathways using altered kinetics and different requirements for cellular components.30,31 PMA-induced NETosis relies on the production of ROS and translocation of elastase and MPO to the nucleus, causing chromatin decondensation,32 as indicated by a lack of PMA-induced NETosis in NADPH oxidase–defective CGD neutrophil or cells from MPO-deficient patients.30,44 A recent study in a patient with Papillon-Lefèvre syndrome, caused by mutations in the CTSC gene, found that the proteases elastase and cathepsin G normally found in AGs were absent.45 NET formation was also absent in this patient, which reinforces the combined requirement of ROS and several azurophilic granule components for PMA-induced NETosis to occur under the in vitro conditions that are used to test this process. Notably, in patients wtih GPS, neutrophil MPO levels were found to be normal. Moreover, ROS production as well as proteolytic activity by elastase and cathepsin G were also found to be completely unaffected by NBEAL2 deficiency. The fact that NBEAL2-deficient blood neutrophils show a partial SG deficiency suggests a role for SG-derived components in the in vitro process of NETosis. The finding of a very limited release of SG from healthy NBEAL2-expressing CD34+ HSC-derived neutrophils supports the hypothesis that one or more of the SG proteins have a previously unidentified impact in this cellular process. Even though these cells produce ROS and can release their azurophilic content required for PMA-induced NET formation,30,32 the reason for the lack of NETosis of CD34+ HSC-derived neutrophils remains to be explained. It does suggest an important role for the SG compartment and not the absence of NBEAL2 per se in case of GPS neutrophils. In fact, neutrophils from patients with SGD also lacked NET formation upon PMA stimulation (E.G.G.S., A.T.J.T, Floris P.J. van Alphen, Mikko Seppänen, Eira Leinonen, R.v.B., A.B.M., T.W.K., manuscript in preparation), which is further proof for the role of SGs in NETosis. Based on our live-cell imaging (supplemental Videos 1-2), it seems that the SG-derived components may be important in the final stage of NETosis, since GPS neutrophils did show chromatin swelling and cell rounding, but the cell membrane did not rupture.
Whereas PMA is a very potent inducer of NETosis, it is not a physiological stimulus. When GPS neutrophils were activated by more physiological stimuli, such as opsonized C albicans, these cells did show some NET formation in our confocal slides, indicating that these cells are not completely inert and NETosis is not totally absent. Nonetheless, when we objectively quantified the NET formation via a DNA release assay, neutrophils from patients with GPS were significantly less able to form NETs as compared with neutrophils from healthy donors. This suggests a more general role for SGs in the process of NETosis. The lack of clinical recurrent or severe infections in NBEAL2-deficient patients with GPS raises the question whether in vitro NETosis reflects a physiological antimicrobial activity in innate immunity. Although a neutrophil defect has been suggested in Nbeal2-deficient mice, granule formation in murine neutrophils is different form the human counterpart and hence cannot be easily translated to human disease.46-48
In summary, we show that neutrophils from NBEAL2-deficient patients with GPS have a normal distribution and mobilization of AGs. However, we observed a reduced amount of SGs and their content. As previously suggested for platelets from NBEAL2-deficient patients with GPS and studies with Nbeal2−/− mice, there is no reduced granule formation during development, where it is indicated that NBEAL2 acts as an important regulator of granule retention/release.4,5,49 Our findings in human primary neutrophils support this notion and show the requirement of NBEAL2 in SG retention instead of SG protein synthesis. In addition, our data also suggest that the reduced presence of one or more SG proteins prevents the formation of proper NET formation in neutrophils upon their activation without major clinical consequences for innate host defense.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Dirk Roos for critical reading of the manuscript. They are grateful to Ilenia Simeoni for the sequence analysis of the patients with GPS using the ThromboGenomics platform. They thank Harriet McKinney, Carly Kempster, Joana Batista, and Annelotte Mudde for their help with neutrophil function tests. Furthermore, the authors thank Floris van Alphen, Martin de Boer, Erik Mul, Simon Tol, and Mark Hoogenboezem for their technical support.
The patients with GPS were enrolled in the National Institute for Health Research (NIHR) BioResource Rare Diseases and their DNA analyzed by the associated BRIDGE genome sequencing projects. Both initiatives are supported by the NIHR (http://www.nihr.ac.uk). Research in the Ouwehand laboratory is supported by grants from the European Commission, NIHR, and the British Heart Foundation (RP-PG-0310-1002 and RG/09/12/28096). The laboratory also receives funding from NHS Blood and Transplant. K.D. is funded as a Higher Specialist Scientist Training (HSST) trainee by Health Education England. R.P.G. was supported by the Landsteiner Foundation for Blood Transfusion Research (LSBR 1706) awarded to T.W.K. C.E.M. was supported by the Sanquin Blood Supply Product and Process Development Cellular Products Fund (PPOC 2089).
Footnotes
Send data sharing requests via e-mail to the corresponding author, Cathelijn Aarts; e-mail: c.aarts@sanquin.nl.
The .raw MS files and search/identification files obtained with MaxQuant have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD017784.
Authorship
Contribution: T.W.K., W.H.O., R.v.B., and T.K.v.d.B. are the principal investigators who conceived and designed the study; C.E.M.A., R.P.G., E.G.G.S., K.D., J.L.v.H., A.T.J.T., K.W., M.A.K., and H.J. performed the experiments; R.F., M.F., B.Z., B.A.v.d.R., S.M.C.L., and M.G.J.M.v.B. provided patient material and advice; and A.B.M. supervised the MS analysis performed by A.J.H.; and C.E.M.A., R.P.G., and K.D. initiated many of the experiments, performed the analyses, and wrote the manuscript together with T.W.K.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Cathelijn Aarts, Sanquin, Department of Blood Cell Research, Plesmanlaan 125, 1066 CX Amsterdam, The Netherlands; e-mail: c.aarts@sanquin.nl.
References
- 1.Albers CA, Cvejic A, Favier R, et al. Exome sequencing identifies NBEAL2 as the causative gene for gray platelet syndrome. Nat Genet. 2011;43(8):735-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kahr WH, Hinckley J, Li L, et al. Mutations in NBEAL2, encoding a BEACH protein, cause gray platelet syndrome. Nat Genet. 2011;43(8):738-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cullinane AR, Schäffer AA, Huizing M. The BEACH is hot: a LYST of emerging roles for BEACH-domain containing proteins in human disease. Traffic. 2013;14(7):749-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guerrero JA, Bennett C, van der Weyden L, et al. Gray platelet syndrome: proinflammatory megakaryocytes and α-granule loss cause myelofibrosis and confer metastasis resistance in mice. Blood. 2014;124(24):3624-3635. [DOI] [PubMed] [Google Scholar]
- 5.Lo RW, Li L, Leung R, Pluthero FG, Kahr WHA. NBEAL2 (neurobeachin-like 2) Is required for retention of cargo proteins by α-granules during their production by megakaryocytes. Arterioscler Thromb Vasc Biol. 2018;38(10):2435-2447. [DOI] [PubMed] [Google Scholar]
- 6.Kahr WH, Lo RW, Li L, et al. Abnormal megakaryocyte development and platelet function in Nbeal2(-/-) mice. Blood. 2013;122(19):3349-3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo RW, Li L, Pluthero FG, Leung R, Eto K, Kahr WHA. The endoplasmic reticulum protein SEC22B interacts with NBEAL2 and is required for megakaryocyte α-granule biogenesis. Blood. 2020;136(6):715-725. [DOI] [PubMed] [Google Scholar]
- 8.Mayer L, Jasztal M, Pardo M, et al. Nbeal2 interacts with Dock7, Sec16a, and Vac14. Blood. 2018;131(9):1000-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Kostadima M, Martens JHA, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014;345(6204):1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sowerby JM, Thomas DC, Clare S, et al. NBEAL2 is required for neutrophil and NK cell function and pathogen defense. J Clin Invest. 2017;127(9):3521-3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drouin A, Favier R, Massé JM, et al. Newly recognized cellular abnormalities in the gray platelet syndrome. Blood. 2001;98(5):1382-1391. [DOI] [PubMed] [Google Scholar]
- 12.White JG, Brunning RD. Neutrophils in the gray platelet syndrome. Platelets. 2004;15(5):333-340. [DOI] [PubMed] [Google Scholar]
- 13.Chedani H, Dupuy E, Massé JM, Cramer EM. Neutrophil secretory defect in the gray platelet syndrome: a new case. Platelets. 2006;17(1):14-19. [DOI] [PubMed] [Google Scholar]
- 14.Gunay-Aygun M, Zivony-Elboum Y, Gumruk F, et al. Gray platelet syndrome: natural history of a large patient cohort and locus assignment to chromosome 3p. Blood. 2010;116(23):4990-5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011;29(1):1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532-1535. [DOI] [PubMed] [Google Scholar]
- 17.Bianchi M, Hakkim A, Brinkmann V, et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood. 2009;114(13):2619-2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Branzk N, Lubojemska A, Hardison SE, et al. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol. 2014;15(11):1017-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gazendam RP, van Hamme JL, Tool AT, et al. Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. Blood. 2014;124(4):590-597. [DOI] [PubMed] [Google Scholar]
- 20.Herrero-Turrión MJ, Calafat J, Janssen H, Fukuda M, Mollinedo F. Rab27a regulates exocytosis of tertiary and specific granules in human neutrophils. J Immunol. 2008;181(6):3793-3803. [DOI] [PubMed] [Google Scholar]
- 21.Hoogendijk AJ, Pourfarzad F, Aarts CEM, et al. Dynamic transcriptome-proteome correlation networks reveal human myeloid differentiation and neutrophil-specific programming. Cell Rep. 2019;29(8):2505-2519.e4. [DOI] [PubMed] [Google Scholar]
- 22.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367-1372. [DOI] [PubMed] [Google Scholar]
- 23.Tyanova S, Temu T, Sinitcyn P, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731-740. [DOI] [PubMed] [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rørvig S, Østergaard O, Heegaard NH, Borregaard N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: correlation with transcriptome profiling of neutrophil precursors. J Leukoc Biol. 2013;94(4):711-721. [DOI] [PubMed] [Google Scholar]
- 26.Sims MC, Mayer L, Collins JH, et al. ; NIHR BioResource . Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood. 2020;136(17):1956-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Boer M, van Leeuwen K, Geissler J, et al. Hermansky-Pudlak syndrome type 2: Aberrant pre-mRNA splicing and mislocalization of granule proteins in neutrophils. Hum Mutat. 2017;38(10):1402-1411. [DOI] [PubMed] [Google Scholar]
- 28.Zhao XW, Gazendam RP, Drewniak A, et al. Defects in neutrophil granule mobilization and bactericidal activity in familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) syndrome caused by STXBP2/Munc18-2 mutations. Blood. 2013;122(1):109-111. [DOI] [PubMed] [Google Scholar]
- 29.Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134-147. [DOI] [PubMed] [Google Scholar]
- 30.Kenny EF, Herzig A, Krüger R, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. 2017;6:e24437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Linden M, Westerlaken GHA, van der Vlist M, van Montfrans J, Meyaard L. Differential Signalling and Kinetics of Neutrophil Extracellular Trap Release Revealed by Quantitative Live Imaging. Sci Rep. 2017;7(1):6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677-691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neubert E, Meyer D, Rocca F, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9(1):3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20(5):511-517. [DOI] [PubMed] [Google Scholar]
- 35.Grassi L, Pourfarzad F, Ullrich S, et al. Dynamics of transcription regulation in human bone marrow myeloid differentiation to mature blood neutrophils. Cell Rep. 2018;24(10):2784-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calafat J, Kuijpers TW, Janssen H, Borregaard N, Verhoeven AJ, Roos D. Evidence for small intracellular vesicles in human blood phagocytes containing cytochrome b558 and the adhesion molecule CD11b/CD18. Blood. 1993;81(11):3122-3129. [PubMed] [Google Scholar]
- 37.de Haas M, Kerst JM, van der Schoot CE, et al. Granulocyte colony-stimulating factor administration to healthy volunteers: analysis of the immediate activating effects on circulating neutrophils. Blood. 1994;84(11):3885-3894. [PubMed] [Google Scholar]
- 38.Kuijpers TW, Hoogerwerf M, Roos D. Neutrophil migration across monolayers of resting or cytokine-activated endothelial cells. Role of intracellular calcium changes and fusion of specific granules with the plasma membrane. J Immunol. 1992;148(1):72-77. [PubMed] [Google Scholar]
- 39.Göös H, Fogarty CL, Sahu B, et al. Gain-of-function CEBPE mutation causes noncanonical autoinflammatory inflammasomopathy. J Allergy Clin Immunol. 2019;144(5):1364-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serwas NK, Huemer J, Dieckmann R, et al. CEBPE-mutant specific granule deficiency correlates with aberrant granule organization and substantial proteome alterations in neutrophils. Front Immunol. 2018;9:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khanna-Gupta A, Sun H, Zibello T, et al. Growth factor independence-1 (Gfi-1) plays a role in mediating specific granule deficiency (SGD) in a patient lacking a gene-inactivating mutation in the C/EBPepsilon gene. Blood. 2007;109(10):4181-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wada T, Akagi T, Muraoka M, et al. A novel in-frame deletion in the leucine zipper domain of C/EBPε leads to neutrophil-specific granule deficiency. J Immunol. 2015;195(1):80-86. [DOI] [PubMed] [Google Scholar]
- 43.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784-2794. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sørensen OE, Clemmensen SN, Dahl SL, et al. Papillon-Lefèvre syndrome patient reveals species-dependent requirements for neutrophil defenses. J Clin Invest. 2014;124(10):4539-4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassatella MA, Östberg NK, Tamassia N, Soehnlein O. Biological roles of neutrophil-derived granule proteins and cytokines. Trends Immunol. 2019;40(7):648-664. [DOI] [PubMed] [Google Scholar]
- 47.Eruslanov EB, Singhal S, Albelda SM. Mouse vs human neutrophils in cancer: a major knowledge gap. Trends Cancer. 2017;3(2):149-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hidalgo A, Chilvers ER, Summers C, Koenderman L. The neutrophil life cycle. Trends Immunol. 2019;40(7):584-597. [DOI] [PubMed] [Google Scholar]
- 49.Maynard DM, Heijnen HF, Gahl WA, Gunay-Aygun M. The α-granule proteome: novel proteins in normal and ghost granules in gray platelet syndrome. J Thromb Haemost. 2010;8(8):1786-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rensing-Ehl A, Pannicke U, Zimmermann S-Y, et al. Gray platelet syndrome can mimic autoimmune lymphoproliferative syndrome. Blood. 2015;126(16):1967-1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.