

Abstract
The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of Coronavirus Disease 2019 (COVID-19), was declared a pandemic infection in March 2020. As of December 2020, two COVID-19 vaccines have been authorized for emergency use by the U.S. Food and Drug Administration, but there are no effective drugs to treat COVID-19 and pandemic mitigation efforts like physical distancing have had acute social and economic consequences. In this perspective, we discuss how the proteomic research community can leverage technologies and expertise to address the pandemic by investigating four key areas of study in SARS-CoV-2 biology. Specifically, we discuss how (1) mass spectrometry-based structural techniques can overcome limitations and complement traditional structural approaches to inform the dynamic structure of SARS-CoV-2 proteins, complexes, and virions; (2) viral-host protein-protein interaction mapping can identify the cellular machinery required for SARS-CoV-2 replication; (3) global protein abundance and post-translational modification profiling can characterize signaling pathways that are rewired during infection; and (4) proteomic technologies can aid in biomarker identification, diagnostics, and drug development in order to monitor COVID-19 pathology and investigate treatment strategies. Systems-level high-throughput capabilities of proteomic technologies can yield important insights into SARS-CoV-2 biology that are urgently needed during the pandemic, and more broadly, can inform coronavirus virology and host biology.
Keywords: SARS-CoV-2, COVID-19, proteomics, mass spectrometry, virus-host interactions, structural proteomics, protein-protein interactions, host response, drug discovery, biomarker discovery
Graphical Abstract
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel coronavirus that causes Coronavirus Disease 2019 (COVID-19), and its outbreak in 2019 and subsequent pandemic devastated global economies and human health. The coronaviridae family of viruses, named for their crown-like appearance under an electron microscope, includes alpha, beta, gamma, and delta subgroups that infect a wide variety of mammals and birds, but mutations facilite cross-species infections and human-to-human transmission of the viruses1. Seven coronaviruses are known to infect humans, two alpha coronaviruses (HCoV-229E and HCoV-NL63), and five beta coronaviruses (HCoV-OC43, HCoV-HKU1, MERS-CoV, SARS-CoV-1, and SARS-CoV-2)2. HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 regularly infect humans and cause common cold symptoms that are typically cleared but can progress to bronchiolitis or pneumonia3,4. In contrast, MERS-CoV, SARS-CoV-1, and SARS-CoV-2 are more likely to cause severe respiratory disease5–7. While the SARS-CoV-1 (2002/2003) and the MERS-CoV (2012) outbreaks had respective mortality rates of ~10% and 36%8,9, the outbreaks were contained within specific geographic regions with only 8,098 and 2,494 verified infections10,11. SARS-CoV-2 has a lower fatality rate but is more wide-spread than SARS-CoV-1 or MERS-CoV9, with 75,704,857 confirmed cases worldwide as of December 21, 202012. There are currently over 50 COVID-19 vaccines in clinical trials13, and the US Food and Drug Administration (FDA) has authorized the Pfizer-BioNTech and Moderna COVID-19 mRNA vaccines for emergency use14,15. This marks the fastest vaccine development in history, and widespread distribution of an effective vaccine would allow a safer end to stringent, long-term physical distancing that has had profound social and economic consequences16. However, for people not protected by vaccination, there is an urgent need for effective COVID-19 treatment options. At this time, Remdesivir is the only drug approved by the FDA, however, the World Health Organization recommends against its use due to a lack of evidence for its efficacy17,18.
During this unprecedented global crisis, the scientific community mobilized research efforts probing the mechanisms of SARS-CoV-2 infection and replication (Table 1)19–21. SARS-CoV-2 has a ~30kb positive-sense RNA genome with as many as 14 open reading frames (Orfs) encoding 16 non-structural proteins (Nsp 1–16), 4 structural proteins (Spike (S), envelope (E), membrane (M), and nucleocapsid (N)), and 9 accessory proteins (Orf3a, Orf3b, Orf6, Orf7a, Orf7b, Orf8, Orf9b, Orf9c, and Orf10)7,22. In the SARS-CoV-2 life cycle (Figure 1A), viral entry is initiated by the binding of S protein to the human angiotensin-converting enzyme 2 (ACE2) receptor on the cell surface23,24. This is followed by cleavage of S protein by the cellular serine protease TMPRSS2 which is required for fusion of viral and host cell membranes23,24. After entry, the virus’s positive sense RNA genome is immediately ready for polycistronic translation by the host ribosome, and ribosomal frameshifts allow for the expression of the Orf1a and Orf1b polyproteins25. Auto-proteolytic cleavage of Orf1a and Orf1b by viral proteases produces 16 Nsps25. The viral genome is replicated by a viral RNA-dependent RNA polymerase holoenzyme, consisting of Nsp7, Nsp8, Nsp12, and Nsp14 for RNA transcription, capping, and proofreading25,26. Replication takes place on double-membrane structures called replication and transcription complexes (RTCs) derived from and sometimes contiguous with the endoplasmic reticulum (ER)25,26. RTCs are thought to both protect the virus from innate immune responses and concentrate the necessary viral components required for replication. Full length genomic RNA is replicated via a negative-sense intermediate, and a nested set of subgenomic mRNAs encoding viral structural and accessory proteins are synthesized by continuous transcription and then translated either at the ER or in the cytoplasm. Virion assembly takes place in the ER-Golgi intermediate compartment (ERGIC), where N protein binds the RNA genome, virions bud from ER and Golgi membranes, and mature virions are released through a process similar to exocytosis23. Understanding the underlying biology of SARS-CoV-2 infection, more specifically the host proteins and cellular processes that are essential for SARS-CoV-2 infection and replication, will identify targets for both drug repurposing and development of novel host-directed therapies.
Table 1.
Proteomic studies on SARS-CoV-2 highlighted in the perspective.
| Proteomic Technique | Sample Type | Study Objective | Reference |
|---|---|---|---|
| H/DX-MS | Purified protein | N protein structure | Ye et al. Architecture and Self-Assembly of the SARS-CoV-2 Nucleocapsid Protein. bioRxiv 2020. https://doi.org/10.1101/2020.05.17.100685. |
| nMS | Purified protein | S-ACE2 virus-host protein complex structure; drug mechanism (heparin) | Yang et al. The Utility of Native MS for Understanding the Mechanism of Action of Repurposed Therapeutics in COVID-19: Heparin as a Disruptor of the SARS-CoV-2 Interaction with Its Host Cell Receptor. Anal. Chem. 2020. https://doi.org/10.1021/acs.analchem.0c02449. |
| AP-MS | HEK293T cells expressing SARS-CoV-2 proteins | Virus-host protein-protein interactions; drug candidates | Gordon et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020. https://doi.org/10.1038/s41586-020-2286-9. |
| AP-MS | HEK293T cells expressing SARS-CoV-2, SARS-CoV-1, and MERS-CoV proteins | Virus-host protein-protein interactions; drug candidates | Gordon et al. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370 (6521). https://doi.org/10.1126/science.abe9403 |
| AP-MS; phosphoproteomics; ubiquitylation profiling | A549 cells expressing SARS-CoV-2 proteins; ACE2-expressing A549 cells infected with SARS-CoV-2 | Virus-host protein-protein interactions; transcriptome, proteome, phosphoproteome, and ubiquitome during infection; drug candidates | Stukalov et al. Multi-Level Proteomics Reveals Host-Perturbation Strategies of SARS-CoV-2 and SARS-CoV, bioRxiv 2020, 2020.06.17.156455. https://doi.org/10.1101/2020.06.17.156455. |
| PDL | A549 cells expressing SARS-CoV-2 proteins | Virus-host proximal protein interactions | Samavarchi-Tehrani et al. A SARS-CoV-2 - Host Proximity Interactome. bioRxiv 2020, https://doi.org/10.1101/2020.09.03.282103 |
| Phosphoproteomics | Vero E6 cells infected with SARS-CoV-2 | Phosphoproteome during infection; drug candidates | Bouhaddou et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182 (3), 685-712.e19. |
| Abundance proteomics; phosphoproteomics | Vero E6 cells infected with SARS-CoV-2 | Transcriptome, proteome, and phosphoproteome during infection | Davidson et al. Characterisation of the Transcriptome and Proteome of SARS-CoV-2 Reveals a Cell Passage Induced in-Frame Deletion of the Furin-like Cleavage Site from the Spike Glycoprotein. Genome Med. 2020, 12 (1), 68. |
| Phosphoproteomics | Caco-2 cells infected with SARS-CoV-2 | Phosphoproteome during infection; drug candidates | Klann et al. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 2020. https://doi.org/10.1016/j.molcel.2020.08.006. |
| Abundance proteomics | Caco-2 cells infected with SARS-CoV-2 | Translatome and proteome during infection; drug candidates | Bojkova et al. Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets. Nature 2020, 583 (7816), 469–472. |
| Targeted proteomics | Vero E6 cells infected with SARS-CoV-2 |
Diagnostic methods | Bezstarosti et al. Targeted Proteomics for the Detection of SARS-CoV-2 Proteins. bioRxiv, 2020, 2020.04.23.057810. https://doi.org/10.1101/2020.04.23.057810. |
| Targeted proteomics | Patient samples (gargle) | Diagnostic methods | Ihling et al. Mass Spectrometric Identification of SARS-CoV-2 Proteins from Gargle Solution Samples of COVID-19 Patients. bioRxiv, 2020, 2020.04.18.047878. |
| Targeted proteomics | Vero E6 cells infected with SARS-CoV-2; patient samples (nasopharyngeal swabs, bronchoalveolar lavage) |
Diagnostic methods | Zecha et al. Data, Reagents, Assays and Merits of Proteomics for SARS-CoV-2 Research and Testing. Mol. Cell. Proteomics 2020, 19 (9), 1503–1522. |
| Abundance proteomics | Patient samples (sera and plasma) | Biomarkers of COVID-19 disease severity | Messner et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst 2020. https://doi.org/10.1016/j.cels.2020.05.012. |
| TPP | HepG2 cells treated with compounds | Off-target effects of COVID-19 drug candidates (remdesivir, hydroxychloroquine, and more) | Friman et al. CETSA MS Profiling for a Comparative Assessment of FDA Approved Antivirals Repurposed for COVID-19 Therapy Identifies Trip13 as a Remdesivir off-Target. bioRxiv 2020. https://doi.org/10.1101/2020.07.19.210492 |
Figure 1.

SARS-CoV-2 life cycle (A). Open questions to further our understanding of SARS-CoV-2 biology and proteomic techniques that can be leveraged to address these questions (B).
Large-scale “omics” approaches, including genomics, transcriptomics, and proteomics are powerful technologies that could yield essential biological information about SARS-CoV-2 virology and COVID-19 pathology. Genomic approaches have been essential to investigate SARS-CoV-2 genome structure and similarities with related coronaviruses, among other foundational contributions to our understanding of this novel virus. With an understanding of the SARS-CoV2 genome, the field is now equipped to probe the actionable components of the virus: the proteins that the viral genome encodes. Proteomic approaches applied to SARS-CoV-2 allow the investigation of open questions of varying size and scale (Figure 1B). Proteomics can inform the structure of a single viral protein, the composition of a complete virion, and a global view of the host proteome during infection. Proteomic methods provide unique insight into the interaction between virus and host, including the host machinery co-opted for viral replication and signaling pathways that characterize the host response. Proteomic tools can also be used to probe interactions between compounds and proteins, and represent a powerful strategy for drug discovery. This perspective will discuss how proteomics can be leveraged to answer the following open and fundamental questions about SARS-CoV-2 biology. How do the dynamic structures of SARS-CoV-2 proteins and virions inform pathogenicity? What cellular machinery does SARS-CoV-2 utilize during infection? Which signaling pathways are rewired during SARS-CoV-2 infection? What are strategies to monitor COVID-19 pathology and investigate treatment strategies?
How do the dynamic structures of SARS-CoV-2 proteins and virions inform pathogenicity?
Viral proteins dictate the virion’s structure and shape, and carry out activities essential for viral replication. Studying the dynamic structures of SARS-CoV-2 proteins and intact virions is essential not only to understand their molecular functions, but also to facilitate the design of effective small molecule therapies that can disrupt virions, viral entry, and virus replication and egress. In addition to atomic-resolution structural approaches that are effective for smaller proteins and protein complexes (e.g. x-ray crystallography and NMR), advances in approaches like cryo-electron microscopy (cryo-EM) have made the analysis of larger, flexible, multi-state proteins and protein complexes more feasible. However, these approaches still require reconstituted complexes often expressed and purified from non-native bacterial or other expression systems, and can require non-native buffers and conditions to achieve structure determination. In contrast, diverse and complementary proteomic approaches including cross-linking mass spectrometry (XL-MS), Hydrogen/Deuterium exchange mass spectrometry (H/DX-MS), and intact protein mass spectrometry can illuminate structural features of proteins and protein complexes under near physiological conditions or even inside biologically-relevant living cells or intact virions (Figure 2)27. Unlike NMR or X-ray crystallography, these mass spectrometry (MS)-based proteomic structure techniques require relatively lower amounts of protein samples. Despite their benefits, XL-MS, H/DX-MS and nMS have been widely considered to be very niche with their applications requiring a high degree of specialized expertise and equipment not generally applied in standard MS experiments. Though there have been great advances in each field opening the technology to non-specialists, this has limited their wide-spread adoption, particularly in rapid-response research. Still, each methodology provides a unique perspective, and when integrated with other structural techniques can provide a more comprehensive understanding of the dynamic complex structures essential for SARS-CoV-2 replication.
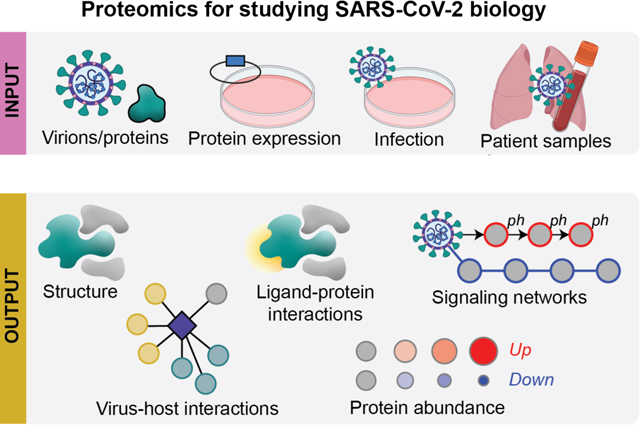
Figure 2.

Overview of MS-based proteomics techniques proposed to study SARS-CoV-2, including sample types that can be used as input, molecular insights that can be obtained as output, and how the technologies can be integrated to inform SARS-CoV-2 biology and COVID-19 pathology.
Cross-linking mass spectrometry (XL-MS)
XL-MS is a powerful approach that can probe protein-protein interaction (PPI) networks and interfaces while overcoming a number of structural biology limitations28,29. Primarily this includes the ability to: 1) capture structural information from transient and dynamic PPIs and PPI binding interfaces; 2) accommodate not only sample impurities but also structural heterogeneity, flexibility and subunit composition; and 3) be applied to live cell and in vivo applications29. When combined with integrative modeling approaches (see below), XL-MS can provide distance restraints for dynamic structure determination. In recent years, a number of diverse XL-MS strategies have emerged though they all generally rely on chemical cross-linkers to covalently bridge adjacent proteins via reactive amino acid residues, followed by MS-based identification of cross-linked peptides29–31. Cross-linked peptide searching and identification is complicated by two main challenges: 1) the lower frequency of cross-linked peptides in a complex mixture of mainly unmodified peptides; and 2) the complexity of cross-linked peptide spectra which has two covalently linked contributing peptide sequences32–34. The majority of XL-MS strategies include some form of enrichment step to prioritize identification of cross-linked peptides either by their size, hydrophobicity, ion mobility, or charge. Improved software and the development of functionalized chemical cross-linkers that are isotope-coded, MS-cleavable, or contain a reporter ion, have made the identification of cross-linked peptides more accurate and straightforward. While the scope of this perspective is not a comprehensive review of XL-MS approaches, recent in depth reviews of these methods can be found in29–31.
The practical applications for XL-MS vary from informing large macromolecular complexes35,36, to conformationally flexible complexes37,38, to more recent applications probing dynamic interfaces of challenging pathogen-host complexes39 and even virus-like particles (VLPs)40. While cryo-tomography and cryo-EM have become very powerful techniques for visualizing viral protein interactions and intact viral particle structures, atomic resolution of dynamic interactions is still challenging. Cryo-electron tomography (cryo-ET) shows ordered binding of part of the flexible receptor to the viral surface, with distal domains in multiple conformations. Using cryo-ET, Meyer et al. demonstrated the binding of adeno-associated virus 2 (AAV2) VLPs to the cell surface receptor AAVR40 and by complementing with XL-MS data they were able to identify regions of AAVR in close proximity to AAV2 proteins. XL-MS data validated the localization of the PKD2 domain of AAVR, improved the placement of the PKD1 domain of AAVR, and explained the disordered EM density in the structure. By combining cryo-ET, cryo-EM, and XL-MS, collective limitations in size, scale, and resolution could be overcome to determine the structure of AAV2 bound to AAVR soluble domains and thus make biological predictions for the effects of neutralizing antibodies. Prchal et al., combined affinity purification, XL-MS, and NMR data to determine the structure and map interaction interfaces between the cytoplasmic tail of Mason-Pfizer monkey virus (M-PMV) envelope and matrix proteins41. Similarly, XL-MS was recently leveraged to study how the HIV protein Nef targets surface receptor CD4 for endocytosis to promote HIV infection39. XL-MS data captured flexible and unresolved components of the Nef-CD4-AP2 crystal structure, and confirmed the observation that Nef serves as a flexible connector between CD4 and clathrin AP2 to promote endocytosis and downregulation of CD439. Finally, XL-MS allows unbiased structure characterization and identification of unknown structural features including additional protein components or post-translational modifications. In Yu et al. the thiol-cleavable cross-linker 3,3’-dithiobis(sulfo-succinimidylpropionate) (DTSSP) was used to identify vimentin as a transient interactor of the SARS-CoV-1 Spike (S)-ACE2 virus-host protein complex. Vimentin expressed on the cell surface was found to be important for SARS-CoV-1 virus entry and Vero E6 cells pretreated with antivimentin antibodies showed > 40% reduction in SARS-CoV-1 VLP uptake42.
Though to date there have been no published studies using XL-MS to characterize SARS-CoV-2 viruses or viral proteins, the studies above demonstrate some of the potential applications for XL-MS. With the advent of newer and faster instruments, improved XL-MS identification software, and optimized design of new cross-linkers for intra-cellular applications, the reality of large-scale unbiased cross-link peptide identification of whole cell networks is approaching. This type of network and structure data collection is feasible only with XL-MS experiments. One particularly appealing application would be the identification of cross-linked peptides from SARS-CoV-2 infected cells. Comparison of XL-MS data of mock versus SARS-CoV-2 infected cells could illuminate not only the viral-host protein interactions, but also the viral-viral protein complexes and the changes in host-host interactions that occur during infection. By unifying this data with the more standard AP-MS approach, scientists could build not only a network model of SARS-CoV-2 infected cells, but also provide PPI interface data to inform protein complex structures. In addition to exploring SARS-CoV-2 infected cells, researchers could look at the virus itself. The same global XL-MS application to purified SARS-CoV-2 virions could, in one experiment, characterize the virion proteome, identifying both viral and host proteins that make up the virion, as well as provide PPI interface data on virus-virus and virus-host interactions of the virion structure. Combined with sophisticated cryo-EM and cryo-ET images, including recent discoveries made by Yao et al.43 and Liu et al.44, these studies could enhance our understanding of and development of chemotherapies against SARS-CoV-2.
Hydrogen/deuterium exchange mass spectrometry (H/DX-MS)
H/DX-MS measures changes in mass associated with the exchange between protein backbone amide hydrogen and deuterium from the surrounding D2O. The rate of exchange is dependent on the conformation, surface accessibility, inductive effect of the neighboring groups, stability of hydrogen bonding networks, and the intrinsic chemical properties of the underlying amino acid sequence45–48. It is used to examine conformations of individual proteins or large protein complexes49, locate protein sites directly or indirectly involved in binding50, probe for allosteric effects51, monitor the folding dynamics of protein domains52, examine intrinsic disorder53 and provide insights into protein-membrane interactions54. H/DX-MS is unique in probing conformational states with single-residue resolution. To perform residue-level measurements by H/DX-MS, suitable gas-phase fragmentation of the deuterated peptides by electron transfer dissociation (ETD) or electron-capture dissociation (ECD) is generally applied55,56. Both ETD and ECD generate fragment ions with vibrationally cold energy, minimizing hydrogen scrambling, a phenomenon seen using other fragmentation strategies like collision-induced dissociation (CID)57– 59. In 2013, Resetca et al. developed a method called Time-Resolved Electrospray Ionization Mass Spectrometry (TRESI-MS) which uses a microfluidic chip in-line with all the steps involved in a ‘bottom-up’ HDX workflow60. This development provided faster sample preparation times and improved reproducibility to the point that is now feasible to characterize rapid structural transitions that occur during protein folding61, ligand binding62 or post-translational modification63, applications mostly inaccessible to conventional techniques.
More recently, H/DX-MS has emerged as a potential tool in structural virology exploring the Hepatitis C E2 glycoprotein64, influenza hemagglutinin65, HIV envelope glycoprotein66–69, and Ebola GP1/GP270. Using H/DX-MS, Ye et al., were able to explore the architecture and self-assembly of SARS CoV-2 N protein and showed that the addition of the C-terminal spacer B/N3 domain to the N2b domain mediates the formation of a homotetramer71. These types of studies provide insights into the conformational dynamics of proteins in-solution that directly reflect the structural changes, mechanism of viral glycoprotein recognition, and virus neutralization caused by the binding of antibodies and small molecules. Further applications of H/DX-MS for SARS-CoV-2 could help to characterize (1) the effect of post-translational modifications on SARS-CoV-2 protein structures such as glycosylation of S protein or phosphorylation of Orf9b and (2) the binding of small molecules or monoclonal antibodies targeting SARS-CoV-2 proteins, such as S protein. This could not only inform therapeutics, but also provide essential information on host immunity, and potentially the development of different types of neutralizing antibodies.
Intact protein mass spectrometry
While XL-MS and H/DX-MS rely on digesting protein complexes into peptides, intact protein MS enables the analysis of intact proteins and protein complexes. Native MS (nMS) is a unique intact protein-MS methodology that maintains non-covalent interactions and stoichiometry of protein complexes in gas phase72,73 while denaturing top-down MS (TDMS) approaches identify proteoforms and PTMs. nMS combined with collision-induced dissociation (CID) disrupts non-covalent interactions between protein subunits based on their strength, and can help decipher stoichiometry and topology of protein complexes. Furthermore, applications with ion mobility-MS (IMMS) have allowed the field to explore greater structural details of large macromolecular assemblies (for detailed review see74). Importantly, IMMS allows researchers to separate and characterize protein complexes and protein subunits in gas-phase based on their size and shape. It opens up avenues for structural analysis of heterogeneous protein complexes allowing assessment of stoichiometry, topology, and cross-section of the assemblies and their subunits. IMMS can aid in generating hypotheses about complex structures, conformations, assembly, and disassembly and offers complementarity in structural biology74.
nMS can capture transition events in protein complex assembly and disassembly, and more recently in proteolytic cleavage events like those that are essential to coronavirus replication and infection74. As described above and shown in Figure 1, the translated coronavirus polyproteins are processed to produce Nsp proteins. In order to investigate the processing of the SARS-CoV-1 polyprotein nsp7–10 region by Mpro (the main protease), as well as the formation of intermediate products and complexes, Krichel et al. used nMS to demonstrate how the in vitro cleavage efficiencies resemble limited proteolysis of a folded protein and are influenced by tertiary polyprotein structure75. In addition, they identify the hetero-tetrameric Nsp7 and Nsp8 complex as the predominant product, thus providing not only the order of proteolytic cleavage, but the formation of post-cleavage structures75. While SARS-CoV-2 S protein and ACE2 are challenging to study by nMS given their extensive glycosylation, nMS with limited charge reduction76 provided meaningful information about the complex between ACE2 and the receptor-binding domain (RBD) of the S protein as well as the role of heparin in destabilizing the ACE2/RBD association77. Yang et al. showed that both short (pentasaccharide) and long (eicosasaccharide) heparin oligomers form a stoichiometric complex with the RBD, indicating a single binding site which alters the protein conformation and subsequently results in a decrease in its ability to associate with ACE277. This study suggests that nMS might be a powerful method for studying the interaction between drugs and their therapeutic target. In addition to these studies, nMS has been applied to investigate viral particle assembly. While the multistep assembly of multiple identical proteins into an icosahedral virus capsid is poorly understood, using charge detection MS, Pierson et al. were able to detect trapped intermediates during the assembly of the hepatitis B virus capsid78. Subsequent cryo-EM analysis indicated incomplete capsids rather than aberrant structures suggesting that the observed structures are on-path intermediates78. Application of these types of experiments to SARS-CoV-2, particularly if complemented with additional structural MS techniques like in vivo XL-MS, could characterize the assembly and structure of SARS-CoV-2 virions.
Monoclonal antibodies (mAbs) that target viral proteins are a promising class of therapeutics against infectious diseases like COVID-1979, Ebola80, Hendra and Nipah virus infection81. High-quality characterization of intact mAbs is essential and is commonly performed by top-down mass spectrometry (TDMS). Similar to nMS, TDMS strategies avoid the use of proteolytic enzymes, providing intact masses of the molecules, which can determine the presence of multiple proteoforms82,83. MS/MS fragmentation of the intact proteins and large protein subunits can provide amino acid resolution for interpretation of sequence heterogeneity and presence of PTMs83–85. TDMS-associated technologies have significantly advanced over the past decades83,86, mainly focusing on two areas of development, instrumentation and fragmentation approaches84,87. Newer methods such as ultraviolet photodissociation (UVPD)88, ECD89, ETD90 have radically increased the sequence coverage and PTM information for TDMS experiments. As a result, TDMS has emerged as a powerful means in basic, translational, and clinical research for protein identification and different proteoforms elucidation. Though no studies thus far have capitalized on TDMS for studying SARS-CoV-2 antibodies purified from convalescent sera, the potential exists for this application to aid in the identification of proteins and proteoforms that have higher specificity or neutralization potential which could help in design of monoclonal antibody therapy, or in predicting reaction to new mutations.
Integrating complementary structural data
Complementary data collected from these diverse structural proteomic methodologies can be combined to maximize structural insight through integrative modeling. Integrative modeling synthesizes experimental data from structural, biochemical, proteomic and genetic studies to optimize a comprehensive and accurate model of protein complexes91–93. In this way, integrative modeling represents the most complete understanding of a structure that accounts for all data types. Integrative modeling is particularly powerful when defining macromolecular complexes for which atomic resolution might be feasible for individual or small, inflexible, subunits and sub-complexes, but not for the intact holocomplex. For example, integrative modeling was essential to synthesize cryo-EM, XL-MS, and crystal structure data to determine the dynamic structure of the yeast nuclear pore complex (NPC), a challenging macromolecular membrane bound structure consisting of 500+ proteins91. While cryo-EM and XL-MS heavily feature in integrative modeling, few studies have capitalized on the combined strengths of XL-MS, H/DX, and nMS. Particularly useful is the ability to define in-solution structures that are more representative of their native state, and in the case of XL-MS, can be performed in living cells.
To date, there are no integrative structures provided for SARS-CoV-2 viral proteins or complexes. Given the recent emergence of the virus, this can likely be attributed to the specialization and time required by each structure technology as well as for integrative modeling. However, applied structural proteomic technologies could be very powerful in studying SARS-CoV-2, as a number of SARS-CoV-2 proteins and virus-host complexes present structural challenges including size, cellular localization, and flexibility. Full-length SARS-CoV-2 Nsp3 is a large, multi-domain, multi-functional, enzymatic, essential protein, that has yet to be fully structurally characterized. Expression, in cell cross-linking, and purification of Nsp3 from human cells could provide structural information pertaining to adjacent Nsp3 domain residue contacts and could capture Nsp3-host interactors that may be transient in nature. Additional TDMS experiments of Nsp3 purified from human cells could identify essential proteoforms that exist in cells. By combining these data with high resolution X-ray diffraction data of individual domains and H/DX-MS studies probing individual domain interactions, integrative modeling could determine a comprehensive and dynamic structure for Nsp3 as it looks in human cells. Investigating SARS-CoV-2 viral entry with integrative modeling informed by structural proteomics like XL-MS could also be particularly powerful to study S protein interactions and SARS-CoV-2 VLP interactions with the ACE2 receptor and the TMPRSS2 protease, as was demonstrated for AAV2 and AAVR in the example above40. This type of XL-MS data would pair well with the nMS study by Yang et al. described above that focused on defining the ACE2-RBD S protein interaction. Combined with H/DX-MS studies that help identify solute exposed surfaces of free S protein, and protected interfaces of S bound to ACE2 or TMPRSS2, an integrative model could provide useful information about the structure in solution. Understanding the structure of these viral-host interaction interfaces will be critical for developing drugs that disrupt and prevent SARS-CoV-2 entry into host cells.
What cellular machinery does SARS-CoV-2 utilize during infection?
Like all viruses, SARS-CoV-2 does not encode all of the machinery required for its replication, and must co-opt host machinery for the production of progeny virions. Studying viral-host PPIs can identify essential host factors and provide mechanistic details into the viral life cycle (Figure 2). Affinity purification mass spectrometry (AP-MS) is the most widely applied proteomics method to systematically characterize host-viral PPIs by expressing and purifying individual affinity tagged viral proteins in host cells. As a complementary method to classical AP-MS, proximity-dependent labeling (PDL) coupled to quantitative MS (PDL-MS) has emerged to study proximal PPIs, particularly those that are more transient or weaker interactions94. While AP-MS and PDL are powerful approaches to define host interactions of single viral proteins, recent developments in complex centric proteome profiling (CCPP) allow global mapping of cellular macromolecular complexes95,96.
Affinity purification-mass spectrometry (AP-MS)
AP-MS is an established method that has been widely applied to systematically characterize host-viral PPIs by expressing individual affinity tagged viral proteins in host cells, purifying stably-bound host protein interactors, and identifying those interactors by MS97–103. In a recent AP-MS study, Gordon et al. expressed and purified 26 of the 29 SARS-CoV-2 proteins from HEK293T cells, which led to the discovery of 332 virus-host PPIs104. Subsequent chemoinformatic analysis identified 69 drugs and small molecules that target the SARS-CoV-2-human PPI network, thus having the potential to disrupt host factor-dependent viral processes. Of 47 compounds tested, two classes of small molecules emerged that exhibited strong antiviral effects: those that modulate the activity of Sigma receptors, and those that inhibit mRNA translation. In a subsequent study, AP-MS efforts in HEK293T cells were expanded to map the full interactomes of SARS-CoV-1 and MERS-CoV105. Virus-host interactions for each of the three highly pathogenic human coronavirus strains were compared to identify and understand pan-coronavirus molecular mechanisms which revealed a high number of shared interactions between SARS-CoV-1 and SARS-CoV-2 and higher divergence comparing SARS-CoV-1 and 2 to MERS105. One notable example is the mitochondrial outer membrane protein Tom70 which interacts with the mitochondria localized Orf9b for both SARS-CoV-1 and SARS-CoV-2105. MERS does not have an Orf9b protein and thus did not show an interaction with Tom70. Interestingly, in the same study Tom70 was validated to be a host-dependency factor for SARS-CoV-2105.
In another impressive large-scale study that integrated AP-MS and global proteomic data, Stukalov et al. systematically mapped the virus-host interactions for both SARS-CoV-2 and SARS-CoV-1 in A549 lung carcinoma cells106. Performing global protein abundance, phosphorylation, and ubiquitylation profiling of cells overexpressing individual viral proteins, and integrating this data with SARS-CoV-2 PPI networks identified cellular pathways regulated by viral proteins106. This integrated PPI network was then used to predict well-characterized selective drugs that could be targeted for host-directed therapies and identified drugs targeting AKT and matrix metalloproteases as having anti-SARS-CoV-2 activity106. These studies demonstrate the potential of rapidly translating AP-MS data into druggable host factors with subsequent identification of repurposable drugs that have potent antiviral activity. Beyond the translational potential of global host-pathogen interaction maps, these studies also show the impact and breadth of work that can be carried out by rapid response of international collaborations between interdisciplinary scientists.
Proximity-dependent labeling (PDL)
Proximity-dependent labeling (PDL) relies on enzymes which catalyze covalent transfer of biotin or biotin derivatives to proteins in proximity of the enzyme, including promiscuous biotin protein ligases (BirA/BioID/TurboID)107,108 and engineered ascorbic acid peroxidase (APEX)109. To map PPIs using PDL, the enzyme is fused to a protein of interest, such as a viral protein, and upon addition of a substrate the proximal proteins will become biotinylated, followed by their enrichment using Streptavidin resin, and analysis using quantitative MS. The covalent labeling of proximal proteins in cells allows for the capture and identification of not only stable, but also transient and weaker interactions, allows purification under harsh lysis conditions, and provides additional spatial information about the subcellular location of the PPIs through proximal labeling. This was demonstrated for example by applying the APEX-based PDL approach to study agonistinduced changes of G-protein coupled receptors (GPCRs) interaction networks, which successfully characterized functionally relevant GPCR interactors with temporal and spatial resolution110.
PDL has also been extended to study host-pathogen interactions. Coyaud et al. applied a combined AP-MS and BioID approach to systematically map the Zika-host interactome composed of over 1200 Zika-host interactions, which revealed extensive organellar targeting by Zika virus and a role of peroxisomes for Zika virus infection111. A recent study introduced BirA into the viral genome of the MHV (murine coronavirus) to define the microenvironment of the RTC112. The study identified more than 500 proteins in proximity to the RTC and discovered a close association of viral RNA synthesis sites with the host translation machinery112. Another PDL-MS study performed BioID for 27 SARS-CoV-2 proteins fused to the miniTurbo enzyme in a lung adenocarcinoma cell line113, which revealed proximal interactions with 2242 host proteins. The inclusion of several host subcellular markers as baits provided testable hypotheses regarding the functional consequences of virus-host interactions. For example the proximal interactome of the N protein revealed the depletion of proteins critical to the formation of stress granules, suggesting that the interaction between N protein and G3BP1 might prevent stress granule formation113.
These studies demonstrate the power of PDL-MS as a complementary approach to AP-MS to map SARS-CoV-2-host interactions, particularly to characterize transient interactions and inform cellular location. SARS-CoV-2 relies on organellar targeting with replication taking place in membrane bound vesicles derived from the ER and Golgi. Application of PDL to key viral proteins such as members of the RTC (Nsp7, Nsp8, Nsp12, and Nsp14), or key host factors such as the Nsp6 interactor Sigma-1 receptor in the context of SARS-CoV-2 infection could track PPIs and their cellular location, essentially providing an intra-cellular GPS for virus-host PPIs throughout the viral lifecycle. As a recent extension of proximity labeling approaches, split enzymes have been engineered which enable contact-specific PDL. The two fragments of the split enzyme remain inactive apart, and become active upon reconstitution when they are driven together by PPIs114,115. These sophisticated approaches could be particularly powerful in validating and functionally characterizing high-confidence host-viral PPIs identified by AP-MS and/or PDL-MS. A split enzyme PDL experiment could simultaneously validate a putative interaction, determine additional protein complex-specific subunits, and derive intracellular spatial information for the specific interaction. For example, splitting the labeling enzyme between a viral protein and an ER protein can inform protein interactions that take place on the surface of double-membrane vesicles of SARS-CoV-2 RTCs116.
Complex centric proteome profiling (CCPP)
In complex centric proteome profiling (CCPP), samples are gently lysed to preserve native protein complexes which are then: (1) separated into fractions using size exclusion chromatography; (2) digested into peptides and analyzed by highly sensitive data-independent acquisition (DIA) quantitative MS; and (3) protein patterns across the fractions correlated to determine the composition of protein complexes. This approach has recently been utilized to quantitatively compare protein complexes in two distinct cell-cycle states, suggesting a model for disassembly of the nuclear pore complex during the transition from interphase to mitosis96. Host complexes are similarly disassembled and rearranged during viral infection, and CCPP would allow the capture of those changes during SARS-CoV-2 infection in a systematic fashion. Indeed, one of the limitations with AP-MS and PDL-MS strategies, which rely on individually tagged bait proteins, is that the captured PPIs are missing the context of the full suite of viral proteins in infected cells. Therefore, virus-host interactions dependent on multiple viral proteins, nucleic acids, or signaling will be absent in the final analysis. Characterizing SARS-CoV-2 infected cells using the CCPP would avoid the necessity for tagged components, and can provide a global picture of the complexes viral proteins form and interact with, as well as how they manipulate cellular machinery through recruitment or dissociation of specific components.
The integration of CCPP with organelle fractionation could illuminate additional spatial information of host-pathogen interactions117–119. Organelle fractionation alone was recently applied to determine targeting of viral proteins to distinct organelles and to define alterations in organellar proteome composition during Cytomegalovirus infection117. Proteins localized to one compartment in uninfected cells and a different compartment during infection were identified as translocated proteins, with most translocations occurring between the plasma membrane, ER, golgi, and lysosome. Notably, this approach identified MYO18A as a protein that translocates from the plasma membrane to the lysosome during Cytomegalovirus infection, and siRNA knockdown of MYO18A significantly decreased virion production. Because SARS-CoV-2 replication relies on trafficking throughout cellular compartments, especially the ER and Golgi, CCPP combined with organelle fractionation could represent a particularly powerful approach to identify how host proteins are globally hijacked and translocated throughout the SARS-CoV-2 lifecycle.
Towards an integrated virus-host PPI network
Complementary data from various viral-host PPI mapping approaches can be integrated to provide a model for how SARS-CoV-2 hijacks host machinery to replicate inside of cells. A model that accounts for the molecular players involved as well as the timing and localization of events throughout the viral life cycle will aid in the rational design of novel drugs, the repurposing of existing drugs, and the development of combinatorial therapies (Figure 2). Given the initial successes of host-pathogen interaction mapping in standard, though less physiologically-relevant cell lines like HEK293T cells, it is imperative to expand these efforts to clinically-relevant cell models that mimic the infection in human lung epithelia. While expression of individual affinity-tagged viral proteins allows high-throughput discovery of stable virus-host interactions, this approach will miss highly transient or less stable interactions, as well as those interactions that rely on simultaneous expression of multiple viral proteins, such as the viral RNA-dependent RNA polymerase holoenzyme responsible for viral genome replication. Therefore, host-pathogen interaction studies should be performed in the context of viral infection. In addition, cross-linking coupled with AP-MS, or the application of global XL-MS experiments, can capture transient and weaker interactions from SARS-CoV-2 infected cells (see XL-MS section above). These experiments can provide PPI network data and inform virus-host protein complex structures. Furthermore, exciting work could be done to investigate not only SARS-CoV-2, but other human coronaviruses (i.e. HCoV-OC43, SARS-CoV-1 and MERS-CoV) in reservoir and cell lines from cross-species, such as bats. Through identification of conserved virus-host interactions across different coronavirus species, host factors that increase or mitigate pathogenicity, severity, infectivity, and cross-species barriers could be identified. These data would be a starting point for designing pan-Coronavirus treatment strategies.
Which signaling pathways are rewired during SARS-CoV-2 infection?
During infection a complicated battle takes place wherein viruses must simultaneously rewire cellular pathways they need for replication while evading the host cell’s innate immune defenses. Perturbations to post-translational modifications (PTMs) allow viruses to quickly manipulate the host environment to control cell cycle, prioritize transcription and translation of viral products, and evade the immune response. For example, HIV-1 Vif degrades regulatory subunits of the key cellular phosphatase PP2A, which results in hyperphosphorylation of many cellular proteins, including substrates of the aurora kinases, and causes cell cycle arrest in G2120–122. Nuclear factor kB (NF-kB) is an innate immune pathway regulated by phosphorylation and ubiquitylation and has been co-opted by viruses such as HIV-1, which uses NF-kB as a transcription factor to express viral mRNA123. Phosphorylation is also key for viral trafficking throughout the viral life cycle124. For example, Ebola entry requires phosphoinositide-3 kinase signaling125, Influenza replication relies on nuclear-cytoplasmic shuttling regulated by multiple phosphorylation sites on its nucleoprotein126, and the phosphorylation of HIV protein p6 is required for virion budding and release from the cell127. Given the important role of perturbing phosphorylation for viral infection, mapping phosphorylation events to kinases and phosphatases could allow for the identification of druggable targets and repurposing of FDA approved drugs128.
Ubiquitin signaling is another important PTM for viral replication, and inhibition of the host ubiquitin-proteasome system (UPS) can lead to restriction of viral entry and expression of viral proteins, as it has been shown for coronaviruses including mouse hepatitis virus and SARS-CoV-1129. Viruses often exploit the host UPS is to target restriction factors, or host factors that inhibit viral infection, for degradative ubiquitylation. One such example is HIV-1 Vif, which together with the transcriptional cofactor CBFβ hijacks the Cul5-RING E3 ubiquitin ligase and acts as a non-native substrate adaptor to target cellular APOBEC3 restriction factors for polyubiquitylation and proteasomal degradation130–137. Unbiased proteomic approaches that selectively enrich and identify specific PTMs like phosphorylation and ubiquitylation can study global perturbations during infection and highlight cellular signaling pathways that are critical for SARS-CoV-2 replication (Figure 2).
Global phosphoproteomics
Phosphorylation is a quick, reversible PTM that viruses use to (1) regulate viral proteins and (2) alter the stability, subcellular location, and enzymatic activity of host proteins to aid viral replication. Protein phosphorylation is regulated by kinases and phosphatases, which phosphorylate and dephosphorylate substrates, respectively. While serine, threonine and tyrosine are the most commonly phosphorylated and studied amino acids, other amino acids such as arginine, lysine, aspartic acid, glutamic acid, histidine and cysteine are also phosphorylated and though harder to study, have been implicated in various host biology138,139.
MS-based proteomics has emerged as the method of choice to systematically study protein phosphorylation and its dynamics. To obtain higher sensitivity in identifying phosphorylation sites in complex protein samples such as cell lysates, several approaches have been developed to enrich phosphorylated peptides based on ion metal affinity chromatography, ion exchange chromatography, and antibody-based immunoprecipitation140. Combined with developments in MS and computational approaches to identify, quantify and localize phosphorylation sites, tens of thousands of phosphosites can routinely be accurately quantified in a single experiment141–143. The ability to subsequently map the quantified phosphorylation events to kinases might reveal druggable targets and the repurposing of kinase inhibitors that have been approved for the treatment of other diseases such as cancer144–146. These inhibitors can then be readily tested for efficacy against viral infection and contribute to a rapid development of efficient treatment strategies for the SARS-CoV-2 pandemic.
The feasibility of this concept was recently demonstrated by applying global phosphorylation analysis to SARS-CoV-2 infected ACE2-expressing A549, Vero E6, and Caco-2 cells106,128,147,148. The study in Vero E6 cells identified amongst others p38, CK2, CDKs, AXL, and PIKFYVE as dysregulated kinases and demonstrated that their pharmacological inhibition restricted SARS-CoV-2 replication128. Bouhaddou et al. discovered that CK2 is activated by the N protein resulting in upregulation of CK2 cytoskeleton-related targets, which contributed to the formation of filopodial protrusions where virus particles seem to be budding from128. Klann et al. discovered not only extensive rearrangements of growth factor receptor (GFR) signaling, but also validated antiviral efficacy for multiple GFR inhibitors, thus demonstrating the central function of GFP signaling in SARS-CoV-2 infection148. Stukalov et al. followed a conceptually similar approach integrating global protein abundance, phosphorylation and ubiquitination data measured throughout SARS-CoV-2 infection in ACE2-expressing A549 lung carcinoma cells to identify regulation of cellular pathways perturbed by the virus106. Targeting selected pathways with drugs they identified several kinase and Matrix metallopeptidase inhibitors with significant antiviral effects against SARS-CoV-2106.
Global ubiquitylation profiling
Ubiquitylation is another reversible PTM which is essential for the replication of many different virus families. As viruses do not typically encode their own ubiquitin machinery, members of most virus families exploit their host’s UPS to ubiquitylate or de-ubiquitylate proteins to target them for proteasomal degradation, alter their trafficking, or to change their activity149,150. Ubiquitylation requires E1 enzymes for activation, E2 enzymes for conjugation, and E3 ligases that recruit the protein substrate. Substrates can be mono- or poly-ubiquitylated, and poly-ubiquitin chains can be connected by any of ubiquitin’s seven lysines (K6, K11, K27, K29, K33, K48, and K63) or N terminus. Different linkages allow for different signaling outcomes, for example, K48 linkage is associated with degradation by the proteasome, while K63 linkage is associated with non-degradative signaling including DNA damage repair, innate immunity, and intracellular trafficking. Cells encode a large number of deubiquitylating enzymes (DUBs) that catalyze the removal of ubiquitin from target substrates, and are important for regulating a number of cellular processes and have been implicated in a number of human diseases151–154. Viruses may hijack E3 ligases to establish novel ubiquitylation events or hijack DUBs to remove established ubiquitylation events in order to aid their replication.
Since enrichment increases the identification of ubiquitination sites, the most commonly used method for assessing global ubiquitination relies on immunoprecipitation of peptides containing a lysine residue modified by diglycine, an adduct left at sites of ubiquitination after trypsin digestion155,156. To distinguish between ubiquitylation events that target proteins for degradation or change protein activity, global ubiquitylation enrichment can be combined with (1) proteasome inhibition which should stabilize proteins targeted for degradation and (2) global abundance proteomics which should identify proteins that are targeted for degradation as downregulated. In contrast to phosphorylation where many enzyme-substrate relationships are well characterized, ubiquitin ligase-substrate relationships are less defined. This complicates the mapping of ubiquitin ligases to ubiquitylation events, and hampers the identification of ubiquitin-related druggable targets. However, ubiquitylation involves a physical interaction between a virus and one or more host proteins, therefore global ubiquitin profiling can be combined with AP-MS and/or proximity labeling to identify relevant interactions with ligases, representing druggable target candidates.
Global ubiquitylation combined with global protein abundance analyses of ACE2-expressing A549 cells infected with SARS-CoV-2 identified virus-mediated ubiquitylation events and host proteins targeted for degradation106. In this study, 1053 host and viral proteins, including the ACE2 receptor protein, were identified as having SARS-CoV-2-regulated ubiquitylation site. In addition to global ubiquitylation analysis, complementary approaches have suggested ubiquitin signaling as particularly important for SARS-CoV-2 infection. It has been shown by AP-MS that multiple E3 ubiquitin ligases physically interact with viral proteins, which have been subsequently validated to be functionally relevant for SARS-CoV-2 infection. ORF10 was found to interact with the CUL2ZYG11B E3 ligase complex104 and knockdown of ZYG11B in Huh-7.5 hepatoma cells resulted in decreased SARS-CoV-2-induced cytopathic effect157, suggesting that ZYG11B is a dependency factor for SARS-CoV-2 infection. Another example is the E3 ligase MIB1, which was discovered as an interactor of Nsp9104, and interestingly knockout of MIB1 increased the virusinduced cytopathic effect not only for SARS-CoV-2, but also for HCoV-229E, HCoV-NL63, HCoV-OC43157. These results suggest that MIB1, which is known to function in TBK1 polyubiquitination, which in turn is a signal integrator of multiple RIG-like receptors and positive regulator of IRF3, might establish an antiviral state that broadly controls coronavirus infection157. Finally, the viral protein Nsp3 has DUB activity, and its inhibition by small molecules decreases SARS-CoV-2 replication158.
Global protein abundance profiling and integrating signaling data
Viral perturbations in ubiquitin and phosphorylation signaling have downstream consequences at the protein level, and abundance proteomics is the method of choice to investigate how viral infection globally rewires the host proteome. Host proteins that are upregulated and downregulated during SARS-CoV-2 infection can be key host dependency and restriction factors that the virus utilizes or evades respectively to promote viral replication. Integrating protein abundance data with changes to protein ubiquitylation and phosphorylation can be an incredibly powerful way to identify not only individual proteins, but cellular pathways targeted by the virus, allowing us to build testable hypotheses of the molecular mechanisms of SARS-CoV-2 infection. Bojkova et al. analyzed the proteome of SARS-CoV-2 infected Caco-2 cells compared to mock infection, identified significantly perturbed pathways, and tested the effect of drug inhibition of those pathways on viral replication159. In one notable example, 25 spliceosome proteins were found to be upregulated during infection, and the spliceosome inhibitor pladienolide B showed antiviral activity159. To better understand the mechanism of how SARS-CoV-2 rewires signaling pathways, PTM and abundance analysis can be performed after infection with genetic mutant viruses to identify which viral proteins are responsible for rewiring a given pathway. This approach has been applied to HIV to study phosphorylation and ubiquitin signaling associated with accessory proteins Vif, Vpr, and Vpu120,122,160. In applying this approach to SARS-CoV-2, Nsp3 is a promising protein for this type of study due to its DUB activity. PTM and abundance changes identified during infection with wild type SARS-CoV-2 but not with a ΔNsp3 virus may represent proteins and PTMs regulated by Nsp3. Taken together, phosphorylation and ubiquitylation signaling integrated with protein abundance in SARS-CoV-2 infection is an important area of study to understand how the virus rewires host cells and identify druggable targets.
Despite the promise of data integration, its application for host-pathogen signaling has been difficult in part due to variability in and complexity of the individual datasets and in part due to the challenge of measuring both host and pathogen components (reviewed in 161). While identical time points or even split samples might be used to reduce reproducibility issues across data types, there will still be missing values that contribute to noise and inconsistency. In addition, a single protein might have multiple isoforms and multiple phosphorylation or ubiquitylation sites, with each potentially being unchanged, up- or down-regulated, in opposing ways. How to address this nonlinear integration is just the first step. Interpreting integrated data can also be challenging, as tracking meaningful changes across thousands of proteins, and forming testable hypotheses can lead to over- or under-interpretation. Finally, testing the importance of identified proteins or PTMs can be difficult. Genetic manipulation through knock-out, knock-down, or mutation can be complicated by cellular and molecular redundancies. Additionally, to validate the specific modified site, site directed mutagenesis of endogenous genes needs to be performed, which is not established widely and in high-throughput fashion. Drug treatment can also complicate interpretation since there can be nonspecific interactions or cell-type related biology involved. Thus a multi-pronged approach combining multiple functional validations can be key, though are often much lower in throughput. For instance, in Bouhaddou et al. parallel siRNA knockdowns and inhibitor treatments targeting the same p38 pathway were performed to demonstrate the p38 pathway as an essential signaling pathway for SARS-CoV-2128.
What are strategies to monitor COVID-19 pathology and investigate treatment strategies?
Managing the SARS-CoV-2 pandemic requires rapid and reliable diagnostics to trace disease spread, biomarkers to monitor disease severity, efficacious drug treatment to improve patient outcomes, and the development of vaccines to prevent the spread of disease. In addition to genomic-based approaches, proteomics might aid the high volume of diagnostic testing required for contact-tracing or be utilized to identify and monitor biomarkers of disease severity or treatment response (Figure 2). Improving patient outcomes also requires the identification of safe and effective drug treatments. As mentioned above, studying SARS-CoV-2 replication inside host cells can identify druggable targets. Proteomic technologies can also be employed subsequently to investigate binding of drugs to those targets and reveal potential off-target binding. These technologies include limited proteolysis-coupled mass spectrometry (LiP-MS), thermal proteome profiling (TPP), and activity-based protein profiling (ABPP) (Figure 2).
Targeted proteomics for monitoring disease progression and treatment response
Most biological samples taken from individuals suspected with COVID-19 are nasopharyngeal swabs, bronchoalveolar lavages, gargle samples, and blood samples, all of which represent complex proteomes with large dynamic range of protein concentrations. Thus a highly sensitive method is required to allow for detection of SARS-CoV-2 proteins in complex diagnostic testing samples. Targeted proteomics approaches in which a predefined set of proteins and their corresponding peptides are selectively and recursively isolated and then fragmented over their chromatographic elution time, offers highest sensitivity and quantitative accuracy162. Several studies developed parallel reaction monitoring (PRM) assays for the detection of SARS-CoV-2 proteins163–166, which were subsequently tested in dilute patient gargle samples164, nasopharyngeal swab166 and bronchoalveolar lavage samples165. While these studies demonstrated that it is theoretically possible to detect viral proteins in patient samples using targeted proteomics, compared to PCR-based assays that can experimentally amplify signal and have high-throughput efficiency, the proteomic approach is limited by low sample throughput and sensitivity. Given these limitations proteomics approaches are to date not suitable for diagnostic purposes of COVID-19.
However, proteomics approaches might be powerful to study the biological factors contributing to COVID-19 severity and patient outcomes and identify potential predictive biomarkers. Patients with more severe COVID-19 infections accompanied with rapid deterioration of lung function are distinguished by significant immune dysregulation. The biological pathways that drive disease severity and lead to immune dysregulation remain poorly understood. The identification of differentially regulated proteins and pathways in plasma samples derived from uninfected and infected patient groups with varying disease severity (i.e. asymptomatic, moderate, and severe) would not only provide more information about the biological nature of the dysregulation, but might deliver biomarkers for predicting disease severity that can be measured non-invasively. The complexity and dynamic concentration range of the plasma proteome has posed challenges for reproducible and sensitive MS-based plasma proteome profiling162. However, the performance of MS-based proteomics has matured reaching a sensitivity and dynamic range which allows quantifying 500–700 proteins routinely and reproducibly across large patient cohorts which makes it interesting for biomarker studies167,168. These molecular measurements can then be correlated with clinical parameters to identify predictive biomarkers167,168. Using unbiased proteomics to discover biomarkers for disease severity has the advantage that it might lead to more specific biomarkers for COVID-19 compared to established serological assays for cytokine and inflammatory proteins.
To identify potential biomarkers of COVID-19 severity, Messner et al. developed a lowcost and high-throughput platform which can handle 180 plasma samples within a single day169. Using patient sera and plasma from hospitalized patients they identified 27 putative proteins that are differentially expressed depending on the WHO severity grade of COVID-19169. These proteins include complement factors, proteins of the coagulation system, inflammatory modulators, and pro-inflammatory signaling molecules, thus capturing the host response to SARS-CoV-2 infection169. If validated in large independent patient cohorts, targeted proteomics assays which in contrast to immunoassays do not rely on specific antibodies could be developed rapidly for the proteomic signature to support clinical decision making and monitor treatment response162.
Limited proteolysis-mass spectrometry (LiP-MS)
Limited proteolysis (LiP) coupled with quantitative mass spectrometry (LiP-MS) is a powerful approach for systematically characterizing protein conformational changes resulting from ligand or protein binding. In LiP, a broad specificity protease used at low enzyme to substrate ratio for a short time digests native protein extracts to generate large polypeptide fragments dependent on the protein’s structural properties170. The structure-specific protein fragments are then denatured and subjected to tryptic digestion to generate peptides that are amenable for MS analysis170. An aliquot of the protein extract prior to LiP is fully digested with trypsin as a control and compared to the LiP sample171. When analyzed by quantitative MS, the tryptic peptides containing the LiP sites, so called conformotypic peptides, should have a lower abundance in the LiP digested sample compared to the trypsin-only control. Comparison of proteolytic signatures from samples that have been subjected to different conditions allows the identification of protein regions that underwent structural changes as a response to the perturbation171.
Piazza et al. developed a chemoproteomics approach to systematically determine metabolite-protein interactions by combining LiP with quantitative MS on a cell extract in the presence and absence of metabolites to assess metabolite-induced structural alterations on a proteome-wide scale172. The LiP-small molecule mapping (LiP-SMap) approach was applied to Eschericia coli to map the metabolite-protein interactions of 20 metabolites in an unbiased manner. This allowed not only the identification of 1,678 protein-metabolite interactions with the majority being novel interactions, but also the determination of the structural regions that are affected by metabolite binding172 which were found to be in close proximity to the binding site. Recently, the LiP-SMap approach was extended to enable systematic investigation of protein-small molecule interactions in complex eukaryotic proteomes173. Due to the higher complexity of the eukaryotic proteomes resulting in the initial discovery of multiple drug target candidates, Piazza et al. developed the machine-learning based LiP-Quant workflow which performs LiP on lysate treated with a drug dilution series to score and prioritize target candidates173. While the initial LiP-SMap identified 37 putative drug targets for cells treated with Rapamycin, the additional LiP-Quant scoring method could confirm FKBP1A as the highest-ranking candidate protein target173. As of this publication, there have been no published studies using LiP-MS to study SARS-CoV-2 or any recently identified drugs that are in or being considered for clinical trials. However, in light of the ongoing large scale drug discovery efforts for COVID-19, the relatively simple experimental design of LiP-MS and its broad applicability make it an ideal technique to identify cellular targets of existing drugs in clinical trials or for prioritizing drugs or antibodies based on their off-target reactivity.
Thermal proteome profiling (TPP)
Thermal protein profiling (TPP) combines cellular thermal shift assay (CETSA) with quantitative MS174,175. The basic principle of TPP relies on denaturation and aggregation of proteins in cells at their intrinsic melting temperature, which results in solubility changes of proteins with increasing temperatures. By quantifying the abundance of the protein remaining in solution after subjecting cells to heat spanning a predetermined temperature gradient, a melting profile for a protein can be established. Protein conformational changes upon binding of small molecules or other ligands change the thermal stability of a protein and alter the melting profile. The combination of TPP with multiplexed quantitative MS-based proteomics can systematically determine melting profiles and melting temperature shifts upon drug treatment for thousands of expressed proteins simultaneously to discover drug targets and off-target binding.
As a proof of concept, TPP was applied to the promiscuous kinase inhibitors staurosporine and GSK3182571 with a known spectrum of targets, which induced shifts in the melting temperatures of many kinase targets174. Interestingly, in addition to the proteins that are directly bound by the ligand, downstream pathway members like regulatory subunits of kinase complexes displayed thermal stability shifts, likely as a result of altered post-translational modifications, thus demonstrating that TPP could inform the drug’s mechanism of action174. The application of TPP was initially limited to soluble, mainly cytosolic proteins. However, since many ligand-binding receptors and drug targets represent transmembrane proteins, the method was extended to profile membrane-protein targets of small molecules by addition of a mild detergent during cell lysis which allows solubilization of membranes but does not solubilize protein aggregates176. While initially only applied to cultured cells, most recently the method has been extended to tissue (tissue-TPP) and blood (blood-TPP) specimens177. Therefore, target and off-target binding could be characterized in primary in vivo studies, which is critical to predict adverse reactions of drugs. Following intravenous administration of the HDAC inhibitor panobinostat, TPP identified Hdac1, Hdac2 and Ttc38 as known targets across all analyzed tissues, including spleen, liver, kidney, and lung. However, different off-target profiles were obtained across the different tissues which could be explained by heterogeneous levels of protein expression and drug exposure comparing the different tissues177. A recent study applied TPP to understand off-target effects of Remdesivir, a repurposed drug for treating COVID-19 patients, in uninfected HepG2 cells and identified the hexameric AAA+ ATPase Trip13 as a target of Remdesivir178. Further experiments are necessary to investigate the relevance of the Remdesivir-Trip13 interaction for SARS-CoV-2, and in fact there is currently a lack of evidence that Remdesivir is effective against SARS-CoV-218.
Activity-based protein profiling (ABPP)
Activity-based protein profiling (ABPP) is a sophisticated chemical proteomic strategy that can be used to systematically interrogate cellular enzymes and discover in vivo inhibitors of enzymatic activity (reviewed by Niphakis et al.179 and Kahler et al.180). In ABPP, activity-based probes target a specific activity or structural feature of enzyme active sites in order to covalently modify the target with a reporter tag. The activity-based probe will have: 1) a reactive group that forms the covalent bond to the target protein; 2) a binding group that directs the probe to active sites and typically mimics natural substrates; and 3) a reporter/detection tag that allows measurement of the probe labeling. Depending on the assay and experimental design, labeled events can be visualized by fluorescence microscopy, flow cytometry, or SDS-PAGE, detected in vivo by radio-isotope detection, or can be enriched by affinity purification and analyzed by MS181,182. Since probe labeling is dependent on active site accessibility, it can differentiate between inactive and active states of selective targets that would otherwise react to the probe, thus providing a unique opportunity to profile cells in various environmental, genetic, or conditional backgrounds. A number of enzymatic activities can be targeted, including cysteine proteases, cathepsins, kinases, metalloproteases, serine proteases, and oxidoreductases179,180,183.
There are two main ABPP strategies that can be employed, those that are chemocentric and focused on the small molecule(s), and a target-centric approach which focuses on the enzyme. Chemocentric ABPP strategies can be used to discover cellular enzymes that are targets of specific drugs. This type of strategy is exemplified in an ABPP-based study that determined prodrug dimethyl fumarate (DMF), and not monomethyl fumarate (MMF) that DMF is converted to, is responsible for blocking activation of primary T cells in human and mice184. Activity-based probes have also been used to screen for drugs against specific targets. In an example of a target-based strategy, ABPP was used to identify targets of cellular ABHD2, a serine hydrolase involved in immune response, virus replication, and fertility185. In this study, novel inhibitors of ABHD2 that could be used to modulate sperm fertility were discovered and probed for off-target profiles. As of this publication, there have been no published studies using ABPP to study SARS-CoV-2 or any recently identified drugs that are in or being considered for clinical trials. One of the strengths of ABPP approaches is their applicability to detecting these events in cell models, primary cells, or in vivo animal models. Taken together, these approaches are particularly powerful for identifying protein/enzymatic cellular targets of drugs, characterizing and screening drugs against specific targets, and informing drug efficacy and safety as they are equipped to assess interaction and activity of compounds as well as identify off-target effects. While ABPP methods are still very specialized in their broad application, they could provide unique insight to help identify cellular targets of existing drugs in clinical trials or for prioritizing drugs based on their off-target reactivity for treatments of SARS-CoV-2.
Prioritizing candidates from large-scale SARS-CoV-2 drug screens
Large drug screening efforts are ongoing to identify drug candidates that could be repurposed as SARS-CoV-2 antivirals186. To prioritize drugs that showed promising results in in vitro systems for in vivo testing, it is imperative to assess target and off-target binding of promising small molecules from large-scale screens before moving them to animal models and testing them in humans. In light of the broad applicability of TPP, LiP-MS and ABPP, these represent individually or in combination promising strategies for small molecule prioritization. To assess the effectiveness of novel or repurposed drugs for SARS-CoV-2 treatment in a non-invasive manner, drug discovery efforts could be accompanied with biomarker discovery and validation studies relying on global protein abundance and targeted proteomics approaches to identify biomarkers for monitoring treatment response in body fluids.
Conclusion
The SARS-CoV-2 pandemic necessitates the urgent study of viral protein structure, viral replication, viral-host interaction, and host response to gain molecular understanding of pathogenicity and to investigate strategies for treatment options. The field of proteomics is wellpositioned to inform open questions as discussed in this perspective, and indeed there is a precedent of important proteomic contributions in the study of other viral infections including HIV and Influenza, among others. The pandemic has also impressed upon the need for cross-platform collaborations to quickly investigate, translate, and apply proteomic research into clinical outcomes. This type of rapid-response requires cross-discipline expertise and cooperation to not only generate, analyze and integrate the data, but to interpret, functionally validate, and translate findings into actionable hypotheses that can influence meaningful clinical studies161.
Proteomic techniques are strikingly dynamic in the size and scale of the questions they can answer, including the study of viral infection from the structure of a single viral protein to tracking global changes in the human proteome during infection. Beyond size and scale, proteomics can also provide insight into timing and cellular location, which are critical to understanding how to combat SARS-CoV-2 infection and manage COVID-19 pathology. The application of existing proteomic tools to questions of SARS-CoV-2 biology is pressingly important, and this unprecedented pandemic also invokes an opportunity for creativity in developing new tools, and applying and integrating tools in new ways. As proteomic scientists, it is imperative that we contribute our unique proteomic perspective to open questions about SARS-CoV-2 biology, so we can provide an essential complement to studies in other fields as the scientific community works together to meet this monumental challenge. In fact, a number of studies have already demonstrated the impact and breadth of work that can be carried out by rapid response of international collaborations between interdisciplinary scientists.
ACKNOWLEDGMENT
We would like to thank Kirsten Obernier, Alicia Richards, Helene Foussard, and Jeffrey R. Johnson for critical reading and editing the manuscript and the whole Krogan lab for feedback on the structure of the perspective and figure design. Figure 1 was adapted from “Coronavirus Replication Cycle”, by BioRender.com (2020). Retrieved from https://app.biorender.com/biorender-templates.
Funding Sources
This research was supported by grants from the National Institutes of Health (P50AI150476, U19AI135990, U19AI135972, R01AI143292 and R01AI120694 to N.J.K., R01AI122747 to R.M.K.), Defense Advance Research Projects Agency HR0011-19-2-0020 (to N.J.K. and R.H.), by the Laboratory for Genomics Research (LGR) Excellence in Research Award (ERA) from the Innovative Genomics Institute at UC Berkeley (Grantnumber 133122P), and by funding from F. Hoffmann-La Roche and Vir Biotechnology and gifts from The RonConway Family to N.J.K.
ABBREVIATIONS
- ABPP
activity-based protein profiling
- AP-MS
affinity purification-mass spectrometry
- CCPP
complex centric proteome profiling
- CETSA
cellular thermal shift assay
- CID
collision-induced dissociation
- COVID-19
Coronavirus Disease 2019
- cryo-EM
cryo-electron microscopy
- cryo-ET
cryo-electron tomography
- DIA
data-independent acquisition
- DMF
dimethyl fumarate
- DTSSP
3,3’-dithiobis(sulfo-succinimidylpropionate)
- E
envelope protein
- ER
endoplasmic reticulum
- ERGIC
ER-Golgi intermediate compartment
- FDA
US Food and Drug Administration
- GFR
growth factor receptor
- GPCRs
G-protein coupled receptors
- H/DX-MS
hydrogen/deuterium exchange mass spectrometry
- LiP-MS
limited proteolysis-coupled mass spectrometry
- LiP-SMap
limited proteolysis-small molecule mapping
- LiP
limited proteolysis
- M-PMV
Mason-Pfizer monkey virus
- M
membrane protein
- MMF
monomethyl fumarate
- N
nucleocapsid protein
- nMS
native mass spectrometry
- NPC
nuclear pore complex
- Nsps
non-structural proteins
- Orfs
open reading frames
- PDL-MS
proximity-dependent labeling mass spectrometry
- PDL
proximity-dependent labeling
- PPIs
protein-protein interactions
- PRM
parallel reaction monitoring
- PTMs
post-translational modifications
- RBD
receptor-binding domain
- RTCs
replication and transcription complexes
- S
Spike protein
- SARS-CoV-2
severe acute respiratory syndrome coronavirus 2
- TDMS
top-down mass spectrometry
- TPP
thermal proteome profiling
- UPS
ubiquitin-proteasome system
- VLPs
virus-like particles
- XL-MS
cross-linking mass spectrometry
REFERENCES
- (1).Su S; Wong G; Shi W; Liu J; Lai ACK; Zhou J; Liu W; Bi Y; Gao GF Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24 (6), 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Center for Disease Control and Prevention Human Coronavirus Types https://www.cdc.gov/coronavirus/types.html (accessed December 17, 2020).
- (3).Centers for Disease Control and Prevention Common Human Coronaviruses https://www.cdc.gov/coronavirus/general-information.html (accessed December 17, 2020).
- (4).van der Hoek L. Human Coronaviruses: What Do They Cause? Antivir. Ther. 2007, 12 (4 Pt B), 651–658. [PubMed] [Google Scholar]
- (5).Drosten C; Günther S; Preiser W; van der Werf S; Brodt H-R; Becker S; Rabenau H; Panning M; Kolesnikova L; Fouchier RAM; Berger A; Burguière A-M; Cinatl J; Eickmann M; Escriou N; Grywna K; Kramme S; Manuguerra J-C; Müller S; Rickerts V; Stürmer M; Vieth S; Klenk H-D; Osterhaus ADME; Schmitz H; Doerr HW Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med 2003, 348 (20), 1967–1976. [DOI] [PubMed] [Google Scholar]
- (6).Zaki AM; van Boheemen S; Bestebroer TM; Osterhaus ADME; Fouchier RAM Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med 2012, 367 (19), 1814–1820. [DOI] [PubMed] [Google Scholar]
- (7).Wu F; Zhao S; Yu B; Chen Y-M; Wang W; Song Z-G; Hu Y; Tao Z-W; Tian J-H; Pei Y-Y; Yuan M-L; Zhang Y-L; Dai F-H; Liu Y; Wang Q-M; Zheng J-J; Xu L; Holmes EC; Zhang Y-Z A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579 (7798), 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).de Wit E; van Doremalen N; Falzarano D; Munster VJ SARS and MERS: Recent Insights into Emerging Coronaviruses. Nat. Rev. Microbiol 2016, 14 (8), 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Petersen E; Koopmans M; Go U; Hamer DH; Petrosillo N; Castelli F; Storgaard M; Al Khalili S; Simonsen L. Comparing SARS-CoV-2 with SARS-CoV and Influenza Pandemics. Lancet Infect. Dis 2020. 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Centers for Disease Control and Prevention SARS Basics Fact Sheet https://www.cdc.gov/sars/about/fs-sars.html (accessed September 28, 2020).
- (11).World Health Organization Middle East Respiratory Syndrome Coronavirus https://www.who.int/emergencies/mers-cov/en/ (accessed September 28, 2020).
- (12).World Health Organization Coronavirus Disease (COVID-19) Dashboard https://covid19.who.int/ (accessed December 22, 2020).
- (13).World Health Organization COVID-19 Vaccines https://www.who.int/emergencies/diseases/novel-coronavirus-2019/covid-19-vaccines (accessed December 17, 2020).
- (14).US Food and Drug Administration FDA Takes Key Action in Fight Against COVID-19 By Issuing Emergency Use Authorization for First COVID-19 Vaccine https://www.fda.gov/news-events/press-announcements/fda-takes-key-action-fight-against-covid-19-issuing-emergency-use-authorization-first-covid-19 (accessed December 17, 2020).
- (15).US Food & Drug Administration Moderna COVID-19 Vaccine https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019covid-19/moderna-covid-19-vaccine (accessed December 21, 2020).
- (16).Ashraf BN Economic Impact of Government Interventions during the COVID-19 Pandemic: International Evidence from Financial Markets. J Behav Exp Finance 2020, 27, 100371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Grein J; Ohmagari N; Shin D; Diaz G; Asperges E; Castagna A; Feldt T; Green G; Green ML; Lescure F-X; Nicastri E; Oda R; Yo K; Quiros-Roldan E; Studemeister A; Redinski J; Ahmed S; Bernett J; Chelliah D; Chen D; Chihara S; Cohen SH; Cunningham J; D’Arminio Monforte A; Ismail S; Kato H; Lapadula G; L’Her E; Maeno T; Majumder S; Massari M; Mora-Rillo M; Mutoh Y; Nguyen D; Verweij E; Zoufaly A; Osinusi AO; DeZure A; Zhao Y; Zhong L; Chokkalingam A; Elboudwarej E; Telep L; Timbs L; Henne I; Sellers S; Cao H; Tan SK; Winterbourne L; Desai P; Mera R; Gaggar A; Myers RP; Brainard DM; Childs R; Flanigan T. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med 2020, 382 (24), 2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).World Health Organization WHO recommends against the use of remdesivir in COVID-19 patients https://www.who.int/news-room/feature-stories/detail/who-recommends-against-the-use-of-remdesivir-in-covid-19-patients (accessed December 17, 2020).
- (19).Kumar S; Nyodu R; Maurya VK; Saxena SK Morphology, Genome Organization, Replication, and Pathogenesis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). In Coronavirus Disease 2019 (COVID-19): Epidemiology, Pathogenesis, Diagnosis, and Therapeutics; Saxena SK, Ed.; Springer Singapore: Singapore, 2020; pp 23–31. [Google Scholar]
- (20).Lu R; Zhao X; Li J; Niu P; Yang B; Wu H; Wang W; Song H; Huang B; Zhu N; Bi Y; Ma X; Zhan F; Wang L; Hu T; Zhou H; Hu Z; Zhou W; Zhao L; Chen J; Meng Y; Wang J; Lin Y; Yuan J; Xie Z; Ma J; Liu WJ; Wang D; Xu W; Holmes EC; Gao GF; Wu G; Chen W; Shi W; Tan W. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395 (10224), 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wan Y; Shang J; Graham R; Baric RS; Li F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol 2020, 94 (7). 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chan JF-W; Kok K-H; Zhu Z; Chu H; To KK-W; Yuan S; Yuen K-Y Genomic Characterization of the 2019 Novel Human-Pathogenic Coronavirus Isolated from a Patient with Atypical Pneumonia after Visiting Wuhan. Emerg. Microbes Infect 2020, 9 (1), 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fung TS; Liu DX Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol 2019, 73, 529–557. [DOI] [PubMed] [Google Scholar]
- (24).Hoffmann M; Kleine-Weber H; Schroeder S; Krüger N; Herrler T; Erichsen S; Schiergens TS; Herrler G; Wu N-H; Nitsche A; Müller MA; Drosten C; Pöhlmann S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181 (2), 271–280.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).V’kovski P; Kratzel A; Steiner S; Stalder H; Thiel V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol 2020. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sola I; Almazán F; Zúñiga S; Enjuanes L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu Rev Virol 2015, 2 (1), 265–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Dülfer J; Kadek A; Kopicki J-D; Krichel B; Uetrecht C. Structural Mass Spectrometry Goes Viral. Adv. Virus Res 2019, 105, 189–238. [DOI] [PubMed] [Google Scholar]
- (28).Iacobucci C; Götze M; Sinz A. Cross-Linking/mass Spectrometry to Get a Closer View on Protein Interaction Networks. Curr. Opin. Biotechnol 2020, 63, 48–53. [DOI] [PubMed] [Google Scholar]
- (29).Piotrowski C; Sinz A. Structural Investigation of Proteins and Protein Complexes by Chemical Cross-Linking/Mass Spectrometry. Adv. Exp. Med. Biol 2018, 1105, 101–121. [DOI] [PubMed] [Google Scholar]
- (30).Chavez JD; Bruce JE Chemical Cross-Linking with Mass Spectrometry: A Tool for Systems Structural Biology. Curr. Opin. Chem. Biol 2019, 48, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).O’Reilly FJ; Rappsilber J. Cross-Linking Mass Spectrometry: Methods and Applications in Structural, Molecular and Systems Biology. Nat. Struct. Mol. Biol 2018, 25 (11), 1000–1008. [DOI] [PubMed] [Google Scholar]
- (32).Rappsilber J. The Beginning of a Beautiful Friendship: Cross-Linking/mass Spectrometry and Modelling of Proteins and Multi-Protein Complexes. J. Struct. Biol 2011, 173 (3), 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yılmaz Ş; Shiferaw GA; Rayo J; Economou A; Martens L; Vandermarliere E. Cross-Linked Peptide Identification: A Computational Forest of Algorithms. Mass Spectrom. Rev 2018, 37 (6), 738–749. [DOI] [PubMed] [Google Scholar]
- (34).Liu F; Heck AJR Interrogating the Architecture of Protein Assemblies and Protein Interaction Networks by Cross-Linking Mass Spectrometry. Curr. Opin. Struct. Biol 2015, 35, 100–108. [DOI] [PubMed] [Google Scholar]
- (35).Lin DH; Hoelz A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem 2019, 88, 725–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Fernandez-Martinez J; Kim SJ; Shi Y; Upla P; Pellarin R; Gagnon M; Chemmama IE; Wang J; Nudelman I; Zhang W; Williams R; Rice WJ; Stokes DL; Zenklusen D; Chait BT; Sali A; Rout MP Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell 2016, 167 (5), 1215–1228.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tüting C; Iacobucci C; Ihling CH; Kastritis PL; Sinz A. Structural Analysis of 70S Ribosomes by Cross-Linking/mass Spectrometry Reveals Conformational Plasticity. Sci. Rep 2020, 10 (1), 12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Klykov O; van der Zwaan C; Heck AJR; Meijer AB; Scheltema RA Missing Regions within the Molecular Architecture of Human Fibrin Clots Structurally Resolved by XL-MS and Integrative Structural Modeling. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (4), 1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kwon Y; Kaake RM; Echeverria I; Suarez M; Karimian Shamsabadi M; Stoneham C; Ramirez PW; Kress J; Singh R; Sali A; Krogan N; Guatelli J; Jia X. Structural Basis of CD4 Downregulation by HIV-1 Nef. Nat. Struct. Mol. Biol 2020. 10.1038/s41594-020-0463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Meyer NL; Hu G; Davulcu O; Xie Q; Noble AJ; Yoshioka C; Gingerich DS; Trzynka A; David L; Stagg SM; Chapman MS Structure of the Gene Therapy Vector, Adeno-Associated Virus with Its Cell Receptor, AAVR. Elife 2019, 8. 10.7554/eLife.44707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Prchal J; Sýs J; Junková P; Lipov J; Ruml T. The Interaction Interface of Mason-Pfizer Monkey Virus Matrix and Envelope Proteins. J. Virol 2020. 10.1128/JVI.01146-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yu YT-C; Chien S-C; Chen I-Y; Lai C-T; Tsay Y-G; Chang SC; Chang M-F Surface Vimentin Is Critical for the Cell Entry of SARS-CoV. J. Biomed. Sci 2016, 23, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Yao H; Song Y; Chen Y; Wu N; Xu J; Sun C; Zhang J; Weng T; Zhang Z; Wu Z; Cheng L; Shi D; Lu X; Lei J; Crispin M; Shi Y; Li L; Li S. Molecular Architecture of the SARS-CoV-2 Virus. Cell 2020, 183 (3), 730–738.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Liu C; Mendonça L; Yang Y; Gao Y; Shen C; Liu J; Ni T; Ju B; Liu C; Tang X; Wei J; Ma X; Zhu Y; Liu W; Xu S; Liu Y; Yuan J; Wu J; Liu Z; Zhang Z; Liu L; Wang P; Zhang P. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure 2020, 28 (11), 1218–1224.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Konermann L; Pan J; Liu Y-H Hydrogen Exchange Mass Spectrometry for Studying Protein Structure and Dynamics. Chem. Soc. Rev 2011, 40 (3), 1224–1234. [DOI] [PubMed] [Google Scholar]
- (46).Hvidt A; Nielsen SO Hydrogen Exchange in Proteins. Adv. Protein Chem 1966, 21, 287–386. [DOI] [PubMed] [Google Scholar]
- (47).Englander SW; Kallenbach NR Hydrogen Exchange and Structural Dynamics of Proteins and Nucleic Acids. Q. Rev. Biophys 1983, 16 (4), 521–655. [DOI] [PubMed] [Google Scholar]
- (48).Masson GR; Burke JE; Ahn NG; Anand GS; Borchers C; Brier S; Bou-Assaf GM; Engen JR; Englander SW; Faber J; Garlish R; Griffin PR; Gross ML; Guttman M; Hamuro Y; Heck AJR; Houde D; Iacob RE; Jørgensen TJD; Kaltashov IA; Klinman JP; Konermann L; Man P; Mayne L; Pascal BD; Reichmann D; Skehel M; Snijder J; Strutzenberg TS; Underbakke ES; Wagner C; Wales TE; Walters BT; Weis DD; Wilson DJ; Wintrode PL; Zhang Z; Zheng J; Schriemer DC; Rand KD Recommendations for Performing, Interpreting and Reporting Hydrogen Deuterium Exchange Mass Spectrometry (HDX-MS) Experiments. Nat. Methods 2019, 16 (7), 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Harrison RA; Engen JR Conformational Insight into Multi-Protein Signaling Assemblies by Hydrogen-Deuterium Exchange Mass Spectrometry. Curr. Opin. Struct. Biol 2016, 41, 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chalmers MJ; Busby SA; Pascal BD; West GM; Griffin PR Differential Hydrogen/deuterium Exchange Mass Spectrometry Analysis of Protein-Ligand Interactions. Expert Rev. Proteomics 2011, 8 (1), 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Englander JJ; Del Mar C; Li W; Englander SW; Kim JS; Stranz DD; Hamuro Y; Woods VL Jr. Protein Structure Change Studied by Hydrogen-Deuterium Exchange, Functional Labeling, and Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (12), 7057–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Englander SW; Mayne L. The Nature of Protein Folding Pathways. Proc. Natl. Acad. Sci. U. S. A 2014, 111 (45), 15873–15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Balasubramaniam D; Komives EA Hydrogen-Exchange Mass Spectrometry for the Study of Intrinsic Disorder in Proteins. Biochim. Biophys. Acta 2013, 1834 (6), 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Vadas O; Jenkins ML; Dornan GL; Burke JE Using Hydrogen-Deuterium Exchange Mass Spectrometry to Examine Protein-Membrane Interactions. Methods Enzymol. 2017, 583, 143–172. [DOI] [PubMed] [Google Scholar]
- (55).Rand KD; Adams CM; Zubarev RA; Jørgensen TJD Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. J. Am. Chem. Soc 2008, 130 (4), 1341–1349. [DOI] [PubMed] [Google Scholar]
- (56).Zehl M; Rand KD; Jensen ON; Jørgensen TJD Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc 2008, 130 (51), 17453–17459. [DOI] [PubMed] [Google Scholar]
- (57).Jørgensen TJD; Gårdsvoll H; Ploug M; Roepstorff P. Intramolecular Migration of Amide Hydrogens in Protonated Peptides upon Collisional Activation. J. Am. Chem. Soc 2005, 127 (8), 2785–2793. [DOI] [PubMed] [Google Scholar]
- (58).Ferguson PL; Pan J; Wilson DJ; Dempsey B; Lajoie G; Shilton B; Konermann L. Hydrogen/deuterium Scrambling during Quadrupole Time-of-Flight MS/MS Analysis of a Zinc-Binding Protein Domain. Anal. Chem 2007, 79 (1), 153–160. [DOI] [PubMed] [Google Scholar]
- (59).Sterling HJ; Williams ER Real-Time Hydrogen/deuterium Exchange Kinetics via Supercharged Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem 2010, 82 (21), 9050–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Resetca D; Wilson DJ Characterizing Rapid, Activity-Linked Conformational Transitions in Proteins via Sub-Second Hydrogen Deuterium Exchange Mass Spectrometry. FEBS J. 2013, 280 (22), 5616–5625. [DOI] [PubMed] [Google Scholar]
- (61).Pan J; Wilson DJ; Konermann L. Pulsed Hydrogen Exchange and Electrospray Charge-State Distribution as Complementary Probes of Protein Structure in Kinetic Experiments: Implications for Ubiquitin Folding. Biochemistry 2005, 44 (24), 8627–8633. [DOI] [PubMed] [Google Scholar]
- (62).Resetca D; Haftchenary S; Gunning PT; Wilson DJ Changes in Signal Transducer and Activator of Transcription 3 (STAT3) Dynamics Induced by Complexation with Pharmacological Inhibitors of Src Homology 2 (SH2) Domain Dimerization. J. Biol. Chem 2014, 289 (47), 32538–32547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zhu S; Shala A; Bezginov A; Sljoka A; Audette G; Wilson DJ Hyperphosphorylation of Intrinsically Disordered Tau Protein Induces an Amyloidogenic Shift in Its Conformational Ensemble. PLoS One 2015, 10 (3), e0120416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Khan AG; Whidby J; Miller MT; Scarborough H; Zatorski AV; Cygan A; Price AA; Yost SA; Bohannon CD; Jacob J; Grakoui A; Marcotrigiano J. Structure of the Core Ectodomain of the Hepatitis C Virus Envelope Glycoprotein 2. Nature 2014, 509 (7500), 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Garcia NK; Guttman M; Ebner JL; Lee KK Dynamic Changes during Acid-Induced Activation of Influenza Hemagglutinin. Structure 2015, 23 (4), 665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Davenport TM; Guttman M; Guo W; Cleveland B; Kahn M; Hu S-L; Lee KK Isolate-Specific Differences in the Conformational Dynamics and Antigenicity of HIV-1 gp120. J. Virol 2013, 87 (19), 10855–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Guttman M; Garcia NK; Cupo A; Matsui T; Julien J-P; Sanders RW; Wilson IA; Moore JP; Lee KK CD4-Induced Activation in a Soluble HIV-1 Env Trimer. Structure 2014, 22 (7), 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Guttman M; Váradi C; Lee KK; Guttman A. Comparative Glycoprofiling of HIV gp120 Immunogens by Capillary Electrophoresis and MALDI Mass Spectrometry. Electrophoresis 2015, 36 (11–12), 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Kim M; Sun Z-YJ; Rand KD; Shi X; Song L; Cheng Y; Fahmy AF; Majumdar S; Ofek G; Yang Y; Kwong PD; Wang J-H; Engen JR; Wagner G; Reinherz EL Antibody Mechanics on a Membrane-Bound HIV Segment Essential for GP41-Targeted Viral Neutralization. Nat. Struct. Mol. Biol 2011, 18 (11), 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Bale S; Liu T; Li S; Wang Y; Abelson D; Fusco M; Woods VL Jr; Saphire EO Ebola Virus Glycoprotein Needs an Additional Trigger, beyond Proteolytic Priming for Membrane Fusion. PLoS Negl. Trop. Dis 2011, 5 (11), e1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Ye Q; West AMV; Silletti S; Corbett KD Architecture and Self-Assembly of the SARS-CoV-2 Nucleocapsid Protein. bioRxiv 2020. 10.1101/2020.05.17.100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chandler SA; Benesch JL Mass Spectrometry beyond the Native State. Curr. Opin. Chem. Biol 2018, 42, 130–137. [DOI] [PubMed] [Google Scholar]
- (73).Brogan WC 3rd; Kemp PM; Bost RO; Glamann DB; Lange RA; Hillis LD Collection and Handling of Clinical Blood Samples to Assure the Accurate Measurement of Cocaine Concentration. J. Anal. Toxicol 1992, 16 (3), 152–154. [DOI] [PubMed] [Google Scholar]
- (74).Uetrecht C; Rose RJ; van Duijn E; Lorenzen K; Heck AJR Ion Mobility Mass Spectrometry of Proteins and Proteinassemblies. Chem. Soc. Rev 2010, pp 1633–1655. 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- (75).Krichel B; Falke S; Hilgenfeld R; Redecke L; Uetrecht C. Processing of the SARS-CoV pp1a/ab nsp7–10 Region. Biochem. J 2020, 477 (5), 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Abzalimov RR; Kaltashov IA Electrospray Ionization Mass Spectrometry of Highly Heterogeneous Protein Systems: Protein Ion Charge State Assignment via Incomplete Charge Reduction. Anal. Chem 2010, 82 (18), 7523–7526. [DOI] [PubMed] [Google Scholar]
- (77).Yang Y; Du Y; Kaltashov IA The Utility of Native MS for Understanding the Mechanism of Action of Repurposed Therapeutics in COVID-19: Heparin as a Disruptor of the SARS-CoV-2 Interaction with Its Host Cell Receptor. Anal. Chem 2020. 10.1021/acs.analchem.0c02449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Pierson EE; Keifer DZ; Selzer L; Lee LS; Contino NC; Wang JC-Y; Zlotnick A; Jarrold MF Detection of Late Intermediates in Virus Capsid Assembly by Charge Detection Mass Spectrometry. J. Am. Chem. Soc 2014, 136 (9), 3536–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Wang C; Li W; Drabek D; Okba NMA; van Haperen R; Osterhaus ADME; van Kuppeveld FJM; Haagmans BL; Grosveld F; Bosch B-J A Human Monoclonal Antibody Blocking SARS-CoV-2 Infection. Nat. Commun 2020, 11 (1), 2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Saphire EO; Schendel SL; Gunn BM; Milligan JC; Alter G. Antibody-Mediated Protection against Ebola Virus. Nat. Immunol 2018, 19 (11), 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Prabakaran P; Zhu Z; Xiao X; Biragyn A; Dimitrov AS; Broder CC; Dimitrov DS Potent Human Monoclonal Antibodies against SARS CoV, Nipah and Hendra Viruses. Expert Opin. Biol. Ther 2009, 9 (3), 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Srzentić K; Fornelli L; Tsybin YO; Loo JA; Seckler H; Agar JN; Anderson LC; Bai DL; Beck A; Brodbelt JS; van der Burgt YEM; Chamot-Rooke J; Chatterjee S; Chen Y; Clarke DJ; Danis PO; Diedrich JK; D’Ippolito RA; Dupré M; Gasilova N; Ge Y; Goo YA; Goodlett DR; Greer S; Haselmann KF; He L; Hendrickson CL; Hinkle JD; Holt MV; Hughes S; Hunt DF; Kelleher NL; Kozhinov AN; Lin Z; Malosse C; Marshall AG; Menin L; Millikin RJ; Nagornov KO; Nicolardi S; Paša-Tolić L; Pengelley S; Quebbemann NR; Resemann A; Sandoval W; Sarin R; Schmitt ND; Shabanowitz J; Shaw JB; Shortreed MR; Smith LM; Sobott F; Suckau D; Toby T; Weisbrod CR; Wildburger NC; Yates JR 3rd; Yoon SH; Young NL; Zhou M. Interlaboratory Study for Characterizing Monoclonal Antibodies by Top-Down and Middle-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom 2020, 31 (9), 1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Lermyte F; Tsybin YO; O’Connor PB; Loo JA Top or Middle? Up or Down? Toward a Standard Lexicon for Protein Top-Down and Allied Mass Spectrometry Approaches. J. Am. Soc. Mass Spectrom 2019, 30 (7), 1149–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Fornelli L; Toby TK; Schachner LF; Doubleday PF; Srzentić K; DeHart CJ; Kelleher NL Top-down Proteomics: Where We Are, Where We Are Going? J. Proteomics 2018, 175, 3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).McLafferty FW; Breuker K; Jin M; Han X; Infusini G; Jiang H; Kong X; Begley TP Top-down MS, a Powerful Complement to the High Capabilities of Proteolysis Proteomics. FEBS J. 2007, 274 (24), 6256–6268. [DOI] [PubMed] [Google Scholar]
- (86).Resemann A; Jabs W; Wiechmann A; Wagner E; Colas O; Evers W; Belau E; Vorwerg L; Evans C; Beck A; Suckau D. Full Validation of Therapeutic Antibody Sequences by Middle-up Mass Measurements and Middle-down Protein Sequencing. MAbs 2016, 8 (2), 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B; Lantz C; Jung W; Johnson KR; Koller A; Wolff JJ; Campuzano IDG; Auclair JR; Ivanov AR; Whitelegge JP; Paša-Tolić L; Chamot-Rooke J; Danis PO; Smith LM; Tsybin YO; Loo JA; Ge Y; Kelleher NL; Agar JN Best Practices and Benchmarks for Intact Protein Analysis for Top-down Mass Spectrometry. Nat. Methods 2019, 16 (7), 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Brodbelt JS Photodissociation Mass Spectrometry: New Tools for Characterization of Biological Molecules. Chem. Soc. Rev 2014, 43 (8), 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).McLafferty FW; Horn DM; Breuker K; Ge Y; Lewis MA; Cerda B; Zubarev RA; Carpenter BK Electron Capture Dissociation of Gaseous Multiply Charged Ions by Fourier-Transform Ion Cyclotron Resonance. J. Am. Soc. Mass Spectrom 2001, 12 (3), 245–249. [DOI] [PubMed] [Google Scholar]
- (90).Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (26), 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Kim SJ; Fernandez-Martinez J; Nudelman I; Shi Y; Zhang W; Raveh B; Herricks T; Slaughter BD; Hogan JA; Upla P; Chemmama IE; Pellarin R; Echeverria I; Shivaraju M; Chaudhury AS; Wang J; Williams R; Unruh JR; Greenberg CH; Jacobs EY; Yu Z; de la Cruz MJ; Mironska R; Stokes DL; Aitchison JD; Jarrold MF; Gerton JL; Ludtke SJ; Akey CW; Chait BT; Sali A; Rout MP Integrative Structure and Functional Anatomy of a Nuclear Pore Complex. Nature 2018, 555 (7697), 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Rout MP; Sali A. Principles for Integrative Structural Biology Studies. Cell 2019, 177 (6), 1384–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Gutierrez C; Chemmama IE; Mao H; Yu C; Echeverria I; Block SA; Rychnovsky SD; Zheng N; Sali A; Huang L. Structural Dynamics of the Human COP9 Signalosome Revealed by Cross-Linking Mass Spectrometry and Integrative Modeling. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (8), 4088–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Samavarchi-Tehrani P; Samson R; Gingras A-C Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell. Proteomics 2020, 19 (5), 757–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Heusel M; Bludau I; Rosenberger G; Hafen R; Frank M; Banaei-Esfahani A; van Drogen A; Collins BC; Gstaiger M; Aebersold R. Complex-Centric Proteome Profiling by SEC-SWATH-MS. Mol. Syst. Biol 2019, 15 (1), e8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Heusel M; Frank M; Köhler M; Amon S; Frommelt F; Rosenberger G; Bludau I; Aulakh S; Linder MI; Liu Y; Collins BC; Gstaiger M; Kutay U; Aebersold R. A Global Screen for Assembly State Changes of the Mitotic Proteome by SEC-SWATH-MS. Cell Syst 2020, 10 (2), 133–155.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Batra J; Hultquist JF; Liu D; Shtanko O; Von Dollen J; Satkamp L; Jang GM; Luthra P; Schwarz TM; Small GI; Arnett E; Anantpadma M; Reyes A; Leung DW; Kaake R; Haas P; Schmidt CB; Schlesinger LS; LaCount DJ; Davey RA; Amarasinghe GK; Basler CF; Krogan NJ Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175 (7), 1917–1930.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Eckhardt M; Zhang W; Gross AM; Von Dollen J; Johnson JR; Franks-Skiba KE; Swaney DL; Johnson TL; Jang GM; Shah PS; Brand TM; Archambault J; Kreisberg JF; Grandis JR; Ideker T; Krogan NJ Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus-Host Protein Network. Cancer Discov. 2018, 8 (11), 1474–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Jäger S; Cimermancic P; Gulbahce N; Johnson JR; McGovern KE; Clarke SC; Shales M; Mercenne G; Pache L; Li K; Hernandez H; Jang GM; Roth SL; Akiva E; Marlett J; Stephens M; D’Orso I; Fernandes J; Fahey M; Mahon C; O’Donoghue AJ; Todorovic A; Morris JH; Maltby DA; Alber T; Cagney G; Bushman FD; Young JA; Chanda SK; Sundquist WI; Kortemme T; Hernandez RD; Craik CS; Burlingame A; Sali A; Frankel AD; Krogan NJ Global Landscape of HIV-Human Protein Complexes. Nature 2011, 481 (7381), 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Jean Beltran PM; Federspiel JD; Sheng X; Cristea IM Proteomics and Integrative Omic Approaches for Understanding Host-Pathogen Interactions and Infectious Diseases. Mol. Syst. Biol 2017, 13 (3), 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Luo Y; Jacobs EY; Greco TM; Mohammed KD; Tong T; Keegan S; Binley JM; Cristea IM; Fenyö D; Rout MP; Chait BT; Muesing MA HIV-Host Interactome Revealed Directly from Infected Cells. Nat Microbiol 2016, 1 (7), 16068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Shah PS; Link N; Jang GM; Sharp PP; Zhu T; Swaney DL; Johnson JR; Von Dollen J; Ramage HR; Satkamp L; Newton B; Hüttenhain R; Petit MJ; Baum T; Everitt A; Laufman O; Tassetto M; Shales M; Stevenson E; Iglesias GN; Shokat L; Tripathi S; Balasubramaniam V; Webb LG; Aguirre S; Willsey AJ; Garcia-Sastre A; Pollard KS; Cherry S; Gamarnik AV; Marazzi I; Taunton J; FernandezSesma A; Bellen HJ; Andino R; Krogan NJ Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175 (7), 1931–1945.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Watanabe T; Kawakami E; Shoemaker JE; Lopes TJS; Matsuoka Y; Tomita Y; Kozuka-Hata H; Gorai T; Kuwahara T; Takeda E; Nagata A; Takano R; Kiso M; Yamashita M; Sakai-Tagawa Y; Katsura H; Nonaka N; Fujii H; Fujii K; Sugita Y; Noda T; Goto H; Fukuyama S; Watanabe S; Neumann G; Oyama M; Kitano H; Kawaoka Y. Influenza Virus-Host Interactome Screen as a Platform for Antiviral Drug Development . Cell Host Microbe 2014, 16 (6), 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Gordon DE; Jang GM; Bouhaddou M; Xu J; Obernier K; White KM; O’Meara MJ; Rezelj VV; Guo JZ; Swaney DL; Tummino TA; Huettenhain R; Kaake RM; Richards AL; Tutuncuoglu B; Foussard H; Batra J; Haas K; Modak M; Kim M; Haas P; Polacco BJ; Braberg H; Fabius JM; Eckhardt M; Soucheray M; Bennett MJ; Cakir M; McGregor MJ; Li Q; Meyer B; Roesch F; Vallet T; Mac Kain A; Miorin L; Moreno E; Naing ZZC; Zhou Y; Peng S; Shi Y; Zhang Z; Shen W; Kirby IT; Melnyk JE; Chorba JS; Lou K; Dai SA; Barrio-Hernandez I; Memon D; Hernandez-Armenta C; Lyu J; Mathy CJP; Perica T; Pilla KB; Ganesan SJ; Saltzberg DJ; Rakesh R; Liu X; Rosenthal SB; Calviello L; Venkataramanan S; Liboy-Lugo J; Lin Y; Huang X-P; Liu Y; Wankowicz SA; Bohn M; Safari M; Ugur FS; Koh C; Savar NS; Tran QD; Shengjuler D; Fletcher SJ; O’Neal MC; Cai Y; Chang JCJ; Broadhurst DJ; Klippsten S; Sharp PP; Wenzell NA; Kuzuoglu D; Wang H-Y; Trenker R; Young JM; Cavero DA; Hiatt J; Roth TL; Rathore U; Subramanian A; Noack J; Hubert M; Stroud RM; Frankel AD; Rosenberg OS; Verba KA; Agard DA; Ott M; Emerman M; Jura N; von Zastrow M; Verdin E; Ashworth A; Schwartz O; d’Enfert C; Mukherjee S; Jacobson M; Malik HS; Fujimori DG; Ideker T; Craik CS; Floor SN; Fraser JS; Gross JD; Sali A; Roth BL; Ruggero D; Taunton J; Kortemme T; Beltrao P; Vignuzzi M; García-Sastre A; Shokat KM; Shoichet BK; Krogan NJ A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Gordon DE; Hiatt J; Bouhaddou M; Rezelj VV; Ulferts S; Braberg H; Jureka AS; Obernier K; Guo JZ; Batra J; Kaake RM; Weckstein AR; Owens TW; Gupta M; Pourmal S; Titus EW; Cakir M; Soucheray M; McGregor M; Cakir Z; Jang G; O’Meara MJ; Tummino TA; Zhang Z; Foussard H; Rojc A; Zhou Y; Kuchenov D; Hüttenhain R; Xu J; Eckhardt M; Swaney DL; Fabius JM; Ummadi M; Tutuncuoglu B; Rathore U; Modak M; Haas P; Haas KM; Naing ZZC; Pulido EH; Shi Y; Barrio-Hernandez I; Memon D; Petsalaki E; Dunham A; Marrero MC; Burke D; Koh C; Vallet T; Silvas JA; Azumaya CM; Billesbølle C; Brilot AF; Campbell MG; Diallo A; Dickinson MS; Diwanji D; Herrera N; Hoppe N; Kratochvil HT; Liu Y; Merz GE; Moritz M; Nguyen HC; Nowotny C; Puchades C; Rizo AN; Schulze-Gahmen U; Smith AM; Sun M; Young ID; Zhao J; Asarnow D; Biel J; Bowen A; Braxton JR; Chen J; Chio CM; Chio US; Deshpande I; Doan L; Faust B; Flores S; Jin M; Kim K; Lam VL; Li F; Li J; Li Y-L; Li Y; Liu X; Lo M; Lopez KE; Melo AA; Moss FR 3rd; Nguyen P; Paulino J; Pawar KI; Peters JK; Pospiech TH Jr; Safari M; Sangwan S; Schaefer K; Thomas PV; Thwin AC; Trenker R; Tse E; Tsui TKM; Wang F; Whitis N; Yu Z; Zhang K; Zhang Y; Zhou F; Saltzberg D; QCRG Structural Biology Consortium; Hodder AJ; Shun-Shion AS; Williams DM; White KM; Rosales R; Kehrer T; Miorin L; Moreno E; Patel AH; Rihn S; Khalid MM; Vallejo-Gracia A; Fozouni P; Simoneau CR; Roth TL; Wu D; Karim MA; Ghoussaini M; Dunham I; Berardi F; Weigang S; Chazal M; Park J; Logue J; McGrath M; Weston S; Haupt R; Hastie CJ; Elliott M; Brown F; Burness KA; Reid E; Dorward M; Johnson C; Wilkinson SG; Geyer A; Giesel DM; Baillie C; Raggett S; Leech H; Toth R; Goodman N; Keough KC; Lind AL; Zoonomia Consortium; Klesh RJ; Hemphill KR; Carlson-Stevermer J; Oki J; Holden K; Maures T; Pollard KS; Sali A; Agard DA; Cheng Y; Fraser JS; Frost A; Jura N; Kortemme T; Manglik A; Southworth DR; Stroud RM; Alessi DR; Davies P; Frieman MB; Ideker T; Abate C; Jouvenet N; Kochs G; Shoichet B; Ott M; Palmarini M; Shokat KM; García-Sastre A; Rassen JA; Grosse R; Rosenberg OS; Verba KA; Basler CF; Vignuzzi M; Peden AA; Beltrao P; Krogan NJ Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science 2020, 370 (6521). 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Stukalov A; Girault V; Grass V; Bergant V; Karayel O; Urban C; Haas DA; Huang Y; Oubraham L; Wang A; Hamad SM; Piras A; Tanzer M; Hansen FM; Enghleitner T; Reinecke M; Lavacca TM; Ehmann R; Wölfel R; Jores J; Kuster B; Protzer U; Rad R; Ziebuhr J; Thiel V; Scaturro P; Mann M; Pichlmair A. Multi-Level Proteomics Reveals Host-Perturbation Strategies of SARS-CoV-2 and SARS-CoV, 2020, 2020.06.17.156455. 10.1101/2020.06.17.156455. [DOI] [PubMed] [Google Scholar]
- (107).Roux KJ; Kim DI; Raida M; Burke B. A Promiscuous Biotin Ligase Fusion Protein Identifies Proximal and Interacting Proteins in Mammalian Cells. J. Cell Biol 2012, 196 (6), 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Branon TC; Bosch JA; Sanchez AD; Udeshi ND; Svinkina T; Carr SA; Feldman JL; Perrimon N; Ting AY Efficient Proximity Labeling in Living Cells and Organisms with TurboID. Nat. Biotechnol 2018, 36 (9), 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hung V; Zou P; Rhee H-W; Udeshi ND; Cracan V; Svinkina T; Carr SA; Mootha VK; Ting AY Proteomic Mapping of the Human Mitochondrial Intermembrane Space in Live Cells via Ratiometric APEX Tagging. Mol. Cell 2014, 55 (2), 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Lobingier BT; Hüttenhain R; Eichel K; Miller KB; Ting AY; von Zastrow M; Krogan NJ An Approach to Spatiotemporally Resolve Protein Interaction Networks in Living Cells. Cell 2017, 169 (2), 350–360.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Coyaud E; Ranadheera C; Cheng D; Gonçalves J; Dyakov BJA; Laurent EMN; St-Germain J; Pelletier L; Gingras A-C; Brumell JH; Kim PK; Safronetz D; Raught B. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell. Proteomics 2018, 17 (11), 2242–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).V’kovski P; Gerber M; Kelly J; Pfaender S; Ebert N; Braga Lagache S; Simillion C; Portmann J; Stalder H; Gaschen V; Bruggmann R; Stoffel MH; Heller M; Dijkman R; Thiel V. Determination of Host Proteins Composing the Microenvironment of Coronavirus Replicase Complexes by Proximity-Labeling. Elife 2019, 8. 10.7554/eLife.42037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Samavarchi-Tehrani P; Abdouni H; Knight JDR; Astori A; Samson R; Lin Z-Y; Kim D-K; Knapp JJ; St-Germain J; Go CD; Larsen B; Wong CJ; Cassonnet P; Demeret C; Jacob Y; Roth FP; Raught B; Gingras A-C A SARS-CoV-2 – Host Proximity Interactome. 10.1101/2020.09.03.282103. [DOI] [Google Scholar]
- (114).Schopp IM; Amaya Ramirez CC; Debeljak J; Kreibich E; Skribbe M; Wild K; Béthune J. Split-BioID a Conditional Proteomics Approach to Monitor the Composition of Spatiotemporally Defined Protein Complexes. Nat. Commun 2017, 8, 15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Han Y; Branon TC; Martell JD; Boassa D; Shechner D; Ellisman MH; Ting A. Directed Evolution of Split APEX2 Peroxidase. ACS Chem. Biol 2019, 14 (4), 619–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Cho KF; Branon TC; Rajeev S; Svinkina T; Udeshi ND; Thoudam T; Kwak C; Rhee H-W; Lee I-K; Carr SA; Ting AY Split-TurboID Enables Contact-Dependent Proximity Labeling in Cells. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (22), 12143–12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Jean Beltran PM; Mathias RA; Cristea IM A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst 2016, 3 (4), 361–373.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Itzhak DN; Tyanova S; Cox J; Borner GH Global, Quantitative and Dynamic Mapping of Protein Subcellular Localization. Elife 2016, 5. 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Geladaki A; Kočevar Britovšek N; Breckels LM; Smith TS; Vennard OL; Mulvey CM; Crook OM; Gatto L; Lilley KS Combining LOPIT with Differential Ultracentrifugation for High-Resolution Spatial Proteomics. Nat. Commun 2019, 10 (1), 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Greenwood EJ; Matheson NJ; Wals K; van den Boomen DJ; Antrobus R; Williamson JC; Lehner PJ Temporal Proteomic Analysis of HIV Infection Reveals Remodelling of the Host Phosphoproteome by Lentiviral Vif Variants. Elife 2016, 5. 10.7554/eLife.18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Marelli S; Williamson JC; Protasio AV; Naamati A; Greenwood EJ; Deane JE; Lehner PJ; Matheson NJ Antagonism of PP2A Is an Independent and Conserved Function of HIV-1 Vif and Causes Cell Cycle Arrest. Elife 2020, 9. 10.7554/eLife.53036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Naamati A; Williamson JC; Greenwood EJ; Marelli S; Lehner PJ; Matheson NJ Functional Proteomic Atlas of HIV Infection in Primary Human CD4+ T Cells. Elife 2019, 8. 10.7554/eLife.41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Roulston A; Lin R; Beauparlant P; Wainberg MA; Hiscott J. Regulation of Human Immunodeficiency Virus Type 1 and Cytokine Gene Expression in Myeloid Cells by NFKappa B/Rel Transcription Factors. Microbiol. Rev 1995, 59 (3), 481–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Keating JA; Striker R. Phosphorylation Events during Viral Infections Provide Potential Therapeutic Targets. Rev. Med. Virol 2012, 22 (3), 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Saeed MF; Kolokoltsov AA; Freiberg AN; Holbrook MR; Davey RA Phosphoinositide-3 Kinase-Akt Pathway Controls Cellular Entry of Ebola Virus. PLoS Pathog. 2008, 4 (8), e1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Zheng W; Li J; Wang S; Cao S; Jiang J; Chen C; Ding C; Qin C; Ye X; Gao GF; Liu W. Phosphorylation Controls the Nuclear-Cytoplasmic Shuttling of Influenza A Virus Nucleoprotein. J. Virol 2015, 89 (11), 5822–5834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Müller B; Patschinsky T; Kräusslich H-G The Late-Domain-Containing Protein p6 Is the Predominant Phosphoprotein of Human Immunodeficiency Virus Type 1 Particles. J. Virol 2002, 76 (3), 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Bouhaddou M; Memon D; Meyer B; White KM; Rezelj VV; Correa Marrero M; Polacco BJ; Melnyk JE; Ulferts S; Kaake RM; Batra J; Richards AL; Stevenson E; Gordon DE; Rojc A; Obernier K; Fabius JM; Soucheray M; Miorin L; Moreno E; Koh C; Tran QD; Hardy A; Robinot R; Vallet T; Nilsson-Payant BE; Hernandez-Armenta C; Dunham A; Weigang S; Knerr J; Modak M; Quintero D; Zhou Y; Dugourd A; Valdeolivas A; Patil T; Li Q; Hüttenhain R; Cakir M; Muralidharan M; Kim M; Jang G; Tutuncuoglu B; Hiatt J; Guo JZ; Xu J; Bouhaddou S; Mathy CJP; Gaulton A; Manners EJ; Félix E; Shi Y; Goff M; Lim JK; McBride T; O’Neal MC; Cai Y; Chang JCJ; Broadhurst DJ; Klippsten S; De Wit E; Leach AR; Kortemme T; Shoichet B; Ott M; Saez-Rodriguez J; tenOever BR; Mullins RD; Fischer ER; Kochs G; Grosse R; García-Sastre A; Vignuzzi M; Johnson JR; Shokat KM; Swaney DL; Beltrao P; Krogan NJ The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182 (3), 685–712.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Raaben M; Posthuma CC; Verheije MH; te Lintelo EG; Kikkert M; Drijfhout JW; Snijder EJ; Rottier PJM; de Haan CAM The Ubiquitin-Proteasome System Plays an Important Role during Various Stages of the Coronavirus Infection Cycle. J. Virol 2010, 84 (15), 7869–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Yu X; Yu Y; Liu B; Luo K; Kong W; Mao P; Yu X-F Induction of APOBEC3G Ubiquitination and Degradation by an HIV-1 Vif-Cul5-SCF Complex. Science 2003, 302 (5647), 1056–1060. [DOI] [PubMed] [Google Scholar]
- (131).Mehle A; Goncalves J; Santa-Marta M; McPike M; Gabuzda D. Phosphorylation of a Novel SOCS-Box Regulates Assembly of the HIV-1 Vif-Cul5 Complex That Promotes APOBEC3G Degradation. Genes Dev. 2004, 18 (23), 2861–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Mehle A; Strack B; Ancuta P; Zhang C; McPike M; Gabuzda D. Vif Overcomes the Innate Antiviral Activity of APOBEC3G by Promoting Its Degradation in the Ubiquitin-Proteasome Pathway. J. Biol. Chem 2004, 279 (9), 7792–7798. [DOI] [PubMed] [Google Scholar]
- (133).Sheehy AM; Gaddis NC; Choi JD; Malim MH Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418 (6898), 646–650. [DOI] [PubMed] [Google Scholar]
- (134).Sheehy AM; Gaddis NC; Malim MH The Antiretroviral Enzyme APOBEC3G Is Degraded by the Proteasome in Response to HIV-1 Vif. Nat. Med 2003, 9 (11), 1404–1407. [DOI] [PubMed] [Google Scholar]
- (135).Conticello SG; Harris RS; Neuberger MS The Vif Protein of HIV Triggers Degradation of the Human Antiretroviral DNA Deaminase APOBEC3G. Curr. Biol 2003, 13 (22), 2009–2013. [DOI] [PubMed] [Google Scholar]
- (136).Stopak K; de Noronha C; Yonemoto W; Greene WC HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12 (3), 591–601. [DOI] [PubMed] [Google Scholar]
- (137).Jäger S; Kim DY; Hultquist JF; Shindo K; LaRue RS; Kwon E; Li M; Anderson BD; Yen L; Stanley D; Mahon C; Kane J; Franks-Skiba K; Cimermancic P; Burlingame A; Sali A; Craik CS; Harris RS; Gross JD; Krogan NJ Vif Hijacks CBF-β to Degrade APOBEC3G and Promote HIV-1 Infection. Nature 2011, 481 (7381), 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Hardman G; Perkins S; Brownridge PJ; Clarke CJ; Byrne DP; Campbell AE; Kalyuzhnyy A; Myall A; Eyers PA; Jones AR; Eyers CE Strong Anion Exchange-Mediated Phosphoproteomics Reveals Extensive Human Non-Canonical Phosphorylation. EMBO J. 2019, 38 (21), e100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Fuhs SR; Meisenhelder J; Aslanian A; Ma L; Zagorska A; Stankova M; Binnie A; Al-Obeidi F; Mauger J; Lemke G; Yates JR 3rd; Hunter T. Monoclonal 1- and 3Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell 2015, 162 (1), 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Li XS; Yuan BF; Feng YQ Recent Advances in Phosphopeptide Enrichment: Strategies and Techniques. Trends Analyt. Chem 2016. [Google Scholar]
- (141).Bekker-Jensen DB; Bernhardt OM; Hogrebe A; Martinez-Val A; Verbeke L; Gandhi T; Kelstrup CD; Reiter L; Olsen JV Rapid and Site-Specific Deep Phosphoproteome Profiling by Data-Independent Acquisition without the Need for Spectral Libraries. Nat. Commun 2020, 11 (1), 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Searle BC; Lawrence RT; MacCoss MJ; Villén J. Thesaurus: Quantifying Phosphopeptide Positional Isomers. Nat. Methods 2019, 16 (8), 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Humphrey SJ; Azimifar SB; Mann M. High-Throughput Phosphoproteomics Reveals in Vivo Insulin Signaling Dynamics. Nat. Biotechnol 2015, 33 (9), 990–995. [DOI] [PubMed] [Google Scholar]
- (144).Hernandez-Armenta C; Ochoa D; Gonçalves E; Saez-Rodriguez J; Beltrao P. Benchmarking Substrate-Based Kinase Activity Inference Using Phosphoproteomic Data. Bioinformatics 2017, 33 (12), 1845–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Ochoa D; Jonikas M; Lawrence RT; El Debs B; Selkrig J; Typas A; Villén J; Santos SD; Beltrao P. An Atlas of Human Kinase Regulation. Mol. Syst. Biol 2016, 12 (12), 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Krug K; Mertins P; Zhang B; Hornbeck P; Raju R; Ahmad R; Szucs M; Mundt F; Forestier D; Jane-Valbuena J; Keshishian H; Gillette MA; Tamayo P; Mesirov JP; Jaffe JD; Carr SA; Mani DR A Curated Resource for Phosphosite-Specific Signature Analysis. Mol. Cell. Proteomics 2019, 18 (3), 576–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Davidson AD; Williamson MK; Lewis S; Shoemark D; Carroll MW; Heesom KJ; Zambon M; Ellis J; Lewis PA; Hiscox JA; Matthews DA Characterisation of the Transcriptome and Proteome of SARS-CoV-2 Reveals a Cell Passage Induced in-Frame Deletion of the Furin-like Cleavage Site from the Spike Glycoprotein. Genome Med. 2020, 12 (1), 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Klann K; Bojkova D; Tascher G; Ciesek S; Münch C; Cinatl J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 2020. 10.1016/j.molcel.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Mahon C; Krogan NJ; Craik CS; Pick E. Cullin E3 Ligases and Their Rewiring by Viral Factors. Biomolecules 2014, 4 (4), 897–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Viswanathan K; Früh K; DeFilippis V. Viral Hijacking of the Host Ubiquitin System to Evade Interferon Responses. Curr. Opin. Microbiol 2010, 13 (4), 517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Shin JY; Muniyappan S; Tran N-N; Park H; Lee SB; Lee B-H Deubiquitination Reactions on the Proteasome for Proteasome Versatility. Int. J. Mol. Sci 2020, 21 (15). 10.3390/ijms21155312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Li T; Zou C. The Role of Deubiquitinating Enzymes in Acute Lung Injury and Acute Respiratory Distress Syndrome. Int. J. Mol. Sci 2020, 21 (14). 10.3390/ijms21144842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Chakraborty J; Ziviani E. Deubiquitinating Enzymes in Parkinson’s Disease. Front. Physiol 2020, 11, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Celebi G; Kesim H; Ozer E; Kutlu O. The Effect of Dysfunctional Ubiquitin Enzymes in the Pathogenesis of Most Common Diseases. Int. J. Mol. Sci 2020, 21 (17). 10.3390/ijms21176335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Xu G; Paige JS; Jaffrey SR Global Analysis of Lysine Ubiquitination by Ubiquitin Remnant Immunoaffinity Profiling. Nat. Biotechnol 2010, 28 (8), 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Kim W; Bennett EJ; Huttlin EL; Guo A; Li J; Possemato A; Sowa ME; Rad R; Rush J; Comb MJ; Harper JW; Gygi SP Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44 (2), 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Hoffmann H-H; Schneider WM; Sánchez-Rivera FJ; Luna JM; Ashbrook AW; Soto-Feliciano YM; Leal AA; Le Pen J; Ricardo-Lax I; Michailidis E; Hao Y; Stenzel AF; Peace A; Allis CD; Lowe SW; MacDonald MR; Poirier JT; Rice CM Functional Interrogation of a SARS-CoV-2 Host Protein Interactome Identifies Unique and Shared Coronavirus Host Factors. bioRxiv 2020. 10.1101/2020.09.11.291716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Klemm T; Ebert G; Calleja DJ; Allison CC; Richardson LW; Bernardini JP; Lu BG; Kuchel NW; Grohmann C; Shibata Y; Gan ZY; Cooney JP; Doerflinger M; Au AE; Blackmore TR; van der Heden van Noort GJ; Geurink PP; Ovaa H; Newman J; Riboldi-Tunnicliffe A; Czabotar PE; Mitchell JP; Feltham R; Lechtenberg BC; Lowes KN; Dewson G; Pellegrini M; Lessene G; Komander D. Mechanism and Inhibition of the Papain-like Protease, PLpro, of SARS-CoV-2. EMBO J. 2020, e106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Bojkova D; Klann K; Koch B; Widera M; Krause D; Ciesek S; Cinatl J; Münch C. Proteomics of SARS-CoV-2-Infected Host Cells Reveals Therapy Targets. Nature 2020, 583 (7816), 469–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Johnson JR; Crosby DC; Hultquist JF; Li D; Marlett J; Swann J; Hüttenhain R; Verschueren E; Johnson TL; Newton BW; Shales M; Beltrao P; Frankel AD; Marson A; Fregoso OI; Young JAT; Krogan NJ Global Post-Translational Modification Profiling of HIV-1-Infected Cells Reveals Mechanisms of Host Cellular Pathway Remodeling, 2020, 2020.01.06.896365. 10.1101/2020.01.06.896365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Eckhardt M; Hultquist JF; Kaake RM; Hüttenhain R; Krogan NJ A Systems Approach to Infectious Disease. Nat. Rev. Genet 2020, 21 (6), 339–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Hüttenhain R; Soste M; Selevsek N; Röst H; Sethi A; Carapito C; Farrah T; Deutsch EW; Kusebauch U; Moritz RL; Niméus-Malmström E; Rinner O; Aebersold R. Reproducible Quantification of Cancer-Associated Proteins in Body Fluids Using Targeted Proteomics. Sci. Transl. Med 2012, 4 (142), 142ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Bezstarosti K; Lamers MM; Haagmans BL; Demmers JAA Targeted Proteomics for the Detection of SARS-CoV-2 Proteins. bioRxiv, 2020, 2020.04.23.057810. 10.1101/2020.04.23.057810. [DOI] [Google Scholar]
- (164).Ihling C; Tänzler D; Hagemann S; Kehlen A; Hüttelmaier S; Sinz A. Mass Spectrometric Identification of SARS-CoV-2 Proteins from Gargle Solution Samples of COVID-19 Patients. bioRxiv, 2020, 2020.04.18.047878. 10.1101/2020.04.18.047878. [DOI] [PubMed] [Google Scholar]
- (165).Zecha J; Lee C-Y; Bayer FP; Meng C; Grass V; Zerweck J; Schnatbaum K; Michler T; Pichlmair A; Ludwig C; Kuster B. Data, Reagents, Assays and Merits of Proteomics for SARS-CoV-2 Research and Testing. Mol. Cell. Proteomics 2020, 19 (9), 1503–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Cardozo KHM; Lebkuchen A; Okai GG; Schuch RA; Viana LG; Olive AN; Lazari CDS; Fraga AM; Granato CFH; Pintão MCT; Carvalho VM Establishing a Mass Spectrometry-Based System for Rapid Detection of SARS-CoV-2 in Large Clinical Sample Cohorts. Nat. Commun 2020, 11 (1), 6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Geyer PE; Voytik E; Treit PV; Doll S; Kleinhempel A; Niu L; Müller JB; Buchholtz M-L; Bader JM; Teupser D; Holdt LM; Mann M. Plasma Proteome Profiling to Detect and Avoid Sample-Related Biases in Biomarker Studies. EMBO Mol. Med 2019, 11 (11), e10427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Geyer PE; Holdt LM; Teupser D; Mann M. Revisiting Biomarker Discovery by Plasma Proteomics. Mol. Syst. Biol 2017, 13 (9), 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Messner CB; Demichev V; Wendisch D; Michalick L; White M; Freiwald A; Textoris-Taube K; Vernardis SI; Egger A-S; Kreidl M; Ludwig D; Kilian C; Agostini F; Zelezniak A; Thibeault C; Pfeiffer M; Hippenstiel S; Hocke A; von Kalle C; Campbell A; Hayward C; Porteous DJ; Marioni RE; Langenberg C; Lilley KS; Kuebler WM; Mülleder M; Drosten C; Suttorp N; Witzenrath M; Kurth F; Sander LE; Ralser M. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst 2020. 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Fontana A; de Laureto PP; Spolaore B; Frare E; Picotti P; Zambonin M. Probing Protein Structure by Limited Proteolysis. Acta Biochim. Pol 2004, 51 (2), 299–321. [PubMed] [Google Scholar]
- (171).Feng Y; De Franceschi G; Kahraman A; Soste M; Melnik A; Boersema PJ; de Laureto PP; Nikolaev Y; Oliveira AP; Picotti P. Global Analysis of Protein Structural Changes in Complex Proteomes. Nat. Biotechnol 2014, 32 (10), 1036–1044. [DOI] [PubMed] [Google Scholar]
- (172).Piazza I; Kochanowski K; Cappelletti V; Fuhrer T; Noor E; Sauer U; Picotti P. A Map of Protein-Metabolite Interactions Reveals Principles of Chemical Communication. Cell 2018, 172 (1–2), 358–372.e23. [DOI] [PubMed] [Google Scholar]
- (173).Piazza I; Beaton N; Bruderer R; Knobloch T; Barbisan C; Chandat L; Sudau A; Siepe I; Rinner O; de Souza N; Picotti P; Reiter L. A Machine Learning-Based Chemoproteomic Approach to Identify Drug Targets and Binding Sites in Complex Proteomes. Nat. Commun 2020, 11 (1), 4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Savitski MM; Reinhard FBM; Franken H; Werner T; Savitski MF; Eberhard D; Martinez Molina D; Jafari R; Dovega RB; Klaeger S; Kuster B; Nordlund P; Bantscheff M; Drewes G. Tracking Cancer Drugs in Living Cells by Thermal Profiling of the Proteome. Science 2014, 346 (6205), 1255784. [DOI] [PubMed] [Google Scholar]
- (175).Martinez Molina D; Jafari R; Ignatushchenko M; Seki T; Larsson EA; Dan C; Sreekumar L; Cao Y; Nordlund P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science 2013, 341 (6141), 84–87. [DOI] [PubMed] [Google Scholar]
- (176).Reinhard FBM; Eberhard D; Werner T; Franken H; Childs D; Doce C; Savitski MF; Huber W; Bantscheff M; Savitski MM; Drewes G. Thermal Proteome Profiling Monitors Ligand Interactions with Cellular Membrane Proteins. Nat. Methods 2015, 12 (12), 1129–1131. [DOI] [PubMed] [Google Scholar]
- (177).Perrin J; Werner T; Kurzawa N; Rutkowska A; Childs DD; Kalxdorf M; Poeckel D; Stonehouse E; Strohmer K; Heller B; Thomson DW; Krause J; Becher I; Eberl HC; Vappiani J; Sevin DC; Rau CE; Franken H; Huber W; Faelth-Savitski M; Savitski MM; Bantscheff M; Bergamini G. Identifying Drug Targets in Tissues and Whole Blood with Thermal-Shift Profiling. Nat. Biotechnol 2020, 38 (3), 303–308. [DOI] [PubMed] [Google Scholar]
- (178).Friman T; Chernobrovkin A; Molina DM; Arnold LH CETSA MS Profiling for a Comparative Assessment of FDA Approved Antivirals Repurposed for COVID-19 Therapy Identifies Trip13 as a Remdesivir off-Target. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Niphakis MJ; Cravatt BF Enzyme Inhibitor Discovery by Activity-Based Protein Profiling. Annu. Rev. Biochem 2014, 83, 341–377. [DOI] [PubMed] [Google Scholar]
- (180).Kahler JP; Vanhoutte R; Verhelst SHL Activity-Based Protein Profiling of Serine Proteases in Immune Cells. Arch. Immunol. Ther. Exp 2020, 68 (4), 23. [DOI] [PubMed] [Google Scholar]
- (181).Bar-Peled L; Kemper EK; Suciu RM; Vinogradova EV; Backus KM; Horning BD; Paul TA; Ichu T-A; Svensson RU; Olucha J; Chang MW; Kok BP; Zhu Z; Ihle NT; Dix MM; Jiang P; Hayward MM; Saez E; Shaw RJ; Cravatt BF Chemical Proteomics Identifies Druggable Vulnerabilities in a Genetically Defined Cancer. Cell 2017, 171 (3), 696–709.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Kok BP; Ghimire S; Kim W; Chatterjee S; Johns T; Kitamura S; Eberhardt J; Ogasawara D; Xu J; Sukiasyan A; Kim SM; Godio C; Bittencourt JM; Cameron M; Galmozzi A; Forli S; Wolan DW; Cravatt BF; Boger DL; Saez E. Discovery of Small-Molecule Enzyme Activators by Activity-Based Protein Profiling. Nat. Chem. Biol 2020. 10.1038/s41589-020-0555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Fuerst R; Breinbauer R. Activity-Based Protein Profiling (ABPP) of Oxidoreductases. Chembiochem 2020. 10.1002/cbic.202000542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Blewett MM; Xie J; Zaro BW; Backus KM; Altman A; Teijaro JR; Cravatt BF Chemical Proteomic Map of Dimethyl Fumarate–sensitive Cysteines in Primary Human T Cells. Science Signaling. 2016, pp rs10–rs10. 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Baggelaar MP; den Dulk H; Florea BI; Fazio D; Perruzza D; Bernab N; Raspa M; Janssen APA; Scavizzi F; Barboni B; Overkleeft HS; Maccarrone M; van der Stelt M. ABHD2 Inhibitor Identified by Activity-Based Protein Profiling Reduces Acrosome Reaction. ACS Chem. Biol 2019, 14 (12), 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Riva L; Yuan S; Yin X; Martin-Sancho L; Matsunaga N; Pache L; Burgstaller-Muehlbacher S; De Jesus PD; Teriete P; Hull MV; Chang MW; Chan JF-W; Cao J; Poon VK-M; Herbert KM; Cheng K; Nguyen T-TH; Rubanov A; Pu Y; Nguyen C; Choi A; Rathnasinghe R; Schotsaert M; Miorin L; Dejosez M; Zwaka TP; Sit K-Y; Martinez-Sobrido L; Liu W-C; White KM; Chapman ME; Lendy EK; Glynne RJ; Albrecht R; Ruppin E; Mesecar AD; Johnson JR; Benner C; Sun R; Schultz PG; Su AI; García-Sastre A; Chatterjee AK; Yuen K-Y; Chanda SK Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020. 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]