

Abstract
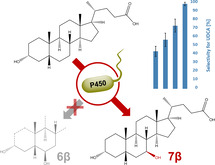
We engineered the cytochrome P450 monooxygenase CYP107D1 (OleP) from Streptomyces antibioticus for the stereo‐ and regioselective 7β‐hydroxylation of lithocholic acid (LCA) to yield ursodeoxycholic acid (UDCA). OleP was previously shown to hydroxylate testosterone at the 7β‐position but LCA is exclusively hydroxylated at the 6β‐position, forming murideoxycholic acid (MDCA). Structural and 3DM analysis, and molecular docking were used to identify amino acid residues F84, S240, and V291 as specificity‐determining residues. Alanine scanning identified S240A as a UDCA‐producing variant. A synthetic “small but smart” library based on these positions was screened using a colorimetric assay for UDCA. We identified a nearly perfectly regio‐ and stereoselective triple mutant (F84Q/S240A/V291G) that produces 10‐fold higher levels of UDCA than the S240A variant. This biocatalyst opens up new possibilities for the environmentally friendly synthesis of UDCA from the biological waste product LCA.
Keywords: 7β-hydroxylation, cytochrome P450 monooxygenase, lithocholic acid, protein engineering, Ursodeoxycholic acid
We report engineering of a P450 monooxygenase for the stereo‐ and regioselective 7β‐hydroxylation of lithocholic acid to produce ursodeoxycholic acid (UDCA). Structural and 3DM analysis, and molecular docking, identified selectivity‐influencing residues. A “small but smart” mutant library was then screened with a selective colorimetric assay. The best mutant has nearly perfect regio‐ and stereoselectivity, enabling a new route for UDCA synthesis.
Ursodeoxycholic acid (UDCA) is a valuable bile acid frequently prescribed for the treatment of cholecystitis as it can solubilize cholesterol gallstones with fewer side effects than chenodeoxycholic acid (CDCA). [1] UDCA also has anti‐inflammatory properties [2] and is applied in the therapy of cystic fibrosis [3] and liver diseases like primary biliary cholangitis. [4] The major natural source of UDCA is bear bile, [5] a popular traditional medicine obtained by biliary catheterization of farmed bears. Alternatively, semi‐synthetic UDCA can be produced from cholic acid (CA) [6] or CDCA.[ 7 , 8 ] The synthesis route starting from CA forms CDCA within 5 steps, including a Wolff–Kishner reduction, and an epimerization at C7 to produce UDCA (Scheme 1 a, Scheme S1). [9] The yields of this pathway do not exceed 30 %. To overcome these limitations, a shorter synthesis route based on the biocatalytic epimerization of CDCA to UDCA (Scheme 1 a) has been developed.[ 7 , 10 ]
Scheme 1.
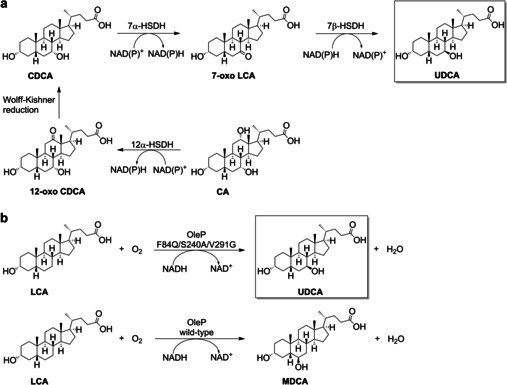
a) Chemoenzymatic synthesis routes towards UDCA starting from CA or CDCA. b) The desired 7β‐hydroxylation of LCA towards UDCA by an engineered OleP variant and wild‐type OleP mediated formation of MDCA, starting from LCA.
LCA is an abundant and inexpensive waste product of meat production [11] as this bile acid is found in farmed animals like sheep, [12] cattle, [12] and pigs. [13] Currently, no biotechnological process [14] utilizing LCA originating from these sources is known, making it a desirable starting material for the synthesis of UDCA. A few microbial organisms have been reported to form UDCA from LCA. [15] For example, the fungus Fusarium equiseti converts LCA to a number of products, including UDCA at 35 % yield. [16]
However, there is currently no enzyme known to selectively hydroxylate LCA at the 7β‐position to form UDCA and its synthesis pathway in microbial organisms, starting from LCA, remains enigmatic. An enzyme for direct 7β‐hydroxylation would be a valuable tool for direct conversion of LCA to UDCA without involving the complex metabolism of fungi that invariably [16] produce multiple undesired side products, [15] complicating downstream processing.
A major challenge in the enzymatic conversion of LCA to UDCA is the hydrophobicity and thus, extremely low water solubility of LCA compared to the more widely used CA or CDCA. [17]
P450s are heme‐containing enzymes capable of stereo‐ and regioselective hydroxylation reactions of a wide variety of substrates, using molecular oxygen as oxidant. [18]
This enzyme class has been intensively investigated due to its potential for late‐stage hydroxylation of industrially relevant precursor compounds. [19] Protein engineering is often employed to produce P450 monooxygenases that meet the requirements of excellent regio‐ and stereoselectivity for the desired applications. [20]
The monooxygenase CYP107D1 (OleP), originally described for an epoxidation[ 21 , 22 ] in the oleandomycin biosynthesis pathway, [23] was found to accept 12‐membered macrolactone substrates [24] as well. OleP can also hydroxylate testosterone at the positions 6β, 7β, 12β, and 15β. [25] However, bile acids like LCA and deoxycholic acid (DCA) are hydroxylated exclusively at the 6β‐position, forming MDCA (Scheme 1 b) and 3α‐, 6β‐, 12α‐trihydroxy‐5β‐cholan‐24‐oic acid, respectively. [26]
To open up a novel reaction pathway towards UDCA, as no enzyme is known for selective 7β‐hydroxylation, and to avoid using whole cell fungi, wherein the action of multiple P450s frequently results in side‐product formation, we decided to engineer OleP and create an Escherichia coli (E. coli)‐based whole‐cell system for the regio‐ and stereoselective hydroxylation of LCA at the 7β‐position to form UDCA (Scheme 1 b).
Initially, we determined which amino acid residues could influence the positioning of LCA in the OleP active site. Using crystal structures[ 21 , 22 ] of OleP with bound inhibitors and a natural substrate analog, we were able to dock LCA into the OleP active site in multiple conformations (Figure 1). In this evaluation process, a docked structure was found matching three desired criteria. First, LCA had to be positioned at most 5 Å away from the heme iron. Second, the 6β‐hydrogen had to be oriented towards the heme, mimicking the binding of LCA for MDCA formation. This resulted in the elimination of perpendicular orientations. Finally, since the steroid nucleus can flip in the horizontal orientation, we used the most frequently occurring conformations in our docking experiment (Figure 1 a). Based on this docking pose, residues in a zone of 5–14 Å around the heme were chosen. Important P450 specificity‐determining residues known from literature[ 21 , 24 , 27 ] were also selected. Finally, residues interacting with the substrate analog [24] were also selected. In total, 24 active site residues were identified for further investigation.
Figure 1.
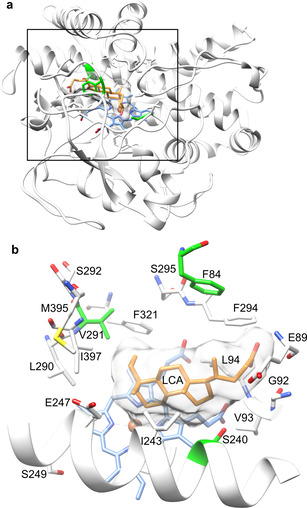
a) Structure of OleP (PDB ID: 4XE3) with docked LCA. b) Active site residues surrounding the docked LCA. The heme cofactor is colored blue and the iron atom in red, LCA is orange with its surface shown in white. Residues selected for alanine scanning are colored white and the selectivity influencing residues F84, S240, and V291 are green.
Alanine scanning was used to generate space in the active site to allow LCA to shift from the 6β‐ towards 7β‐hydroxylation orientation and to investigate the contributions of residues to hydroxylation activity.
Of the 24 residues selected (Figure 1 b), four were already alanine or glycine. The remaining 20 were exchanged to alanine by site‐directed mutagenesis. The variants were co‐expressed with the redox partner proteins Putidaredoxin (PdX) and Putidaredoxin reductase (PdR), employing a two‐plasmid system in E. coli C43(DE3). Whole‐cell biotransformations were performed with 5 mM LCA. HPLC analysis revealed five UDCA‐producing variants (F84A, V93A, L94A, S240A, and V291A). L179A and S295A produced no product and the remaining variants produced exclusively MDCA, like wild‐type OleP.
The variants F84A, S240A, and V291A were most promising as they produced mostly MDCA and UDCA, compared to V93A and L94A, which produced several unidentified products.
The residue F84 of the BC loop is associated with substrate recognition. [22] S240 is part of the I‐helix and coordinates parts of the hydrogen‐bond network, which connects OleP with the substrate. V291 is within a β‐hairpin (β3) and part of a hydrophobic bulge facilitating coordination of the substrate by van der Waals interactions. [22] This suggested that manipulation of the water network and taking advantage of the hydrophobic interactions, which have significant effects in the positioning of the substrate, could enhance the 7β‐hydroxylation of LCA to form UDCA.
A 3DM [28] database of P450 monooxygenases was used to evaluate the most frequently occurring amino acids at each of the residues altered in the initial UDCA‐forming variants (F84A, V93A, L94A, S240A, and V291A). Each residue was mutated to the four most frequently occurring amino acids. The aim was to diversify these positions without the introduction of amino acids which are evolutionarily unfavored and likely to result in inactive variants. With these variants created, UDCA formation and the occurrence of side products were analyzed by HPLC. Substitutions of F84, S240, and V291 resulted in increased UDCA production. We then designed a “small but smart” 3DM library [29] focused on F84, S240, and V291 (Table S2). The library, containing 4,480 unique variants, was ordered from Twist Bioscience, thereby eliminating bias, and cloned into the pET‐28a vector. [30] E. coli C43(DE3) was co‐transformed with the library and the redox partner system pACYCDuet‐1‐pdR/pdX. Sequencing a random sample revealed only the desired codons. Cultivation and whole‐cell biotransformations took place in 24 deep‐well plates, which allowed us to standardize the process. Detection of the product initially depended on HPLC measurements.
Screening of libraries towards desired regio‐ and stereoselective hydroxylation of steroids and bile acids has—with some exceptions [31] —mostly been done by chromatographic methods. A colorimetric UDCA assay, employing an NADP+‐dependent 7β‐hydroxysteroid dehydrogenase, [32] allowed us to screen 600 variants per round (Scheme S2).
The S240A variant, which was the most active variant resulting from the initial alanine scanning, was used as a positive control during screening (Figure S5). Screening a set of 3,400 clones actually led to the identification of S240A as a hit. In total, 34 clones, with up to 18‐fold higher absorbance than that of the S240A variant, were identified (Figure S5). Of these 34 hits, 32 were confirmed to produce UDCA by HPLC in comparison to a commercial standard.
We observed a clear correlation between the colorimetric UDCA assay signals and the number of side products formed. An increased signal in the 7β‐HSDH assay correlated with fewer side products as confirmed by HPLC (Figure 2). The variants showing the highest colorimetric signal mostly produced MDCA and UDCA in different ratios ranging from 25.9 % UDCA formation (F84Q/S240A), through 31.9 % UDCA formation (F84C/S240A/V291A), up to 72.8 % UDCA formation (F84Q/S240A/V291D) (Table S3). The specificity of F84Q/S240A/V291D for UDCA formation was remarkable, considering that wild‐type OleP exclusively produces MDCA and the S240A variant forms only 8.2 % UDCA. None of the UDCA‐producing mutants formed the 7α‐hydroxylation product (CDCA), demonstrating perfect stereoselectivity.
Figure 2.

UDCA‐producing variants with increased selectivity. OleP variants show increasing selectivity towards UDCA (blue) and increased UDCA formation (red). UDCA was quantified by HPLC of three replicate reactions.
We continued by sequencing the HPLC‐confirmed UDCA‐producing variants and found very clear amino acid preferences at each of the randomized positions (Figure S6). In UDCA‐producing variants, F84 was most frequently mutated to Q (44 %) or T (19 %). For S240, a clear preference for A (87 %) was observed. For V291, the most common variants were G (34 %), A (14 %) and E (14 %). By the introduction of a polar and more flexible amino acid like Q or T at position F84, the flexibility of the BC‐loop is enhanced. This loop is involved in substrate recognition and stabilization of the bile acid. Since S240 is both a key residue establishing the water network of the active site and associated with the water‐mediated coordination of substrates, we hypothesize that with the introduction of a nonpolar and small residue, the water network in the active site is remodeled, favoring the 7β‐hydroxylation. This concept is in line with recent findings reported for a testosterone hydroxylation using the P450 monooxygenase BM3. [34] As V291 is part of a hydrophobic cleft, its mutation to glycine could be beneficial for the 7β‐hydroxylation by the generation of space to position the more dented bile acid in the active site better compared to flat steroids such as testosterone.
To get further insights into the contribution of each position and to find the most selective variant, we created a set of 37 single, double, and triple mutants at residues F84, S240A, and V291. This selection was based on UDCA:MDCA ratios of the individual variants and sequencing of UDCA producing variants (Figure S6 and Table S3). All newly created variants were analyzed by the colorimetric assay as well as HPLC. Several single, double, and triple mutants (Figure 2) were capable of producing UDCA (Table S3) although the selectivity towards UDCA formation varied significantly. The most striking variant, F84Q/S240A/V291G, produced 67 μM of UDCA and exhibited outstanding selectivity. Only trace amounts of MDCA were formed as the sole side‐product. Interestingly, this mutant combines the most frequently occurring residues in UDCA‐producing variants (Figure S6). By scaling the reaction to 500 mL using OleP F84Q/S240A/V291G, we were able to isolate the product and confirmed it as UDCA by NMR‐spectroscopy (Figures S11 and S12). The side‐product formed only in traces was confirmed as MDCA (Figure S13 and S14).
We were thus able to engineer OleP, which exclusively formed MDCA from LCA, towards UDCA production. The variant F84Q/S240A/V291G showed perfect stereoselectivity and excellent regioselectivity for the 7β‐hydroxylation of LCA. This discovery opens up new synthesis routes towards 7β‐hydroxylated therapeutic agents like UDCA. These results also expand our knowledge about P450‐mediated hydroxylations.
We successfully engineered the P450 monooxygenase OleP for the regio‐ and stereoselective 7β‐hydroxylation of LCA to form UDCA. We started with alanine scanning of important active site residues identified from literature, inspection of crystal structures, and molecular docking. The bioinformatic tool 3DM was then used to design a “small but smart” synthetic library, which was efficiently screened employing a high‐throughput colorimetric UDCA assay. Starting from an enzyme which did not even have trace activity in the formation of the target compound, we were able to create a variant that produces mainly UDCA, with only traces of MDCA formed. The near complete inversion of regioselectivity from 6β‐ (MDCA formation) to 7β‐hydroxylation (UDCA production) demonstrates how protein engineering can be used to create custom biocatalysts, even for reactions where no naturally occurring counterpart has yet been identified.
Conflict of interest
S.K. and H.B. are employees of Enzymicals AG and B.G. is an employee of HERBRAND PharmaChemicals GmbH.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Technologie‐Beratungs‐Institut GmbH for funding of the P450‐OH project (grant number: TBI‐V‐1‐198‐VBW‐068). We are also grateful to Dr. Henk‐Jan Joosten for useful discussions related to the 3DM database used. T.B. was supported by the Austrian Science Fund (FWF) through the Erwin Schrödinger Fellowship (grant number: J4231‐B21). S.W. was supported by a Humboldt research fellowship. Open access funding enabled and organized by Projekt DEAL.
S. Grobe, C. P. S. Badenhorst, T. Bayer, E. Hamnevik, S. Wu, C. W. Grathwol, A. Link, S. Koban, H. Brundiek, B. Großjohann, U. T. Bornscheuer, Angew. Chem. Int. Ed. 2021, 60, 753.
References
- 1. Leuschner U., Leuschner M., Sieratzki J., Kurtz W., Hübner K., Dig. Dis. Sci. 1985, 30, 642–649. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Santiago P., Scheinberg A. R., Levy C., Ther. Adv. Gastroenter. 2018, 11, 1–15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Goossens J.-F., Bailly C., Pharmacol. Ther. 2019, 203, 107396. [DOI] [PubMed] [Google Scholar]
- 3. Cheng K., Ashby D., Smyth R. L., Cochrane Db. Syst. Rev. 2017, 9, CD000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Gulamhusein A. F., Hirschfield G. M., Nat. Rev. Gastro. Hepat. 2020, 17, 93–110; [DOI] [PubMed] [Google Scholar]
- 4b. Goel A., Kim W. R., Clin. Liv. Dis. 2018, 22, 563–578; [DOI] [PubMed] [Google Scholar]
- 4c. Chascsa D. M. H., Lindor K. D., J. Gastroenterol. 2020, 55, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hagey L. R., Crombie D. L., Espinosa E., Carey M. C., Igimi H., Hofmann A. F., J. Lipid Res. 1993, 34, 1911–1917. [PubMed] [Google Scholar]
- 6. He X.-L., Wang L.-T., Gu X.-Z., Xiao J.-X., Qiu W.-W., Steroids 2018, 140, 173–178. [DOI] [PubMed] [Google Scholar]
- 7. Zheng M.-M., Wang R.-F., Li C.-X., Xu J.-H., Process Biochem. 2015, 50, 598–604. [Google Scholar]
- 8. Tonin F., Arends I. W. C. E., Beilstein J. Org. Chem. 2018, 14, 470–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Hofmann A. F., Lundgren G., Theander O., Brimacombe J. S., Cook M. C., Acta Chem. Scand. 1963, 17, 173–186; [Google Scholar]
- 9b. Eggert T., Bakonyi D., Hummel W., J. Biotechnol. 2014, 191, 11–21. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Giovannini P. P., Grandini A., Perrone D., Pedrini P., Fantin G., Fogagnolo M., Steroids 2008, 73, 1385–1390; [DOI] [PubMed] [Google Scholar]
- 10b. Tonin F., Otten L. G., Arends I. W. C. E., ChemSusChem 2019, 12, 3192–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park R. J., Steroids 1981, 38, 383–395. [DOI] [PubMed] [Google Scholar]
- 12. Sheriha G. M., Waller G. R., Chan T., Tillman A. D., Lipids 1968, 3, 72–78. [DOI] [PubMed] [Google Scholar]
- 13. Tsai S.-J. J., Zhong Y.-S., Weng J.-F., Huang H.-H., Hsieh P.-Y., J. Chromatogr. A 2011, 1218, 524–533. [DOI] [PubMed] [Google Scholar]
- 14. Wu S., Snajdrova R., Moore J., Baldenius K., Bornscheuer U. T., Angew. Chem. Int. Ed. 2020, 59, 10.1002/anie.202006648; Angew. Chem 2020, 132, . [DOI] [Google Scholar]
- 15. Kollerov V. V., Monti D., Deshcherevskaya N. O., Lobastova T. G., Ferrandi E. E., Larovere A., Gulevskaya S. A., Riva S., Donova M. V., Steroids 2013, 78, 370–378. [DOI] [PubMed] [Google Scholar]
- 16. Kulprecha S., Ueda T., Nihira T., Yoshida T., Taguchi H., Appl. Environ. Microbiol. 1985, 49, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hofmann A. F., Hagey L. R., Cell. Mol. Life Sci. 2008, 65, 2461–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Urlacher V. B., Girhard M., Trends Biotechnol. 2019, 37, 882–897. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Park H., Park G., Jeon W., Ahn J.-O., Yang Y.-H., Choi K.-Y., Biotechnol. Adv. 2020, 40, 107504; [DOI] [PubMed] [Google Scholar]
- 19b. Wei Y., Ang E. L., Zhao H., Curr. Opin. Chem. Biol. 2018, 43, 1–7. [DOI] [PubMed] [Google Scholar]
- 20. Qu G., Li A., Sun Z., Acevedo-Rocha C. G., Reetz M. T., Angew. Chem. Int. Ed. 2020, 59, 13204–13231; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13304–13333. [Google Scholar]
- 21. Montemiglio L. C., Parisi G., Scaglione A., Sciara G., Savino C., Vallone B., Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 465–475. [DOI] [PubMed] [Google Scholar]
- 22. Parisi G., Montemiglio L. C., Giuffrè A., Macone A., Scaglione A., Cerutti G., Exertier C., Savino C., Vallone B., FASEB J. 2019, 33, 1787–1800. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Shah S., Xue Q., Tang L., Carney J. R., Betlach M., McDaniel R., J. Antibiot. 2000, 53, 502–508; [DOI] [PubMed] [Google Scholar]
- 23b. Olano C., Rodriguez A. M., Michel J. M., Méndez C., Raynal M. C., Salas J. A., Mol. Gen. Genet. 1998, 259, 299–308; [DOI] [PubMed] [Google Scholar]
- 23c. Gaisser S., Lill R., Staunton J., Méndez C., Salas J., Leadlay P. F., Mol. Microbiol. 2002, 44, 771–781. [DOI] [PubMed] [Google Scholar]
- 24. Lee S. K., Basnet D. B., Hong J. S. J., Jung W. S., Choi C. Y., Lee H. C., Sohng J. K., Ryu K. G., Kim D. J., Ahn J. S., et al., Adv. Synth. Catal. 2005, 347, 1369–1378. [Google Scholar]
- 25. Agematu H., Matsumoto N., Fujii Y., Kabumoto H., Doi S., Machida K., Ishikawa J., Arisawa A., Biosci. Biotechnol. Biochem. 2006, 70, 307–311. [DOI] [PubMed] [Google Scholar]
- 26. Grobe S., Wszołek A., Brundiek H., Fekete M., Bornscheuer U. T., Biotechnol. Lett. 2020, 42, 819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gotoh O., J. Biol. Chem. 1992, 267, 81–90. [PubMed] [Google Scholar]
- 28. Kuipers R. K., Joosten H.-J., van Berkel W. J. H., Leferink N. G. H., Rooijen E., Ittmann E., van Zimmeren F., Jochens H., Bornscheuer U., Vriend G., Martins dos Santos V. A. P., Schaap P. J., Proteins Struct. Funct. Bioinf. 2010, 78, 2101–2113. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Jochens H., Bornscheuer U. T., ChemBioChem 2010, 11, 1861–1866; [DOI] [PubMed] [Google Scholar]
- 29b. Jochens H., Hesseler M., Stiba K., Padhi S. K., Kazlauskas R. J., Bornscheuer U. T., ChemBioChem 2011, 12, 1508–1517; [DOI] [PubMed] [Google Scholar]
- 29c. Jochens H., Aerts D., Bornscheuer U. T., Protein Eng. Des. Sel. 2010, 23, 903–909. [DOI] [PubMed] [Google Scholar]
- 30. Li A., Acevedo-Rocha C. G., Sun Z., Cox T., Xu J. L., Reetz M. T., ChemBioChem 2018, 19, 221–228. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Appel D., Schmid R. D., Dragan C.-A., Bureik M., Urlacher V. B., Anal. Bioanal. Chem. 2005, 383, 182–186; [DOI] [PubMed] [Google Scholar]
- 31b. Lisurek M., Kang M.-J., Hartmann R. W., Bernhardt R., Biochem. Biophys. Res. Commun. 2004, 319, 677–682. [DOI] [PubMed] [Google Scholar]
- 32. Liu L., Aigner A., Schmid R. D., Appl. Microbiol. Biotechnol. 2011, 90, 127–135. [DOI] [PubMed] [Google Scholar]
- 33. Gielen F., Hours R., Emond S., Fischlechner M., Schell U., Hollfelder F., Proc. Natl. Acad. Sci. USA 2016, 113, E7383–E7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li A., Acevedo-Rocha C. G., D′Amore L., Chen J., Peng Y., Garcia-Borràs M., Gao C., Zhu J., Rickerby H., Osuna S., Zhou J., Reetz M. T., Angew. Chem. Int. Ed. 2020, 59, 12499–12505; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12599–12605. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary