

Abstract
Background and purpose
Infections with human herpesvirus 6A (HHV‐6A) and Epstein–Barr virus (EBV) have been linked to multiple sclerosis (MS) development. For EBV, late infection has been proposed as a risk factor, but serological support is lacking. The objective of this study was to investigate how age affects the EBV and HHV‐6A associated risks of developing MS.
Methods
In this nested case–control study, Swedish biobanks were accessed to find pre‐symptomatically collected blood samples from 670 individuals who later developed relapsing MS and 670 matched controls. A bead‐based multiplex assay was used to determine serological response against EBV and HHV‐6A. Conditional logistic regression was used to calculate odds ratios and 95% confidence intervals.
Results
Seropositivity against EBV exhibited a pattern where associations switched from a decreased risk of developing MS in the group below 20 years of age to an increased risk amongst individuals aged 20–29 and 30–39 years (p for trend 0.020). The age of transition was estimated to be 18.8 years. In contrast, HHV‐6A was associated with increased MS risk in all age groups (total cohort odds ratio 2.1, 95% confidence interval 1.6–2.7).
Conclusions
This study suggests EBV infection after adolescence and age independent HHV‐6A infection as risk factors for MS.
Keywords: case–control studies, Epstein–Barr virus, human herpesvirus 6A, multiple sclerosis, serology
Epstein–Barr virus infection after adolescence and age independent human herpesvirus 6A infection are risk factors for multiple sclerosis.

Introduction
Despite continued studies of how environmental factors together with genetic susceptibility influence the risk of developing multiple sclerosis (MS), the exact cause of this disease is yet to be explained. Several infectious agents have been implicated in MS etiology over the years, but most findings have been inconsistent. Amongst the more promising candidates are viruses belonging to the Herpesviridae family, specifically the Epstein–Barr virus (EBV), which has shown the most consistent association with MS [1, 2] and human herpesvirus 6A (HHV‐6A) [3].
These viruses share many attributes, such as the ability to establish lifelong latent infection in the host and being successful in evading the immune system, but differ in important aspects such as disease panorama, cell tropism [4] and strength of association to MS [1, 3]. EBV infects primarily B‐lymphocytes and is known to cause infectious mononucleosis (IM) [5] a condition that is associated with increased risk of developing MS [6]. HHV‐6A and HHV‐6B were quite recently recognized as two separate viruses [7]. The latter of the two is the cause of roseola in young children [8, 9] whereas the primary infection symptoms of HHV‐6A are less clear. They both primarily infect CD4+ T‐lymphocytes, although they are known to infect other cells as well [10, 11]. As for the association with MS, the picture is less clear than for EBV which may partly be explained by the fact that HHV‐6A and HHV‐6B are so similar that, until now, it has been hard to distinguish between them serologically [3].
As indicated by migration studies [12] and the association between MS and IM [6] the age at exposure to environmental factors such as viral infections are probably important for their potential role in MS development. Analogous to the association between late infection with the polio virus and poliomyelitis [13] it is possible that only late EBV infection increases MS risk. In a recent study, using a novel assay to differentiate serological response to HHV‐6A and HHV‐6B, it was demonstrated that HHV‐6A serology was associated with an increased MS risk both before and after MS onset [14]. In that study a substantial interaction was also observed between HHV‐6A and EBV serology in relation to MS risk, in an established MS cohort.
In this nested case–control study serological response to EBV and HHV‐6A was analyzed in enlarged pre‐MS material. The objective was to test the hypothesis that the age at infection with these viruses influences the risk of developing MS. Additionally, it was sought to explore whether HHV‐6A interacts with EBV to modulate MS risk. To accomplish these aims pre‐symptomatically drawn blood samples from persons who later developed MS were analyzed along with matched controls. The samples used were stored in six different Swedish biobanks, selected because they contained material from a substantial number of young individuals.
Material and methods
Case ascertainment
This study included serum or plasma drawn from 670 MS patients before onset of symptoms and 670 matched controls. The samples were identified by crosslinking the Swedish MS registry (www.neuroreg.se), containing data on 11,196 MS cases as of February 2012, with five Swedish microbiological biobanks. These biobanks contain the remainders of sera after clinical microbiological analyses performed at the University Hospitals of Skåne, Göteborg, Örebro, Linköping and the Public Health Agency of Sweden (PHAS). For case identification in an additional biobank, located in Umeå, a local registry of MS and possible MS diagnoses was used. Inclusion criteria for the study were that cases had developed relapsing–remitting MS and that samples were drawn before symptom onset and before the age of 40. For every MS case, one control matched for biobank, sex, date of blood sampling and date of birth (decreasing priority) was selected. The controls were generally well matched with an absolute mean difference of 6 days for date of sampling and 152 days for age at sampling. Data on HHV‐6A and HHV‐6B antibody reactivity have been published previously for some individuals (n = 944) included in this study [14].
Laboratory procedures
The samples were analyzed with a bead‐based multiplex assay, described in detail elsewhere [15] to quantify immunoglobulin G antibodies against viral proteins from EBV, HHV‐6A and HHV‐6B by measuring median fluorescence intensity (MFI) with a Luminex 200 analyzer. The EBV antigens were EBNA‐1 trunc (aa 325‐641), EBNA‐1 pep (aa 385‐420) [16] and VCA p18 (aa 1‐175) [17]. HHV‐6 antigens were immediate early 1 (IE1) protein regions derived from HHV‐6A and HHV‐6B (IE1A and IE1B respectively) and a region of the structural protein 101K from HHV‐6B. Samples were analyzed in multiple batches and inter‐batch controls were used to correct for batch‐related variability using standard linear or modified logarithmic models where appropriate [14].
Statistical analysis
Based on the age at sample collection the study population was divided into three age groups, <20, 20–29 and 30–39 years of age. For 29 matched case–control pairs, the case and control were on different sides of an age cut‐off, and these sets were assigned to the group with the least individuals (i.e., the oldest or youngest group). Odds ratios (ORs) for being seropositive against EBV or HHV‐6A were calculated using conditional logistic regression, both in univariable models and in models adjusting for antibody reactivities against the other virus. Conditional logistic regression was also used to test for trends across the three age groups. For distribution comparisons between groups, the Mann–Whitney U test or the Kruskal–Wallis test were used. Calculations were performed with IBM SPSS statistics version 23 (IBM Corporation, New York, NY, USA) or the software R version 3.6.1 (R Core Team, Vienna, Austria). All graphs were constructed in R.
Epstein–Barr virus antigen serostatus was determined using previously published cut‐offs: EBNA‐1 trunc, 1800 MFI; EBNA‐1 pep, 411 MFI; and VCA p18, 2,526 MFI [17]. An individual was considered seronegative for EBV if there was no seroresponse to any of the three EBV antigens. The HHV‐6A and HHV‐6B assays were not previously validated against a serological reference assay. Thus, seropositivity was determined using a cut‐off of 50 MFI to maximize sensitivity whilst also remaining above the technical noise of the assay at 30 MFI. This cut‐off was adjusted in two separate sensitivity analyses, one using >30 MFI and another applying >80 MFI to determine positivity. A sensitivity analysis of the multivariable logistic regression models was also performed where seropositive individuals had their antibody reactivity modeled as quartiles.
The proportion of EBV seropositives was also modeled using logistic regression adjusting for age, group (i.e., case–control status) and the age–group interaction. The age of intersection between groups was calculated from interaction terms and model visualization was made using predicted probabilities. Attributable proportions from interactions were calculated using logistic regression according to Hosmer and Lemeshow [18].
Ethical considerations
This study was performed in accordance with the ethical standards of the Declaration of Helsinki and approved by a Regional Ethical Review Board in Umeå (2011‐198‐31M). Written informed consent was not required. Study participants were informed of the study through a letter in the mail and had a chance to opt out.
Results
A total of 670 case–control sets, from six different biobanks, with a median age of 25 years at the time of sampling were included in this study. A majority were female (84%) and the median time from sampling until disease onset was 8 years (Table 1). Individuals were stratified into three groups based on the age at sampling and analyses were performed separately for these groups. Age at disease onset differed between age groups, where the cases in the older groups had a later age at MS onset (p < 0.001). Seropositivity amongst controls in the three largest biobanks (Skåne, PHAS and Umeå) that together made up 83% of all samples was almost identical for EBV (93%, 92% and 93% respectively) whereas there was more variation regarding HHV‐6A positivity (25%, 32% and 26% respectively).
Table 1.
Characteristics of cases and controls
| n | Cases | n | Controls | p | |
|---|---|---|---|---|---|
| All | |||||
| Sex (M/F), % | 670 | 16/84 | 670 | 16/84 | – |
| Age at sampling, years | 25 (21–29) | 25 (21–29) | 0.97 | ||
| Age at disease onset, years | 33 (28–40) | n.a. | – | ||
| Time from sampling until disease onset, years | 8 (4–13) | n.a. | – | ||
| Age group < 20 years | |||||
| Sex (M/F), % | 143 | 23/77 | 143 | 23/77 | – |
| Age at sampling, years | 18 (14–19) | 18 (14–19) | 0.82 | ||
| Age at disease onset, years | 26 (22–31) | n.a. | – | ||
| Time from sampling until disease onset, years | 10 (6–16) | n.a. | – | ||
| Age group 20–29 years | |||||
| Sex (M/F), % | 376 | 14/86 | 376 | 14/86 | – |
| Age at sampling, years | 25 (22–27) | 25 (23–27) | 0.89 | ||
| Age at disease onset, years | 33 (29–38) | n.a. | – | ||
| Time from sampling until disease onset, years | 8 (3–13) | n.a. | – | ||
| Age group 30–39 years | |||||
| Sex (M/F), % | 151 | 15/85 | 151 | 15/85 | – |
| Age at sampling, years | 33 (31–35) | 33 (31–35) | 0.72 | ||
| Age at disease onset, years | 40 (37–43) | n.a. | – | ||
| Time from sampling until disease onset, years | 6 (3–10) | n.a. | – | ||
| Biobank | |||||
| Umeå | 102 | 15.2 % | 102 | 15.2 % | – |
| PHAS | 139 | 20.8 % | 139 | 20.8 % | – |
| Örebro | 29 | 4.3 % | 29 | 4.3 % | – |
| Göteborg | 47 | 7.0 % | 47 | 7.0 % | – |
| Skåne | 314 | 46.9 % | 314 | 46.9 % | – |
| Linköping | 39 | 5.8 % | 39 | 5.8 % | – |
Values expressed as a percentage for proportions and median (interquartile range) for continuous variables. p values were calculated with the Mann–Whitney U test.
Abbreviation: PHAS, Public Health Agency of Sweden.
For EBNA‐1 trunc, EBNA‐1 pep and VCA p18, there were consistent patterns where seropositivity was associated with a lower risk of developing MS in the youngest age group and with an increased risk of developing MS in the older groups (Table 2). Trend analysis of EBV serostatus and age at sampling was significant (p = 0.020). The age where seropositivity to EBV switched from being a protective factor for MS to a risk factor was 18.8 years (Figure 1). In a multivariable conditional logistic regression analysis of seropositivity to EBNA‐1 trunc, adjusted for HHV‐6A IE1A antibody reactivity on a continuous scale, there was a significantly lower risk of MS in the group < 20 years of age (OR = 0.52, 95% confidence interval [CI] 0.29–0.94) whilst older individuals aged 20–39 had an increased risk (OR = 3.5, 95% CI 1.7–7.1). A sensitivity analysis adjusting for IE1A reactivity modeled as quartiles amongst positive individuals showed similar results, with the addition that EBV positivity against any of the three antigens also became significantly associated with a reduced risk for MS in the youngest group (OR = 0.51, 95% CI 0.26–0.99).
Table 2.
Associations between seropositivity and MS risk
| Viral antigen | Seropositive/total n | Univariable | Multivariable | |||||
|---|---|---|---|---|---|---|---|---|
| Case | Control | OR | 95% CI | p | OR | 95% CI | p | |
| EBNA‐1 trunc | HHV‐6A adj. | |||||||
| All | 613/670 | 600/670 | 1.3 | 0.88–2.0 | 0.19 | 1.2 | 0.83–1.9 | 0.30 |
| <20 | 100/143 | 113/143 | 0.59 | 0.34–1.0 | 0.07 | 0.52 | 0.29–0.94 | 0.03 |
| 20–29 | 364/376 | 349/376 | 2.9 | 1.3–6.4 | 0.01 | 3.0 | 1.3–6.7 | 0.01 |
| 30–39 | 149/151 | 138/151 | 6.5 | 1.5–28.8 | 0.01 | 5.5 | 1.2–24.8 | 0.03 |
| Trend | 0.003 | |||||||
| EBNA‐1 pep | HHV‐6A adj. | |||||||
| All | 600/670 | 568/670 | 1.6 | 1.1–2.2 | 0.01 | 1.5 | 1.1–2.1 | 0.02 |
| <20 | 98/143 | 109/143 | 0.67 | 0.39–1.1 | 0.14 | 0.60 | 0.34–1.0 | 0.07 |
| 20–29 | 357/376 | 331/376 | 2.6 | 1.5–4.7 | 0.001 | 2.6 | 1.5–4.6 | 0.001 |
| 30–39 | 145/151 | 128/151 | 3.8 | 1.6–9.4 | 0.003 | 3.5 | 1.4–8.6 | 0.01 |
| Trend | 0.001 | |||||||
| VCA p18 | HHV‐6A adj. | |||||||
| All | 598/670 | 575/670 | 1.4 | 1.0–2.0 | 0.04 | 1.3 | 0.95–1.9 | 0.10 |
| <20 | 105/143 | 107/143 | 0.91 | 0.51–1.6 | 0.76 | 0.79 | 0.43–1.5 | 0.45 |
| 20–29 | 350/376 | 333/376 | 1.7 | 1.0–2.9 | 0.03 | 1.7 | 1.0–2.8 | 0.0499 |
| 30–39 | 143/151 | 135/151 | 2.1 | 0.87–5.3 | 0.10 | 2.0 | 0.79–4.9 | 0.15 |
| Trend | 0.12 | |||||||
| EBV | HHV‐6A adj. | |||||||
| All | 629/670 | 624/670 | 1.2 | 0.72–1.8 | 0.55 | 1.1 | 0.66–1.7 | 0.81 |
| <20 | 111/143 | 121/143 | 0.60 | 0.32–1.1 | 0.12 | 0.53 | 0.27–1.0 | 0.06 |
| 20–29 | 368/376 | 360/376 | 2.1 | 0.87–5.3 | 0.10 | 2.0 | 0.81–4.9 | 0.13 |
| 30–39 | 150/151 | 143/151 | 8.0 | 1.0–64.0 | 0.0499 | 6.7 | 0.82–54.6 | 0.08 |
| Trend | 0.020 | |||||||
| IE1A | EBV adj. | |||||||
| All | 263/670 | 166/670 | 2.1 | 1.6–2.7 | <0.001 | 2.0 | 1.5–2.6 | <0.001 |
| <20 | 47/143 | 29/143 | 2.0 | 1.1–3.5 | 0.02 | 1.7 | 0.92–3.1 | 0.09 |
| 20–29 | 148/376 | 95/376 | 2.0 | 1.5–2.9 | <0.001 | 2.0 | 1.3–2.8 | <0.001 |
| 30–39 | 68/151 | 42/151 | 2.5 | 1.4–4.4 | 0.001 | 2.4 | 1.3–4.5 | 0.006 |
| Trend: | 0.56 | |||||||
Bold Values are significant to p<0.05
Abbreviations: CI, confidence interval; EBV, positive seroresponse to either EBNA‐1 trunc, EBNA‐1 pep or VCA p18; EBV adj, adjusted for reactivity against all three EBV antigens; HHV‐6A adj, adjusted for reactivity against IE1A; OR, odds ratio; MS, multiple sclerosis.
Figure 1.
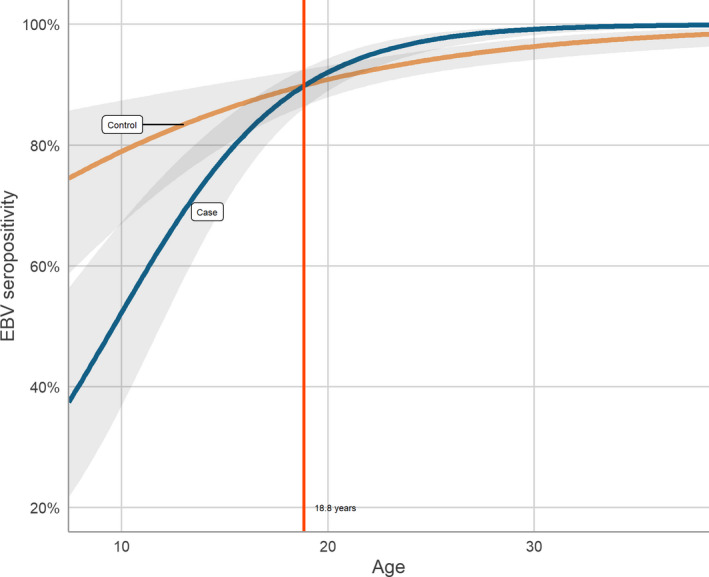
Predicted probability of EBV seropositivity by age for cases and controls with 95% confidence intervals. Curves were estimated using logistic regression. A cut‐off of 18.8 years was calculated as the age at intersection between the two curves. EBV seropositivity is defined as seroresponse to EBNA‐1 trunc, EBNA‐1 pep or VCA p18. [Colour figure can be viewed at wileyonlinelibrary.com]
Seropositivity against IE1A was associated with significantly increased MS risk in the total cohort (OR = 2.1, 95% CI 1.6–2.7) as well as in all three age groups (Table 2). This finding was shown to be robust in sensitivity analyses with both higher and lower cut‐offs, as well as in the multivariable sensitivity analyses that adjusted for EBV antibody reactivity modeled as quartiles amongst seropositives. No significant associations with risk for MS development were seen for either HHV‐6B IE1B or 101K seropositivity (data not shown) and these antigens were excluded from further analyses.
Analysis of interaction on an additive scale between EBV and HHV‐6A seropositivity was not significant although the estimated attributable proportion due to interaction was high (43%). For this analysis, the group with the lowest risk was used as the reference category (EBV+ IE1A for those < 20 years and EBV− IE1A for the age group 20–39 years) (Figure 2).
Figure 2.
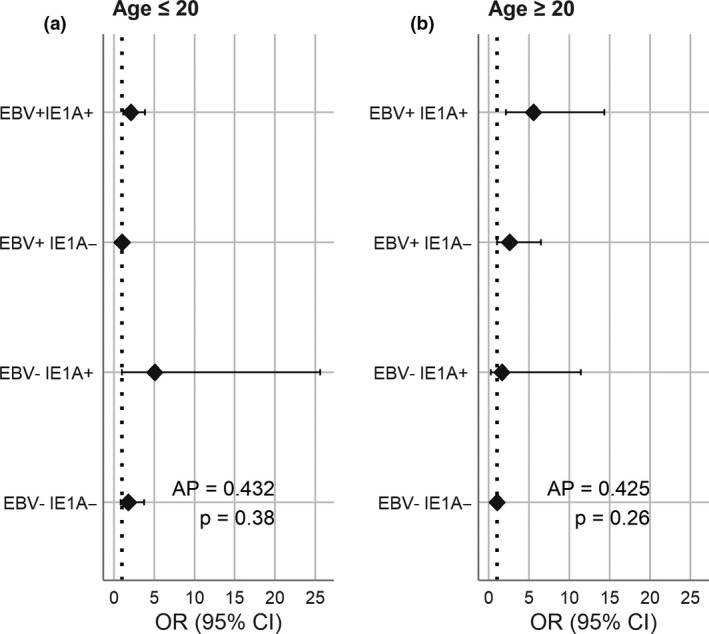
Interaction between EBV and HHV‐6A in relation to MS risk. AP, attributable proportion; OR, odds ratio. EBV seropositivity defined as seroresponse to EBNA‐1 trunc, EBNA‐1 pep or VCA p18. HHV‐6A positivity defined as reactivity against IE1A > 50 median fluorescence intensity.
Discussion
In this study associations between MS and serological response to EBV, HHV‐6A and HHV‐6B antigens were investigated using prospectively collected blood samples stored for up to four decades in Swedish microbiological biobanks. The biobanks provided a unique material of frozen plasma and serum that enabled us to look at associations between MS and serological markers of infection several years before symptom onset, at an age believed to be critical for disease initiation. A substantial number of the analyzed samples came from individuals below 20 years of age at the time of sampling, providing an opportunity to study markers of EBV and HHV‐6A infection during childhood/adolescence amongst those who later developed MS.
Our findings of associations between EBV seropositivity and risk of developing MS support the hypothesis that EBV infection influences MS risk and stress the importance of timing. Depending on the age at infection, the effect of EBV on the risk of developing MS seems to shift from a lowering to an increase in risk. It is important to note, however, that most individuals that were EBV negative below 20 years of age will be infected later in life. Therefore, the apparently protective effect of EBV exposure in the youngest age group should not be interpreted as contradicting the large body of evidence associating EBV negativity with low risk of developing MS. An increased risk, as seen here in individuals aged above 20 years, is an expected result in line with earlier studies [19]. The age dependence of EBV infection in relation to MS has also been suggested earlier based on epidemiology [20] as well as antibody reactivity to EBNA before and after 20 years of age [21]. This, however, to our knowledge is the first time a serological study indicates that individuals who later develop MS become seropositive to EBV later in life compared with controls, further supporting earlier epidemiological studies showing that MS is rare in populations where EBV infection occurs at an early age [22]. Why an infection during or after adolescence, in comparison to an infection during childhood, more often leads to IM is not well understood. Exposure to larger virus volume at transmission, preexisting cross‐reactive CD8+ T cells or a less effective natural killer cell response in adolescence have been suggested as explanations [23]. It is possible that the powerful activation of the immune system during IM increases the risk of a persistent dysregulated immune system, which in turn could be a prerequisite for MS.
Another way to interpret this finding is that late infection is a surrogate marker of increased hygiene, where infections supposedly occur later in life in more affluent societies, which in turn may infer increased MS risk through currently unknown pathways. This is unlikely to be the entire explanation, however, since EBV infection has been shown to precede MS onset [24] and individuals negative to EBV have an extremely low risk, if any, of developing MS [19]. Additionally, it conflicts with another finding in this study that a larger proportion of individuals who develop MS have detectable antibodies toward HHV‐6A compared to controls. It seems unlikely that a high level of hygiene should delay infection with one very prevalent human herpesvirus (EBV) but not another (HHV‐6A).
Contrary to our findings regarding EBV, HHV‐6A positivity was significantly associated with increased MS risk across all age strata. HHV‐6A has repeatedly been implicated in MS etiology [3] but since, until recently, it has been difficult to separate the serological response against HHV‐6A and HHV‐6B, much is still unknown about the epidemiology of these two viruses. However, it is known that they share a broad cell tropism, even though HHV‐6A seems to have greater neurotropism and is acquired later in life [25]. In addition to CD4+ T lymphocytes being the main target for infection and replication, HHV‐6A has been shown, in vitro, to infect both astrocytes [11] and oligodendrocytes [10] implying that it might be a prime candidate for direct involvement in demyelinating autoimmune diseases of the central nervous system (CNS).
In our earlier pre‐symptomatic study investigating serological response against five common childhood infections, HHV‐6 (detected with a method not able to separate A from B) was the only virus that together with EBV was associated with an increased MS risk [26]. There is also evidence that some patients have a subset of oligoclonal bands against HHV‐6, A or B not specified [27]. The present study shows a consistent association exclusively to HHV‐6A when using a species‐specific serological assay. Just as for EBV, the mechanism by which HHV‐6A infection might influence MS risk is currently unknown, but a few hypotheses have been put forward. HHV‐6A has been shown to transactivate HERV‐K18 expression [28] similarly to EBV, which in turn may induce expression of a superantigen. Additionally, HHV‐6A could contribute to demyelinating disease by affecting myelin production through interference with migration of oligodendrocyte progenitor cells [29] or via cytotoxic effect on oligodendrocytes [30].
Two possible mechanisms by which infection with EBV could increase the risk of developing MS are molecular mimicry, supported by our recent study [31] or through direct CNS infection [1]. These mechanisms may also apply to HHV‐6A. Given that HHV‐6A can infect oligodendrocytes, it is possible that it directs the immune system against this host cell by incorporation of host cell proteins and lipids in its membrane [32] and thus specifically directs the immune system against the oligodendrocytes. However, upon inoculation with HHV‐6A the most potent activator of T‐cells (i.e., dendritic cells) lose their capacity to activate T‐cell proliferation [33, 34] suggesting that the hypothesis of host cell protein incorporation and molecular mimicry, which requires that the virus constitutes an adjuvant effect, is somewhat problematic. Fortunately, the virus is not able to completely avoid immune detection and serological responses are indeed mounted against HHV‐6A, as seen in the present study as well as previous studies. EBV might also have a role in this hypothesized disease mechanism of host protein incorporation by HHV‐6A, as EBV immortalization of activated B cells could make an inappropriate immune response against oligodendrocytes difficult to terminate.
An interaction between EBV and HHV‐6A has already been suggested as a causal factor in MS etiopathogenesis [35] through reactivation of latent EBV in the CNS by HHV‐6A infection [36, 37]. One way to investigate such interactions is through testing for departure from additivity, which if present could indicate that two risk factors are part of the same causal pathway [38]. Using this methodology a significant interaction between EBV and HHV‐6A in samples taken after MS onset was found previously [14]. Whilst no significant interaction was found in the present study, possibly due to power limitations, the additive effect observed was large (attributable proportion 43%). However, a possible effect modification was found, where seropositivity based on EBNA‐1 trunc was significantly associated with reduced MS risk only in the multivariable model adjusted for IE1A reactivity.
Our study has limitations that warrant discussion, one being the fact that MS itself may cause a dysregulation of the immune system. Evidence of a preclinical or prodromal phase of MS, lasting many years or even decades, is accumulating [39]. For example, higher levels of serum neurofilament light chain, a marker of axonal damage, was detected a median of 6 years before clinical onset in a recently published study [40]. In light of this, even a pre‐symptomatic approach with samples collected a median of 8 years prior to symptom onset, such as in this study, might not be sufficient to completely mitigate this effect. An additional limitation is that only one sample from each individual was accessed and so the age at which seroconversion had occurred could not be determined. Therefore, older individuals seropositive for EBV could have been infected early in life. This does not explain, however, the differences between cases and controls of similar age seen in this study. Perhaps more problematic is that age at disease onset differs depending on age at sampling, where individuals in the group with samples drawn at a young age had MS onset earlier than those with samples drawn later in life. This is probably a consequence of the case selection process since only cases who had their sample drawn before MS onset were included, but it may still have affected the results. Another limitation is that the assay used to separate immunoglobulin G responses to HHV‐6A and HHV‐6B could not be validated against a gold standard method. Conclusive data on the sensitivity and specificity of the assay to separate infections of these two viruses are therefore currently lacking. However, a specificity study performed in our recent publication [14] suggests that the assay can discriminate IE1B from IE1A. Also, the two sensitivity analyses using different cut‐offs for seropositivity to HHV‐6A found significant associations, indicating clear differences between cases and controls in their immunological response to this antigen.
To conclude, our findings give serological support to the hypothesis that late, in contrast to early, EBV infection increases risk of MS development. It is also shown that antibodies against HHV‐6A are associated with increased MS risk independent of age. To determine whether these viruses interact as risk factors for MS, further prospective studies with sufficient power are needed.
Disclosure of conflicts of interest
M.B. has received a speaker fee from Biogen. T.O. has received grants and personal fees from Biogen, Novartis, Sanofi, Merck, Roche and Almirall, not related to this study. The other authors report no conflicts of interest.
Acknowledgements
Financial support was provided by the Swedish Research Council (2015‐02419). T.O. received grant support from the Swedish Brain Foundation, KAW Foundation and Margaretha af Ugglas Foundation. J.H. and I.K. were supported by Horizon 2020 MultipleMS grant number 733161. J.H. also received a grant from the Multiple Sclerosis Society of Canada (EGID 3045).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Ascherio A, Munger K. Epidemiology of multiple sclerosis: from risk factors to prevention—an update. Semin Neurol 2016; 36: 103–114. [DOI] [PubMed] [Google Scholar]
- 2. Olsson T, Barcellos LF, Alfredsson L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 2017; 13: 25–36. [DOI] [PubMed] [Google Scholar]
- 3. Leibovitch EC, Jacobson S. Evidence linking HHV‐6 with multiple sclerosis: an update. Curr Opin Virol 2014; 9: 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grinde B. Herpesviruses: latency and reactivation—viral strategies and host response. J Oral Microbiol 2013; 5: 22766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thorley‐Lawson DA. EBV persistence—introducing the virus. Curr Top Microbiol Immunol 2015; 390: 151–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Handel AE, Williamson AJ, Disanto G, et al An updated meta‐analysis of risk of multiple sclerosis following infectious mononucleosis. PLoS One 2010; 5: e12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ablashi D, Agut H, Alvarez‐Lafuente R, et al Classification of HHV‐6A and HHV‐6B as distinct viruses. Arch Virol 2014; 159: 863–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamanishi K, Okuno T, Shiraki K, et al Identification of human herpesvirus‐6 as a causal agent for exanthem subitum. Lancet 1988; 1: 1065–1067. [DOI] [PubMed] [Google Scholar]
- 9. Dewhurst S, McIntyre K, Schnabel K, Hall CB. Human herpesvirus 6 (HHV‐6) variant B accounts for the majority of symptomatic primary HHV‐6 infections in a population of U.S. infants. J Clin Microbiol 1993; 31: 416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ahlqvist J, Fotheringham J, Akhyani N, et al Differential tropism of human herpesvirus 6 (HHV‐6) variants and induction of latency by HHV‐6A in oligodendrocytes. J Neurovirol 2005; 11: 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donati D, Martinelli E, Cassiani‐Ingoni R, et al Variant‐specific tropism of human herpesvirus 6 in human astrocytes. J Virol 2005; 79: 9439–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gale CR, Martyn CN. Migrant studies in multiple sclerosis. Prog Neurogibol 1995; 47: 425–448. [PubMed] [Google Scholar]
- 13. Nathanson N, Kew OM. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am J Epidemiol 2010; 172: 1213–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engdahl E, Gustafsson R, Huang J, et al Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol 2019; 10: 2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waterboer T, Sehr P, Michael KM, et al Multiplex human papillomavirus serology based on in situ‐purified glutathione S‐transferase fusion proteins. Clin Chem 2005; 51: 1845–1853. [DOI] [PubMed] [Google Scholar]
- 16. Sundström P, Nyström M, Ruuth K, Lundgren E. Antibodies to specific EBNA‐1 domains and HLA DRB1*1501 interact as risk factors for multiple sclerosis. J Neuroimmunol 2009; 215: 102–107. [DOI] [PubMed] [Google Scholar]
- 17. Brenner N, Mentzer AJ, Butt J, et al Validation of multiplex serology detecting human herpesviruses 1–5. PLoS One 2018; 13: e0209379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hosmer DW, Lemeshow S. Confidence interval estimation of interaction. Epidemiology 1992; 3: 452–456. [DOI] [PubMed] [Google Scholar]
- 19. Pakpoor J, Disanto G, Gerber JE, et al The risk of developing multiple sclerosis in individuals seronegative for Epstein–Barr virus: a meta‐analysis. Mult Scler 2013; 19: 162–166. [DOI] [PubMed] [Google Scholar]
- 20. Warner HB, Carp RI. Multiple sclerosis and Epstein–Barr virus. Lancet 1981; 2: 1290. [DOI] [PubMed] [Google Scholar]
- 21. Levin LI, Munger KL, Rubertone MV, et al Temporal relationship between elevation of Epstein–Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA 2005; 293: 2496–500. [DOI] [PubMed] [Google Scholar]
- 22. Jons D, Sundström P, Andersen O. Targeting Epstein–Barr virus infection as an intervention against multiple sclerosis. Acta Neurol Scand 2015; 131: 69–79. [DOI] [PubMed] [Google Scholar]
- 23. Dunmire SK, Hogquist KA, Balfour HH. Infectious mononucleosis Münz C., In: Current topics in microbiology and immunology. Springer: Cham; 2015:211–240. 10.1007/978-3-319-22822-8_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levin LI, Munger KL, O’Reilly EJ, et al Primary infection with the Epstein–Barr virus and risk of multiple sclerosis. Ann Neurol 2010; 67: 824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hall CB, Caserta MT, Schnabel KC, et al Persistence of human herpesvirus 6 according to site and variant: possible greater neurotropism of variant A. Clin Infect Dis 1998; 26: 132–137. [DOI] [PubMed] [Google Scholar]
- 26. Sundström P, Juto P, Wadell G, et al An altered immune response to Epstein–Barr virus in multiple sclerosis: a prospective study. Neurology 2004; 62: 2277–2782. [DOI] [PubMed] [Google Scholar]
- 27. Virtanen JO, Pietiläinen‐Nicklén J, Uotila L, et al Intrathecal human herpesvirus 6 antibodies in multiple sclerosis and other demyelinating diseases presenting as oligoclonal bands in cerebrospinal fluid. J Neuroimmunol 2011; 237: 93–97. [DOI] [PubMed] [Google Scholar]
- 28. Tai AK, Luka J, Ablashi D, Huber BT. HHV‐6A infection induces expression of HERV‐K18‐encoded superantigen. J Clin Virol 2009; 46: 47–48. [DOI] [PubMed] [Google Scholar]
- 29. Campbell A, Hogestyn JM, Folts CJ, et al Expression of the human herpesvirus 6A latency‐associated transcript U94A disrupts human oligodendrocyte progenitor migration. Sci Rep 2017; 7: 3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kong H, Baerbig Q, Duncan L, et al Human herpesvirus type 6 indirectly enhances oligodendrocyte cell death. J Neurovirol 2003; 9: 539–550. [DOI] [PubMed] [Google Scholar]
- 31. Tengvall K, Huang J, Hellström C, et al Molecular mimicry between anoctamin 2 and Epstein–Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc Natl Acad Sci USA 2019; 116: 16955–16960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nauclér CS, Larsson S, Möller E. A novel mechanism for virus‐induced autoimmunity in humans. Immunol Rev 1996; 152: 175–192. [DOI] [PubMed] [Google Scholar]
- 33. Gustafsson RKL, Engdahl EE, Hammarfjord O, et al Human herpesvirus 6A partially suppresses functional properties of DC without viral replication. PLoS One 2013; 8: e58122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gustafsson R, Svensson M, Fogdell‐Hahn A. Modulatory effects on dendritic cells by human herpesvirus 6. Front. Microbiol 2015; 6: 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fierz W. Multiple sclerosis: an example of pathogenic viral interaction? Virol J. 2017; 14: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cuomo L, Angeloni A, Zompetta C, et al Human herpesvirus 6 variant A, but not variant B, infects EBV‐positive B lymphoid cells, activating the latent EBV genome through a BZLF‐1‐dependent mechanism. AIDS Res Hum Retroviruses 1995; 11: 1241–1245. [DOI] [PubMed] [Google Scholar]
- 37. Flamand L, Menezes J. Cyclic AMP‐responsive element‐dependent activation of Epstein–Barr virus zebra promoter by human herpesvirus 6. J Virol 1996; 70: 1784–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ahlbom A, Alfredsson L. Interaction: A word with two meanings creates confusion. Eur J Epidemiol 2005; 20: 563–564. [DOI] [PubMed] [Google Scholar]
- 39. Giovannoni G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol 2017; 16: 413–414. [DOI] [PubMed] [Google Scholar]
- 40. Bjornevik K, Munger KL, Cortese M, et al Serum neurofilament light chain levels in patients with presymptomatic multiple sclerosis. JAMA Neurol 2019; 77: 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.