

Accelerated drug development for diseases with high unmet need demands patient focus without compromising scientific rigor. Precision dosing informed by physiologically‐based pharmacokinetic (PBPK) models will enable inclusive clinical trials and narrow the gap between clinical development and real‐world use. Early incorporation of the mass balance (MB) study is crucial to maximize the fidelity of a PBPK model‐informed approach to precision dosing across populations and contexts of use in clinical development and therapeutic use.
Critical Determinants of PBPK Models for Precision Dosing
A call to action for inclusive trials and rational dosing recommendations across clinical use settings will require precision dosing solutions enabled by quantitative translational frameworks with a “totality of evidence” mindset. 1 We posit that PBPK models based on quantitative understanding of in vivo human clearance routes (e.g., biliary, renal, and metabolic), pathways (e.g., primary biotransformations), and molecular mechanisms (e.g., enzymes and transporters) are critical enablers. For these models to be applied with confidence, it is essential that the relative contributions of the clearance components are fully elucidated to be able to accurately capture, in simulations, the effects of disease (physiology and biochemistry) or interacting drugs in vivo. As an example, such a high‐fidelity PBPK model, with well‐defined disposition, was developed previously for the antipsychotic olanzapine to assess the effects of smoking and subsequent cessation on the exposure of the drug in schizophrenics. 2
The pivotal role of the human MB study
Two categories of questions rationalize the conduct of human MB studies. 3 Historically, a key focus has been characterizing metabolite profiles to identify and qualify toxicologic coverage of circulating metabolites. Secondly, germane to what we posit here is the pivotal role of the MB study in quantifying clearance mechanisms. The latter is foundationally important to defining sources of variability in pharmacokinetics (absorption, distribution, metabolism, and excretion (ADME) pharmacogenomics, drug–drug interactions (DDIs), and eliminating organ function) and therefore drug response.
Typically, an MB study would be conducted after human pharmacokinetics are characterized and a pharmacologically active dose range is identified. Ideally, it would be done in parallel with proof‐of‐concept (PoC) trials to build knowledge relevant to Metabolites in Safety Testing (MIST) ahead of pivotal trials and to allow development of a high‐fidelity PBPK model to inform the clinical pharmacology plan and support acceleration of development, should emerging data from the PoC study support this. Furthermore, MB data can inform classification under the Biopharmaceutics Classification System for rational development and bridging of formulations.
In therapeutic areas like oncology, it is not uncommon for PoC studies, sometimes integrated within the first‐in‐human study, to start generating evidence of efficacy that propels the development program to the registration‐enabling phase. In such cases, a pragmatic approach to inform the immediate needs of the clinical development plan has been to conduct a DDI study with a strong inhibitor of the drug‐metabolizing enzyme with the highest expected contribution to overall metabolism based solely on in vitro studies. Such a strategy, while practically reasonable, may be at risk of limited translatability given the complexities of enzyme–transport interplay, enterohepatic recirculation, and extrahepatic metabolism often associated with molecules in the current drug development space. It is acknowledged that frontloading (micro)tracer development and clinical mass balance studies ahead of PoC may not always be practically feasible. Accordingly, considerations of uncertainty in quantitative translation of human clearance mechanisms, overall development strategy (e.g., accelerated vs. standard), and the target patient population (e.g., risks for polypharmacy or organ impairment) should guide an optimal plan. Prior to availability of MB data, data from metabolite scouting of human urine and feces using radiocalibrants from preclinical or in vitro metabolism studies for fit‐for‐purpose quantitation should be leveraged for PBPK model development.
MB studies traditionally use the clinically intended route of administration. While sufficient to address MIST objectives, the value of information from this study can be greatly enhanced for PBPK model development if evaluation of absolute bioavailability is built in. 4 If feces are analyzed for parent drug following intravenous (IV) administration, quantification of biliary plus intestinal secretory clearance will be possible.
As a result of high‐throughput ADME screening, early metabolite identification, and exploration of novel chemical entities, metabolically stable compounds, which are more susceptible to transporter‐mediated disposition, continue to increase in drug discovery and development portfolios. Novel in vitro approaches continue to be developed to address the challenges of low clearance, a significant advance that can only enhance the development of high‐fidelity PBPK models and translational approaches. 5 Coupled with in vitro enzyme kinetic studies of uptake and efflux transport, enzyme–transport interplay can be appropriately factored into PBPK models to enhance fidelity of human predictions of DDI and impact of pharmacogenetic polymorphisms. 6
Roadmap for precision dosing based on the totality of evidence
How can one implement precision dosing across the R&D continuum and ultimately in guidance for prescribers at the point of drug approval? Herein, we envision and propose a pyramid view of the roadmap beginning early in development (Figure 1 ). The foundation (Base) of the pyramid relates to quantitative assessment of clearance mechanisms, consisting of MB data and relevant in vitro studies that quantify enzyme/transporter contributions for the pathways informed by the MB study. The middle of the pyramid consists of PBPK modeling within a Predict‐Learn‐Confirm‐Apply (P‐L‐C‐A) framework where data from the Base of the pyramid are crucial to the ”Predict” component. 7 In settings of accelerated development where decisions need to be made early, a “Confirm” opportunity (i.e., a DDI study) may not be available. If based solely on in vitro data, application of the PBPK model for enabling precision dosing and inclusive drug development is likely to be limited. While a purely reductionist approach to PBPK model development is aspirational, complexities in human ADME processes challenge the fidelity of purely bottom‐up models. As such, having the MB data as a core component of the predictive translational framework elevates its fidelity to a point where the P‐L‐C‐A framework takes the form of a P‐LC‐A framework.
Figure 1.
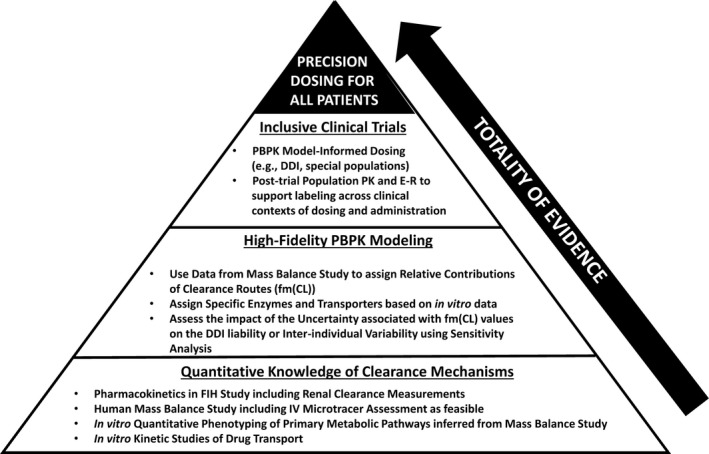
Roadmap for precision dosing based on the totality of evidence. A pyramid view of the roadmap consists of the following key elements: the base of the pyramid relates to quantitative assessment of clearance mechanisms based on MB (mass balance) data and relevant in vitro studies; the middle of the pyramid pertains to the development of a high‐fidelity PBPK model informed by the MB and in vitro data; in the upper level of the pyramid, predictions from a high‐fidelity PBPK model can inform optimal dosing across clinical trials. DDI, drug–drug interactions; E–R, exposure–response; FIH, first‐in‐human; fm(Cl), contributions of routes to overall clearance; IV, intravenous; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetics.
As illustrated in the upper level of the pyramid, predictions from a high‐fidelity PBPK model can inform optimal dosing across clinical contexts of use in later‐phase trials, or potentially even in the postmarketing setting without clinical data (e.g., DDI studies and organ impairment studies; Figure 1 ). Access to patients in clinical trials and access to promising investigational agents by patients is a continuing challenge in drug development for oncologic indications and for rare diseases. Precision dosing in pivotal trials based on high‐fidelity PBPK models and exposure–response model‐informed knowledge of the therapeutic window is a rational approach to patient‐focused drug development. Coupled with population PK assessments across all patients in such inclusive trials employing precision dosing, the “C” of “Confirm” in the P‐L‐C‐A framework can be realized. In some cases, it may even be necessary for such confirmation to emerge post marketing (e.g., as part of postmarketing surveillance), necessitating high confidence in PBPK model‐based predictions. While PBPK model‐informed labeling for DDIs without a single index DDI study is largely unprecedented, one recent example is voxelotor, discussed below as one of two cases. In that case, the pivotal enabler to compress “L” and “C” of the P‐L‐C‐A framework for the intended context of use was the MB study.
Case study 1: Buprenorphine
Neonatal abstinence syndrome, a condition affecting newborns exposed to an opioid in utero, is often treated with buprenorphine. Few pharmacokinetic studies of buprenorphine in children, particularly neonates, are available as conducting clinical trials in this population is challenging. The availability of a high‐fidelity PBPK model allows dosing strategies for buprenorphine in children to be assessed. As described previously, clearance routes (cytochrome P450 (CYP)–mediated and uridine diphosphate (UDP)‐glucuronosyltransferase (UGT)–mediated metabolism and biliary clearance) were assigned based on MB data, including metabolite identification, after IV administration (Figure 2 ). 8 In vitro studies indicate that CYP3A4 and UGT1A1 are the main enzymes involved in the metabolism. About 33% of the parent drug appeared unchanged in the feces; this may represent buprenorphine itself undergoing P‐glycoprotein (P‐gp)–mediated efflux out of the liver or deconjugation of the glucuronidated form of the parent. Both scenarios were assessed using the PBPK model and the relative contributions of the different clearance mechanisms were confirmed using the clinical DDI data (PLCA framework). Application of age‐related physiology and ontogeny of relevant enzymes, demonstrated that the model was able to capture exposures of buprenorphine after sublingual dosing in term neonates with neonatal abstinence syndrome. 8
Figure 2.
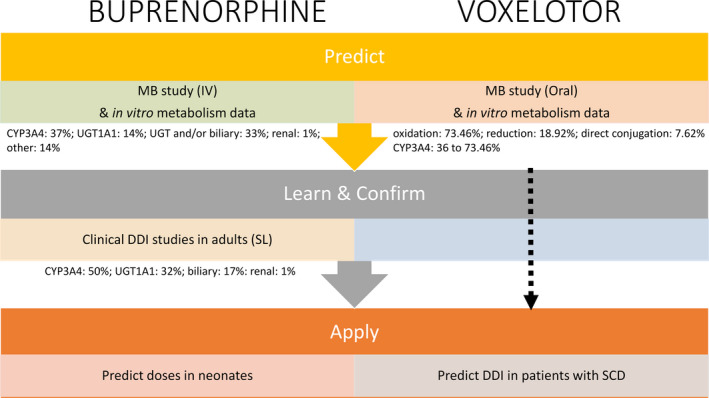
PBPK modeling within a Predict‐Learn‐Confirm‐Apply framework. High‐fidelity PBPK models for buprenorphine and voxelotor were informed by quantitative understanding of clearance mechanisms from the human MB (mass balance) study. The final models were used to assess the exposure of buprenorphine in neonates with NAS (neonatal abstinence syndrome) and investigate DDIs (drug–drug interactions) in patients with SCD (sickle cell disease), respectively. CYP, cytochrome P450; IV, intravenous; SL, sublingual; UGT, uridine diphosphate (UDP)‐glucuronosyltransferase.
The route of administration in clinical DDI studies was sublingual, and therefore elucidation of the metabolic (CYP and UGT) vs. efflux transporter contributions (via P‐gp) was complicated in that nonspecific modulators of both CYP3A4 and P‐gp (ketoconazole and rifampin) were used. Thus, in this case study, exploitation of the MB study data, particularly for IV administration, in conjunction with the in vitro data, was crucial to the integrity of the PBPK model development, as it allowed clearance routes to be defined and differentiated (intestine vs. liver) and constraints to be set around the relative contributions of these route.
Case study 2: Voxelotor
Voxelotor was recently approved by the US Food and Drug Administration (FDA) for treatment of patients with sickle cell disease (SCD). 9 Data from an MB study and in vitro studies involving recombinant enzymes and chemical inhibition in human liver microsomes indicate that voxelotor is extensively metabolized through phase I (oxidation and reduction) and phase II (glucuronidation) metabolism. Oxidation of voxelotor is mediated primarily by CYP3A4, with minor contributions from CYP2C19, CYP2B6, and CYP2C9. A PBPK model based on these data was utilized to quantify the effects of CYP3A4 modulators on the exposure of voxelotor in patients with SCD. The MB study results were used to estimate the contribution of metabolic pathways to the overall clearance of voxelotor (oxidation: 73.46%; reduction: 18.92%; and direct conjugation: 7.62%). Three scenarios were simulated to address the uncertainty associated with the CYP3A4‐mediated metabolism of voxelotor; contributions of 36% (chemical inhibition in human liver microsomes), 56% (recombinant enzymes), and 73.46% (worst‐case scenario assuming that all of the oxidation was mediated by CYP3A4) were considered.
As there were no clinical DDI studies with voxelotor as a victim, the simulations were used to inform labeling; concomitant administration of drugs that are strong CYP3A4 inhibitors (ketoconazole) is predicted to increase voxelotor exposure by 42% to 83%. Strong or moderate inducers of CYP3A4 are predicted to decrease voxelotor exposure by 77% and 60%, respectively. Although it is recommended to avoid coadministration of strong/moderate CYP3A4 modulators in patients with SCD taking daily doses of 1,500 mg, if unavoidable, a dose reduction to 1,000 mg (strong CYP3A4 inhibitor and fluconazole) and dose escalation to 2,500 mg (strong/moderate CYP3A4 inducer) is warranted, as reflected in the United States prescribing information. 10 The proposed dose adjustment (2,500 mg once daily) considered the PBPK model‐predicted effect of rifampin, exposure–response relationships, and the dose range of voxelotor that had been evaluated during clinical development.
Concluding Remarks
Implementation of precision dosing should enable inclusive drug development, thereby resulting in the broadest possible access to medicines and prescribing guidance across populations and clinical contexts of use. High‐fidelity PBPK models informed by quantitative understanding of clearance mechanisms from the human MB study represent critical translational enablers which may realize the promise of precision dosing beginning in early drug development through practice.
Funding
No funding was received for this work.
Conflict of Interest
K.R.Y. is an employee of Certara UK Limited (Simcyp Division). K.V. is an employee of EMD Serono Research & Development Institute, Inc., a business of Merck KGaA. K.R.Y. and K.V. have declared no competing interests for this work.
Disclaimer
As an Associate Editor of Clinical Pharmacology & Therapeutics, Karthik Venkatakrishnan was not involved in the review or decision process for this paper.
Contributor Information
Karen Rowland Yeo, Email: Karen.Yeo@certara.com.
Karthik Venkatakrishnan, Email: karthik.venkatakrishnan@emdserono.com.
References
- 1. Powell, J.R. , Cook, J. , Wang, Y. , Peck, R. & Weiner, D. Drug dosing recommendations for all patients: A roadmap for change. Clin. Pharmacol. Ther. 109, 65–72 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sun, L. , von Moltke, L. & Rowland Yeo, K. Physiologically‐based pharmacokinetic modeling for predicting drug interactions of a combination of olanzapine and samidorphan. CPT Pharmacometrics Syst. Pharmacol. 9, 106–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roffey, S.J. , Obach, R.S. , Gedge, J.I. & Smith, D.A. What is the objective of the mass balance study? A retrospective analysis of data in animal and human excretion studies employing radiolabled drug. Drug Metab. Rev. 39, 17–43 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Spracklin, D.K. , Chen, D. , Bergman, A.J. , Callegari, E. & Obach, R.S. Mini‐review: comprehensive drug disposition knowledge generated in the modern human radiolabeled ADME study. CPT Pharmacometrics Syst. Pharmacol. 9, 428–434 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Di, L. & Obach, R.S. Addressing the challenge of low clearance in drug research. AAPS J. 17, 352–357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Patilea‐Vrana, G. & Unadkat, J.D. Transport vs. metabolism: what determines the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs? Insights from the extended clearance model. Clin. Pharmacol. Ther. 100, 413–418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao, P. et al Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 89, 259–267 (2011). [DOI] [PubMed] [Google Scholar]
- 8. Johnson, T.N. , Jamei, M. & Rowland Yeo, K. How does in vivo biliary elimination of drugs change with age? Evidence from in vitro and clinical data using a systems pharmacology approach. Drug Metab. Dispos. 44, 1090–1098 (2016). [DOI] [PubMed] [Google Scholar]
- 9. US Food and Drug Administration . Oxbryta (voxeletor) clinical pharmacology and biopharmaceutics review: summary basis of approval <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213137Orig1s000OtherR.pdf> (2019). [Google Scholar]
- 10. USFDA . Oxbryta [prescribing information]. (Global Blood Therapeutics, South San Francisco, CA, 2019) <https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213137s000lbl.pdf> [Google Scholar]