

Abstract
Essentials.
Liver diseases are associated with profound hemostatic changes proportional to severity of illness.
Hemostatic changes in acute‐on‐chronic liver failure (ACLF) may in part reflect critical illness.
Hemostatic changes in ACLF partly overlap with those in sepsis, with rebalanced hemostasis in both.
Patients with sepsis had hyperfibrinogenemia, associated with a thrombogenic clot structure.
Abstract
Background
Even the sickest patients with chronic liver disease (CLD), such as those with acute‐on‐chronic liver failure (ACLF) remain in hemostatic balance due to a concomitant decline in pro‐ and antihemostatic factors.
Objectives
We aimed to study whether the hemostatic status in ACLF is merely an exaggeration from the status in patients with compensated and acutely decompensated cirrhosis, or whether sepsis‐associated hemostatic changes contribute.
Methods
We performed extensive hemostatic profiling in 31 adult patients with ACLF, 20 patients with sepsis without underlying CLD, and 40 healthy controls.
Results
We found similarly elevated plasma levels of the platelet adhesive protein von Willebrand factor (VWF) and decreased levels of the VWF‐regulating protease ADAMTS13 in both groups compared to healthy controls. In vivo markers of activation of coagulation (thrombin‐antithrombin III, D‐dimer) were similarly elevated in both groups compared to controls, but ex vivo thrombin‐generating capacity was similar between patients and controls, despite a much more profound international normalized ratio elevation in ACLF. Plasma fibrinogen levels were much higher in septics, which was accompanied by a decreased ex vivo clot permeability and an increase in ex vivo resistance to clot lysis. All hemostatic parameters were remarkably stable over the first 10 days after admission.
Conclusions
We have found hemostatic changes in ACLF to partially overlap with that of patients with sepsis, and evidence of preserved hemostatic capacity in both patient groups. The notable difference was a profound hyperfibrinogenemia, associated with a thrombogenic clot structure and a marked ex vivo resistance to fibrinolysis in patients with sepsis.
Keywords: cirrhosis, coagulation, fibrinogen, fibrinolysis, intensive care
1. INTRODUCTION
Patients with chronic liver disease (CLD) frequently have complex alterations in their hemostatic system. These changes include thrombocytopenia, low levels of coagulation factors and inhibitors, low levels of fibrinolytic proteins, and elevated levels of endothelial‐derived hemostatic proteins including the platelet adhesive molecule von Willebrand factor (VWF). 1 The extent of the hemostatic alterations are proportional to the severity of liver disease, but surprisingly, the net effect of the hemostatic changes appears largely independent of disease severity. Both in patients with compensated cirrhosis, 2 and in patients with acute‐on‐chronic liver failure (ACLF) in which cirrhosis is complicated by multi‐organ failure, 3 the hemostatic system appears to remain in balance due to a concomitant decline in pro‐ and antihemostatic drivers. 4 , 5 However, it may be that the stability of this new hemostatic balance decreases with increasing severity of CLD, which would result in an increased risk for bleeding and thrombotic complications. ACLF is a syndrome in patients with underlying cirrhosis characterized by systemic inflammation, development of organ failure, and high short‐term mortality; 6 , 7 , 8 it can occur at any stage of cirrhosis. Sepsis, often related to spontaneous bacterial peritonitis as well as pneumonia and urinary tract infections, is a frequent precipitant of ACLF, 6 and presence or development of sepsis is associated with a poor prognosis. 9
We recently reported on the hemostatic status of patients with ACLF. We found a progressive decrease in plasma levels of hemostatic proteins from patients with compensated cirrhosis to patients with acute decompensation of cirrhosis to patients with ACLF, and a progressive increase in endothelial‐derived proteins such as VWF. 3 The net result of these changes was a comparable thrombin‐generating capacity, and a mixed fibrinolytic phenotype with both hyper‐ and hypofibrinolysis in patients with ACLF compared to healthy individuals. 10 We studied these hemostatic changes in samples taken at the time of hospital admission or at the time of diagnosis of ACLF. However, because ACLF can have a very dynamic evolution, 11 the hemostatic status may vary significantly over time and therefore longitudinal assessment of these hemostatic parameters would be of value.
At first sight, the hemostatic changes in patients with ACLF are merely an exaggeration of the hemostatic changes in patients with compensated cirrhosis, and appear to reflect the severity of hepatic synthetic failure. However, some of the hemostatic changes we observed in ACLF also occur in patients with critical illness in the absence of underlying CLD. For example, patients with sepsis or disseminated intravascular coagulation (DIC) have been reported to have high circulating levels of VWF with low levels of the VWF‐cleaving protease ADAMTS13, 12 , 13 , 14 low levels of pro‐ and anticoagulant proteins with relatively preserved thrombin generating capacity, 15 , 16 , 17 and a hypofibrinolytic state. 18 Additional similarities between ACLF and septic patients without CLD exist, such as the phenomenon of immuneparesis, 19 which may also have consequences for the hemostatic system. 20
We performed a systematic profiling of hemostatic capacity by using functional hemostasis tests and by quantifying plasma levels of selected hemostatic proteins in patients with ACLF in comparison to patients with sepsis in the absence of underlying CLD to assess the potential contribution of sepsis to the hemostatic changes in ACLF. Hemostatic profiles were determined longitudinally to assess changes in hemostatic status during the course of the disease.
2. METHODS
2.1. Patients
We included 31 adult patients with ACLF and 20 patients with sepsis in the absence of CLD who were admitted to King's College Hospital London (UK) and provided written informed consent in this study. The National Research Ethics Service (NRES) Committee London‐Westminster (12/LO/1417) approved the study protocol, which was in accordance with the Declaration of Helsinki. Informed consent or assent was obtained from participants or their personal consultees in the case of mental incapacity. Exclusion criteria for this study were acute liver failure, known congenital coagulation disorders, pregnancy, human immunodeficiency virus positivity, extrahepatic malignancy, and hepatocellular carcinoma outside the Milan criteria. Cirrhosis was defined by the presence of two or more of the following: (a) histological evidence of cirrhosis on liver biopsy, (b) laboratory abnormalities consistent with cirrhosis, or (c) radiological findings consistent with cirrhosis and portal hypertension. ACLF was defined and graded according to the number of organ failures in concordance with criteria reported in the CANONIC study. 16 For patients with sepsis without CLD, the diagnosis of sepsis was based on the Sepsis‐3 criteria, 21 in which life‐threatening organ dysfunction caused by a dysregulated host response to infection was evident, with organ dysfunction defined by an increase in the sequential (sepsis‐related) organ failure assessment (SOFA) score of 2 points or more. The absence of underlying CLD in this patient group was determined by a combined assessment of biochemical and radiological parameters. Healthy controls aged >18 years (n = 40) were recruited at King's College Hospital to establish reference values for the various laboratory tests performed. Healthy controls also provided informed consent with the protocol approved by the NRES Committee London‐Westminster (12/LO/1417). Exclusion criteria for healthy controls were body mass index below 18 or above 28; pregnancy or active breastfeeding, a personal history of thrombotic or liver disease; untreated medical conditions; chronic medical conditions requiring regular primary or secondary care review; or current use of anticoagulants, platelet function inhibitors, or oral contraceptives.
2.2. Blood samples
Blood samples were collected in sodium citrate‐containing vacutainer tubes (0.129 mol/L) from an arterial line, central venous catheter, or by standard peripheral venous phlebotomy within the first 2 days of admission or after the development of ACLF or sepsis. Additional samples were taken at day 3, between day 5 and day 7, and day 10. Within 2 hours after the blood draw, the sample was centrifuged at 2000 and 10 000 g, respectively, for 10 minutes at ambient temperature to obtain platelet‐poor plasma. Plasma was stored at −80°C until it was used for analyses.
2.3. Hemostatic assays
VWF levels were assessed with an in‐house enzyme‐linked immunosorbent assay (ELISA) using commercially available polyclonal antibodies against VWF (DAKO). Plasma activity of a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) was measured using the FRETS‐VWF73 assay (Peptanova). Plasma samples were pre‐treated with bilirubin oxidase (2.5 U/mL, Sigma‐Aldrich) for 30 minutes at 37°C to avoid interference of bilirubin with the FRETS‐VWF73 assay. 22 Levels of VWF and ADAMTS13 in pooled normal plasma were set at 100%, and values obtained in test plasmas were expressed as a percentage of pooled normal plasma.
The thrombin generation assay (TGA) was performed with the fluorimetric method described by Hemker et al: calibrated automated thrombography (CAT). 23 Coagulation was activated using commercially available reagents containing recombinant tissue factor (final concentration: 5 pmol/L), phospholipids (final concentration: 4 µmol/L), in the presence of soluble thrombomodulin (TM, the concentration of which is not revealed by the manufacturer). Reagents were purchased from Thrombinoscope BV, Maastricht, the Netherlands, and thrombin generation experiments were executed following protocols provided by Thrombinoscope.
The international normalized ratio (INR), D‐dimer levels, and plasma levels of factor VIII (FVIII), factor X (FX), factor II (FII), fibrinogen, antithrombin, and plasminogen were assessed on an automated coagulation analyzer (ACL 300 TOP) according to the protocols from the manufacturer (Werfen). Thrombin‐antithrombin complexes were quantified by a commercially available ELISA (Siemens). Factor XIII activity was determined as described previously. 24 Plasminogen activator inhibitor type 1 (PAI‐1) levels were quantified by commercially available ELISA from R & D systems.
Fibrin clot quality was quantified by estimating the average pore size of the fibrin clot, which was determined by permeation studies as described previously. 25 The permeability coefficient Ks was calculated following Darcy's law.
Plasma fibrinolytic potential was estimated by studying lysis of a tissue‐factor‐‐induced clot by exogenous tissue plasminogen activator (tPA) by monitoring changes in turbidity during clot formation and subsequent lysis as described previously. 26 Clot lysis time was determined as the time from the midpoint of the clear‐to‐maximum‐turbid transition, which characterizes clot formation, to the midpoint of the maximum turbid‐to‐clear transition, which represents clot lysis. Samples that were still clotted at 3 hours after the start of the experiment were arbitrarily assigned a clot lysis time of 180 minutes.
2.4. Statistical analyses
GraphPad Prism (San Diego, CA, USA) was used for data presentation and analysis. Tabulated data are presented as medians and ranges or as absolute numbers. Values of the various analytes were compared between groups, between different time points, and to controls using the Kruskal‐Wallis H test (with Dunn's posttest). A P‐value < .05 was considered significant.
3. RESULTS
3.1. Patient characteristics
We included 31 patients with ACLF and 20 with sepsis without CLD (from hereon referred to as non‐liver septics). Of the 31 ACLF patients, we sampled 17 at day 3, 14 between days 5 and 7, and 9 at day 10. Of the 20 non‐liver septics, we sampled 11 at day 3, 11 between days 5 and 7, and 10 at day 10. Demographic data obtained on admission are shown in Table 1, laboratory data on admission in Table 2, and results of microbiology screening in Table S1 in supporting information.
TABLE 1.
Demographic and clinical data of the study population on admission
| ACLF n = 31 | Non‐liver sepsis n = 20 | |
|---|---|---|
| Age, years | 48 (22–66) | 61 (23–71) |
| Etiology of liver disease | ||
| Alcohol | 21 (68%) | |
| Viral | 1 (3%) | |
| NASH | 3 (10%) | |
| Biliary | 2 (6%) | |
| Other | 4 (13%) | |
| Source of sepsis | ||
| Pulmonary | 13 (65%) | |
| Cellulitis | 2 (10%) | |
| Endocarditis | 1 (5%) | |
| Meningitis | 1 (5%) | |
| Dental | 1 (5%) | |
| Nosocomial vascular catheter | 1 (5%) | |
| Bacteraemia source unclear | 1 (5%) | |
| Male (n) | 21 (68%) | 11 (55%) |
| SOFA score | 8 (2–19) | 4 (1–9) |
| CLIF‐SOFA score | 11 (5–20) | n/a |
| CLIF‐C‐ACLF score | 57 (38–71) | n/a |
| ISTH DIC score | 0 (0–5) | |
| MELD, points | 40 (15–67) | n/a |
| Child‐Pugh, points | 11 (5–20) | n/a |
| Mechanical ventilation (n) | 11 (35%) | 18 (90%) |
| Vasopressors required (n) | 11 (35%) | 11 (55%) |
| RRT (n) | 17 (55%) | 5 (25%) |
| GCS | 15 (3–15) | 7 (2–15) |
| Mortality within 30 days of admission n (%) | 9 (29%) | 5 (25%) |
| Medication on admission | ||
| Antibiotics | ||
| Any | 30 (97%) | 20 (100%) |
| Oral | 26 (84%) | 9 (45%) |
| Parenteral | 6 (19%) | 11 (55%) |
| Antifungal | 8 (26%) | 1 (5%) |
| Betablocker | 17 (55%) | 0 (0%) |
| Rifaximin | 2 (6%) | 0 (0%) |
| Unfractionated heparin | 1 (3%) | 0 (0%) |
| Low molecular weight heparin | 1 (3%) | 3 (15%) |
| Platelet concentrate | 3 (10%) | 1 (5%) |
| Fresh frozen plasma | 10 (32%) | 2 (10%) |
| Cryoprecipitate | 5 (16%) | 0 (0%) |
| Ascites | ||
| No | 7 (23%) | 20 (100%) |
| Minimal | 14 (45%) | 0 (0%) |
| Moderate/Severe | 10 (32%) | 0 (0%) |
Shown are numbers or medians with ranges.
Abbreviations: ACLF, acute‐on‐chronic liver failure; CLIF, chronic liver failure; GCS, Glasgow Coma Score; ISTH DIC, International Society on Thrombosis and Haemostasis Disseminated Intravascular Coagulation; MELD, model of end‐stage liver disease; n/a, not applicable; NASH, non‐alcoholic steatohepatitis; RRT, renal replacement therapy; SOFA, sequential organ failure assessment.
TABLE 2.
Laboratory data of the study population on admission
| ACLF n = 31 | Non‐liver sepsis n = 20 | P value | |
|---|---|---|---|
| Full blood count | |||
| Hemoglobin (g/L) | 84 (66–136) | 93 (79–133) | .02 |
| Plt count (×109/L) | 76 (20–280) | 216 (79–568) | <.0001 |
| WBC count (×109/L) | 11.3 (2.3–25.8) | 13.2 (4.7–22.3) | n.s. |
| Neutrophils (×109/L) | 9.2 (1.3–22.9) | 9.5 (4.0–18.9) | n.s. |
| Lymphocytes (×109/L) | 1.0 (0.3–2.5) | 1.4 (0.5–3.1) | n.s. |
| Monocytes (×109/L) | 0.65 (0.12–2.04) | 0.59 (0.10–1.53) | n.s. |
| Standard coagulation | |||
| INR | 2.0 (1.2–7.7) | 1.2 (1.1–2.2) | <.0001 |
| APTT ratio | 1.6 (1.1–2.4) | 1.2 (1.0–3.4) | <.001 |
| Biochemistry | |||
| Na, mmol/L | 135 (121–149) | 140 (101–152) | .0076 |
| Urea, mmol/L | 10.2 (2.2–18.7) | 8.7 (3.5–22.4) | n.s. |
| Creatinine, µmol/L | 92 (2–329) | 90 (32–365) | n.s. |
| Bilirubin, µmol/L | 164 (10–714) | 7 (2–32) | <.0001 |
| Gamma glutamyl transaminase, IU/L | 76 (15–661) | 64 (13–58) | n.s. |
| Alkaline phosphatase, IU/L | 114 (32–363) | 92 (42–531) | n.s. |
| Aspartate transaminase, IU/L | 80 (13–662) | 64 (13–3718) | n.s. |
| Albumin, g/L | 29 (19–43) | 28 (21–38) | n.s. |
| CRP, mg/L | 35 (2–558) | 216 (23–381) | <.0001 |
| Interleukin 6 (ng/mL) | 77 (35–138) | 60 (41–238) | n.s. |
| Tumor necrosis factor alpha (pg/mL) | 14 (8–53) | 12 (8–444) | n.s. |
| Arterial | |||
| pH | 7.44 (7.33–7.56) | 7.39 (7.27–7.53) | n.s. |
| NH4+, µmol/L | 69 (24–124) | 30 (12–53) | <.0001 |
| Lactate, mmol/L | 1.7 (0.9–4.2) | 1.0 (0.7–1.9) | <.001 |
Shown are medians with ranges.
Abbreviations: APTT, activated partial thromboplastin time; CRP, C‐reactive protein; INR, international normalized ratio; n.s., not significant; plt, platelet; WBC, white blood cell.
3.2. A similar VWF and ADAMTS13 unbalance in ACLF and non‐liver septics
Plasma levels of VWF were substantially elevated in patients with ACLF and non‐liver sepsis compared to controls at all time points examined (Figure 1A). Although some patients with ACLF had much higher VWF levels particularly on day 1 compared to the non‐liver septics, there were no statistically significant differences between the patient groups at any time point. Also, there were no significant changes in VWF levels over time in both groups. ADAMTS13 levels were substantially decreased compared to controls in both groups at all time points examined with no significant changes over time (Figure 1B). Three of the non‐liver septics had undetectable ADAMTS13 at day 1 and 3. Platelet counts were substantially lower in patients with ACLF compared to non‐liver septics with the majority of patients below the normal range, and in non‐liver septics, platelet counts appeared to improve over time (Figure 1C).
FIGURE 1.
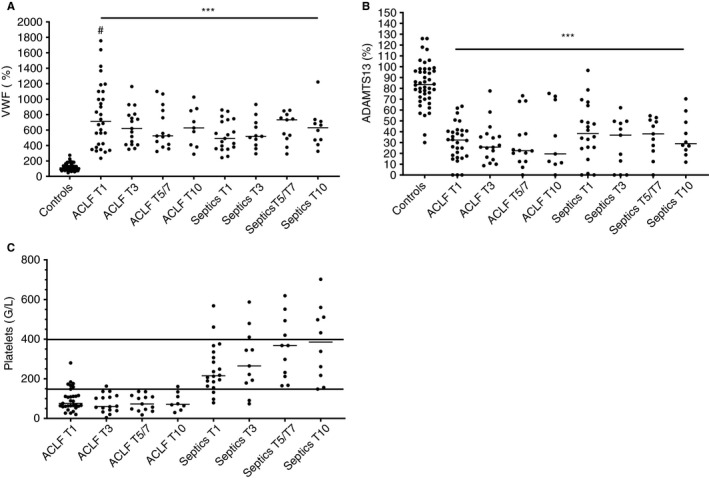
Plasma levels of von Willebrand factor (A), ADAMTS13 (B), and platelet count (C) in patients with ACLF and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. *** = P < .001 versus controls, # = P < .05 versus sepsis
3.3. Similar in vitro and in vivo hemostatic potential in ACLF and non‐liver septics despite much better preserved factor levels in non‐liver septics
The INR was substantially elevated in ACLF patients, but only slightly higher than controls in non‐liver septics (Figure 2). Accordingly, plasma levels of FX and FII and of antithrombin were substantially decreased in patients with ACLF but only marginally lower in non‐liver septics (Figure 3A‐C). Plasma levels of FVIII were slightly elevated in ACLF, and more profoundly elevated in non‐liver septics (Figure 3D). None of these analytes changed appreciably over time, although the INR appeared to decrease in ACLF patients after day 1, with an increase in plasma levels of FX and FII.
FIGURE 2.
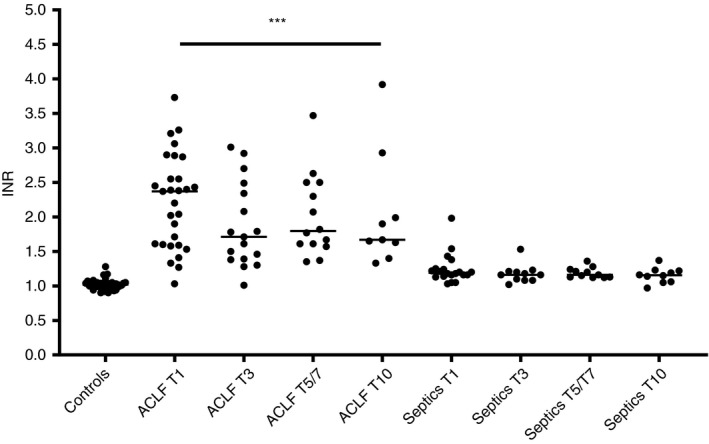
INR values in patients with ACLF and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. *** = P < .001 vs controls
FIGURE 3.
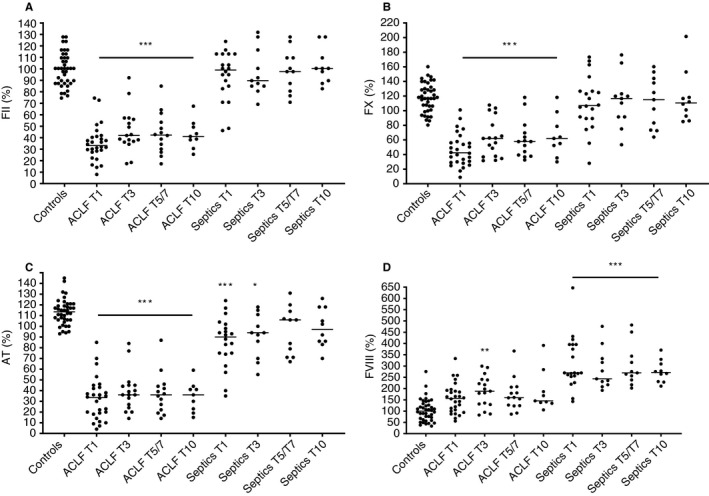
Plasma levels of FII (A), FX (B), AT (C), and FVIII (D) in patients with acute‐on‐chronic liver failure and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. * P < .05, *** = P < .001 vs controls
Despite profound differences in plasma levels of procoagulant proteins, in vitro thrombin‐generating capacity as assessed by thrombomodulin‐modified CAT was not significantly different among controls, ACLF, and non‐liver septics (Figure 4A). However, individual patients had endogenous thrombin potentials both below and above the reference ranges. Levels of D‐dimer and thrombin‐antithrombin complexes, which are markers of in vivo activation of coagulation, were elevated in both ACLF and non‐liver septics, but levels were similar between the patient groups with few changes over time (Figure 4B‐C).
FIGURE 4.
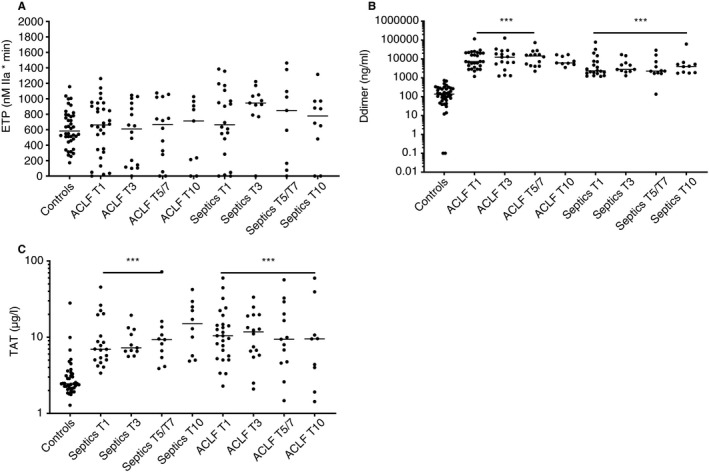
Endogenous thrombin potential values (A), D‐dimer levels (B), and thrombin‐antithrombin complex levels (C) in patients with acute‐on‐chronic liver failure and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. *** = P < .001 versus controls
3.4. Near normal fibrinogen parameters in ACLF but severe hyperfibrinogenemia with reduced clot permeability in non‐liver septics
Fibrinogen plasma levels were lower than the control group in the majority of patients with ACLF, although the difference did not reach statistical significance (Figure 5A). In contrast, fibrinogen levels were substantially elevated in non‐liver septics, with a mild decrease in levels over time. We assessed permeability of plasma clots as a measure of clot quality in day 1 samples primarily because the permeation assay is very labor intensive. Clot permeability was comparable between controls and ACLF patients, but a substantially decreased permeability was observed in the non‐liver septics (Figure 5B).
FIGURE 5.
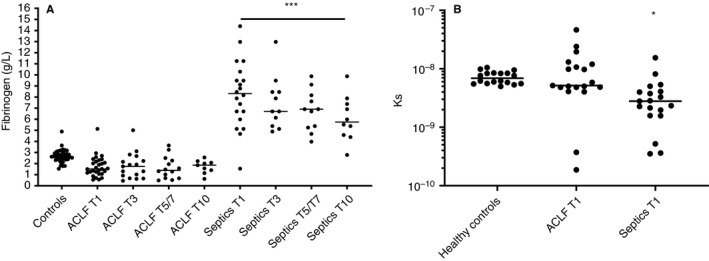
Plasma levels of fibrinogen (A) and fibrin clot permeability (B) in patients with acute‐on‐chronic liver failure and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. * P < .05, *** = P < .001 versus controls
3.5. Mixed in vitro fibrinolytic capacity in ACLF, but uniform hypofibrinolysis in non‐liver septics but similar in vivo activation of fibrinolysis in both groups
Plasma fibrinolytic potential was mixed in the ACLF group with some patients showing substantially prolonged, and some clearly shortened clot lysis times (Figure 6A). In contrast, the non‐liver septics all had normal to severely prolonged clot lysis times. Although PAI‐1 and plasminogen levels are the main determinants of clot lysis time in the general population, variation in these proteins did not explain the differences in CLT between ACLF and non‐liver septics (Figure 6B‐C). Plasminogen levels were substantially decreased in ACLF, but largely within the normal range in non‐liver septics, whereas PAI‐1 levels were increased in both groups, with virtually identical levels on day 1. Despite the difference in ex vivo fibrinolytic capacity, plasma levels of plasmin‐2‐antiplasmin (PAP) complexes, which indicate in vivo activation of fibrinolysis, were elevated to a similar extent in ACLF and non‐liver septics (Figure 6D). All fibrinolytic parameters varied at most only mildly over time.
FIGURE 6.
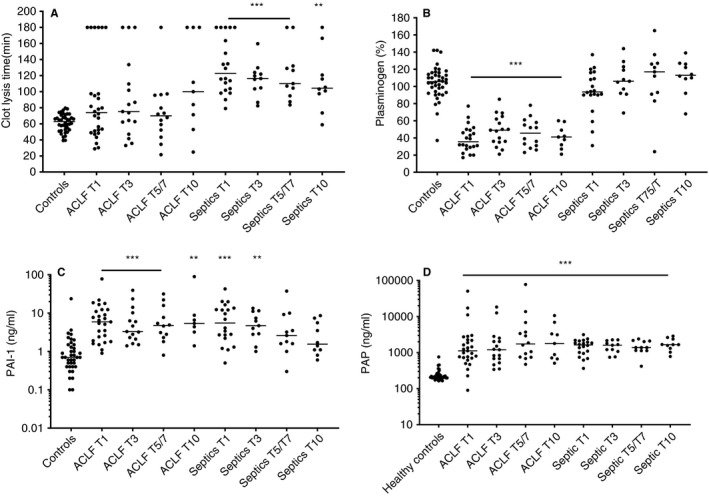
Clot lysis times (A), plasminogen levels (B), plasminogen activator inhibitor type 1 levels (C), and plasmin‐2‐antiplasmin complex levels in patients with acute‐on‐chronic liver failure and non‐liver septics sampled at various time points after admission in comparison to healthy controls. Horizontal lines indicate medians. ** P < .01, *** = P < .001 versus controls
4. DISCUSSION
Here we have performed extensive hemostatic profiling of a well‐phenotyped cohort of patients with ACLF in comparison with patients with sepsis without underlying CLD. We found a similar VWF/ADAMTS13 imbalance, and similar in vitro and in vivo coagulation potential despite much lower plasma levels of pro‐ and anticoagulant factors in ACLF patients. The notable difference between ACLF and non‐liver septics was a profound hyperfibrinogenemia with a thrombogenic clot structure and a profound in vitro resistance to fibrinolysis in the latter group. The non‐liver septics thus have a more hypercoagulable profile compared to ACLF patients. Notably, although both ACLF and sepsis are clinically very dynamic syndromes, we observed remarkably little change in hemostatic parameters over time.
The ACLF and non‐liver septics are remarkably similar in VWF/ADAMTS13 parameters, which may reflect similarities in inflammatory status leading to endothelial activation with release of reactive ultralarge VWF, which requires processing by ADAMTS13. 27 Interestingly, FVIII levels were lower in ACLF than in sepsis, which may be explained by the difference in VWF multimeric size between patients with ACLF and those with sepsis. Whereas VWF multimeric size is normal or enhanced in patients with sepsis, 28 patients with liver disease have a clear reduction in higher molecular weight multimers, despite decreased plasma levels of ADAMTS13. 29 In addition, modification of VWF by plasma proteases could potentially also directly disrupt the FVIII binding site on VWF in patients with ACLF. 30 Major differences in the plasma levels of hepatocyte‐derived hemostatic proteins are apparent, with much lower levels in ACLF, reflecting defective hepatic synthetic capacity. In our non‐liver sepsis patients, who did not have overt disseminated intravascular coagulation evidenced by the near normal INR and factor levels, preserved platelet count, and elevated fibrinogen, 31 , 32 coagulation status appears preserved. Nevertheless, global coagulation potential as assessed by CAT revealed individual patients with very low and others with very high thrombin generating capacity. Patients with very low thrombin generation had documented administration of unfractionated or low molecular weight heparin and detectable anti‐Xa levels. Why some patients had clearly elevated thrombin generation, however, is less clear. In addition to preserved coagulation capacity, non‐liver septics had substantially elevated fibrinogen levels, which reflects the physiological acute phase response. These unusually high levels were associated with a very thrombogenic clot structure, which in combination with a profound hypofibrinolytic state may constitute a thrombotic risk. We have previously demonstrated that the combination of hypofibrinolysis as detected with our in‐house plasma‐based assay acts synergistically with hypercoagulable features such as carriership of factor V Leiden in increasing the risk of a first venous thrombosis in the general population. 26 Extrapolating from these data, the combination of hypofibrinolysis with hyperfibrinogenemia may form a significant thrombotic risk.
Despite these specific prothrombotic features in the non‐liver septics, we found no differences in in vivo markers of coagulation (TAT complexes), coagulation with subsequent fibrinolysis (D‐dimers), and activation of fibrinolysis (PAP complexes) between the ACLF and non‐liver septic cohorts. Although it may be that in vivo activation of coagulation and fibrinolysis is similarly increased compared to controls in the ACLF and non‐liver septic cohorts, an alternative explanation is that the increase in these markers in the non‐liver septics is real, but that the increase in ACLF is due to accumulation as the liver is the site of clearance of these markers.
Our data on hypercoagulable features in non‐liver septics are consistent with clinical observations of an increased risk of both venous and arterial thrombosis in these patients, even in patients receiving optimal pharmacological thromboprophylaxis. 33 , 34 , 35 , 36 In addition, the hypercoagulable features might contribute to microvascular thrombotic events that have been linked to organ failure. 37 The usefulness of anticoagulant therapy to treat the hypercoagulable features of sepsis and sepsis‐associated disseminated intravascular coagulation have been debated given the mixed results of clinical studies. 38 , 39 Our data suggest that a VWF/ADAMTS13 imbalance, hyperfibrinogenemia, and hypofibrinolysis are the prime hypercoagulable features of sepsis, which could indicate that anticoagulant drugs are not optimally suited to reduce micro‐ and macrovascular thrombotic events. Rather, interventions aimed at restoring the VWF/ADAMTS13 imbalance 40 or decreasing the thrombogenicity of the fibrin clot may be more suitable.
Although we have demonstrated remarkable similarities between non‐liver septics and patients with ACLF, there are notable differences, particularly in plasma levels of coagulation factors and fibrinogen. Perhaps the hemostatic status of patients with ACLF is even more comparable to the patients with sepsis‐associated DIC, and a comparison in hemostatic mechanisms between these groups would therefore be of value. The application of anticoagulant therapy in patients with sepsis in particular seems to be associated with a mortality reduction in patients with sepsis‐associated DIC. 41 In addition, a prothrombotic state (notably hypofibrinolysis) is related to organ failure and poor outcome in ACLF. 10 Therefore, interventions that improve outcome in sepsis‐associated DIC may also be beneficial for patients with ACLF.
The VWF/ADAMTS13 imbalance that likely compensates for thrombocytopenia, and preserved thrombin‐generating capacity, argues against correction of hemostasis by fresh frozen plasma (FFP) and/or platelet concentrates in non‐bleeding patients with ACLF or non‐liver sepsis. 42 , 43 Importantly, blood products fail to improve hemostatic capacity in critically ill patients, 17 and have major side effects. 42 Interestingly, in the present series, hemostatic support with FFP and cryoprecipitate was substantial on admission and during further hospitalization in patients with ACLF, but virtually absent in the patients with non‐liver sepsis. Although we did not record bleeding events, these observations suggest that prohemostatic therapy frequently is a response to abnormal routine diagnostic tests of hemostasis, rather than a response to true hemostatic failure, which is more difficult to routinely assess given the complexity of the aforementioned assays. We purposely did not exclude patients receiving pro‐ or anticoagulant therapy as our aim was to assess the hemostatic status of patients with ACLF and sepsis without underlying CLD in a real‐life clinical setting, in which (prophylactic) administration of both pro‐ and anticoagulants are common. Procoagulant therapy, notably FFP, that was given to a third of ACLF patients, does not change hemostatic potential in patients with liver disease, 44 but does change plasma levels of individual coagulation factors, which should be considered when interpreting our results. Similarly, anticoagulants, which were only rarely given in our cohort, alter global tests of hemostasis.
In both ACLF and non‐liver septics, the hemostatic profile remains remarkably stable over time, although both diseases are known for a very dynamic clinical evolution. Whether this implies that the risk for bleeding or thrombotic complications does not substantially change over time is unknown. It might be very useful to perform global hemostatic screens in these patients at baseline in order to determine who will benefit most from prophylactic pro‐ or antihemostatic therapy, and studies assessing predictive value of global screening utilizing viscoelastic tests or thrombin generation tests are needed to ascertain whether a personalized approach to hemostatic management in these patients could be established with clinically available assays.
In summary, here we have documented hemostatic changes in ACLF to partially overlap with that of patients with sepsis without underlying CLD, and found evidence of preserved hemostatic capacity in both patient groups. However, patients with non‐liver sepsis were characterized by a profound hyperfibrinogenemia with a thrombogenic clot structure and a profound ex vivo resistance to fibrinolysis. In‐depth hemostatic profiling as we have applied here of these acutely ill patients may eventually facilitate a more rational approach to hemostatic management.
CONFLICTS OF INTEREST
None of the authors have a conflict of interest to report.
AUTHOR CONTRIBUTIONS
Ton Lisman: conceptualization, formal analysis, funding acquisition, supervision, visualization, writing original draft. Bethlehem Arefaine: data curation, resources, project administration, writing review, and editing. Jelle Adelmeijer: investigation, methodology, project administration, visualization, writing review, and editing. Ane Zamalloa: data curation, resources, writing review, and editing. Eleanor Corcoran: data curation, resources, writing review, and editing. John G. Smith: data curation, resources, writing review, and editing. William Bernal: conceptualization, data curation, supervision, resources, writing review, and editing. Vishal C. Patel: conceptualization, data curation, supervision, resources, writing review, and editing.
Supporting information
Table S1
ACKNOWLEDGMENTS
We are very grateful to all the patient participants and healthy volunteers for agreeing to take part in this study, and to the clinical and liver research teams at King's College Hospital London for facilitating recruitment, collecting metadata, and sample collection. Particular thanks go to the Liver Research Team and to all involved members of the anaesthetics, critical care, emergency and trauma (ACET) research team who assisted in screening of and recruiting study participants, undertaking biological sampling, and collating clinical metadata. We thank the King's College Hospital Institute of Liver Studies and Transplantation Charitable Research for providing funding for some of the consumable costs.
Lisman T, Arefaine B, Adelmeijer J, et al. Global hemostatic status in patients with acute‐on‐chronic liver failure and septics without underlying liver disease. J Thromb Haemost. 2021;19:85–95. 10.1111/jth.15112
Manuscript handled by: Patricia Liaw
Final decision: Patricia Liaw, 22 September 2020
William Bernal and Vishal C. Patel are joint senior authors
Funding informationThe study was financially supported by Departmental Funds from TL.
REFERENCES
- 1. Lisman T, Porte RJ. Pathogenesis, prevention, and management of bleeding and thrombosis in patients with liver diseases. Res Pract Thromb Haemost. 2017;1:150‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bos S, van den Boom B, Kamphuisen PW, et al. Haemostatic profiles are similar across all aetiologies of cirrhosis. Thromb Haemost. 2019;119:246‐253. [DOI] [PubMed] [Google Scholar]
- 3. Fisher C, Patel VC, Stoy SH, et al. Balanced haemostasis with both hypo‐ and hyper‐coagulable features in critically ill patients with acute‐on‐chronic‐liver failure. J Crit Care. 2017;43:54‐60. [DOI] [PubMed] [Google Scholar]
- 4. Lisman T, Porte RJ. Rebalanced hemostasis in patients with liver disease: evidence and clinical consequences. Blood. 2010;116:878‐885. [DOI] [PubMed] [Google Scholar]
- 5. Tripodi A, Mannucci PM. The coagulopathy of chronic liver disease. N Engl J Med. 2011;365:147‐156. [DOI] [PubMed] [Google Scholar]
- 6. Moreau R, Jalan R, Gines P, et al. Acute‐on‐chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology. 2013;144:1426‐1437. [DOI] [PubMed] [Google Scholar]
- 7. Mahmud N, Kaplan DE, Taddei TH, Goldberg DS. Incidence and mortality of acute on chronic liver failure using two definitions in patients with compensated cirrhosis. Hepatology. 2019;69:2150‐2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hernaez R, Kramer JR, Liu Y, et al. Prevalence and short‐term mortality of acute‐on‐chronic liver failure: a national cohort study from the USA. J Hepatol. 2019;70:639‐647. [DOI] [PubMed] [Google Scholar]
- 9. Fernandez J, Acevedo J, Wiest R, et al. Bacterial and fungal infections in acute‐on‐chronic liver failure: prevalence, characteristics and impact on prognosis. Gut. 2018;67:1870‐1880. [DOI] [PubMed] [Google Scholar]
- 10. Blasi A, Patel VC, Adelmeijer J, et al. Mixed fibrinolytic phenotypes in decompensated cirrhosis and ACLF with hypofibrinolysis in those with complications and poor survival. Hepatology. 2019;71:1381‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gustot T, Fernandez J, Garcia E, et al. Clinical course of acute‐on‐chronic liver failure syndrome and effects on prognosis. Hepatology. 2015;62:243‐252. [DOI] [PubMed] [Google Scholar]
- 12. Kremer Hovinga JA, Zeerleder S, Kessler P, et al. ADAMTS‐13, von willebrand factor and related parameters in severe sepsis and septic shock. J Thromb Haemost. 2007;5:2284‐2290. [DOI] [PubMed] [Google Scholar]
- 13. Ono T, Mimuro J, Madoiwa S, et al. Severe secondary deficiency of von willebrand factor‐cleaving protease (ADAMTS13) in patients with sepsis‐induced disseminated intravascular coagulation: Its correlation with development of renal failure. Blood. 2006;107:528‐534. [DOI] [PubMed] [Google Scholar]
- 14. Habe K, Wada H, Ito‐Habe N, et al. Plasma ADAMTS13, von willebrand factor (VWF) and VWF propeptide profiles in patients with DIC and related diseases. Thromb Res. 2012;129:598‐602. [DOI] [PubMed] [Google Scholar]
- 15. Van Dreden P, Woodhams B, Rousseau A, Dreyfus JF, Vasse M. Contribution of procoagulant phospholipids, thrombomodulin activity and thrombin generation assays as prognostic factors in intensive care patients with septic and non‐septic organ failure. Clin Chem Lab Med. 2013;51:387‐396. [DOI] [PubMed] [Google Scholar]
- 16. Carlier L, Hunault G, Lerolle N, Macchi L. Ex vivo thrombin generation patterns in septic patients with and without disseminated intravascular coagulation. Thromb Res. 2015;135:192‐197. [DOI] [PubMed] [Google Scholar]
- 17. Muller MC, Arbous MS, Spoelstra‐de Man AM, et al. Transfusion of fresh‐frozen plasma in critically ill patients with a coagulopathy before invasive procedures: a randomized clinical trial (CME). Transfusion. 2015;55:26‐35. [DOI] [PubMed] [Google Scholar]
- 18. Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemost. 2013;39:392‐399. [DOI] [PubMed] [Google Scholar]
- 19. Wasmuth HE, Kunz D, Yagmur E, et al. Patients with acute on chronic liver failure display "sepsis‐like" immune paralysis. J Hepatol. 2005;42:195‐201. [DOI] [PubMed] [Google Scholar]
- 20. Stoy S, Patel VC, Sturgeon JP, et al. Platelet‐leucocyte aggregation is augmented in cirrhosis and further increased by platelet transfusion. Aliment Pharmacol Ther. 2018;47:1375‐1386. [DOI] [PubMed] [Google Scholar]
- 21. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis‐3). JAMA. 2016;315:801‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eckmann CM, De Laaf RT, Van Keulen JM, Van Mourik JA, De Laat B. Bilirubin oxidase as a solution for the interference of hyperbilirubinemia with ADAMTS‐13 activity measurement by FRETS‐VWF73 assay. J Thromb Haemost. 2007;5:1330‐1331. [DOI] [PubMed] [Google Scholar]
- 23. Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4‐15. [DOI] [PubMed] [Google Scholar]
- 24. Ariens RA, Kohler HP, Mansfield MW, Grant PJ. Subunit antigen and activity levels of blood coagulation factor XIII in healthy individuals. Relation to sex, age, smoking, and hypertension. Arterioscler Thromb Vasc Biol. 1999;19:2012‐2016. [DOI] [PubMed] [Google Scholar]
- 25. Hugenholtz GC, Mccrae FL, Adelmeijer J, et al. Procoagulant changes in fibrin clot structure in patients with cirrhosis are associated with oxidative modifications of fibrinogen. J Thromb Haemost. 2016;15:1054‐1066. [DOI] [PubMed] [Google Scholar]
- 26. Meltzer ME, Lisman T, Doggen CJ, de Groot PG, Rosendaal FR. Synergistic effects of hypofibrinolysis and genetic and acquired risk factors on the risk of a first venous thrombosis. PLoS Med. 2008;5:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell‐derived ultralarge von willebrand factor multimers under flow. Blood. 2004;104:100‐106. [DOI] [PubMed] [Google Scholar]
- 28. Bockmeyer CL, Claus RA, Budde U, et al. Inflammation‐associated ADAMTS13 deficiency promotes formation of ultra‐large von willebrand factor. Haematologica. 2008;93:137‐140. [DOI] [PubMed] [Google Scholar]
- 29. Lisman T, Bongers TN, Adelmeijer J, et al. Elevated levels of von willebrand factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology. 2006;44:53‐61. [DOI] [PubMed] [Google Scholar]
- 30. Hollestelle MJ, Lai KW, van Deuren M, et al. Cleavage of von willebrand factor by granzyme M destroys its factor VIII binding capacity. PLoS One. 2011;6:e24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British committee for standards in haematology. Br J Haematol. 2009;145:24‐33. [DOI] [PubMed] [Google Scholar]
- 32. Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective validation of the international society of thrombosis and haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004;32:2416‐2421. [DOI] [PubMed] [Google Scholar]
- 33. Kaplan D, Casper TC, Elliott CG, et al. VTE incidence and risk factors in patients with severe sepsis and septic shock. Chest. 2015;148:1224‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levine RL, LeClerc JR, Bailey JE, Monberg MJ, Sarwat S. Venous and arterial thromboembolism in severe sepsis. Thromb Haemost. 2008;99:892‐898. [DOI] [PubMed] [Google Scholar]
- 35. Rhodes A, Evans LE, Alhazzani W, et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. 2017;43:304‐377. [DOI] [PubMed] [Google Scholar]
- 36. Reynolds PM, Van Matre ET, Wright GC, et al. Evaluation of prophylactic heparin dosage strategies and risk factors for venous thromboembolism in the critically ill patient. Pharmacotherapy. 2019;39:232‐241. [DOI] [PubMed] [Google Scholar]
- 37. Levi M, van der Poll T. Coagulation and sepsis. Thromb Res. 2017;149:38‐44. [DOI] [PubMed] [Google Scholar]
- 38. Meziani F, Gando S, Vincent JL. Should all patients with sepsis receive anticoagulation? Yes. Intensive Care Med. 2017;43:452‐454. [DOI] [PubMed] [Google Scholar]
- 39. van der Poll T, Opal SM. Should all septic patients be given systemic anticoagulation? No. Intensive Care Med. 2017;43:455‐457. [DOI] [PubMed] [Google Scholar]
- 40. Levi M, Scully M, Singer M. The role of ADAMTS‐13 in the coagulopathy of sepsis. J Thromb Haemost. 2018;16:646‐651. [DOI] [PubMed] [Google Scholar]
- 41. Iba T, Levy JH, Raj A, Warkentin TE. Advance in the management of sepsis‐induced coagulopathy and disseminated intravascular coagulation. J Clin Med. 2019;8(5):728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gajic O, Dzik WH, Toy P. Fresh frozen plasma and platelet transfusion for nonbleeding patients in the intensive care unit: benefit or harm? Crit Care Med. 2006;34:S170‐S173. [DOI] [PubMed] [Google Scholar]
- 43. de Bruin S, Scheeren TWL, Bakker J, van Bruggen R, Vlaar APJ. Cardiovascular dynamics section and transfusion guideline task force of the ESICM . Transfusion practice in the non‐bleeding critically ill: An international online survey‐the TRACE survey. Crit Care. 2019;23:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rassi AB, d'Amico EA, Tripodi A, et al. Fresh frozen plasma transfusion in patients with cirrhosis and coagulopathy: effect on conventional coagulation tests and thrombomodulin‐modified thrombin generation. J Hepatol. 2020;72:85‐94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1