

Abstract
The efficient production of many medicinally or synthetically important starting materials suffers from wasteful or toxic precursors for the synthesis. In particular, the aromatic non‐protected primary amine function represents a versatile synthetic precursor, but its synthesis typically requires toxic oxidizing agents and transition metal catalysts. The twofold electrochemical amination of activated benzene derivatives via Zincke intermediates provides an alternative sustainable strategy for the formation of new C−N bonds of high synthetic value. As a proof of concept, we use our approach to generate a benzoxazinone scaffold that gained attention as a starting structure against castrate‐resistant prostate cancer. Further improvement of the structure led to significantly increased cancer cell line toxicity. Thus, exploiting environmentally benign electrooxidation, we present a new versatile and powerful method based on direct C−H activation that is applicable for example the production of medicinally relevant compounds.
Keywords: benzoxazinone, drug scaffold, electrochemistry, sustainable chemistry, twofold amination
Efficient and sustainable access to medicinally and synthetically important C−N bonds is in high demand. However, aromatic primary amine functions via direct C−H amination still remain the major challenge in synthetic organic chemistry. Herein, a new environmentally benign twofold electrochemical C−H amination procedure to provide versatile primary amines and valuable heterocycles for biomedical applications is reported.
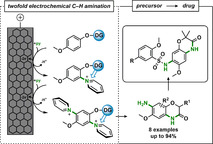
Introduction
The carbon–nitrogen functionality represents an important basic element in synthetic organic chemistry. This structural motif has been utilized for the synthesis of manifold natural products, [1] pharmaceuticals, [2] dyes, [3] as well as polymers and functional materials. [4] Thus, efficient and selective synthetic approaches for new ways to form C−N bonds and especially towards aromatic primary non‐protected amine functions are highly desired. However, common synthesis strategies suffer from excessive use of toxic chemicals including reagents for nitration, pre‐functionalization such as aryl halides, or transition‐metal catalysts. In addition, nitration reactions are highly exothermic processes that frequently lead to regioisomeric mixtures [5] and the subsequent separation of such mixtures is often tedious. The major challenge regarding new synthetic approaches for primary amines remains unsolved. Recently, ambitious efforts have been made by using electrophilic nitrogen sources such as ammonium mesylate salts, [6] or photocatalytically generated N‐centered pyridinium radical cations. [7] However, such innate amination procedures also suffer from low regioselectivity and the use of transition metal catalysts. Modern approaches employ ammonia as the nitrogen source for the synthesis of aromatic primary amines (Scheme 1). [8] However, all these methods suffer from high catalyst loading, excessive use of toxic and expensive chemicals, pre‐functionalized substrates and/or harsh reaction conditions such as strong bases and elevated temperatures. For instance, DeBoef and co‐workers established a synthesis of primary amines via stoichiometric amounts of hypervalent iodine reagents (Scheme 1). Unfortunately, this strategy generates significant waste and is highly unfavorable with regard to the ecological and sustainable footprint of organic synthesis. [9]
Scheme 1.

Common synthesis strategies for the synthesis of aromatic unprotected primary amine functions in comparison with the single and twofold electrochemical C−H‐amination procedures.
Yoshida and co‐workers established a remarkable electrochemical C−N bond formation with electron‐rich arenes 1 a (Scheme 1).[ 10 , 11 ] Based on these reports Waldvogel and co‐workers could demonstrate the electrochemical synthesis of benzoxazinones, which gained interest as basic heterocycle motifs in potential anti‐cancer drugs.[ 12 , 13 , 14 ]
Recently, electrosynthetic conversions have gained more interest in the academic community as the use of inexpensive, environmentally benign reactants such as simple electrons and recyclable electrodes is superior to expensive and toxic reactants like elaborate transition metal complexes. [15] Waldvogel and co‐workers were able to establish versatile electrochemical approaches for direct C−C coupling reactions,[ 16 , 17 ] C−N functionalizations,[ 12 , 17 , 18 , 19 ] and intramolecular C−N, N−N, and S−S bond formations. [20]
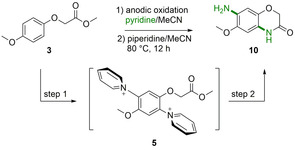
Due to the importance of aromatic primary amines as fundamental scaffolds in natural products and pharmaceutically relevant substances, such as benzoxazinones, we thus aimed to further extend our electrochemical approaches toward the direct twofold amination of methyl phenoxyacetate structure 3 (Scheme 2). Besides providing access to a valuable heterocycle, the ester functionality plays a crucial role in stabilizing intermediates. Anodic oxidation of electron‐rich aromatic compounds 1 a in the presence of pyridine usually leads to relatively stable and positively charged pyridinium intermediates (Zincke‐type salts 2 a, Scheme 1). Upon treatment with piperidine these cationic species liberate the aromatic primary amine function in a second step. Yoshida et al. postulated that due to the electron withdrawing effect (EWG) of the pyridinium intermediate and the electrostatic repulsion from the anode, further oxidation and pyridination processes were suppressed. [11] However, Waldvogel et al. recently reported a twofold amination process of polycyclic aromatic compounds, albeit with low yields since electron donating substituents were found to play a crucial role in stabilizing the pyridinium intermediates. Furthermore, the regioselectivity of the electrochemical amination procedure is associated with electron releasing functional groups, thus the aminated products consisted of regioisomers. [19]
Scheme 2.

a) Proposed masking effect of the pyridinium moiety via a stabilizing ester function by n–π, π–π or n–cation interactions, b) the absence of a side chain on substrate 4 results in the lack of desired product formation.
Results and Discussion
Based on Yoshida's hypothesis regarding the reaction intermediate dependent suppression of oxidation processes, we aimed to evaluate whether overoxidation of the Zincke‐type salt is truly suppressed and the first pyridinium intermediate accumulates. We thus designed substrates capable of efficiently masking the electron‐withdrawing effect of the first pyridinium moiety. In contrast to the prior hypothesis, these compounds were capable of further oxidation and consequently provide an expeditious route to valuable product synthesis. Efficient electrochemical C−H amination requires electron‐rich arenes with electron donating and radical cation stabilizing substituents such as methoxy groups. [11] Initially, we compared activated compounds such as 1,4‐dimethoxybenzene (4) and methyl 2‐(4‐methoxyphenoxy)‐acetate (3) with regard to their ability to undergo a second pyridination process (Scheme 2). We hypothesized that the ester functionality of the phenoxy acetate derivative 3 might be able to stabilize the pyridinium moiety via n–π, π–π or n–cation interactions. Stabilization of the electron‐deficient substituent can be beneficial for further oxidation. These interactions act as shielding effects to disguise the positive charge, thus the electron withdrawing effect diminishes and further oxidation of 3’ at the anode can occur (Scheme 2).
Initially, we conducted various cyclic voltammetry experiments to evaluate the phenoxy acetate 3 and to observe whether the ester moiety provides the expected beneficial impact (Figure 2). The electron‐rich arene 1,4‐dimethoxybenzene (4) was used for comparison (Figure 1).
Figure 2.

Cyclic voltammograms of 0.1 m Et4NBF4/CH3CN with methyl 2‐(4‐methoxyphenoxy)acetate (3); scan rate: 50 mV s−1 (blue line); scan rate: 500 mV s−1 (purple line); and with additive pyridine (py) (4 equiv.); scan rate: 50 mV s−1 (orange line); scan rate: 500 mV s−1 (green line); working electrode: BDD; counter electrode: glassy carbon; reference electrode: Ag/AgNO3.
Figure 1.

Cyclic voltammograms of 0.1 m Et4NBF4/CH3CN with 1,4‐dimethoxybenzene (4) (purple line) and with pyridine (py) as additive (4 equiv.) (green line); working electrode: boron‐doped diamond (BDD); counter electrode: glassy carbon; reference electrode: Ag/AgNO3; scan rate 500 mV s−1 (for more information: see Supporting Information).
In the cyclic voltammogram of 1,4‐dimethoxybenzene (4) (Figure 1, purple line), one quasi‐reversible oxidation step and another oxidation peak were observed (E Ox1a=0.94 V, E Ox2a=1.61 V vs. FcH/FcH+, respectively). With the addition of pyridine (Figure 1, green line) the second oxidation peak is shifted to elevated values (E Ox1b=0.94 V, E Ox2b=2.43 V vs. FcH/FcH+, respectively). These data are in line with the mechanism postulated by Yoshida, where initially a radical cation is formed and then trapped in situ by pyridine. This molecule is stabilized by deprotonation and further oxidation to establish the first pyridinium intermediate (for more details on the mechanism see Supporting Information). The trapping mechanism is related to the absence of a reduction wave (E Red1’a=0.90 V vs. FcH/FcH+, Figure 1, green line). Subsequently, radical cations are generated and consumed by a follow‐up reaction with pyridine. The presence of pyridine (Figure 1, green line) shifts the second pyridination peak close to the electrochemical window of the electrolyte (E Ox2b=2.43 V vs. FcH/FcH+). This suggests that the electron withdrawing effect of the positively charged scaffold destabilizes the benzene system. Our data thus strongly support Yoshida's original proposal that the pyridinium intermediate and its electron withdrawing nature will prevent further oxidation processes.
In the cyclic voltammogram of methyl 2‐(4‐methoxyphenoxy)‐acetate (3) (Figure 2, purple line), three distinct oxidation peaks are observed (E Ox1=1.05 V, E Ox2=1.24 V, and E Ox3=1.61 V vs. FcH/FcH+, respectively). The first oxidation peak is quasi‐reversible at high scan rates (Figure 2, purple line, peak 1). By reducing the scan rate from 500 mV s−1 to 50 mV s−1, the first two oxidation peaks unite to one broad peak (E Ox1+2=1.12 V, Figure 2, blue line). This suggests that the resolution of this experiment on a fast time scale corresponds approximately to two narrow single‐electron transfers (SETs). At a slow time scale chemical reactions become observable and might interfere with emerging radical cations. Consequently, the two peaks merge and appear almost indistinguishable. With the addition of pyridine, the time scale of the electron transfer process becomes inconsequential for the reaction progress (Figure 2, orange and green lines). Hence, three oxidation peaks are observed at slow and fast scan rates. The formation of any radical cation species will be trapped in situ by pyridine. The presence of pyridine also influences and shifts the equilibrium of the electron transfer towards the oxidized substrate, thus the peak current is increased substantially (E Ox1=1.05 V vs. FcH/FcH+, Figure 2, green line). Interestingly, no significant elevated oxidation peaks appear in contrast to what is observed with the 1,4‐dimethoxybenzene derivative 4. This indicates a shielding and stabilizing effect provided by the dangling ester function (for more details see Supporting Information).
The promising results obtained by cyclic voltammetry experiments in combination with our earlier successful electrochemical synthesis of benzoxazinones [12] prompted us to test further amination procedures, in particular with regard to the synthesis of aromatic non‐protected primary amines with benzoxazinone scaffolds. [12] The cyclic voltammetry data revealed beneficial properties with respect to a twofold electrochemical C−H amination (Scheme 3).
Scheme 3.
Electrochemical twofold amination of 3 as the test substrate and the proposed Zincke‐type intermediate 5.
The optimization reactions were carried out in divided Teflon cells with a porous glass‐frit or an anion exchange membrane like Thomapor® as separators (Supporting Information, Figure 1 and Figure 2). [21] This screening technique is time and cost efficient and allows the simultaneous variation of several electrochemical parameters, for example current density and applied charge (for the experimental setup see Supporting Information). The results of the screening experiments were determined via 1H NMR analysis with 1,1,2,2‐tetrachloroethane as an internal standard (ISTD). Tetraethylammonium tetrafluoroborate (NEt4BF4) was used as the supporting electrolyte. The increased salt character of tetraethyl ammonium compared to the corresponding butyl salts facilitated the workup procedure by column chromatography.
Previous work has shown that isostatic graphite anodes can be used in electrochemical oxidation reaction for electron‐rich phenoxyacetates to efficiently generate new C−N functionalities. [12] To determine the optimal diamination conditions of phenoxy acetate 3, we systematically investigated the influence of the current density, the amount of charge and different membranes with isostatic graphite as anode material (Table 1). In principle, the current density is directly correlated to the concentration of radical cations close to the electrode surface, thus higher current densities could favor further oxidation of relative inert Zincke‐type salt 3’. Recently, when using naphthalene as the starting material we observed that low current densities 1–6 mA cm−2 benefit the single amination process, whereas higher current densities lead to competing pathways. [19] Based on these results, it was envisioned that a two‐step electrolysis might be feasible.
Table 1.
Screening of different electrolysis conditions for the twofold amination process. 0.5 mmol substrate, 0.2 m supporting electrolyte, 12 mmol pyridine, 5 mmol trifluoromethanesulfonic acid (TfOH), isostatic graphite anode, platinum cathode, divided cell.
|
# |
Separator |
Supporting electrolyte |
Current density [mA cm−2] |
Applied charge [F] |
Yield [%][a] |
|---|---|---|---|---|---|
|
1 |
glass frit |
NBu4BF4 |
10 |
4.2 |
0[b] |
|
2 |
glass frit |
NEt4BF4 |
10 |
4.2 |
30 |
|
3 |
glass frit |
NEt4BF4 |
10 |
5.0 |
29 |
|
4 |
glass frit |
NEt4BF4 |
10 |
6.0 |
25 |
|
5 |
glass frit |
NEt4BF4 |
10 |
7.0 |
0 |
|
6 |
Thomapor® |
NEt4BF4 |
1 |
4.2 |
34 |
|
7 |
Thomapor® |
NEt4BF4 |
3 |
4.2 |
40 |
|
8 |
Thomapor® |
NEt4BF4 |
7 |
4.2 |
50 |
|
9 |
Thomapor® |
NEt4BF4 |
10 |
4.2 |
49 |
|
10 |
Thomapor® |
NEt4BF4 |
13 |
4.2 |
49 |
|
11 |
Thomapor® |
NEt4BF4 |
16 |
4.2 |
46 |
|
12 |
Thomapor® |
NEt4BF4 |
10 |
4.5 |
56 |
|
13 |
Thomapor® |
NEt4BF4 |
10 |
5.0 |
57 |
|
14 |
Thomapor® |
NEt4BF4 |
10 |
5.5 |
59 |
|
15 |
Thomapor® |
NEt4PF6 |
10 |
5.5 |
50 |
|
16 |
Thomapor® |
NEt4BF4 |
i) 5 ii) 10 |
i) 3.4 ii) 2.1 |
69[c] |
[a] Determined via 1H NMR with 1,1,2,2‐tetrachloroethane as ISTD. [b] Workup procedure did not fully lead to isolation of desired product. [c] Achieved via two‐step electrolysis.
Optimal reaction conditions were achieved when Thomapor®, an anion exchange membrane, was applied as a separator. Due to its superior properties such as permeaselectivity and low electrical resistivity to glass frits, the envisioned two‐step electrolysis led to optimal results (for more details on the condition screening see Table 1 and Table S2, Supporting Information). Previous studies on C−H amination have shown that the actual demand of charge is significantly higher compared to theory. [12] Therefore, the electrochemical conditions for the two‐step electrolysis were set to 3.4 F with 5 mA cm−2 and 2.1 F with 10 mA cm−2. This approach resulted in high yields of 69 % with compound 10 (entry 16, Table 1).
The optimized electrolysis conditions consist of a divided cell setup with isostatic graphite as the anode and platinum as the cathodic material, Thomapor® as the separator and a two‐step‐electrolysis of 3.4 F with 5 mA cm−2 and 2.1 F with 10 mA cm−2 at 25 °C. These optimized reaction conditions were then used on a collection of activated arene systems (Figure 3).
Figure 3.

Scope of diaminated products with various substitution patterns obtained via optimized reaction conditions. Divided cell; separator: Thomapor®.
Importantly, our improved electrochemical protocol for a twofold amination process is applicable to a broad scope of different 1,4‐activated phenoxy systems as seen in Figure 3. Other substitution patterns including 1,3‐activated phenoxy systems were also investigated. However, even with an increased amount of charge only a single aminated product 18 of 54 % yield was generated (Figure 3). By attaching two fluoride functions, the desired conversion was not observed and instead only a single amination product 19 in 57 % yield was obtained. This is presumably due to the electron withdrawing nature of fluorine, stereoelectronic effects, and the consequently destabilized benzene system.
1,4‐Activated phenoxy systems like compound 10 could be isolated with a yield of 57 %. For specific scaffolds such as highly activated benzene systems, we still obtained a relatively high yield of 56 %, but regioisomers were formed (see 11 a and 11 b). Based on our results, we then focused on the effect of larger aromatic systems including naphthyl derivatives. For instance, the 2,6‐substituted naphthyl derivative was successfully transformed into the desired product 12 with yields of up to 47 %.
Next, we investigated the influence of the electron withdrawing effect of the positive charged pyridinium moiety in comparison to the electrostatic repulsion by applying the electrochemical twofold amination protocol to a separated π‐system. Surprisingly, this resulted in an almost quantitative yield of 94 % for 13. This suggests that the electrostatic repulsion has a less adverse effect on the second pyridination process than the electron‐withdrawing effect of the Zincke‐type intermediate. The best reactivity and selectivity were observed with 1,4‐substituted aromatic compounds, due to the low steric demands of two attached pyridinium intermediates and their respective electrostatic properties. Thus, other substituent patterns close to the ring system were investigated to assess whether they may show reduced influence on the electron‐density of the aromatic compound. By attaching two methyl groups to the side chain, the resulting product 14 was formed in satisfactory yields of up to 68 %. Next, a phenyl‐substituted scaffold was transformed into the diaminated product 15 with 33 % yield. Surprisingly, even a halo function such as a chloro‐substituent was compatible with this methodology, obtaining 16 with a yield of 48 %. Furthermore, our protocol was even capable of transforming phenyl ketones intramolecularly into cyclic aminated imine structures with a yield of 19 % for 17. These lower yields may be explained by the inevitably occurring enamine formation with piperidine during the transformation. Nevertheless, the electro‐conversion of these phenyl ketones provides a novel synthetic access route towards precursors of asymmetric imine reduction. [22]
Recently, benzoxazinones similar to compound 20 gained attention as general structures for novel drugs against so‐called castrate‐resistant prostate cancer (CRPC).[ 13 , 14 ] Currently, patients that display CRPC ultimately always succumb to this disease. Androgen deprivation therapy (ADT) drugs are being heavily investigated for their potential to counteract the proliferative effects of the androgen receptor in prostate cancer cells. However, cancer patients develop the above‐mentioned resistance due to various biochemical survival adaptions of the cancer.[ 23 , 24 ] Therefore, there is an urgent need for the development of alternative ADT therapeutics.
Xu et al. recently demonstrated via X‐ray crystallography (PDB:5Z1S) that benzoxazinone 20 directly interacts with bromodomain containing protein 4 (BRD4) and that this compound 20 can be used as an efficient cell viability inhibitor (IC50=4.41 μm) of the prostate cancer cell line LnCAP. [14] BRD4 belongs to the so‐called bromodomain and extra‐terminal domain (BET) protein family and interacts with the N‐terminus of the androgen receptor. Inhibiting this protein interaction with BRD4 inhibitors ultimately leads to an anti‐proliferative effect, which is especially pronounced in prostate cancer cells. [23]
To evaluate the effect of our electrochemically generated benzoxazinone 20 and select derivatives against prostate‐cancer cell lines, we carried out cell‐viability assays with LnCAP and non‐cancerous HEK293 cells as a control. In contrast to what was observed previously, [14] we did not observe a complete cell growth inhibition at increasing concentrations of compound 20 (Figure 4 a). Rather, the dose‐response curve is bell‐shaped. Bell‐shaped dose‐response curves could indicate the formation of colloidal drug aggregates and consequently a decreased ability of the compound to penetrate cellular membranes and to ultimately interact with their cellular target such as the nuclear protein BRD4. [25] Intriguingly, our crystal structure of benzoxazinone 20 indeed shows a distinct 3D linkage via H‐bond formation of the bromine (for more information see Supporting Information, Figure S8). To examine the role of the halide moiety for compound oligomerization and cell toxicity, we thus substituted bromine with fluorine, resulting in benzoxazinone 21. Fluorine as substituent possesses a higher electronegativity but simultaneous less polarizability than bromine, which could thus avoid H‐bond formation. Indeed, the crystal structure of the fluoro compound 21 (for more information see Supporting Information, Figure S7) revealed the loss of the 3D linkage previously observed for 20. At the same time, however, cell toxicity of 21 was lost (Figure 4 b).
Figure 4.
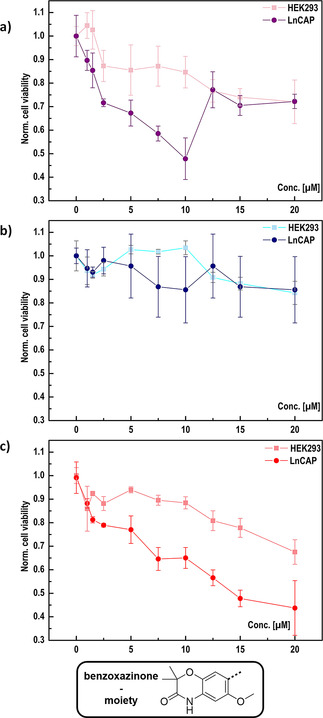
Effects of compound 20 (a), 21 (b), 22 (c) on cell viability of the prostate cancer cell line LnCAP and HEK293 as the control cell line. Cells were treated with increasing concentrations of compounds, with a final DMSO concentration of 0.5 vol %. Cell viability was measured by MTT assays. Results represent three independent experiments with four technical replicates each. Boxed molecule shows the electrochemically generated benzoxazinone backbone precursor for the synthesis of compounds 20, 21 and 22.
The crystal structure of BRD4 in complex with benzoxazinone 20 [14] indicates that the 5‐bromo‐2‐methoxybenzene moiety of benzoxazinone 20 interacts with the WPF shelf of BRD4, a hydrophobic cavity within BRD4 (see Supporting Information, Figure S9). The substitution of bromine with fluorine could thus lead to an impaired BRD4 binding behavior and consequently a decreased inhibitory effect on LnCAP cell growth. On the other hand, this encouragingly indicates that the overall toxicity of this compound for human cells is low and that off‐target effects are minimal thus fulfilling important requirements for a putative drug. To evaluate whether the inhibitory effect of compound 20 relies on the polarizability of bromine or its space requirement, we substituted the bromine with a methyl group to yield compound 22. With increasing compound 22 concentrations, LnCAP viability decreases (Figure 4 c). Compared to sulfonamide 20, derivative 22 shows a clear dependence of cell viability for LnCAP cells on compound concentration, indicating no compound aggregation at higher concentrations. This is in agreement with our crystal structure of 22 (for more information see Supporting Information, Figure S10) which does not show H‐bond acceptor abilities such as bromine and therefore no 3D linking and aggregation. With the methyl derivative 22 obtained using our optimized electrochemical protocol, we provide a compound that displays a significant boost in solubility and, in contrast to related molecules, a dose‐dependent toxicity against a prostate cancer cell line. Our findings thus combine the design of an anti‐prostate cancer drug precursor with improved properties with a significantly enhanced electrochemical protocol for the selective amination of important molecular scaffolds for many downstream applications.
Conclusions
In summary, the application of the phenoxy‐acetate moiety revealed a new and interesting reactivity towards twofold amination products. We introduced a versatile and powerful method to electrochemically generate doubly positive charged Zincke intermediates. These pyridinium intermediates liberate one aromatic non‐protected primary amine function and one amine which induces the formation of an intramolecular heterocycle. We conducted detailed cyclic voltammetry studies of activated arene systems to elucidate their reactivity and behavior towards a twofold amination protocol. With our new electrochemical C−H amination procedure, twofold aminated products with more than 90 % yields were accessible. This process does not require leaving groups and transition metal catalysts and thus represents a new and powerful approach for C−N bond formation. Furthermore, we also could show a new way to synthesize pharmaceutical key compounds with sustainable electrochemistry, which will be highly valuable for future pharmaceutical productions against the background of environment protection and shortage of raw materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (DFG: Wa1276/17‐2) is highly appreciated. E.D. acknowledges a TransMED PhD Fellowship and the Sibylle Kalkhof‐Rose‐Stiftung. This work was supported by the Center for Biomolecular Magnetic Resonance (BMRZ), Frankfurt University funded by the state of Hesse. Open access funding enabled and organized by Projekt DEAL.
L. J. Wesenberg, E. Diehl, T. J. B. Zähringer, C. Dörr, D. Schollmeyer, A. Shimizu, J.-i. Yoshida, U. A. Hellmich, S. R. Waldvogel, Chem. Eur. J. 2020, 26, 17574.
References
- 1. Nicolaou K. C., Li A., Edmonds D. J., Angew. Chem. Int. Ed. 2006, 45, 7086–7090; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7244–7248. [Google Scholar]
- 2.
- 2a. Hili R., Yudin A. K., Nat. Chem. Biol. 2006, 2, 284; [DOI] [PubMed] [Google Scholar]
- 2b. Czarnik A. W., Acc. Chem. Res. 1996, 29, 112; [DOI] [PubMed] [Google Scholar]
- 2c. Bikker J. A., Brooijmans N., Wissner A., Mansour T. S., J. Med. Chem. 2009, 52, 1493; [DOI] [PubMed] [Google Scholar]
- 2d. Wissner A., Berger D. M., Boschelli D. H., Floyd M. B., Greenberger L. M., Gruber B. C., Johnson B. D., Mamuya N., Nilakantan R., Reich M. F., Shen R., Tsou H.-R., Upeslacis E., Wang Y. F., Wu B., Ye F., Zhang N., J. Med. Chem. 2000, 43, 3244. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Meth-Cohn O., Smith M., J. Chem. Soc. Perkin Trans. 1 1994, 5; [Google Scholar]
- 3b. Sousa M. M., Melo M. J., Parola A. J., Morris P. J. T., Rzepa H. S., de Melo J. S. S., Chem. Eur. J. 2008, 14, 8507; [DOI] [PubMed] [Google Scholar]
- 3c. Perkin W. H., Q. J. Chem. Soc. 1862, 14, 230. [Google Scholar]
- 4.
- 4a. Schlüter A. D., Rabe J. P., Angew. Chem. Int. Ed. 2000, 39, 864–883; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 860–880; [Google Scholar]
- 4b. Green A. G., Woodhead A. E., J. Chem. Soc. Trans. 1910, 97, 2388; [Google Scholar]
- 4c. Letheby H., J. Chem. Soc. 1862, 15, 161; [Google Scholar]
- 4d. Yang Q.-L., Wang X.-Y., Lu J.-Y., Zhang L.-P., Fang P., Mei T.-S., J. Am. Chem. Soc. 2018, 140, 11487; [DOI] [PubMed] [Google Scholar]
- 4e. Yella A., Lee H.-W., Tsao H. N., Yi C., Chandiran A. K., Nazeeruddin M. K., Diau E. W.-G., Yeh C.-Y., Zakeeruddin S. M., Grätzel M., Science 2011, 334, 629. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Downing R. S., Kunkeler P. J., van Bekkum H., Catal. Today 1997, 37, 121; [Google Scholar]
- 5b. Olah G. A., Malhotra R., Narang S. C., Nitration. Methods and Mechanisms, Wiley, New York, 1989. [Google Scholar]
- 6. Legnani L., Prina Cerai G., Morandi B., ACS Catal. 2016, 6, 8162. [Google Scholar]
- 7.
- 7a. Ham W. S., Hillenbrand J., Jacq J., Genicot C., Ritter T., Angew. Chem. Int. Ed. 2019, 58, 532–536; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 542–546; [Google Scholar]
- 7b. Rössler S. L., Jelier B. J., Tripet P. F., Shemet A., Jeschke G., Togni A., Carreira E. M., Angew. Chem. Int. Ed. 2019, 58, 526; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 536. [Google Scholar]
- 8.
- 8a. Xia N., Taillefer M., Angew. Chem. Int. Ed. 2009, 48, 337–339; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 343–345; [Google Scholar]
- 8b. Rao H., Fu H., Jiang Y., Zhao Y., Angew. Chem. Int. Ed. 2009, 48, 1114–1116; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1134–1136. [Google Scholar]
- 9.
- 9a. Schäfer H. J., C. R. Chim. 2011, 14, 745; [Google Scholar]
- 9b. Frontana-Uribe B. A., Little R. D., Ibanez J. G., Palma A., Vasquez-Medrano R., Green Chem. 2010, 12, 2099; [Google Scholar]
- 9c. Kantak A. A., Potavathri S., Barham R. A., Romano K. M., DeBoef B., J. Am. Chem. Soc. 2011, 133, 19960; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Kim H. J., Kim J., Cho S. H., Chang S., J. Am. Chem. Soc. 2011, 133, 16382; [DOI] [PubMed] [Google Scholar]
- 9e. Steckhan E., Arns T., Heineman W. R., Hilt G., Hoormann D., Jörissen J., Kröner L., Lewall B., Pütter H., Chemosphere 2001, 43, 63. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Hayashi R., Shimizu A., Song Y., Ashikari Y., Nokami T., Yoshida J.-i., Chem. Eur. J. 2017, 23, 61; [DOI] [PubMed] [Google Scholar]
- 10b. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2014, 136, 4496; [DOI] [PubMed] [Google Scholar]
- 10c. Morofuji T., Shimizu A., Yoshida J.-i., Chem. Eur. J. 2015, 21, 3211; [DOI] [PubMed] [Google Scholar]
- 10d. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2015, 137, 9816. [DOI] [PubMed] [Google Scholar]
- 11. Morofuji T., Shimizu A., Yoshida J.-i., J. Am. Chem. Soc. 2013, 135, 5000. [DOI] [PubMed] [Google Scholar]
- 12. Wesenberg L. J., Herold S., Shimizu A., Yoshida J.-i., Waldvogel S. R., Chem. Eur. J. 2017, 23, 12096. [DOI] [PubMed] [Google Scholar]
- 13. Xue X., Zhang Y., Wang C., Zhang M., Xiang Q., Wang J., Wang A., Li C., Zhang C., Zou L., Wang R., Wu S., Lu Y., Chen H., Ding K., Li G., Xu Y., Eur. J. Med. Chem. 2018, 152, 542. [DOI] [PubMed] [Google Scholar]
- 14. Xiang Q., Zhang Y., Li J., Xue X., Wang C., Song M., Zhang C., Wang R., Li C., Wu C., Zhou Y., Yang X., Li G., Ding K., Xu Y., ACS Med. Chem. Lett. 2018, 9, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Horn E. J., Rosen B. R., Baran P. S., ACS Cent. Sci. 2016, 2, 302; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Kärkäs M. D., Chem. Soc. Rev. 2018, 47, 5786; [DOI] [PubMed] [Google Scholar]
- 15d. Wiebe A., Gieshoff T., Möhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721; [Google Scholar]
- 15e. Möhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 15f. Waldvogel S. R., Lips S., Selt M., Riehl B., Kampf C. J., Chem. Rev. 2018, 118, 6706. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Wiebe A., Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2017, 56, 14727–14731; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14920–14925; [Google Scholar]
- 16b. Lips S., Wiebe A., Elsler B., Schollmeyer D., Dyballa K. M., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 10872–10876; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11031–11035; [Google Scholar]
- 16c. Lips S., Frontana-Uribe B. A., Dörr M., Schollmeyer D., Franke R., Waldvogel S. R., Chem. Eur. J. 2018, 24, 6057; [DOI] [PubMed] [Google Scholar]
- 16d. Lips S., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 13325–13329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13509–13513; [Google Scholar]
- 16e. Lipp A., Ferenc D., Gütz C., Geffe M., Vierengel N., Schollmeyer D., Schäfer H. J., Waldvogel S. R., Opatz T., Angew. Chem. Int. Ed. 2018, 57, 11055–11059; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11221–11225; [Google Scholar]
- 16f. Röckl J. L., Schollmeyer D., Franke R., Waldvogel S. R., Angew. Chem. Int. Ed. 2020, 59, 315–319; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 323–327. [Google Scholar]
- 17. Herold S., Möhle S., Zirbes M., Richter F., Nefzger H., Waldvogel S. R., Eur. J. Org. Chem. 2016, 1274. [Google Scholar]
- 18. Waldvogel S. R., Möhle S., Angew. Chem. Int. Ed. 2015, 54, 6398–6399; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6496–6497. [Google Scholar]
- 19. Möhle S., Herold S., Richter F., Nefzger H., Waldvogel S. R., ChemElectroChem 2017, 4, 2196. [Google Scholar]
- 20.
- 20a. Gieshoff T., Schollmeyer D., Waldvogel S. R., Angew. Chem. Int. Ed. 2016, 55, 9437–9440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9587–9590; [Google Scholar]
- 20b. Kehl A., Breising V. M., Schollmeyer D., Waldvogel S. R., Chem. Eur. J. 2018, 24, 17230; [DOI] [PubMed] [Google Scholar]
- 20c. Breising V. M., Gieshoff T., Kehl A., Kilian V., Schollmeyer D., Waldvogel S. R., Org. Lett. 2018, 20, 6785. [DOI] [PubMed] [Google Scholar]
- 21. Gütz C., Klöckner B., Waldvogel S. R., Org. Process Res. Dev. 2016, 20, 26. [Google Scholar]
- 22.
- 22a. Fleischer S., Zhou S., Werkmeister S., Junge K., Beller m., Chem. Eur. J. 2013, 19, 4997; [DOI] [PubMed] [Google Scholar]
- 22b. Xie J.-H., Zhu S.-F., Zhou Q.-L., Chem. Rev. 2011, 111, 1713. [DOI] [PubMed] [Google Scholar]
- 23. Asangani I. A., Dommeti V. L., Wang X., Malik R., Cieslik M., Yang R., Escara-Wilke J., Wilder-Romans K., Dhanireddy S., Engelke C., Iyer M. K., Jing X., Wu Y.-M., Cao X., Qin Z. S., Wang S., Feng F. Y., Chinnaiyan A. M., Nature 2014, 510, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen C. D., Welsbie D. S., Tran C., Baek S. H., Chen R., Vessella R., Rosenfeld M. G., Sawyers C. L., Nat. Med. 2004, 10, 33. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Owen S. C., Doak A. K., Ganesh A. N., Nedyalkova L., McLaughlin C. K., Shoichet B. K., Shoichet M. S., ACS Chem. Biol. 2014, 9, 777; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Owen S. C., Doak A. K., Wassam P., Shoichet M. S., Shoichet B. K., ACS Chem. Biol. 2012, 7, 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary