

Abstract
In a time with decreasing biodiversity, especially among insects, a detailed understanding about specific resource utilization strategies is crucial. The physiological and behavioural responses to host switches in phytophagous insects are poorly understood. Earlier studies indicate that a host plant switch might be associated with distinctive molecular and physiological responses in different lineages. Expanding the assessment of such associations across Lepidoptera will reveal if there are general patterns in adaptive responses, or if each switch event is more of a unique character. We investigated host plant preference, fitness consequences, effects on expression profiles and gut microbiome composition in two common wood white (Leptidea sinapis) populations with different host plant preferences from the extremes of the species distribution area (Sweden and Catalonia). Our results show that female Catalonian wood whites lack preference for either host plant (Lotus corniculatus or L. dorycnium), while Swedish females laid significantly more eggs on L. corniculatus. Individuals from both populations reared on L. dorycnium had longer developmental times and smaller body size as adults. This indicates that both environmental and genetic factors determine the choice to use a specific host plant. Gene expression analysis revealed a more pronounced response to host plant in the Catalonian compared to the Swedish population. In addition, host plant treatment resulted in a significant shift in microbiome community structure in the Catalonian population. Together, this suggests that population specific plasticity associated with local conditions underlies host plant utilisation in wood whites.
Keywords: gene expression, host plant, Lepidoptera, microbiome, speciation, wood white
1. INTRODUCTION
In a time with decreasing biodiversity, especially among insects, it is important to increase our understanding of the mechanisms underlying resource utilization and the ability to shift to novel resources. Insects feeding on plants (phytophagous) are generally specialized on particular species or groups of host plants (Ehrlich & Raven, 1964; Janz, 2011). This can be a result of genetic, physiological, ecological and geographical restrictions in the specialists (Schoonhoven et al., 2005; Steward et al., 2019; Weingartner et al., 2006) and, crucially, the ability to overcome host plant defence mechanisms involving toxic metabolites (Ehrlich & Raven, 1964; Govind et al., 2010; Kirsch et al., 2011). Female butterflies generally identify the appropriate host plant species via chemoreception. To use a specific plant, butterflies have thus developed mechanisms both for recognition of plant‐specific chemical cues and for detoxification of, or tolerance to, potentially harmful plant metabolites (Renwick & Chew, 1994). Chemical cues can be unique to specific host plants, but similarity in composition is expected in closely related plant species (Renwick & Chew, 1994). The potential for utilization of novel host plants thus depends on (i) the similitude of plant chemical composition; (ii) its recognition by adult females; and (iii) the fitness of the larvae feeding on the plant (Pearse et al., 2013; Wiklund, 1975). Costs associated with host plant switches, and the specific adaptations needed by phytophagous species (Govind et al., 2010), imply that host plant expansions are unlikely, unless they include plant taxa with similar compositions of chemical cues and toxic metabolites (Janz & Nylin, 1998). Consequently, butterflies that overcome the recognition and detoxification barriers associated with novel host plant use may enter an unexplored “adaptive realm”, which could lead to comparatively rapid diversification, if the novel host plant is part of a diverse plant lineage where most species have similar chemical composition (Ehrlich & Raven, 1964; Janz, 2011). Host plant expansions can also result in so‐called (eco)evolutionary traps, when adult female butterflies start to prefer invasive or anthropogenic plant species for egg‐laying that are suboptimal for larval development or survival (Singer & Parmesan, 2018; Steward et al., 2019). Furthermore, female host preference and larval performance are not necessarily coupled and females may also oviposit on unsuitable host plants (Wiklund, 1975).
Although the theoretical foundations for host plant switch effects on butterfly diversity have been laid out in some detail (Ehrlich & Raven, 1964; Janz, 2011; Janz & Nylin, 1998; Pearse et al., 2013; Weingartner et al., 2006), the physiological and behavioural responses to such food resource switches in phytophagous insects, is poorly understood. The immediate response to a host plant switch is commonly governed by gene expression tuning, mutualistic interactions with gut microbiota, or other plastic mechanisms not directly associated with changes in the genome sequence per se (Mack & Nachman, 2017; Romero et al., 2012). Groundwork gene expression analyses in Lepidoptera suggest that host plant shifts are accompanied by differential regulation of genes involved in transcription and translation, membrane transport and detoxification (Alon et al., 2012; Celorio‐Mancera et al., 2012, 2013; Zhong et al., 2017). Interestingly, the relatedness of utilized host plants is associated with expression profiles in the comma butterfly (Polygonia c‐album) (Heidel‐Fischer et al., 2009), but not in the painted lady (Vanessa cardui) (Celorio‐Mancera et al., 2016). Feeding on closely related host plants can also result in gene expression changes, which depend on the concentration of secondary metabolites, as has been shown in the monarch butterfly (Danaus plexippus) and the small cabbage white (Pieris rapae) (Okamura et al., 2019; Tan et al., 2019). Such a response is generally more pronounced in a generalist, like the tobacco budworm (Heliothis virescens), as compared to a specialist, like the large (cabbage) white (Pieris brassicae) (Schweizer et al., 2017). The potential for host plant expansion hence depends on caterpillar plasticity. However, whether this is underpinned by standing variation or de novo selection when exposed to novel hosts is generally unknown.
Many herbivores depend on microbial function to process their food. Changes in the diversity and composition of the microbiome are probably important for diet shifts and/or expansions. In leaf eating insects in general, overall effects of microbiome composition on fitness might be limited (Hammer et al., 2017), and diet has been shown to have a negligible effect on the microbiome diversity in many butterflies (Chaturvedi et al., 2017; Minard et al., 2019; Phalnikar et al., 2018). However, detoxification of secondary metabolites and other functions associated with adaptation to specific host plants could be mediated by a rather limited set of functionally important microbial species (Xia et al., 2013, 2017). Both transcription profiling and microbiome quantification studies hence indicate that host plant switches might be associated with distinctive molecular and physiological responses in different lineages. Expanding the assessment of such associations across the butterfly tree of life will reveal if there are general patterns in adaptive responses or if each switch event is more of a unique character.
The common wood white (Leptidea sinapis) is a widespread butterfly species that occurs over most of Eurasia (Dincă et al., 2011). In Europe, karyotype, mating behaviour, and host plant choice differs between populations in what can be described as a northeast‐southwest gradient. Adult female wood whites preferentially lay eggs on vetches and trefoils (pea family; Fabaceae) with some regional preference differences being present. In Catalonia, wood white females preferentially lay eggs on the locally abundant, woody perennial shrub Lotus dorycnium (until recently classified as Dorycnium pentaphyllum), while Swedish L. sinapis females generally oviposit on herbaceous perennials such as Lotus corniculatus and different Lathyrus species (Friberg & Wiklund, 2009). The wood whites hence constitute an ideal system that we use in this study to investigate; (i) whether female host plant preference differs between the two populations; (ii) whether host plant diet affects larval growth; (iii) whether host plant diet results in population specific alterations of gene expression; and (iv) whether diet and population origin shape gut microbiome composition.
2. MATERIALS AND METHODS
2.1. Host plant preference test
Wild caught, recently mated females (only females with no or very limited wing wear were used as dissection of deceased females showed a single spermatophore present in the bursa copulatrix) of both populations were used for preference and acceptance tests using two experimental set‐ups. In the first experiment, females were placed in small cages (40 × 40 × 40 cm) with one stand of each host plant (Lotus corniculatus [from Sweden] and L. dorycnium [from Catalonia]), randomly placed at either end of the cage. This experiment ran for 48 hr with an 18 hr light + 6 hr darkness light regime at constant temperature (23°C) for females from both populations (nCatalonia = 15; nSweden = 10). A follow‐up experiment was conducted for Swedish females (n = 10) where they were exposed to either L. corniculatus or L. dorycnium for 48 hr using the same light settings. The number of eggs laid on each host plant was used for assessment of host plant acceptance among females and the two experimental set‐ups were treated as separate tests for the Swedish females. The reason for only using females from Sweden for the second test was that we had few survivors after the initial test and we needed more eggs laid on L. dorycnium for the follow‐up analysis.
2.2. Scoring of developmental time and size of adult F1
All offspring (F1) resulting from the eggs laid by the females on either L. dorycnium or L. corniculatus were kept under identical environmental conditions (+23°C, 18 hr daylight + 6 hr darkness) and monitored daily. As a measure of growth rate/developmental time, the timespan (in days) from egg laying to emerging adult was assessed for all offspring individuals. In total, we measured the developmental time of 537 specimens, represented by 310 Catalonian and 227 Swedish individuals (Table S1). Adults (F1) were marked when emerging from the chrysalis, and kept in large cages until they died. After drying individuals in + 50°C for 36 hr, wings from all adults were removed by pulling both forewings simultaneously towards the anterior end of the butterfly, using a stork‐bill fine blunt forceps. This method ensures that wings break off at the same spot for all samples, right at the joint between the forewing and thorax. The right forewing of each sample was photographed with a Nikon Digital Sight DS‐VR camera (Nikon Corp.) mounted on a Nikon SMZ800N stereo microscope (Nikon Corp.), using standardized distance (160 mm), zoom (4×) and light settings, and pictures were stored as high‐resolution JPEG files. Wing size was estimated using two different measurements. First, the distance between the base (centre of joint) and tip (edge of wing where vein R4 ends) of the wing was measured to get the total length. Second, we counted the total number of pixels covered by the entire wing. Both of these measures were done in Photoshop CC 2017 (Adobe Systems Inc.). In total, we measured the wing size (length + area) of 509 specimens, represented by 292 Catalonian and 217 Swedish individuals (Table S1). It should be noted that neither body size nor development time is a direct measure of fitness, especially in controlled laboratory conditions. However, there is ample evidence that body size is positively associated with female fecundity and negatively associated with stress in several butterfly species (Carnicer et al., 2019; Johnson et al., 2014; Niitepõld, 2019), including Leptidea sp. (Friberg & Wiklund, 2009; Lukhtanov et al., 2018). Using development time as a proxy for fitness is perhaps less straightforward for a specific system, although we do expect an association in general. Trade‐offs of using the plant producing the largest body size and shortest development time may exist, for example predation pressure at different life stages, synchrony with host plant availability and a match between generation time and regional climate conditions.
2.3. Rearing of larvae for gene expression profiling and microbiome characterization
Eggs from a subset of L. sinapis females were used in a split brood design to assess gene expression variation associated with the type of host plant used by larvae. Specifically, eggs from each of seven Swedish and seven Catalonian females were divided into cohorts where each cohort was raised on either L. dorycnium or L. corniculatus and larvae were harvested immediately after entering larval stages instar III and instar V. The aim was to get at least four biological replicates, i.e., one offspring from at least four different females in each respective cohort and treatment. This set‐up allows for assessing biological variance for each population and host plant treatment category and control for maternal effects. The number of larvae for each developmental stage, treatment and population ranged between four and seven (Table S2).
2.4. RNA extraction, library preparation and sequencing
RNA was extracted from instar III and instar V larvae. Before the extractions the cuticle was disinfected by sequentially immersing the sample in 500 µl 1% bleach, molecular grade (double‐deionized) water (ddH2O) and 70% ethanol to reduce the risk of contamination from surface microorganisms in the characterization of the gut microbiome. Each larva (n = 66 in total) was dissected to separate the head, gut and abdomen. The abdomens were homogenized in RNeasy lysis buffer with added dithiothreitol (DTT, Acros Organics) using a micropestle and QiaShredder. RNA was extracted using the RNeasy Mini Kit according to the manufacturer's protocol (Qiagen, Inc.). After quality control on a Bioanalyzer (Agilent) to verify the integrity of the RNA, total RNA was sent for library preparation (TruSeq RNA with poly‐A selection) and multiplex sequencing on a single Illumina NovaSeq6000 S1 lane with 150 base pairs (bp) paired‐end reads at the National Genomics Infrastructure (NGI), Science for Life Laboratory (SciLife) in Stockholm. The sequencing of one sample failed (Catalonian III‐instar on L. dorycnium) and 65 sample libraries were used in all downstream analyses.
2.5. DNA extraction, library preparation and sequencing
DNA was extracted from the guts of the same 66 samples used for expression profiling. The gut was homogenized in TRIzol Reagent (Thermo Fischer Scientific) with glass beads (∅ = 1 mm) using the TissueLyser II (Qiagen, Inc.) at 30 Hz for 1 min. The organic phase of the lysate was re‐extracted using standard Proteinase K digestion and a modified phenol chloroform/chloroform purification protocol (Green & Sambrook, 2017). Negative extraction controls were performed for every eight samples (n = 8 in total). The amount and purity of the extracts were controlled with Qubit (Thermo Fischer Scientific) and NanoDrop (Thermo Fischer Scientific), respectively, and the extracts were stored at −20°C. Custom amplicon primers (Eurofins Genomics) were used to obtain a single amplicon of 460 bp from the V3 and V4 region of the 16S ribosomal RNA gene from the extracted DNA (Klindworth et al., 2013):
forward primer = TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG;
reverse primer = GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC.
We performed the amplification in triplicates with the following PCR‐protocol; 2.5 µl sample DNA (approximately 5 ng/µl), a final concentration of 0.2 µM of each primer, and 12.5 µl 2X KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Roche Inc.) for a total volume of 25 µl. The PCR was performed in a 2720 Thermal Cycler (Applied Biosystems) using the following settings: initial denaturation at 95°C for 3 min, 30 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final elongation step at 72°C for 5 min. PCR products were run on a 1.5% agarose gel to ensure accurate fragment size (550 bp) and yield, and cleaned with 20 µl AMPure XP beads per sample, according to the manufacturer's recommendations (Beckman Coulter Life Sciences). Finally, unique dual‐index barcodes and Illumina sequencing adapters, Nextera XT Index Kit (FC‐131–1002, Illumina) were added to each amplicon target for individual identification after pooling (Table S3). The individual libraries for each sample were prepared using 5 µl PCR product, 5 µl Nextera XT Index Primer 1 (N7xx), 5 µl Nextera XT Index Primer 2 (S5xx), 10 µl 2x KAPA HiFi HotStart ReadyMix (KAPA Biosystems) and 25 µl ddH2O, for a total volume of 50 µl. A limited cycle PCR was performed with initial denaturation at 95°C for 3 min, eight cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 30 s, and a final elongation step at 72°C for 5 min. The final cleaning step of the libraries was performed with AMPure XP beads (56 µl per sample) according to the protocol provided by the manufacturer (Beckman Coulter Life Sciences). Libraries were pooled in equimolar concentrations and sequenced on one lane using a 300 bp paired‐end read approach on an Illumina MiSeq platform (Illumina, Inc.) at NGI, SciLifeLab, Stockholm.
2.6. Gene expression profiling
Overall quality of the RNA‐seq reads was assessed with fastqc version 0.11.5 (Andrews, 2016). Initial filtering of raw RNA‐seq reads was performed using trimgalore version 0.4.4 (Krueger, 2017). This step included trimming 12 nucleotide bases from the 5’ end of each sequence, trimming sequences with overall Phred score < 30, filtering out adapter sequences and sequences shorter than 30 bp. fastq masker (http://hannonlab.cshl.edu/fastx_toolkit/; accessed 2019‐05‐01) was used to mask (replace with N) low quality (threshold = 10) nucleotides in the reads. prinseq version 0.20.4 (Schmieder & Edwards, 2011) was then used to trim remaining poly‐A tails and cutadapt version 2.5 (Martin, 2011) was applied to filter out long stretches of A/T nucleotides (threshold = 10 bp) inside reads. condetri (Smeds & Künstner, 2011) was then used to filter out reads with Phred Score < 30 in more than 80% of the read. Remaining sequences of ribosomal origin (rRNA) were removed using sortmerna version 2.1 (Kopylova et al., 2012). Finally, fastq Screen (Wingett, 2017) with bowtie2 version 2.3.5 (Langmead & Salzberg, 2012) was used to identify and screen for contaminants. The most likely contaminants were identified based on previous gene expression studies in L. sinapis (Höök et al., 2019; Leal et al., 2018). The filtered data set was screened for contaminants of the following origin: rRNA, human, Drosophila melanogaster, Wolbachia sp., L. corniculatus, L. dorycnium, L. japonicus, Illumina adapters and primers. The fraction of potential contaminant reads detected was <0.01% in all libraries and therefore not a concern for subsequent analytical steps. The entire pipeline for treatment of RNA‐seq reads before expression profiling is provided in Figure S1.
Filtered reads were indexed and mapped with star version 2.7.2b (Dobin et al., 2013), using previously available L. sinapis genome (Talla et al., 2017) and transcriptome (Höök et al., 2019; Leal et al., 2018) assemblies. Gene specific counts of mapped RNA‐seq reads were obtained using stringtie version 1.3.6 (Pertea et al., 2015). The raw gene counts were used as input for a standardized differential gene expression analyses carried out using deseq2 version 3.6 (Love et al., 2014) as implemented in r version 3.4.3 (R Core Team, 2013). Genes for which only one sample in the entire sample set had nonzero read counts and genes with zero counts in all samples in a specific cohort were removed. A count of one (1) was added to every gene/sample, to stabilize variance at low expressed genes. Low coverage genes with baseMean (count average across all samples) <2 (to account for the 1 count added to every gene/sample) were removed before carrying out the differential expression analysis.
deseq2 was run with default settings. This protocol normalizes counts per gene by library size (the number of reads in a specific library) and carries out significance testing for individual genes using the Wald test (Love et al., 2014). The analysis implements the method of Benjamini and Hochberg (1995) to account for multiple testing, and generates false discovery rate (FDR) adjusted significance levels (padj) for each gene. In order to investigate the effect of host plant diet on gene expression in the different populations and assess potential population specific effects, the contrasts were analysed using (i) the Swedish and Catalonian samples together; (ii) Catalonian samples only; and (iii) Swedish samples only. Each of these data sets was also divided into instar III, instar V male and instar V female samples, to characterize differences between ontogenetic stages and sexes. The deseq2 analysis outputs four result values for each gene: baseMean, log2FoldChange (ratio of gene expression values across treatments, in log 2 scale), p‐value, and FDR‐adjusted p‐value (p adj). Genes with a baseMean > 10, absolute log2FoldChange > 1.0 and p adj < 0.05 were considered significantly differentially expressed between treatments. Enrichment of specific gene functions in differentially expressed gene sets was assessed with the Bioconductor package topgo version 2.38.1 (Alexa & Rahnenfuhrer, 2016) in r version 3.4.3 (R Core Team, 2013) using the database org.Dm.eg.db from the r‐package “AnnotationDbi” (Pagès et al., 2019) for orthologous genes for the gene ontology (GO) categories biological process, cellular component and molecular function.
2.7. Processing of microbiome data and characterization of microbiome composition
Raw sequence data (fastq files) were imported to “dada2” version 1.14.0 (Callahan et al., 2016) and processed as described in the following. Forward reads were clipped to 295 bp (maxEE = 1, maxN = 0, truncQ = 2 and phiX removal activated) and backward reads were omitted due to quality issues in the 3’ region of the backward reads. Chimeric sequences were removed by applying the “removeBimeraDenovo” function as implemented in “dada2”. Taxonomy was assigned to each amplicon sequence variant (ASV) using “idtaxa” (Murali et al., 2018) as implemented in the r package decipher version 2.14.0 (Wright, 2016) with GTDB (r89; Parks et al., 2018) as reference database. The ASVs were aligned using “AlignSeqs” from the DECIPHER package and phylogenetic tree reconstruction was performed applying a general time reversible model with gamma optimization and stochastic rearrangement (“optim.pml” command) as implemented in “phanghorn” version 2.5.5 (Schliep, 2011). Next, ASVs with kingdom not belonging to bacteria were removed as well as all ASVs with unknown phylum assignment. To clean the data further from spurious taxa assignments and potential contaminations, all ASVs belonging to family Mitochondria or genus Chloroplast were removed. In a final step, samples with less than 500 contigs were removed and singleton ASVs (occurring with one contig in only one sample) were removed. Additionally, one sample (sample ID 1024) was identified as a clear outlier (using unweighted “UniFrac” as measure, described below) and removed completely from the data set. Alpha diversity (Shannon, 1948) was estimated using phyloseq version 1.30.0 (McMurdie & Holmes, 2013) and further evaluated in a linear mixed model framework (“lme” package version 3.1–143) with a nonparametric test (Wilcoxon test). Beta diversity was assessed using the unweighted “UniFrac” (function in phyloseq) distance (Lozupone & Knight, 2005) on subsampled data (rarefied to 500 contigs per sample), as suggested by Weiss et al. (2017) Permutational multivariate analysis of variance using distance matrices (PERMANOVA) was performed using the “adonis” command in “vegan” version 2.5–6 (Oksanen et al., 2019) with 99,999 bootstrap permutations. Differences in taxonomic abundances were investigated using nonparametric testing (Wilcoxon test). All analysis were performed in r version 3.6.1 and p‐values were corrected using Benjamini‐Hochberg correction (“p.adjust” function in r).
3. RESULTS
3.1. Host plant preference and effects on growth in Leptidea sinapis
Females from the Catalonian population did not show preference for either of the two host plants (Wilcoxon test: W = 115, p‐value = 0.93). Swedish females, however, laid significantly fewer eggs on L. dorycnium than on L. corniculatus in both the direct choice (mean 0.9 ± SD 1.9 on L. dorycnium versus 23.2 ± 11.2 on L. corniculatus; W = 0, p‐value = 1.3 × 10−4) and the single exposure acceptance test (4.2 ± 5.9 versus 15.2 ± 10.7; W = 21, p‐value = 9.9 × 10−3; Figure 1).
Figure 1.
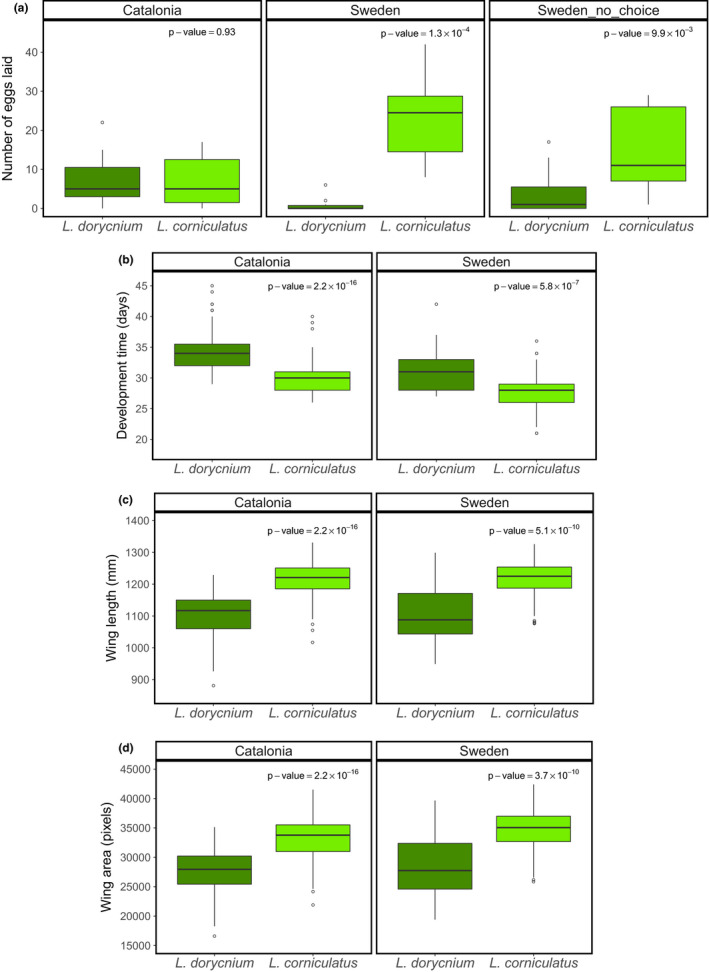
Boxplots showing the number of eggs laid (y‐axis) on the two different host plants Lotus dorycnium (dark green) and L. corniculatus (light green) in the different preference tests (a). Females from Catalonia are represented by the two leftmost panels and Swedish females are represented by the remaining panels. The results from the direct choice test in the middle panels (Sweden) and when exposed to either L. dorycnium or L. corniculatus in the rightmost two panels (Sweden_no_choice). Boxplots depict the development time (b), wing length distribution (c) and wing area distribution (d) in the Catalonian and Swedish L. sinapis individuals reared on L. corniculatus (dark green) or L. dorycnium (light green). p‐values were obtained with a Wilcoxon test for each cohort [Colour figure can be viewed at wileyonlinelibrary.com]
Both the Catalonian and the Swedish individuals had a significantly reduced developmental rate when feeding on L. dorycnium than on L. corniculatus (Figure 1). The mean developmental time for Catalonian individuals was 34.1 ± 3.1 days on L. dorycnium and 29.8 ± 2.4 days on L. corniculatus (W = 21,425, p‐value < 2.2 × 10−16) and the corresponding values were 30.8 ± 3.4 and 27.9 ± 2.6 days, respectively, for the Swedish individuals (W = 5,967.5, p‐value = 5.8 × 10−7; Figure 1). These results also show that wood whites from the Swedish population in general had shorter developmental time than the Catalonian individuals (see numbers above), a statistically significant rate difference both on L. dorycnium (W = 1541, p‐value = 1.2 × 10−8) and L. corniculatus (W = 7,910, p‐value = 1.3 × 10−11) (Figure 1). The individuals reared on L. dorycnium also had significantly shorter wing length and smaller wing area, indicating overall smaller body size, than the individuals utilizing L. corniculatus; this was observed in both the Catalonian and the Swedish cohorts (Figure 1).
3.2. Gene expression profiling
In total, we obtained >336 million reads from the 65 samples. The proportion of reads with Q‐value >30 was 89.03%–94.11% across samples. The number of reads per sample varied from 8.32 to 18.44 million which corresponds to an estimated per site coverage of ≈248×, assuming an entire coding gene set in L. sinapis consisting of 15,000 genes with an average length of 1,000 bp (Leal et al., 2018). To visually inspect the variance associated with population, developmental stage, sex and host plant diet, the global gene expression profiles of individual samples was used in a PCA. This showed that developmental stage was the only determinant resulting in visible clustering, separating instar III and instar V samples along PC1 (Figure S2). The PCA also revealed a larger variance in the Catalonian than in the Swedish samples, indicating a more variable global gene expression profile in Catalonian larvae (Figure S2).
Differences in gene expression profiles between treatment groups were quantified to characterize the expression response to host plant utilisation. The analyses aimed at assessing both general and population specific effects, as well as potential variation across developmental stages and sexes. Initially, individuals from both populations were merged to detect potential general effects between developmental stages and sexes. The numbers of significantly differentially expressed genes between the two host plant treatments in instar III, instar V male and instar V female were 50, 58 and 42, respectively (Figure 2, Table 1, Table S4). Among these genes, a significantly larger fraction of genes showed higher expression levels in cohorts fed on L. dorycnium than on L. corniculatus in instar V males and instar V females (84% and 90%, respectively) but not in instar III larvae (52%) (Figure 2, Table 1, Table S4). To obtain a better insight into population‐specific effects of host plant usage with respect to gene expression, we analysed the Catalonian and Swedish samples separately. In the Swedish samples, the number of differentially expressed genes was lower in general (Figure 2, Table 1, Table S4). The number of differentially expressed genes in the Catalonian contrasts was, however, at the same level or higher than the numbers observed in the joint analysis of all individuals (Figure 2, Table 1, Table S4). In the samples from Catalonia, but not from Sweden, there was also a larger fraction of genes with higher expression levels in response to L. dorycnium than to L. corniculatus (Table 1).
Figure 2.
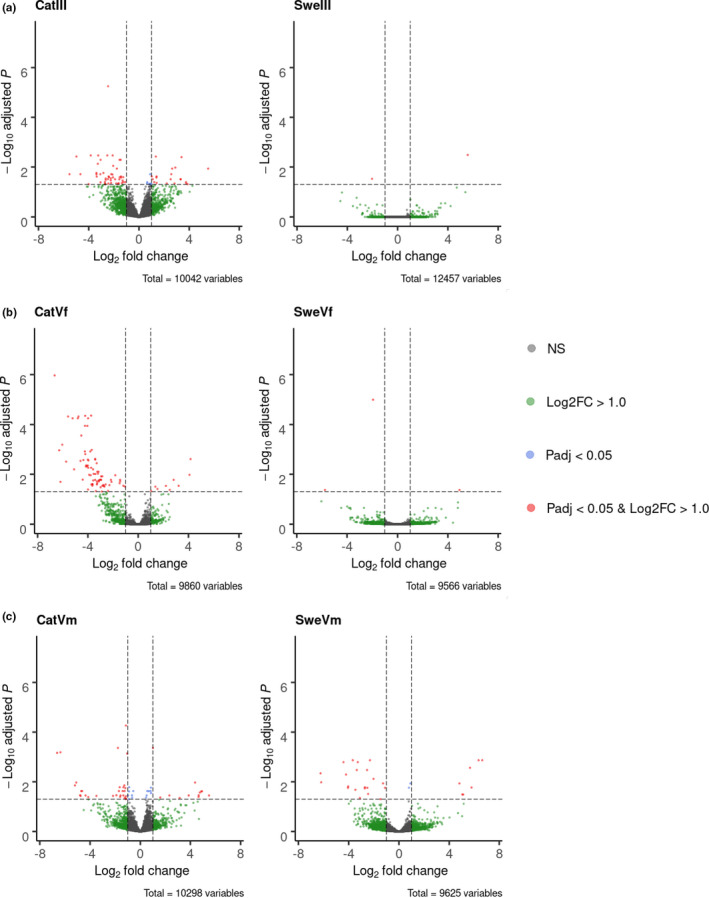
Volcano plots showing the gene expression differential (log2 fold change, x‐axis) and corresponding adjusted p‐value (padj, y‐axis) for cross‐treatment (L. dorycnium versus. L. corniculatus as host plants) comparisons of (a) instar III (III); (b) instar V female (V f); and (c) instar V male (V m) L. sinapis larvae. Each panel is divided according to origin of samples, Catalonia (Cat) and Sweden (Swe) [Colour figure can be viewed at wileyonlinelibrary.com]
Table 1.
The number of differentially expressed genes in the contrasts between treatment groups that were fed on either L. dorycnium or L. corniculatus. The number of upregulated genes for each treatment (host plant) and the total number of differentially expressed genes in each cohort are given
| Population/stage and sex | Total number | L. dorycnium | L. corniculatus | p‐value |
|---|---|---|---|---|
| All III | 50 | 26 | 24 | NS |
| All Vf | 42 | 38 | 4 | *** |
| All Vm | 58 | 49 | 9 | *** |
| CatIII | 57 | 43 | 14 | ** |
| CatVf | 72 | 66 | 6 | *** |
| CatVm | 39 | 29 | 10 | * |
| SweIII | 2 | 1 | 1 | NS |
| SweVf | 3 | 2 | 1 | NS |
| SweVm | 25 | 18 | 7 | NS |
Abbreviations for population, developmental stages and sexes are Cat (Catalonia), Swe (Sweden), III (instar III), V m (instar V male) and V f (instar V female).
Significance levels for differences in the number of upregulated genes between treatments are: NS, p‐value > .05, ***p‐value < .001, **p‐value < .01, *p‐value < .05.
3.3. Gene ontology (GO)/functional inference
To get information about functional categories associated with differentially expressed genes, a GO enrichment analysis was performed on each experimental group (instar III, instar V male and instar V female) for each population independently. In total we found 104 significantly enriched terms (p adj < 5.0 × 10−2) associated with 63 differentially expressed genes: biological process = 61 terms and 26 genes, cellular component = 17 terms and 15 genes, molecular function = 26 terms and 22 genes (Table S5). For biological process, the most common ontology terms (i.e., terms associated with more than one gene or experimental group) included sarcomere organisation, neurotransmitter regulation, metabolic process, translational regulation, larval lymph gland haematopoiesis, cell‐cell adhesion and compound eye development. For cellular component, enriched ontology terms included integral component of plasma membrane, extracellular region, Golgi cis cisterna and Z disc. The molecular function category contained enriched terms associated to transmembrane transporter activity, calcium ion binding, spectrin binding, actin filament binding and hydrolase activity (Table S5). Genes that were differentially expressed in more than one cohort (n = 8) were explored for functionality using BLAST (Table S5). Only two genes (lava lamp, cadherin) had functional annotation included in the GO‐analysis. Three of the genes matched annotated genes in other taxa, a neurotransmitter‐receptor (synaptic vesicle glycoprotein 2A‐like), a histone modification enzyme (histone‐lysine‐N‐metyltransferase) and a lipid binding protein, possibly involved in cell‐cell adhesion in epidermis (filaggrin‐2like) (Table S5).
3.4. Host plant effects on microbiome composition
The total number of contigs remaining after preprocessing ranged from 519 to 199,575 (mean and median = 3,901 and 30,266) between samples. The fraction of chloroplast contigs ranged from 0% to 99% and the fraction of mitochondrial contigs between 0% and 5%. After both preprocessing, quality filtering and exclusion of nonbacterial contigs, the final data set for microbiome diversity and compositional analysis comprised of 178 amplicon sequence variants (ASVs) across 54 samples, with N Cat = 36 and N Swe = 18. We found that Wolbachia sp. was the most common taxon in all samples, with a proportional abundance ranging between 0.83–1.00. There was a significant difference in relative abundance of two bacterial phyla (Wilcoxon test: Proteobacteria, p adj = 1.35 × 10−3; Actinobacteria, p adj = 1.19 × 10−3) when comparing the Catalonian cohorts feeding on different host plants. This pattern was consistent with observations at family level (Anaplasmataceae, p adj = 1.71 × 10−3; Micrococcaceae, p adj = 1.79 × 10−2). At genus level, the difference between treatments was significantly different for Wolbachia (p adj = 2.23 × 10−3), but not for Kocuria (p adj = 8.79 × 10−2) (Figure 3). A more detailed analysis of the distribution showed that the difference was mostly due to an increase in relative abundance of Micrococcaceae (Kocuria; 0.2% to 1.1%), and a reduction in the relative abundance of Anaplasmataceae (Wolbachia; 99.5% to 96.8%) in the Catalonian larvae reared on L. dorycnium compared to larvae reared on L. corniculatus (Figure 3). A similar trend (Kocuria 0.5% to 1.2%; Wolbachia 98.4% to 96.7%) was observed in the samples from the Swedish population, but the differences were not statistically significant (Figure 3). No overall differences in relative abundance were observed between populations (p adj > 5.0 × 10−2).
Figure 3.
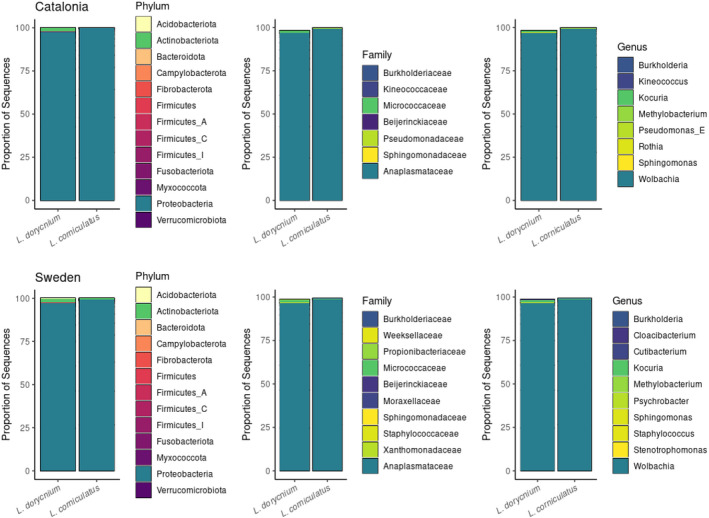
Taxonomic level comparison of gut bacteriome between larvae reared on different host plants, L. dorycnium versus L. corniculatus, separated by population origin, Catalonia and Sweden. Note that only the top 25 taxa are included which explains why the bars do not reach 100% [Colour figure can be viewed at wileyonlinelibrary.com]
The α‐diversity metrics were consistent between the different populations, and there was a trend towards higher diversity in larvae reared on L. dorycnium compared to L. corniculatus in both populations (Figure 4). A mixed effect model revealed a significant association between larval developmental stage and α‐diversity (p‐value = 2.08 × 10−2, population and host plant as random factors), with higher Shannon diversity metrics in the gut microbiome of instar III compared to instar V larvae. This difference was consistent regardless of treatment (Figure 4). The multivariate analysis based on the unweighted UniFrac dissimilarity matrix revealed a significant effect of host plant on microbiome β‐diversity (Figure 4; permanova: r2 = 7.57 × 10−2, p‐value = 2.23 × 10−3). The effect of population was smaller and marginally significant (r2 = 3.70 × 10−2, p‐value = 5.05 × 10−2). We also found a significant interaction effect between host plant and population (r2 = 7.69 × 10−2, p‐value = 1.79 × 10−3). The associations between effects were further explored by modelling each population separately, which revealed a shift in community structure in the Catalonian samples when reared on different host plants (r2 = 2.21 × 10−2, p‐value = 1.00 × 10−4) (Figure 4). No such host plant effect could be detected in the Swedish samples (Figure 4), and there was no effect of developmental stage on the composition of the microbiome in either population (p‐values > 5.0 × 10−2).
Figure 4.
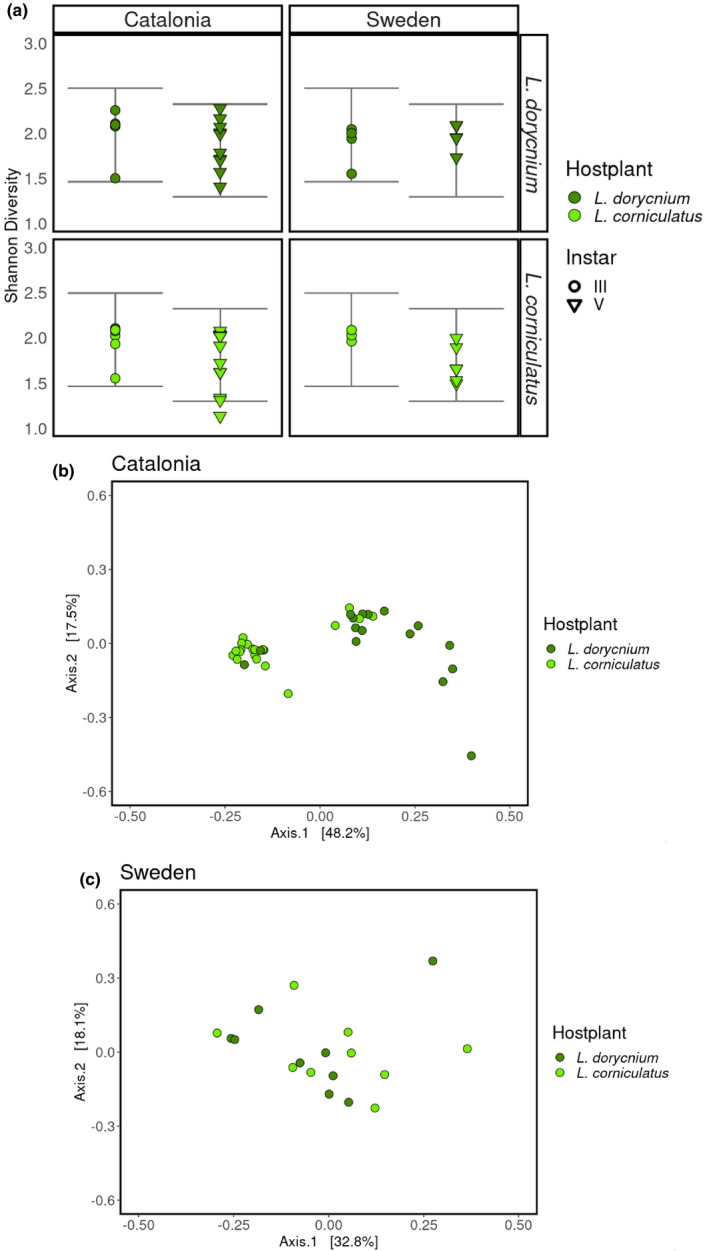
(a) Gut bacteriome α‐diversity. Error bars represent variance of predicted values by the model. Host plant and population effects on β‐diversity visualized by PCoA based on unweighted UniFrac dissimilarity matrix for samples from Catalonia (b) and Sweden (c); dots in b and c are jittered [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
4.1. General
In this study we combined experimental assessment of female egg‐laying preference and larval development with current molecular techniques to investigate the genetic and physiological underpinnings of host plant utilisation in the wood white butterfly, which exhibits geographical variation in host plant use across the distribution range (Friberg & Wiklund, 2009). By combining experimental assays with gene expression profiling and microbiome analysis, we advance our knowledge about the causes and consequences of host plant shifts in phytophagous insects.
4.2. Host plant preference and effects on developmental rate and adult size
Our results showed that female Catalonian wood whites did not show preference for either host plant, while Swedish females preferred L. corniculatus. This observation is interesting since both Spanish and Swedish larvae showed a significantly reduced growth when feeding on L. dorycnium, which is consistent with the hypothesis that female host preference is largely decoupled from larval performance (Wiklund, 1975). While reduced growth might be expected in Swedish wood whites, which have not encountered L. dorycnium in the wild and only marginally recognize it as a potential host, it is notable that Catalonian females utilise L. dorycnium even when L. corniculatus is present, despite the effects on developmental rate and imago size when feeding on this plant. This shows that other factors than nutritional value being involved in female choice of host plants for oviposition. Host plant choice is evidently affected by interactions of phylogenetic, ecological, chemical and environmental factors as well as relationships with predators and/or parasites (Ehrlich & Raven, 1964). In our case, a potential explanation for the difference in host plant preference between Swedish and Catalonian wood white females could be different climates at the sampling sites and the characteristics of the two host plant species. While L. corniculatus is a typical herbaceous plant, L. dorycnium is a woody shrub with needle‐like leaves. Consequently, L. dorycnium is substantially more drought resistant than L. corniculatus. The different populations of L. sinapis have most likely diverged in allopatry in the different refugia during recurrent Pleistocene glaciations and current continuous distribution is due to range expansion and secondary contact (Talla et al., 2019). The use of L. dorycnium could possibly have emerged as a beneficial resource for the Iberian populations during the aridification of the Mediterranean basin during late Pleistocene (Nieto Feliner, 2014), as this plant is more likely to withstand extended drought periods during larval growth. Host plant recognition could have evolved as a local adaptation of the population in dry Mediterranean refugia where L. dorycnium was a more steadfast food source over the longer reproductive season. Therefore, although L. corniculatus may be more easily digestible, and lead to a faster growth rate and larger overall body size, utilization of the hardier L. dorycnium might lead to increased larval survival in a drier climate. An alternative explanation is that there is a trade‐off between developmental time and survival or fecundity, similar to what has previously been observed in Edith's checkerspot butterfly (Euphydryas editha) (Singer & Parmesan, 2018). Other benefits of utilising L. dorycnium may include less intra‐ and/or interspecific competition. Also, L. dorycnium is currently widely distributed in the Mediterranean and it seems a reasonable hypothesis that the use of this plant significantly extends the potential habitat of L. sinapis, even more so given recent summer drought episodes (Meehl & Tebaldi, 2004). In Catalonia, L. sinapis uses mainly L. dorycnium, but also, less frequently, L. corniculatus and L. hirsutus (Vila et al., 2018). All three plants are closely related phylogenetically and the latter is also not present in Sweden. Given that L. sinapis is relatively widespread and common in the Mediterranean, and that it is present during virtually all spring and summer in up to three generations, it is evident that longer development time and smaller adult size when feeding on L. dorycnium does not have a negative impact in natural populations, or that it is compensated by other benefits. An interesting parallelism apparently takes place in the sibling butterfly species Leptidea reali, which frequently uses Onobrychis sp. as host plant in Catalonia, in addition to the presumed core host plant Lathyrus pratensis (Vila et al., 2018). Onobrychis plants are also more drought resistant and widespread than L. pratensis, which is restricted to relatively humid and shady patches. It is thus possible that host plant shifts/expansions have extended the niche in both Leptidea species and allowed them to cope with climate change effects on regional host plant communities.
We found a significantly shorter developmental time in Swedish compared to Catalonian individuals on both host plants. Whether this pattern is consistent across a wider range of conditions (e.g., temperature, day length) is a matter of speculation, since we only tested developmental time at a single temperature and a single day length. It is possible that the wood whites from Catalonia have adapted to develop at higher temperatures or shorter day lengths, so that the difference in development times between populations would be smaller with other settings. An alternative, but not exclusive explanation, could be that the natal host plant (L. dorycnium) in Catalonia, which is a woody shrub, is a less suitable food source for fast development than L. corniculatus, and that Catalonian wood whites have adapted to a slower development rate.
Previous studies on transgenerational effects in butterflies show mixed results (Woestmann & Saastamoinen, 2016). While host plant preference seems largely uncoupled from conditions during female larval development in some cases (e.g., Melitaea cinxia; Salgado & Saastamoinen, 2019), other studies show that host plant quality cues experienced by parents can induce phenotypic adjustments in offspring (Coenonympha pamphilus; Cahenzli & Erhardt, 2013) and female host plant preference can be positively associated with offspring preference (Euphydryas editha; Singer et al., 1988). The impact of natal effects seems to be dependent on the studied insect taxonomic group. There was, for example, a limited effect of the maternal rearing environment on behaviour in pea aphids (Slater et al., 2019), but a significant association between natal origin and oviposition preference in water lily beetles (Verschut et al., 2017). It should be noted that we used wild‐caught females for the preference test and we therefore have no control of potential natal effects – i.e., we do not know which host plants the females fed on as larvae. The Swedish females have obviously not grown up on L. dorycnium, since the plant is not present in Scandinavia and it is therefore difficult to judge the impact of a potential natal effect in this cohort. The females from Catalonia most likely grew up on L. dorycnium, since this is the preferred host plant in natural conditions (Vila et al., 2018), but could of course also have fed on L. corniculatus as larvae since this plant is present in the natal range. Still, Catalonian females did not discriminate between the two host plants and there was no detectable difference in the number of eggs laid on either host plant species. This indicates that natal effects should have limited impact on the preference results. In addition, the maternal microbiome can potentially influence offspring growth, as shown in the large cabbage white (Pieris brassicae) (Paniagua Voirol et al., 2020), but since we used wild caught females this effect cannot be estimated here. However, if anything, we would expect to see a larger diet‐dependent effect on larval growth in the Swedish population since those females did not feed on L. dorycnium as larvae.
4.3. Gene expression variation across populations and experimental cohorts
The main factor affecting global expression profiles across all samples was developmental stage, whereas no effect of sex could be detected. This is in line with previous studies that found considerable differences in gene expression profiles between ontogenetic stages (Leal et al., 2018), but only a small proportion of sex‐biased genes in the larval stages (Höök et al., 2019). We did not observe any obvious clustering by either population or host plant treatment, in contrast to other studies where different host plant use results in clearly distinguishable expression profiles (Celorio‐Mancera et al., 2013). This is somewhat surprising considering the apparent differences in growth for larvae fed on the different host plants. An explanation could be that L. dorycnium and L. corniculatus are relatively closely related (recently merged into the same genus), and may have similar chemical properties that induce only limited differences in gene expression profiles. This is a pattern similar to what was observed in the polyphagous V. cardui, where a distinct gene expression profile was found when feeding on a core set compared to an extended range of host plants (Celorio‐Mancera et al., 2016). It should also be noted that larvae were harvested at fixed developmental stages rather than at specific time points, which has probably reduced the effect of overall growth rate differences on gene expression patterns.
In the differential gene expression analyses across treatments, several general patterns were observed. First, more genes were differentially expressed between treatment groups in the Catalonian than in the Swedish population. Second, a larger fraction of the differentially expressed genes were upregulated in response to the use of L. dorycnium than of L. corniculatus, especially for the contrasts involving Catalonian wood whites. Third, GO terms relating to protein biosynthesis, membrane transport and transporter activity, and various metabolic processes occurred repeatedly. Below, each of these observations is discussed in more detail.
The number of differentially expressed genes between treatments was higher in the Catalonian than in the Swedish contrast. Previous studies suggest that phytophagous insects may attain polyphagy via an increase in expression breadth and regulation of ribosomal, digestion and detoxification related genes (Celorio‐Mancera et al., 2013). Thus, the more flexible expression response in Catalonian individuals may be an adaptation that allows them to metabolise a wider suite of host plants. The use of L. dorycnium in Catalonia could hence be an example of ecological fitting, where plasticity allows for colonization of novel hosts compositionally similar to the ancestral host (Janzen, 1985), but with different habitat preference and ecology (Heidel‐Fischer et al., 2009). In contrast, Swedish L. sinapis, which do not encounter L. dorycnium in the wild, do not appear to have developed the same plastic response. The mechanistic underpinning of this difference between the populations is so far unknown. We do not know whether the gene expression plasticity is genetically determined, or whether encounters with novel host plants generate a transgenerational higher plasticity, for example via maternal effects. Since we neither detected a difference in adult size between the populations nor a significantly reduced growth rate or body size in the Swedish individuals reared on L. dorycnium, it is unclear whether the expression response in the Catalonian population is beneficial. We used development time and adult size as proxies for fitness, and natural selection on these traits seems to be multifaceted and extremely complex for butterflies (Breuker & Brakefield, 2002; Dennis et al., 2012). Moreover, it is likely that other aspects are important for the success of individuals in natural settings, for example population‐specific standing genetic variants, microhabitat choice, appealing to the other sex or degree of toxicity of the butterfly, all potentially influenced by host plant choice.
There was also a consistent overrepresentation of upregulated genes in response to L. dorycnium, most pronounced in the Catalonian contrast. This observation is in line with a general increase in gene expression as a response to a novel host plant in the Asiatic rice borer (Chilo suppressalis; Zhong et al., 2017). The response could also reflect the defence abilities of the host with higher number of upregulated genes in Heliconius melpomene larvae reared on their native host plant containing larger amounts of adverse secondary metabolites than a less defended close relative (Yu et al., 2016). Catalonian females also showed the highest proportion of differentially expressed genes being upregulated in response to L. dorycnium, which could indicate that they are particularly plastic in response to this host plant.
4.4. Functional categories of differentially expressed genes
Enriched GO terms were mostly associated with protein biosynthesis, metabolism and nutrition, detoxification, neuronal development and cuticle integrity. Differential expression of genes associated with protein biosynthesis is in line with increased plasticity allowing for utilization of a broader range of host plants (Celorio‐Mancera et al., 2012; Govind et al., 2010) and altered expression of ribosomal genes, which can mediate the effects of ribosome‐inactivating proteins from the host plant (Zhong et al., 2017; Zhu et al., 2018). Several genes associated with metabolic processes, growth, autophagy and starvation, were also differentially expressed. This indicates a difference in nutritional value between the two host plants, probably associated with the overall longer developmental time and smaller adult size in cohorts feeding on L. dorycnium.
Differences in host plant chemical composition (e.g., secondary metabolites) can also affect growth of the larvae (Jeschke et al., 2017; Li et al., 2000). Lepidopterans have evolved several defense systems against toxins (Pinheiro de Castro et al., 2019; Zagrobelny et al., 2018), that are present in various amounts in both L. corniculatus and L. dorycnium (Puri et al., 1998; Salgado et al., 2016). Similar to what was previously observed in several generalist insect species (Celorio‐Mancera et al., 2012, 2013; Dermauw et al., 2013; Govind et al., 2010; Puinean et al., 2010; Zhong et al., 2017), there was a general upregulation of genes involved in transport activity and cuticle structure in L. sinapis when feeding on L. dorycnium. This implies that the pathways for managing host plant‐specific chemical compounds are similar across a wide range of insect taxa and that fine‐tuning the expression of genes associated with cell transport and cuticle integrity facilitates polyphagy (Celorio‐Mancera et al., 2013; Dermauw et al., 2013). It should be noted that the GO‐terms are derived from homologues in distantly related model species and the enrichment analysis should be seen as an indication of the range of pathways involved in host plant utilization in wood whites. It is also worth stressing that the gene expression data were based on RNA collected from the abdomen and thorax of L. sinapis larvae. If key genes and pathways involved in response to differential host plant use are confined to a specific organ or tissue, potential differences might have been diluted and therefore not detected with our approach.
4.5. Patterns of microbiome composition
We explored the diversity and composition of the gut microbiome and the associations with host plant diet in the two different wood white populations. The small proportion of bacteria compared to chloroplasts in our study is in accordance with previous studies showing low total abundance and complexity of the gut bacteriome in Lepidoptera caterpillars compared to other herbivores (Hammer et al., 2017; Whitaker et al., 2016). These overall low levels of gut bacteria support the notion that the gut microbiome in butterflies is transient with limited importance for nutrient turnover and larval growth (Hammer et al., 2017; Phalnikar et al., 2019; Staudacher et al., 2016) and that microbiome composition mostly depends on diet (Phalnikar et al., 2018) and/or soil bacteria (Hannula et al., 2019) acquired when feeding. The high level of plant‐derived amplicons and the low level of bacterial reads limits our power to detect signals in the data. Despite that, significant differences in diversity and composition were found in some comparisons. The observed reduction in α‐diversity as ontogeny proceeds is in line with other studies (Chaturvedi et al., 2017; Chen et al., 2016), and could be due to acclimatization of the microbiome to the specific conditions of the caterpillar gut, and/or microbiome competition reducing the diversity to a core set of taxa occupying specific ecological niches (Itoh et al., 2019). An alternative explanation is that different developmental stages require distinctive functions from the microbiome, but it is unclear why this should result in a reduction in diversity but not in the overall composition.
We found a significant effect of host plant diet on both the relative abundance and the composition (ß‐diversity) of the microbiome in the Catalonian cohort. This supports the hypothesis that bacterial communities in the gut are associated with larval diet (Minard et al., 2019; Staudacher et al., 2016; Szenteczki et al., 2019). The relative abundance in the Swedish cohort was also affected by diet, but the difference between groups was not significant. This could potentially be explained by a lack of power due to the smaller sample size in the Swedish cohort. Yet, the population specific effects on ß‐diversity suggest that there is a more complex relationship between population origin and diet, as previously observed in insects (Jones et al., 2019; Phalnikar et al., 2018; Pinto‐Tomás et al., 2011). The main difference in our analysis was a decrease in the relative abundance of Wolbachia, and an increase in Kocuria, in larvae reared on L. dorycnium compared to L. corniculatus. Wolbachia is a common intracellular bacteria found in many arthropods and the prevalence is generally high, for example over 90% in L. sinapis (Solovyev et al., 2015). The high relative abundance of Wolbachia in our samples probably originates from the intestinal wall tissue and mimics similar findings in other butterflies (Chaturvedi et al., 2017; Narita et al., 2007; Phalnikar et al., 2018; Whitaker et al., 2016). The lower relative abundance of Wolbachia in the slowly developing larvae reared on L. dorycnium is most likely due to differences in nutrients or other chemical components between the host plants. Although Wolbachia is predominantly known for affecting the sex ratio in many insects (e.g., Jiggins et al., 2010; Sakamoto et al., 2011), studies in for example bedbugs, mosquitoes and moths show that Wolbachia abundance is positively associated with advantageous effects (Body et al., 2013; Moriyama et al., 2015; Ye et al., 2013; Zhang et al., 2017), and we cannot rule out that Wolbachia has a positive effect on larval growth in L. sinapis. The role of Kocuria is unclear, it has been found in lepidopteran gut microbiomes and in various external environments, both as a commensal and as a pathogen, but information on any functional role is limited (van der Hoeven et al., 2008; Kandi et al., 2016).
The relationship between co‐opted microbiota and host plant shifts has been debated. A change in the microbiome that affects the host capability of overcoming plant defence mechanisms, nutrient uptake or secondary metabolite sequestration could mediate an establishment in novel niches (Paniagua Voirol et al., 2018). In contrast, a specialist microbiome could become an “evolutionary dead end”, where maladaptation could impair shifts to novel host plants. Further, the ability to be self‐sufficient in acquiring essential nutrients could facilitate the adoption of new niches, in particular when the host organism is not limited by the ecological boundaries of microbial symbionts (Bennett & Moran, 2015). This release from mutualistic costs could be related to the radiation in the butterfly lineage (Hammer et al., 2017). Our study confirms that the bacterial community in the butterfly gut is comparatively small and that the complexity is low, supporting a limited general importance of specific gut microbiomes in butterflies. However, the differential response of the microbiome composition between the cohorts feeding on different host plants cannot exclude a genetic, or population specific, element.
In conclusion, our results show that the evolutionary consequences of utilising different or multiple host plants for oviposition and as a food source, is multifaceted. Females from the Catalonian population displayed equal preference for both host plants, despite the reduced growth of the larvae on their natal host. However, the clear distinction between host plants by the Swedish females shows that there are chemical and/or physical differences that the butterflies can detect. This indicates that both environmental and genetic factors underpin the choice to use a specific host plant. Both L. sinapis populations showed reduced growth when reared on L. dorycnium, probably a consequence of differences in host plant composition. This was reflected in the functional pathways associated with differentially expressed genes. The response to different host plants includes both population‐specific regulation of gene expression and microbiome composition, pointing towards increased plasticity connected to local environmental conditions. Further investigation of direct functional effects of such tunings are needed to understand the microevolutionary forces that affect insect host plant specialisation or generalisation and, consequently, generation and maintenance of biodiversity.
AUTHOR CONTRIBUTIONS
N.B., K.N., and V.T. designed research and lead the study. C.W. performed all crosses, rearing of butterflies and fitness assays. K.N., and V.M. carried out the molecular work and the analysis of gene expression. A.K., and H.B. analysed the microbiome composition. R.V. provided support with sampling and input on study design and interpretation of results. K.N., V.M., and N.B. wrote the manuscript with input from all coauthors. All authors approved the final version of the manuscript before submission.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by a junior research grant (VR 2013‐4508) and a project research grant (VR 2019‐04791) from the Swedish Research Council to NB. The authors acknowledge support from the National Genomics Infrastructure in Stockholm and Uppsala funded by the Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council, for assistance with massively parallel sequencing and the bioinformatics support team (WABI). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at Uppsala University partially funded by the Swedish Research Council through grant agreement no. 2016‐07213. AK and HB acknowledge computational support from the OMICS compute cluster at the University of Lübeck. HB acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany`s Excellence Strategy – EXC 22167‐390884018. RV acknowledges support from project PID2019‐107078GB‐I00/AEI/10.13039/501100011033. We thank Luis Leal for support with the gene expression analysis and Jesper Boman and Lars Höök for helpful comments on a previous version of this manuscript.
Näsvall K, Wiklund C, Mrazek V, et al. Host plant diet affects growth and induces altered gene expression and microbiome composition in the wood white (Leptidea sinapis) butterfly. Mol Ecol.2021;30:499–516. 10.1111/mec.15745
DATA AVAILABILITY STATEMENT
RNA‐seq data are available in the European Nucleotide Archive (ENA) under Array‐Express accession number E‐MTAB‐9668. Microbiome sequence reads and metadata have been deposited in the European Nucleotide Archive (ENA) under accession number: PRJEB35538. In house developed scripts and pipelines and additional information are available at https://github.com/karinnasvall/Leptidea_hostplant_project and in File S1.
REFERENCES
- Alexa, A. , & Rahnenfuhrer, J. (2016). topGO: Enrichment analysis for gene ontology. (Version 2.28.0) [R package]. Retrieved from https://www.bioconductor.org/packages/release/bioc/html/
- Alon, M. , Elbaz, M. , Ben‐Zvi, M. M. , Feldmesser, E. , Vainstein, A. , & Morin, S. (2012). Insights into the transcriptomics of polyphagy: Bemisia tabaci adaptability to phenylpropanoids involves coordinated expression of defense and metabolic genes. Insect Biochemistry and Molecular Biology, 42, 251–263. 10.1016/j.ibmb.2011.12.007 [DOI] [PubMed] [Google Scholar]
- Andrews, S. (2016). FastQC, v. 0.11.5: a quality control tool for high throughput sequence data.
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bennett, G. M. , & Moran, N. A. (2015). Heritable symbiosis: The advantages and perils of an evolutionary rabbit hole. Proceedings of the National Academy of Sciences of the United States of America, 112, 10169–10176. 10.1073/pnas.1421388112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Body, M. , Kaiser, W. , Dubreuil, G. , Casas, J. , & Giron, D. (2013). Leaf‐miners co‐opt microorganisms to enhance their nutritional environment. Journal of Chemical Ecology, 39, 969–977. 10.1007/s10886-013-0307-y [DOI] [PubMed] [Google Scholar]
- Breuker, C. J. , & Brakefield, P. M. (2002). Female choice depends on size but not symmetry of dorsal eyespots in the butterfly Bicyclus anynana . Proceedings of the Royal Society B: Biological Sciences, 269, 1233–1239. 10.1098/rspb.2002.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahenzli, F. , & Erhardt, A. (2013). Transgenerational acclimatization in an herbivore ‐ host plant relationship. Proceedings of the Royal Society B: Biological Sciences, 280, 20122856 10.1098/rspb.2012.2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , Rosen, M. J. , Han, A. W. , Johnson, A. J. A. , & Holmes, S. P. (2016). DADA2: High‐resolution sample inference from Illumina amplicon data. Nature Methods, 13, 581–583. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnicer, J. , Stefanescu, C. , Vives‐Ingla, M. , López, C. , Cortizas, S. , Wheat, C. , Vila, R. , Llusià, J. , & Peñuelas, J. (2019). Phenotypic biomarkers of climatic impacts on declining insect populations: A key role for decadal drought, thermal buffering and amplification effects and host plant dynamics. Journal of Animal Ecology, 88, 376–391. 10.1111/1365-2656.12933 [DOI] [PubMed] [Google Scholar]
- Celorio‐Mancera, M. P. , Heckel, D. G. , & Vogel, H. (2012). Transcriptional analysis of physiological pathways in a generalist herbivore: Responses to different host plants and plant structures by the cotton bollworm, Helicoverpa armigera . Entomologia Experimentalis Et Applicata, 144, 123–133. 10.1111/j.1570-7458.2012.01249.x [DOI] [Google Scholar]
- Celorio‐Mancera, M. D. L. P. , Wheat, C. W. , Huss, M. , Vezzi, F. , Neethiraj, R. , Reimegård, J. , Nylin, S. , & Janz, N. (2016). Evolutionary history of host use, rather than plant phylogeny, determines gene expression in a generalist butterfly. BMC Evolutionary Biology, 16, 59 10.1186/s12862-016-0627-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celorio‐Mancera, M. P. , Wheat, C. W. , Vogel, H. , Söderlind, L. , Janz, N. , & Nylin, S. (2013). Mechanisms of macroevolution: Polyphagous plasticity in butterfly larvae revealed by RNA‐seq. Molecular Ecology, 22, 4884–4895. 10.1111/mec.12440 [DOI] [PubMed] [Google Scholar]
- Chaturvedi, S. , Rego, A. , Lucas, L. K. , & Gompert, Z. (2017). Sources of variation in the gut microbial community of Lycaeides melissa caterpillars. Scientific Reports, 7, 11335 10.1038/s41598-017-11781-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B. , Teh, B. S. , Sun, C. , Hu, S. , Lu, X. , Boland, W. , & Shao, Y. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis . Scientific Reports, 6, 29505 10.1038/srep29505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis, L. H. R. , Hodgson, J. G. , Hardy, P. B. , & Dapporto, L. (2012). Strategies for size and growth in butterflies (Insecta: Lepidoptera): Counterintuitive trends and unique solutions to achieving maturity. Journal of Natural History, 46, 2415–2437. 10.1080/00222933.2012.707250 [DOI] [Google Scholar]
- Dermauw, W. , Wybouw, N. , Rombauts, S. , Menten, B. , Vontas, J. , Grbic, M. , Clark, R. M. , Feyereisen, R. , & Van Leeuwen, T. (2013). A link between host plant adaptation and pesticide resistance in the polyphagous spider mite Tetranychus urticae . Proceedings of the National Academy of Sciences of the United States of America, 110, E113–E122. 10.1073/pnas.1213214110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dincă, V. , Lukhtanov, V. A. , Talavera, G. , & Vila, R. (2011). Unexpected layers of cryptic diversity in wood white Leptidea butterflies. Nature Communications, 2, e324 10.1038/ncomms1329 [DOI] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C. A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. , Chaisson, M. , & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics, 29, 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, P. R. , & Raven, P. H. (1964). Butterflies and plants ‐ a study in coevolution. Evolution, 18, 586–608. 10.1111/j.1558-5646.1964.tb01674.x [DOI] [Google Scholar]
- Friberg, M. , & Wiklund, C. (2009). Host plant preference and performance of the sibling species of butterflies Leptidea sinapis and Leptidea reali : A test of the trade‐off hypothesis for food specialisation. Oecologia, 159, 127–137. 10.1007/s00442-008- [DOI] [PubMed] [Google Scholar]
- Govind, G. , Mittapalli, O. , Griebel, T. , Allmann, S. , Bocker, S. , & Baldwin, I. T. (2010). Unbiased transcriptional comparisons of generalist and specialist herbivores feeding on progressively defenseless Nicotiana attenuata plants. PLoS One, 5, e8735 10.1371/journal.pone.0008735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, M. R. , & Sambrook, J. (2017). Isolation of high‐molecular‐weight DNA using organic solvents. Cold Spring Harbor Protocols, 4, 356–359. 10.1101/pdb.prot093450 [DOI] [PubMed] [Google Scholar]
- Hammer, T. J. , Janzen, D. H. , Hallwachs, W. , Jaffe, S. P. , & Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proceedings of the National Academy of Sciences of the United States of America, 114, 9641–9646. 10.1073/pnas.1707186114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannula, S. E. , Zhu, F. , Heinen, R. , & Bezemer, T. M. (2019). Foliar‐feeding insects acquire microbiomes from the soil rather than the host plant. Nature Communications, 10, 1254 10.1038/s41467-019-09284-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidel‐Fischer, H. M. , Freitak, D. , Janz, N. , Söderlind, L. , Vogel, H. , & Nylin, S. (2009). Phylogenetic relatedness and host plant growth form influence gene expression of the polyphagous comma butterfly (Polygonia c‐album). BMC Genomics, 10, 506 10.1186/1471-2164-10-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höök, L. , Leal, L. , Talla, V. , & Backström, N. (2019). Multilayered tuning of dosage compensation and Z‐chromosome masculinization in the wood white (Leptidea sinapis) butterfly. Genome Biology and Evolution, 11, 2633–2652. 10.1093/gbe/evz176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, H. , Jang, S. , Takeshita, K. , Ohbayashi, T. , Ohnishi, N. , Meng, X. Y. , Kikuchi, Y. (2019). Host–symbiont specificity determined by microbe–microbe competition in an insect gut. Proceedings of the National Academy of Sciences, 116(45), 22673–22682. 10.1073/pnas.1912397116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz, N. (2011). Ehrlich and Raven revisited: Mechanisms underlying codiversification of plants and enemies. Annual Review of Ecology, Evolution and Systematics, 42, 71–89. 10.1146/annurev-ecolsys-102710-145024 [DOI] [Google Scholar]
- Janz, N. , & Nylin, S. (1998). Butterflies and plants: A phylogenetic study. Evolution, 52, 486–502. 10.1111/j.1558-5646.1998.tb01648.x [DOI] [PubMed] [Google Scholar]
- Janzen, D. H. (1985). On ecological fitting. Oikos, 45, 308–310. 10.2307/3565565 [DOI] [Google Scholar]
- Jeschke, V. , Kearney, E. E. , Schramm, K. , Kunert, G. , Shekhov, A. , Gershenzon, J. , & Vassao, D. G. (2017). How glucosinolates affect generalist lepidopteran larvae: Growth, development and glucosinolate metabolism. Frontiers in Plant Science, 8, 1995 10.3389/fpls.2017.01995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiggins, F. M. , Hurst, G. D. D. , & Majerus, M. E. N. (2010). Sex‐ratio‐distorting Wolbachia causes sex‐role reversal in its butterfly host. Proceedings of the Royal Society B: Biological Sciences, 267, 69–73. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1690502/pdf/10670955.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, H. , Solensky, M. J. , Satterfield, D. A. , & Davis, A. K. (2014). Does skipping a meal matter to a butterfly's appearance? Effects of larval food stress on wing morphology and color in monarch butterflies. PLoS One, 9, e93492 10.1371/journal.pone.0093492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A. G. , Mason, C. J. , Felton, G. W. , & Hoover, K. (2019). Host plant and population source drive diversity of microbial gut communities in two polyphagous insects. Scientific Reports, 9, 2792 10.1038/s41598-019-39163-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandi, V. , Palange, P. , Vaish, R. , Bhatti, A. B. , Kale, V. , Kandi, M. R. , & Bhoomagiri, M. R. (2016). Emerging bacterial infection: Identification and clinical significance of Kocuria species. Cureus, 8, e731 10.7759/cureus.731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch, R. , Vogel, H. , Muck, A. , Reichwald, K. , Pasteels, J. M. , & Boland, W. (2011). Host plant shifts affect a major defense enzyme in Chrysomela lapponica . Proceedings of the National Academy of Sciences of the United States of America, 108, 4897–4901. 10.1073/pnas.1013846108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klindworth, A. , Pruesse, E. , Schweer, T. , Peplies, J. , Quast, C. , Horn, M. , & Glöckner, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Research, 41, e1 10.1093/nar/gks808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova, E. , Noé, L. , & Touzet, H. (2012). SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics, 28, 3211–3217. 10.1093/bioinformatics/bts611 [DOI] [PubMed] [Google Scholar]
- Krueger, F. (2017). Trim Galore, v. 0.4.4: a wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files, with some extra functionality for MspI‐digested RRBS‐type (Reduced Representation Bisufite‐Seq) libraries.
- Langmead, B. , & Salzberg, S. L. (2012). Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal, L. , Talla, V. , Källman, T. , Friberg, M. , Wiklund, C. , Dincă, V. , & Backström, N. (2018). Gene expression profiling across ontogenetic stages in the wood white (Leptidea sinapis) reveals pathways linked to butterfly diapause regulation. Molecular Ecology, 27, 935–948. 10.1111/mec.14501 [DOI] [PubMed] [Google Scholar]
- Li, Q. , Eigenbrode, S. D. , Stringam, G. R. , & Thiagarajah, M. R. (2000). Feeding and growth of Plutella xylostella and Spodoptera eridania on Brassica juncea with varying glucosinolate concentrations and myrosinase activities. Journal of Chemical Ecology, 26, 2401–2419. 10.1023/A:1005535129399 [DOI] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15, 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, C. , & Knight, R. (2005). UniFrac: A new phylogenetic method for comparing microbial communities. Applied Environmental Microbiology, 71, 8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukhtanov, V. A. , Dincă, V. , Friberg, M. , Šíchová, J. , Olofsson, M. , Vila, R. , Marec, F. , & Wiklund, C. (2018). Versatility of multivalent orientation, inverted meiosis, and rescued fitness in holocentric chromosomal hybrids. Proceedings of the National Academy of Sciences of the United States of America, 115, E9610–E9619. 10.1073/pnas.1802610115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, K. L. , & Nachman, M. W. (2017). Gene regulation and speciation. Trends in Genetics, 33, 68–80. 10.1016/j.tig.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. Embnet Journal, 17, 10–12. 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). Phyloseq: An R package for reproducible interactive analysis and graphics of Mmicrobiome census data. PLoS One, 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehl, G. A. , & Tebaldi, C. (2004). More intense, more frequent, and longer‐lasting heat waves in the 21st century. Science, 305, 994–997. 10.1126/science.1098704 [DOI] [PubMed] [Google Scholar]
- Minard, G. , Tikhonov, G. , Ovaskainen, O. , & Saastamoinen, M. (2019). The microbiome of the Melitaea cinxia butterfly shows marked variation but is only little explained by the traits of the butterfly or its host plant. Environmental Microbiology 21, 4253–4269. 10.1111/1462-2920.14786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama, M. , Nikoh, N. , Hosokawa, T. , & Fukatsu, T. (2015). Riboflavin provisioning underlies Wolbachia’s fitness contribution to its insect host. mBio, 6, e01732‐15 10.1128/mBio.01732-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali, A. , Bhargava, A. , & Wright, E. S. (2018). IDTAXA: A novel approach for accurate taxonomic classification of microbiome sequences. Microbiome, 6, 140 10.1186/s40168-018-0521-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita, S. , Nomura, M. , & Kageyama, D. (2007). Naturally occurring single and double infection with Wolbachia strains in the butterfly Eurema hecabe: Transmission efficiencies and population density dynamics of each Wolbachia strain. FEMS Microbiology Ecology, 61, 235–245. 10.1111/j.1574-6941.2007.00333.x [DOI] [PubMed] [Google Scholar]
- Nieto Feliner, G. (2014). Patterns and processes in plant phylogeography in the Mediterranean basin: A review. Perspectives in Plant Ecology, Evolution and Systematics, 16, 265–278. 10.1016/j.ppees.2014.07.002 [DOI] [Google Scholar]
- Niitepõld, K. (2019). Effects of flight and food stress on energetics, reproduction, and lifespan in the butterfly Melitaea cinxia . Oecologia, 191, 271–283. 10.1007/s00442-019-04489-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura, Y. U. , Sato, A. I. , Tsuzuki, N. , Sawada, Y. , Hirai, M. Y. , Heidel‐Fischer, H. , Reichelt, M. , Murakami, M. , & Vogel, H. (2019). Differential regulation of host plant adaptive genes in Pieris butterflies exposed to a range of glucosinolate profiles in their host plants. Scientific Reports, 9, 7256 10.1038/s41598-019-43703-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , & Wagner, H. (2019). vegan: Community Ecology Package (Version 2.5‐6). https://github.com/vegandevs/vegan [Google Scholar]
- Pagès, H. , Carlson, M. , Falcon, S. , & Li, N. (2019). AnnotationDbi: manipulation of SQLite‐based annotations in Bioconductor R package version 1.48.0. https://bioconductor.org/packages/AnnotationDbi [Google Scholar]
- Paniagua Voirol, L. R. , Frago, E. , Kaltenpoth, M. , Hilker, M. , & Fatouros, N. E. (2018). Bacterial symbionts in lepidoptera: Their diversity, transmission, and impact on the host. Frontiers in Microbiology, 9, 556 10.3389/fmicb.2018.00556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paniagua Voirol, L. R. , Weinhold, A. , Johnston, P. R. , Fatouros, N. E. , & Hilker, M. (2020). Legacy of a butterfly’s parental microbiome in offspring performance. Applied and Environmental Microbiology, 86, e00596–e520 10.1128/AEM [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks, D. H. , Chuvochina, M. , Waite, D. W. , Rinke, C. , Skarshewski, A. , Chaumeil, P. A. , & Hugenholtz, P. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nature Biotechnology, 36, 996–1004. 10.1038/nbt.4229 [DOI] [PubMed] [Google Scholar]
- Pearse, I. S. , Harris, D. J. , Karban, R. , & Sih, A. (2013). Predicting novel herbivore–plant interactions. Oikos, 122, 1554–1564. 10.1111/j.1600-0706.2013.00527.x [DOI] [Google Scholar]
- Pertea, M. , Pertea, G. M. , Antonescu, C. M. , Chang, T.‐C. , Mendell, J. T. , & Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology, 33, 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalnikar, K. , Kunte, K. , & Agashe, D. (2018). Dietary and developmental shifts in butterfly‐associated bacterial communities. Proceedings of the Royal Society B: Biological Sciences, 5, 171559 10.1098/rsos.171559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phalnikar, K. , Kunte, K. , & Agashe, D. (2019). Disrupting butterfly caterpillar microbiomes does not impact their survival and development. Proceedings of the Royal Society B: Biological Sciences, 286, 20192438 10.1098/rspb.2019.2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro de Castro, E. C. , Zagrobelny, M. , Zurano, J. P. , Zikan Cardoso, M. , Feyereisen, R. , & Bak, S. (2019). Sequestration and biosynthesis of cyanogenic glucosides in passion vine butterflies and consequences for the diversification of their host plants. Ecology and Evolution, 9, 5079–5093. 10.1002/ece3.5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto‐Tomás, A. A. , Sittenfeld, A. , Uribe‐Lorío, L. , Chavarría, F. , Mora, M. , Janzen, D. H. , Goodman, R. M. , & Simon, H. M. (2011). Comparison of midgut bacterial diversity in tropical caterpillars (Lepidoptera: Saturniidae) fed on different diets. Environmental Entomology, 40, 1111–1122. 10.1603/EN11083 [DOI] [PubMed] [Google Scholar]
- Puinean, A. M. , Foster, S. P. , Oliphant, L. , Denholm, I. , Field, L. M. , Millar, N. S. , Williamson, M. S. , & Bass, C. (2010). Amplification of a cytochrome P450 gene is associated with resistance to neonicotinoid insecticides in the aphid Myzus persicae . PLOS Genetics, 6, e1000999 10.1371/journal.pgen.1000999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri, B. , Hall, A. , Baxter, H. , Harborne, J. B. , & Moss, G. P. (1998). Phytochemical Dictionary: A Handbook of Bioactive Compounds from Plants, 2nd ed E‐book online: CRC Press. [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing, Vienna, Austria: . R Foundation for Statistical Computing; https://www.R-project.org/ [Google Scholar]
- Renwick, J. A. A. , & Chew, F. S. (1994). Oviposition behavior in Lepidoptera. Annual Review of Entomology, 39, 377–400. 10.1146/annurev.en.39.010194.002113 [DOI] [Google Scholar]
- Romero, I. G. , Ruvinsky, I. , & Gilad, Y. (2012). Comparative studies of gene expression and the evolution of gene regulation. Nature Reviews Genetics, 13, 505–516. 10.1038/nrg3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto, Y. , Hirai, N. , Tanikawa, T. , Yago, M. , & Ishii, M. (2011). Infection by two strains of Wolbachia and sex ratio distortion in a population of the endangered butterfly Zizina emelina (Lepidoptera: Lycaenidae) in northern Osaka prefecture, central Japan. Annals of the Entomological Society of America, 104, 483–487. 10.1603/an09168 [DOI] [Google Scholar]
- Salgado, A. L. , & Saastamoinen, M. (2019). Developmental stage‐dependent response and preference for host plant quality in an insect herbivore. Animal Behaviour, 150, 27–38. 10.1016/j.anbehav.2019.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado, A. L. , Suchan, T. , Pellissier, L. , Rasmann, S. , Ducrest, A. L. , & Alvarez, N. (2016). Differential phenotypic and genetic expression of defence compounds in a plant‐herbivore interaction along elevation. Proceedings of the Royal Society B: Biological Sciences, 3, 160226 10.1098/rsos.160226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep, K. P. (2011). Phangorn: Phylogenetic analysis in R. Bioinformatics, 27, 592–593. 10.1093/bioinformatics/btq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder, R. , & Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics, 27, 863–864. 10.1093/bioinformatics/btr026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonhoven, L. M. , Van Loon, J. J. A. , & Dicke, M. (2005). Insect‐plant biology. Oxford University Press. [Google Scholar]
- Schweizer, F. , Heidel‐Fischer, H. , Vogel, H. , & Reymond, P. (2017). Arabidopsis glucosinolates trigger a contrasting transcriptomic response in a generalist and a specialist herbivore. Insect Biochemistry and Molecular Biology, 85, 21–31. 10.1016/j.ibmb.2017.04.004 [DOI] [PubMed] [Google Scholar]
- Shannon, C. E. (1948). A mathematical theory of communication. Bell System Technical Journal, 27, 379–423. 10.1002/j.1538-7305.1948.tb01338.x [DOI] [Google Scholar]
- Singer, M. C. , Ng, D. , & Thomas, C. D. (1988). Heritability of oviposition preference and its relationship to offspring performance within a single insect population. Evolution, 42, 977–985. 10.1111/j.1558-5646.1988.tb02516.x [DOI] [PubMed] [Google Scholar]
- Singer, M. C. , & Parmesan, C. (2018). Lethal trap created by adaptive evolutionary response to an exotic resource. Nature, 557, 238–241. 10.1038/s41586-018-0074-6 [DOI] [PubMed] [Google Scholar]
- Slater, J. M. , Gilbert, L. , Johnson, D. , & Karley, A. J. (2019). Limited effects of the maternal rearing environment on the behaviour and fitness of an insect herbivore and its natural enemy. PLoS One, 14, e0209965 10.1371/journal.pone.0209965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeds, L. , & Künstner, A. (2011). ConDeTri ‐ a content dependent read trimmer for Illumina data. PLoS One, 6, e26314 10.1371/journal.pone.0026314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev, V. I. , Ilinsky, Y. , & Kosterin, O. E. (2015). Genetic integrity of four species of Leptidea (Pieridae, Lepidoptera) as sampled in sympatry inWest Siberia. Comparative Cytogenetics, 9, 299–324. 10.3897/CompCytogen.v9i3.4636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudacher, H. , Kaltenpoth, M. , Breeuwer, J. A. J. , Menken, S. B. J. , Heckel, D. G. , & Groot, A. T. (2016). Variability of bacterial communities in the moth Heliothis virescens indicates transient association with the host. PLoS One, 11, e0154514 10.1371/journal.pone.0154514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward, R. A. , Fisher, L. M. , & Boggs, C. L. (2019). Pre‐ and post‐ingestive defenses affect larval feeding on a lethal invasive host plant. Entomologia Experimentalis Et Applicata, 167, 292–305. 10.1111/eea.12773 [DOI] [Google Scholar]
- Szenteczki, M. A. , Pitteloud, C. , Casacci, L. P. , Kesnerova, L. , Whitaker, M. R. L. , Engel, P. , & Alvarez, N. (2019). Bacterial communities within Phengaris (Maculinea) alcon caterpillars are shifted following transition from solitary living to social parasitism of Myrmica ant colonies. Ecology and Evolution, 9, 4452–4464. 10.1002/ece3.5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talla, V. , Johansson, A. , Dincă, V. , Vila, R. , Friberg, M. , Wiklund, C. , & Backström, N. (2019). Lack of gene flow: Narrow and dispersed differentiation islands in a triplet of Leptidea butterfly species. Molecular Ecology, 28, 3756–3770. [DOI] [PubMed] [Google Scholar]
- Talla, V. , Suh, A. , Kalsoom, F. , Dincă, V. , Vila, R. , Friberg, M. , & Backström, N. (2017). Rapid increase in genome size as a consequence of transposable element hyperactivity in wood‐white (Leptidea) butterflies. Genome Biology and Evolution, 9, 2491–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, W.‐H. , Acevedo, T. , Harris, E. V. , Alcaide, T. Y. , Walters, J. R. , Hunter, M. D. , & de Roode, J. C. (2019). Transcriptomics of monarch butterflies (Danaus plexippus) reveals that toxic host plants alter expression of detoxification genes and down‐regulate a small number of immune genes. Molecular Ecology, 28, 4845–4863. 10.1111/mec.15219 [DOI] [PubMed] [Google Scholar]
- van der Hoeven, R. , Betrabet, G. , & Forst, S. (2008). Characterization of the gut bacterial community in Manduca sexta and effect of antibiotics on bacterial diversity and nematode reproduction. FEMS Microbiology Letters, 286, 249–256. 10.1111/j.1574-6968.2008.01277.x [DOI] [PubMed] [Google Scholar]
- Verschut, T. A. , Blazyte‐Cereskiene, L. , Apsegaite, V. , Mozuraitis, R. , & Hambäck, P. A. (2017). Natal origin affects host preference and larval performance relationships in a tritrophic system. Ecology and Evolution, 7, 2079–2090. 10.1002/ece3.2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila, R. , Stefanescu, C. , & Sesma, J. M. (2018). Guia de les papallones diürnes de Catalunya. Lynx Edicions. [Google Scholar]
- Weingartner, E. , Wahlberg, N. , & Nylin, S. (2006). Dynamics of host plant use and species diversity in Polygonia butterflies (Nymphalidae). Journal of Evolutionary Biology, 19, 483–491. 10.1111/j.1420-9101.2005.01009.x [DOI] [PubMed] [Google Scholar]
- Weiss, S. , Xu, Z. Z. , Peddada, S. , Amir, A. , Bittinger, K. , Gonzalez, A. , & Knight, R. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome, 5, 27 10.1186/s40168-017-0237-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker, M. R. L. , Salzman, S. , Sanders, J. , Kaltenpoth, M. , & Pierce, N. E. (2016). Microbial communities of lycaenid butterflies do not correlate with larval diet. Frontiers in Microbiology, 7, 1920 10.3389/fmicb.2016.01920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiklund, C. (1975). The evolutionary relationship between adult oviposition preferences and larval host plant range in Papilio machaon L. Oecologia, 18, 185–197. [DOI] [PubMed] [Google Scholar]
- Wingett Steven W., Andrews Simon (2018). FastQ Screen: A tool for multi‐genome mapping and quality control. F1000Research, 7, 1338 10.12688/f1000research.15931.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woestmann, L. , & Saastamoinen, M. (2016). The importance of trans‐generational effects in Lepidoptera. Current Zoology, 62, 489–499. 10.1093/cz/zow029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, E. S. (2016). Using DECIPHER v2.0 to analyze big biological sequence data in R. The R Journal, 8, 352. [Google Scholar]
- Xia, X. , Gurr, G. M. , Vasseur, L. , Zheng, D. , Zhong, H. , Qin, B. , Lin, J. , Wang, Y. , Song, F. Q. , Li, Y. , Lin, H. , & You, M. (2017). Metagenomic sequencing of diamondback moth gut microbiome unveils key holobiont adaptations for herbivory. Frontiers in Microbiology, 8, 663 10.3389/fmicb.2017.00663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, X. , Zheng, D. , Zhong, H. , Qin, B. , Gurr, G. M. , Vasseur, L. , Lin, H. , Bai, J. , He, W. , & You, M. (2013). DNA sequencing reveals the midgut microbiota of diamondback moth, Plutella xylostella (L.) and a possible relationship with insecticide resistance. PLoS One, 8, e68852 10.1371/journal.pone.0068852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, Y. H. , Woolfit, M. , Rancès, E. , O'Neill, S. L. , & McGraw, E. A. (2013). Wolbachia‐associated bacterial protection in the mosquito Aedes aegypti . PLOS Neglected Tropical Diseases, 7(8), e2362 10.1371/journal.pntd.0002362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Q. Y. , Fang, S. M. , Zhang, Z. , & Jiggins, C. D. (2016). The transcriptome response of Heliconius melpomene larvae to a novel host plant. Molecular Ecology, 25, 4850–4865. 10.1111/mec.13826 [DOI] [PubMed] [Google Scholar]
- Zagrobelny, M. , Jensen, M. K. , Vogel, H. , Feyereisen, R. , & Bak, S. (2018). Evolution of the biosynthetic pathway for cyanogenic glucosides in Lepidoptera. Journal of Molecular Evolution, 86, 379–394. 10.1007/s00239-018-9854-8 [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Guigue, A. , Dubreuil, G. , Kisiala, A. , Andreas, P. , Emery, R. J. , & Giron, D. (2017). Dynamics and origin of cytokinins involved in plant manipulation by a leaf‐mining insect. Insect Science, 24, 1065–1078. 10.1111/1744-7917.12500 [DOI] [PubMed] [Google Scholar]
- Zhong, H. , Li, F. , Chen, J. , Zhang, J. , & Li, F. (2017). Comparative transcriptome analysis reveals host‐associated differentiation in Chilo suppressalis (Lepidoptera: Crambidae). Scientific Reports, 7, 13778 10.1038/s41598-017-14137-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, F. , Zhou, Y. K. , Ji, Z. L. , & Chen, X. R. (2018). The plant ribosome‐inactivating proteins play important roles in defense against pathogens and insect pest attacks. Frontiers in Plant Science, 9, 146 10.3389/fpls.2018.00146 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
RNA‐seq data are available in the European Nucleotide Archive (ENA) under Array‐Express accession number E‐MTAB‐9668. Microbiome sequence reads and metadata have been deposited in the European Nucleotide Archive (ENA) under accession number: PRJEB35538. In house developed scripts and pipelines and additional information are available at https://github.com/karinnasvall/Leptidea_hostplant_project and in File S1.