

Abstract
Cell‐cycle interference by small molecules has widely been used to study fundamental biological mechanisms and to treat a great variety of diseases, most notably cancer. However, at present only limited possibilities exist for spatio‐temporal control of the cell cycle. Here we report on a photocaging strategy to reversibly arrest the cell cycle at metaphase or induce apoptosis using blue‐light irradiation. The versatile proteasome inhibitor MG132 is photocaged directly at the reactive aldehyde function effectively masking its biological activity. Upon irradiation reversible cell‐cycle arrest in the metaphase is demonstrated to take place in vivo. Similarly, apoptosis can efficiently be induced by irradiation of human cancer cells. With the developed photopharmacological approach spatio‐temporal control of the cell cycle is thus enabled with very high modulation, as caged MG132 shows no effect on proliferation in the dark. In addition, full compatibility of photo‐controlled uncaging with dynamic microscopy techniques in vivo is demonstrated. This visible‐light responsive tool should be of great value for biological as well as medicinal approaches in need of high‐precision targeting of the proteasome and thereby the cell cycle and apoptosis.
Keywords: apoptosis, cell cycle, chemical biology, MG132, photopharmacology, proteasome
Blue‐light control of the proteasome is achieved using a new photocaging approach. Photolabile protection of the versatile proteasome inhibitor MG132 at the reactive aldehyde function abolished proteasome binding. After irradiation MG132 activity is restored resulting in cell‐cycle arrest at metaphase or apoptosis. This visible‐light‐responsive tool opens up new avenues for spatiotemporal control of the proteasome in basic and medicinal research.
Introduction
The development of versatile molecular tools enabling elucidation of fundamental bio‐chemical processes, detailed mechanistic understanding of diseases, sensitive and selective diagnostics, or efficient treatment of medical conditions is a central goal of chemical biology today. Towards this goal, significant progress has been made with the introduction of photo‐pharmacological concepts introducing spatio‐temporal control to the bioactivity of molecular agents. [1] Such an approach is not only valuable in the context of disease treatment to reduce side‐effects and dosage but also shows high potential in enhancing diagnostics and in fundamental research in biology, medicine, or pharmaceutical sciences. A great variety of bioactive components are currently altered into light‐responsive versions either by introducing photoswitch motives[ 1d , 2 ] or by photolabile protecting groups[ 1f , 3 ] that cage the crucial bioactive function. In general, photoswitchable variants offer intrinsically greater spatial resolution due to the possibility of turning off their activity at places where it is not wanted. Photocaging approaches however, possess the advantage of very high ON/OFF modulation of activity as residual activity of the caged forms is typically extremely small. In biological applications it is further highly desirable to use nondamaging visible or even near infrared light for photocontrol. Consequentially fundamental research developing such photoswitches and cages is currently a very active area in itself (for recent developments see selected ref. [4] for photoswitches and ref. [5] for cages).
Using photoresponsive tools for the photoinduction of cell death in a selective fashion is of particular interest not least for drug development. Different strategies are followed that include photoswitching and photocaging,[ 1b , 2a , 2b , 3d , 6 ] or optogenetics, [7] as well as classic photodynamic therapy approaches (for selected references see [8] ). Photoinduction of apoptosis has been achieved either by controlling cellular uptake of proapoptotic peptides[ 8a , 9 ] or caspase 3, [10] optogenetically modified caspases,[ 7 , 11 ] or photoswitchable BH3 peptides. [12] Light control of the cell cycle has been accomplished by the development of photoswitchable microtubule inhibitors [13] or by optogenetic methods. [14] Despite these efforts, examples for precise control of the cell cycle are very scarce at the moment stressing the need for effective photopharmacological tools in this area. Light‐controlled targeting of specifically the proteasome—a key player in cell cycle regulation—is currently only possible with photoswitchable Bortezomib© variants [15] enabling up to five‐fold modulation of activity. [15a]
MG132 is a well‐established proteasome inhibitor that is used as a versatile biochemical tool to study various cellular processes including the cell‐cycle, apoptosis, [16] proteostasis, or virus life cycle [17] (Figure 1). It consists of a simple tri‐peptide structure that mainly targets the chymotrypsin‐type catalytic centers at the beta‐subunits of the proteasome—a multi‐component enzyme, which is responsible for protein degradation. At higher concentrations MG132 also inhibits the protease calpain. [18] MG132 forms a covalent hemi‐acetal adduct with the catalytically active threonine in the active site of the proteasome efficiently inhibiting its proteolytic activity. [19] Proteasome inhibition severely alters protein turnover of the cell. Of special importance in this regard is inhibition of the proteasomes role in cell‐cycle control leading to cell‐cycle arrest in metaphase. [20] Prolonged inhibition of the proteasome ultimately induces apoptosis, preferentially so in cancer cells as compared to healthy ones. [21] Therefore, MG132 also represents an interesting candidate for cancer research and antidotes although its untamed reactivity and rather fast metabolism currently hampers development into a medical drug. [22] Taken together MG132 can be regarded as a multi‐faceted, versatile, and easily applicable biochemical agent, however control of MG132 activity in time or place is currently not possible. We envisioned that especially a light‐responsive MG132 version would thus represent a highly valuable tool for a plethora of different applications. In general, such tool would enable studying fundamental and highly dynamic biological processes directly linked to proteasome function with precise spatial and temporal resolution. For example, dynamic modulation of the proteasome‐dependent protein turnover would become possible during cell cycle progression, cellular signaling, or immune responses. The dynamic nature and resolution advantage would be even more critical in the context of tissues or organisms to instill local and timed effects—for example, in applications for local cancer treatment, tissue targeting, or apoptosis studies in developing animals, to name a few exciting possibilities. Furthermore, caging could prevent fast metabolism of MG132 during its distribution in an organism and thus would potentially alleviate some of the challenges encountered in its drug development.
Figure 1.
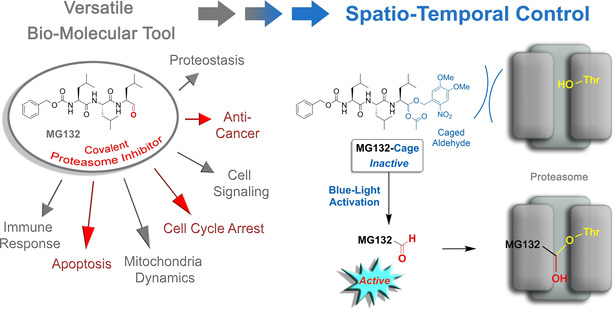
Transformation of the versatile covalent proteasome inhibitor MG132 into a blue‐light‐controlled biomolecular tool MG132‐Cage (1) enabling spatio‐temporal control of various fundamental biological phenomena. The reactive aldehyde anchorpoint of MG132 is caged by a photolabile protecting group in the form of an acetal rendering the resulting compound MG132‐Cage (1) fully inactive. Upon blue‐light irradiation the aldehyde function is restored and covalent proteasome binding and inhibition is triggered.
In this work we present a photocaging strategy allowing us to convert MG132 into a noneffective agent in its protected MG132‐Cage (1) form but trigger full functionality upon irradiation with blue light (Figure 1). For this purpose the covalent binding moiety, that is the aldehyde function of MG132, is protected as mixed acetal of the photolabile 4,5‐dimethoxy‐2‐nitrobenzyl (DMNB) cage, [23] which fully blocks binding to the proteasome. This caging approach therefore is highly effective as it targets the main reactivity of MG132 directly, while also removing potential toxicity of the aldehyde, that is, side effects and unselective reactions with nucleophiles within a cell, before uncaging. Blue‐light irradiation at 405 nm enables full recovery of the MG132 structure at a given time and place and thus allows to instill spatio‐temporal control of the inhibitor function in living cells. The high potential of this new light‐responsive proteasome inhibitor for biological as well as pharmacological research is showcased by light‐induced cell‐cycle arrest as well as apoptosis induction in living human tissue culture cells.
Results and Discussion
Synthesis and Photochemical Assessment
To be able to effectively mask the biological activity of MG132, the main functionality responsible for covalent binding to the proteasome, that is, the aldehyde function, was targeted for protection with a photolabile group. To regain the aldehyde upon irradiation, the oxidation state of its carbon atom should not be altered during uncaging and thus protection as acetal was deemed the most straight‐forward approach. To the best of our knowledge such an aldehyde caging/uncaging method—albeit recently introduced for simple organic aldehydes [24] —has not been used for bioactive compounds or in a biological setting so far. The DMNB photolabile group was chosen for its well established photocaging properties and particularly because of its blue‐light responsiveness, which is favorable for biological applications. [23] MG132‐Cage (1) was synthesized in a convergent and iterative peptide‐coupling sequence as shown in Scheme 1 (for full details see Supporting Information, Scheme S1–S4, and Figure S1–S8). Carboxybenzyl (Z)‐protected leucine 2 was first coupled with leucine methyl ester and after subsequent ester hydrolysis dipeptide 3 was obtained in 57 % yield. The DMNB photolabile protecting group was introduced to another tert‐butyloxycarbonyl (Boc)‐protected leucine (4) via an ester linkage to give the Boc‐protected amine 5 in 69 % yield. After Boc‐deprotection of 5 using trifluoroacetic acid (TFA) the corresponding free amine 7 was joined with 3 to the tripeptide 6 using standard peptide coupling conditions in 77 % yield over the two steps. A final one‐pot ester reduction and mixed acetal formation sequence gave the desired MG132‐Cage (1) in acceptable 17 %. Additionally, the DMNB cage was introduced to propionic acid to obtain the control compound Propionic Acid‐Cage (8, for details see the Supporting Information).
Scheme 1.
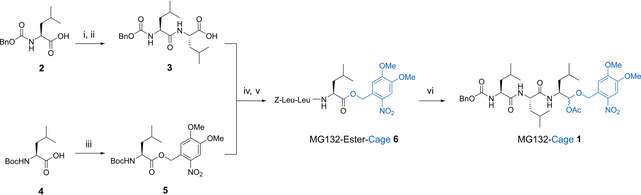
Synthesis of photocaged MG132‐Cage (1). The synthetic precursor MG132‐Ester‐Cage (6) is used as control compound in the biological experiments. The caging group DMNB is introduced via an ester link early in the synthesis. After successive peptide couplings a final reductive step yields MG132‐Cage with the aldehyde protected as acetal. Conditions: i) Leucine methyl ester x HCl, PyBOP, DIPEA; ii) NaOH 57 % yield over two steps; iii) DMNB‐alcohol, DCC, DMAP, 69 % yield; iv) 5, TFA; v) 3, DIPEA, DCC, HOBt, 77 % yield over two steps; vi) DIBAL‐H, acetic anhydride, 17 % yield.
Photodeprotection efficiency of 1 was tested using UV/Vis and especially 1H NMR spectroscopy as quantitative tool (Figure 2 a and Figure S13 and S15). Irradiation with various wavelengths showed 405 nm to be the longest wavelength at which full photodeprotection proceeded in reasonable time scales, that is, within several minutes at high NMR concentrations (approximately 2–5 mm in DMSO solution). Concomitant with the decreasing signals of 1, the known signals of MG132 with the indicative aldehyde signal at 9.5 ppm increased. The responsiveness of the MG132‐Cage towards 405 nm is attributed to its absorption profile, which tails out but does not yet reach zero at this wavelength. Irradiation at UV/Vis concentrations (typically 10−5 m in DMSO solution) with 405 nm light shows the typical spectral changes expected for deprotection of a DMNB group (Figure 2 b). Likewise, photodeprotection of the two control compounds MG132‐Ester‐Cage (6) and Propionic Acid‐Cage (8) in DMSO solution leads to the analogous absorption spectral changes (see Figure S11 and S12, respectively) proving effective uncaging also in these cases. Quantum yield measurements were conducted for the MG132‐Cage in DMSO solution due to solubility issues in buffer media. Consistent with a previous literature report for the same DMNB photocaging group and photorelease of alcohols, [25] we measured a quantum yield of 1.0 % for 405 nm irradiation (for details see Supporting Information and Figure S14–S16). This quantum yield is high enough to warrant effective photodeprotection within minutes under the biological experimental conditions. In a typical biological setup (see below) 1 mL of 5 or 10 μm solutions of caged compound are used (5 or 10 nmol), which are irradiated with a 105 mW 405 nm LED or a 375 mW 405 nm LED positioned in ca. 2 cm distance from the sample solutions.
Figure 2.
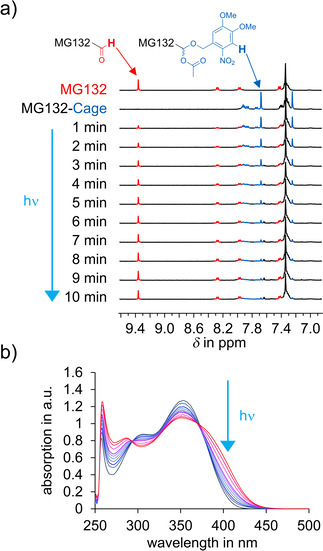
Blue‐light irradiation of MG132‐Cage (1) leads to release of MG132 and restoration of its covalently binding aldehyde function. a) 1H NMR monitoring of the uncaging process shows almost complete photodeprotection, while at the same time the signal of the aldehyde proton of MG132 increases. The spectrum of pure MG132 is shown at the top, and the spectrum of pure MG132‐Cage directly below. Spectra recorded in 1 min intervals during irradiation of a [D6]DMSO solution of MG132‐Cage with a 405 nm LED are shown starting with the third spectrum from the top. b) Changes of the UV/Vis absorption recorded in intervals during 405 nm irradiation of MG132‐Cage in DMSO (blue starting point to red final spectrum).
Light‐Induced Metaphase Arrest in HeLa Cells
After demonstrating a proper photochemical response of the MG132‐Cage, we moved to in vivo studies to showcase its functionality in biological context. For this purpose, light‐induced cell‐cycle control was evidenced using a quantitative imaging assay in HeLa cells (Figure 3 a,b and Figure S17). The cell cycle consists of interphase, during which the cell content including the DNA is duplicated, and the mitotic phase during which the content of the mother cell is distributed to the two daughter cells. The mitotic phase starts with the prometaphase, followed by the metaphase, anaphase, and telophase. During prometaphase the DNA condenses and at metaphase the chromosomes align at the metaphase plate. After chromosome alignment on the metaphase plate the cell proceeds into anaphase and distributes the chromosomes to the two daughter cells. Finally, the mother cell is split into two daughter cells during telophase. During cell division the cell needs to ensure that it only proceeds into anaphase after all chromosomes are correctly attached to the mitotic spindle and aligned at the metaphase plate. Chromosome alignment is monitored by the spindle assembly checkpoint (SAC). [26] In case not all chromosomes are aligned properly at the metaphase plate the SAC keeps the cell in metaphase. To allow metaphase to anaphase transition the proteasome needs to be active to degrade key cell‐cycle regulators for example cyclin B [20] and securin. [27] Our first aim was to arrest cells with light in metaphase by using MG132‐Cage. HeLa cells were synchronized to enrich for mitotic cells. Cells were then incubated with 5 or 10 μm DMSO solutions of MG132‐Cage and either exposed to low power 405 nm LED light for 5 or 10 min or maintained in the dark. After light exposure, cells were incubated for 2 h after which they were fixed and stained for α‐tubulin, filamentous actin (F‐actin), and DNA to determine the cell‐cycle stage by confocal microscopy (Figure 3 b). Over >95 % of the mitotic cells were arrested in metaphase when cells were exposed to 10 min light irradiation (Figure 3 c,d and Figure S17). Similar metaphase arrest was observed when cells were treated directly with MG132 in 5 μm and 10 μm concentrations with and without 10 min LED light irradiation (Figure 3 b and Figure S17b). As a control MG132‐Cage‐treated cells were maintained in the dark for the same time period. We observed that 58 % of those cells were in ana‐/telophase, which was indistinguishable from untreated cells or cells treated with DMSO (Figure 3 d and Figure S17b). Since mitotic cells spend the same amount of time in metaphase as in ana‐ and telophase combined (Figure 3 b, top) a 1:1 ratio of metaphase to ana‐/telophase is expected for non‐disturbing conditions. The control experiments thus confirm that uncaging of MG132‐Cage in the absence of light is neglectable in vivo. To exclude that 405 nm light treatment used for uncaging results in an unspecific metaphase arrest, we also exposed DMSO‐treated and untreated cells to irradiation. We found that DMSO‐treated and untreated cells that were exposed to 405 nm light for 10 min behaved indistinguishably from control cells kept in the dark. In both cases the cells proceeded readily to ana‐/telophase (Figure 3 d and Figure S17b). This demonstrated that 405 nm irradiation does not induce cell‐cycle arrest at metaphase. To test whether the light‐induced arrest in metaphase was reversible, an inhibitor‐washout protocol was applied 2 h after light exposure of MG132‐Cage treated cells (Figure 3 b). After another 2 h waiting time the typical 51 % of the mitotic cells were in ana‐/telophase evidencing light‐induced metaphase arrest can be fully reversed and cells proceed normally into anaphase.
Figure 3.
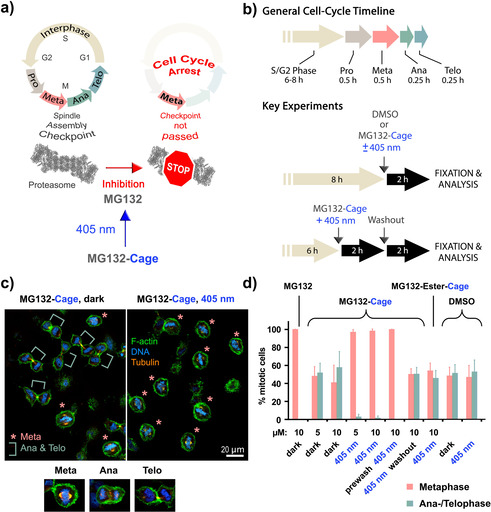
Cell‐cycle control with blue light. a) General mechanism of cell‐cycle arrest induced by photoactivated MG132‐Cage. Blue‐light irradiation releases MG132, which inhibits the proteasome leading to arrest in metaphase. For depiction of the human proteasome crystallographic data (PDB code 5GJR) from Ref. [28] were used. b) Key experimental setups to quantify blue‐light‐induced metaphase arrest. c) Confocal microscopy images of HeLa cells incubated with MG132‐Cage and either maintained in the dark (left) or exposed for 10 min to 405 nm light (right). Cells were stained for DNA (blue), F‐actin (green), and α‐tubulin (orange). Metaphase cells are marked with an asterisk and ana‐/telophase cells with a green bar. Representative confocal images of a metaphase, anaphase, and telophase cell stained for DNA, F‐actin, and α‐tubulin are shown at the bottom. d) Mean percentages of mitotic cells in metaphase and ana‐/telophase for the indicated conditions. The mean of 3–7 independent experiments is shown and error bars represent standard deviations. For the different conditions a minimum of 110 and maximum of 476 cells were analyzed individually.
During photodeprotection MG132‐Cage is split into MG132 and a nitrosobenzaldehyde. To exclude that the nitrosobenzaldehyde induces metaphase arrest we used the synthetic precursor MG132‐Ester‐Cage (compound 6) as well as propionic acid, which was caged with the same photolabile DMNB cage. Upon photoirradiation MG132‐Ester‐Cage as well as Propionic‐Acid‐Cage release the corresponding carboxylic acids (the simple tripeptide Z‐Leu‐Leu‐Leu and propionic acid), which are nontoxic to cells and do not inhibit the proteasome. Both caged control compounds also release the very same nitrosobenzaldehyde. Cells that were incubated with 10 μm MG132‐Ester‐Cage or Propionic‐Acid‐Cage and irradiated for 10 min did not arrest in metaphase (Figure 3 d and Figure S17b). Therefore, light exposed MG132‐Cage‐treated cells arrest in metaphase due to the release of MG132 and not due to the presence of released nitrosobenzaldehyde. Finally, we addressed whether uncaging of MG132‐Cage changes its diffusion across the plasma membrane. To test if uncaging of MG132‐Cage promotes its cellular uptake we incubated cells for 1 h with MG132‐Cage in the dark and subsequently exchanged the MG132‐Cage containing medium with fresh medium without MG132‐Cage just before irradiation with 405 nm light. We found 100 % of the cells were arrested in metaphase after another 2 h incubation in the dark (Figure 3 d, prewash condition). This result was similar to the one from the analogous experiment lacking a prewash step. It shows that MG132‐Cage is taken up by the cells and is present in the cytoplasm prior photoactivation. Light‐induced metaphase arrest is thus due to the release of the active compound MG132 and not due to allowing cellular uptake of released MG132.
Light‐Induced Apoptosis in HeLa Cells
Proteasome inhibitors induce apoptosis in many cell types including cancer cells. Inhibition of the proteasome results in an increase of proapoptotic factors such as p53 and caspases. [29] For this reason proteasome inhibitors are promising candidates in cancer therapies, bortezomib© for example is used to treat multiple myeloma. [30] Our next goal was to induce apoptosis with blue light in HeLa cells using MG132‐Cage (Figure 4 a). HeLa cells were incubated with DMSO or MG132‐Cage and either exposed to 405 nm light for 10 min or kept in the dark. To monitor cell viability over time the number of healthy cells was counted using fixed samples 0 h, 12 h, 18 h, and 24 h after light exposure (Figure 4 b). In the negative control experiments the number of cells doubled within 24 h (200 %). However, for cells incubated with MG132‐Cage and exposed to light for 10 min the cell number was reduced to 41 % after 18 h and 2 % after 24 h (Figure 4 c,d). This decrease in cell number over time was comparable to cells treated with 10 μm MG132 (Figure S18a) suggesting that MG132‐Cage is very effectively uncaged in vivo. Cells incubated with MG132‐Cage but kept in the dark continued to proliferate similar to DMSO‐treated cells (Figure 4 d). This suggests that MG132‐Cage is not uncaged in the absence of light even over prolonged incubation time of several hours. After irradiating control cells with 405 nm light for 10 min we found that light exposure had no effect on cell number suggesting that the amount of light used to uncage MG132‐Cage is not toxic. We also performed an inhibitor‐washout experiment to determine whether the effects of 2 h exposure to uncaged MG132‐Cage can be reversed. Cells treated with MG132‐Cage, DMSO, or MG132 were exposed to 405 nm light for 10 min and incubated for 2 h followed by inhibitor washout. When we counted cell numbers 24 h later we found that MG132‐Cage, DMSO, or MG132 treated cells that had undergone washout displayed similar cell numbers like DMSO‐treated control cells without applied washout (Figure S18b). To exclude that the co‐released nitrosobenzaldehyde reduces cell viability we incubated cells with 10 μm MG132‐Ester‐Cage as well as with 10 μm Propionic Acid‐Cage and exposed them to 405 nm light for 10 min. After 24 h the healthy cell number in these two control experiments were comparable to the ones observed for DMSO‐treated cells indicating that the co‐released nitrosobenzaldehyde does not negatively affect cellular growth (Figure S18b). To corroborate these findings we quantified cell viability using a resazurin‐based assay. In living cells resazurin is reduced to strongly fluorescent resorufin. [31] When we plotted fluorescence intensity for MG132‐Cage (with and without 405 nm light), MG132‐Ester‐Cage (with 405 nm light), Propionic Acid‐Cage (with 405 nm light), MG132 (dark) treated cells against DMSO‐treated control cells similar results were obtained as for the single‐cell fixed analysis (Figure S19).
Figure 4.
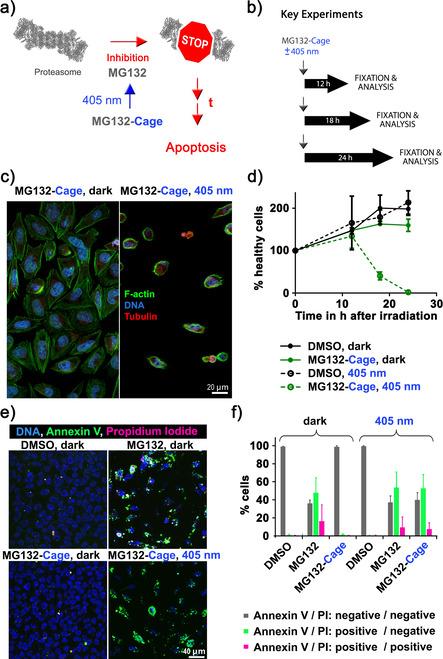
Induction of apoptosis in HeLa cells with blue light. a) Simplified mechanism of apoptosis induction by photoactivated MG132‐Cage. Blue‐light irradiation triggers MG132 release, which inhibits the proteasome leading to apoptosis of the respective cell after prolonged (t) exposure. b) Experimental setups to quantify blue‐light‐induced cell death over time. c) Confocal microscopy images of HeLa cells incubated with 10 μm MG132‐Cage and either maintained in the dark for 24 h (left) or exposed to 405 nm light for 10 min and then cultured for 24 h (right, the few remaining cells display hallmarks of apoptosis such as cell shrinkage and DNA condensation). d) Mean percentages of healthy cells at different time points under the indicated conditions. Photodeprotection of 10 μm MG132‐Cage results in complete cell death 24 hours after irradiation. Mean of two independent experiments is shown and error bars represent standard deviation. e) Maximum z‐projections of five images of HeLa cells stained with Hoechst dye to label all nuclei (DNA, blue), annexin V (green), and PI (red). HeLa cells were treated with 0.1 % DMSO or 10 μm MG132 and maintained in the dark, or treated with 10 μm MG132‐Cage and either exposed to 405 nm light for 10 min or kept in the dark. The 20 h time point was chosen to monitor annexin and PI labeling while cells are dying. f) Percentages of cells that were labeled by annexin V and/or PI for the indicated conditions. The mean of three independent experiments is shown and error bars represent standard deviation. For the different conditions a minimum of 123 and maximum of 409 cells were analyzed individually.
To evaluate whether uncaging of MG132‐Cage kills cells with the same efficiency as MG132 we performed dose‐response experiments. Cells were incubated with different concentrations of MG132 (dark) or MG132‐Cage (irradiated with 405 nm light). After 24 h cells were fixed and stained and the number of healthy cells was counted and plotted relative to DMSO‐treated cells (Figure S20). For both conditions the cell number started to increase at ≈1.5 μm and reached control levels at ≈0.1 μm concentrations. Our dose‐response experiments are in very good agreement with a previous literature report of the dose response of MG132 induced cell death. [32] These experiments confirm that blue‐light irradiation of MG132‐Cage kills cells in a dose‐dependent manner.
To determine whether cells are dying indeed via the apoptosis pathway we also analyzed the cleavage of poly (ADP‐ribose) polymerase‐1 (PARP). In healthy cells PARP mediates DNA damage repair and with the induction of apoptosis PARP is cleaved by caspases. PARP is a 114 kDa protein and upon caspase activation it is cleaved into a 89 and a 24 kDa fragment. [33] PARP cleavage was analyzed by immunoblots and cells that were incubated with MG132‐Cage and exposed to 405 nm light for 10 min showed a reduction in the 114 kDa band and an increase in the 89 kDa fragment, which was similar to cells treated with MG132 (Figure S21). DMSO‐treated cells and MG132‐Cage treated cells that were maintained in the dark showed a prominent band at 114 kDa and were indistinguishable from each other.
During apoptosis phosphatidylserine translocates from the inner to the outer leaflet of the plasma membrane to generate an “eat me” signal for macrophages. [34] To further confirm that MG132‐Cage treated and irradiated cells die by apoptosis, cells were stained with annexin V, which strongly binds to phosphatidylserine. [35] The cells were additionally incubated with propidium iodide (PI), which is a membrane impermeable dye that only labels dead cells. More than 99 % of the DMSO‐treated cells that were either incubated in the dark or irradiated with 405 nm light for 10 min were negative for annexin V and PI (Figure 4 e,f). Treatment of cells with MG132‐Cage and subsequent exposure to 405 nm light for 10 min resulted in 53 % annexin V positive and PI negative cells indicating that these cells were still alive but initiated apoptosis. In addition, 7 % of the cells were positive for both annexin V and PI showing that these cells were dead. Importantly cells incubated with MG132‐Cage but maintained in the dark were indistinguishable from DMSO‐treated control cells. Together with the dose‐response results these experiments confirm that release of MG132 upon blue‐light irradiation induces apoptosis in a dose‐dependent manner.
To dynamically follow the light‐induced cell‐cycle arrest as well as execution of apoptosis upon uncaging of MG132‐Cage, a live‐cell imaging approach was chosen. Transmission and confocal images of mitotic HeLa cells expressing fluorescently labeled α‐tubulin (mKate‐α‐tubulin) were acquired. For transmission images a 470 nm cut‐off filter was placed in the light path to exclude the blue parts of the imaging light and thus to prevent uncaging by image acquisition. Cells were treated with 10 μm MG132‐Cage and either exposed to 405 nm LED light for 5 min or maintained in the dark. After light exposure imaging was resumed and continued for 20 h. As expected from our analysis of fixed cells, we found that 405 nm light exposure resulted in efficient cell cycle arrest at metaphase for several hours (Figure 5 a, see Movie 1 in the Supporting Information). Following 405 nm light exposed cells for longer time revealed that after ≈12 h cells started to shrink and display strong plasma membrane blebbing, which are both hallmarks of apoptosis (Figure 5 b, Movie 2 in the Supporting Information). 20 h post light exposure the majority of cells were undergoing apoptosis. In contrast, cells treated with MG132‐Cage but not exposed to 405 nm LED light continued to proliferate and did not show any signs of apoptosis. To dynamically follow reversibility of blue‐light‐induced metaphase arrest an additional washout experiment was performed using live‐cell imaging. MG132‐Cage treated cells were exposed to 405 nm LED light, which resulted in metaphase arrest (Figure 5 c, Figure S22, and Movie 3 in the Supporting Information). After 2 h cells were washed and imaging was continued. We observed that metaphase cells entered anaphase after the washout and completed cell division. Furthermore, long‐term imaging revealed that the cells continued to proliferate and did not display signs of apoptosis. These results demonstrate that uncaging of MG132‐Cage can easily be combined with live‐cell imaging techniques allowing the investigation of dynamic proteasome‐dependent processes in living cells or organisms at high spatial and temporal resolution.
Figure 5.
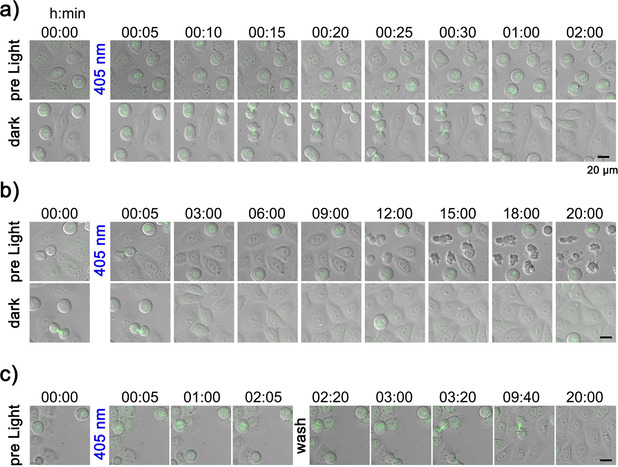
Live‐cell imaging of blue‐light‐induced metaphase arrest and apoptosis in HeLa cells. Cells treated with 10 μm MG132‐Cage were either exposed to 405 nm LED light for 5 min (top row) or maintained in the dark (bottom row). Merged transmission and confocal mKate‐α‐tubulin (maximum z‐projection shown in green) images for indicated time points are shown. Scale bars represent 20 μm. a) Prometaphase cells exposed to 405 nm light arrested in metaphase (top row). Control cells in the dark entered anaphase and completed cell division (bottom row). Selected time frames are taken from Movie 1 (see Supporting Information). b) First apoptotic cells were observed ≈12 h after 405 nm light exposure (top row). Cells maintained in the dark continued proliferation without undergoing apoptosis (bottom row). Selected time frames are taken from Movie 2 (see Supporting Information). c) Prometaphase cells exposed to 405 nm light arrested in metaphase for 2 h. After washing the cells proliferation continues establishing reversibility of the arrest. After 20 h no signs of apoptosis are seen. Selected time frames are taken from Movie 3 (see Supporting Information).
Conclusion
In summary, we present an efficient approach for establishing spatio‐temporal control over the cell‐cycle stage as well as apoptosis of human cancer cells by blue‐light irradiation. To this end a photocaging strategy is used, which effectively masks the reactive and covalently binding aldehyde function of the versatile proteasome inhibitor MG132. In the presence of MG132‐Cage without light, cell viability and proliferation remained unperturbed in vivo. However, after 5–10 min blue‐light irradiation prometaphase cells treated with MG132‐Cage are arrested in metaphase for several hours. This metaphase arrest can be alleviated by applying a washing protocol. If cells are irradiated in the presence of MG132‐Cage and subsequently cultured for prolonged time in the dark almost quantitative apoptosis is induced. Both metaphase arrest as well as apoptosis were followed dynamically with a fluorescent live‐cell imaging approach proving full compatibility of photo‐controlled uncaging with dynamic microscopy techniques in vivo. With this approach a highly promising photopharmacological tool for full spatio‐temporal control of cell cycle, apoptosis, and cancer treatment via simple non‐toxic blue‐light irradiation is presented. We believe that MG132‐Cage will be of great interest for a broad variety of biologically and medically oriented research. Our future efforts are directed at establishing full spatial control over proteasome activity in tissues and organisms as well as studying highly dynamic processes related to the biochemistry of the proteasome.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary
Acknowledgements
E. Zanin thanks the DFG for an Emmy Noether fellowship (ZA619/3). S. Mangal was a member of the International Max Planck Research School for Molecular Life Sciences. H. Dube thanks the Deutsche Forschungsgemeinschaft (DFG) for an Emmy Noether fellowship (DU 1414/1‐1). We further thank the Deutsche Forschungsgemeinschaft (SFB 749, A12) and the Cluster of Excellence “Center for Integrated Protein Science Munich” (CIPSM) for financial support. Microscopy was performed at the center for advanced light microscopy (CALM) at the LMU. We also thank Stefan Wiedemann for help with the synthesis as well as Martin Parniske and Arne Weiberg for access to the plate reader and Constance Tisserant for technical advice. Open access funding enabled and organized by Projekt DEAL.
E. Uhl, F. Wolff, S. Mangal, H. Dube, E. Zanin, Angew. Chem. Int. Ed. 2021, 60, 1187.
In memory of François Diederich
References
- 1.
- 1a. Velema W. A., Szymanski W., Feringa B. L., J. Am. Chem. Soc. 2014, 136, 2178–2191; [DOI] [PubMed] [Google Scholar]
- 1b. Hüll K., Morstein J., Trauner D., Chem. Rev. 2018, 118, 10710–10747; [DOI] [PubMed] [Google Scholar]
- 1c. Fehrentz T., Schonberger M., Trauner D., Angew. Chem. Int. Ed. 2011, 50, 12156–12182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12362–12390; [Google Scholar]
- 1d. Broichhagen J., Frank J. A., Trauner D., Acc. Chem. Res. 2015, 48, 1947–1960; [DOI] [PubMed] [Google Scholar]
- 1e. Morstein J., Trauner D., Curr. Opin. Chem. Biol. 2019, 50, 145–151; [DOI] [PubMed] [Google Scholar]
- 1f. Brieke C., Rohrbach F., Gottschalk A., Mayer G., Heckel A., Angew. Chem. Int. Ed. 2012, 51, 8446–8476; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8572–8604. [Google Scholar]
- 2.
- 2a. Szymański W., Beierle J. M., Kistemaker H. A., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178; [DOI] [PubMed] [Google Scholar]
- 2b. Albert L., Vazquez O., Chem. Commun. 2019, 55, 10192–10213; [DOI] [PubMed] [Google Scholar]
- 2c. Peddie V., Abell A. D., J. Photochem. Photobiol. C 2019, 40, 1–20; [Google Scholar]
- 2d. Kitzig S., Thilemann M., Cordes T., Rück-Braun K., ChemPhysChem 2016, 17, 1252–1263; [DOI] [PubMed] [Google Scholar]
- 2e. Mart R. J., Allemann R. K., Chem. Commun. 2016, 52, 12262–12277. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hansen J. B., Velema W. A., Lerch M. M., Szymanski W., Feringa B. L., Chem. Soc. Rev. 2015, 44, 3358–3377; [DOI] [PubMed] [Google Scholar]
- 3b. Bardhan A., Deiters A., Curr. Opin. Struct. Biol. 2019, 57, 164–175; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Deiters A., ChemBioChem 2010, 11, 47–53; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Ankenbruck N., Courtney T., Naro Y., Deiters A., Angew. Chem. Int. Ed. 2018, 57, 2768–2798; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2816–2848. [Google Scholar]
- 4.
- 4a. Lentes P., Stadler E., Rohricht F., Brahms A., Grobner J., Sonnichsen F. D., Gescheidt G., Herges R., J. Am. Chem. Soc. 2019, 141, 13592–13600; [DOI] [PubMed] [Google Scholar]
- 4b. Klaue K., Garmshausen Y., Hecht S., Angew. Chem. Int. Ed. 2018, 57, 1414–1417; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1429–1432; [Google Scholar]
- 4c. Moreno J., Gerecke M., Grubert L., Kovalenko S. A., Hecht S., Angew. Chem. Int. Ed. 2016, 55, 1544–1547; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1569–1573; [Google Scholar]
- 4d. Dong M., Babalhavaeji A., Collins C. V., Jarrah K., Sadovski O., Dai Q., Woolley G. A., J. Am. Chem. Soc. 2017, 139, 13483–13486; [DOI] [PubMed] [Google Scholar]
- 4e. Dong M., Babalhavaeji A., Samanta S., Beharry A. A., Woolley G. A., Acc. Chem. Res. 2015, 48, 2662–2670; [DOI] [PubMed] [Google Scholar]
- 4f. Bléger D., Schwarz J., Brouwer A. M., Hecht S., J. Am. Chem. Soc. 2012, 134, 20597–20600; [DOI] [PubMed] [Google Scholar]
- 4g. Samanta S., Beharry A. A., Sadovski O., McCormick T. M., Babalhavaeji A., Tropepe V., Woolley G. A., J. Am. Chem. Soc. 2013, 135, 9777–9784; [DOI] [PubMed] [Google Scholar]
- 4h. Lameijer L. N., Budzak S., Simeth N. A., Hansen M. J., Feringa B. L., Jacquemin D., Szymanski W., Angew. Chem. Int. Ed. 2020, 59, 21663–21670; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 21847–21854; [Google Scholar]
- 4i. Konrad D. B., Savasci G., Allmendinger L., Trauner D., Ochsenfeld C., Ali A. M., J. Am. Chem. Soc. 2020, 142, 6538–6547; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4j. Kink F., Collado M. P., Wiedbrauk S., Mayer P., Dube H., Chem. Eur. J. 2017, 23, 6237–6243; [DOI] [PubMed] [Google Scholar]
- 4k. Petermayer C., Thumser S., Kink F., Mayer P., Dube H., J. Am. Chem. Soc. 2017, 139, 15060–15067. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Fichte M. A., Weyel X. M., Junek S., Schafer F., Herbivo C., Goeldner M., Specht A., Wachtveitl J., Heckel A., Angew. Chem. Int. Ed. 2016, 55, 8948–8952; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9094–9098; [Google Scholar]
- 5b. Becker Y., Unger E., Fichte M. A. H., Gacek D. A., Dreuw A., Wachtveitl J., Walla P. J., Heckel A., Chem. Sci. 2018, 9, 2797–2802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Peterson J. A., Wijesooriya C., Gehrmann E. J., Mahoney K. M., Goswami P. P., Albright T. R., Syed A., Dutton A. S., Smith E. A., Winter A. H., J. Am. Chem. Soc. 2018, 140, 7343–7346; [DOI] [PubMed] [Google Scholar]
- 5d. Shrestha P., Dissanayake K. C., Gehrmann E. J., Wijesooriya C. S., Mukhopadhyay A., Smith E. A., Winter A. H., J. Am. Chem. Soc. 2020, 142, 15505–15512; [DOI] [PubMed] [Google Scholar]
- 5e. Olson J. P., Banghart M. R., Sabatini B. L., Ellis-Davies G. C., J. Am. Chem. Soc. 2013, 135, 15948–15954; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Bojtár M., Németh K., Domahidy F., Knorr G., Verkman A., Kállay M., Kele P., J. Am. Chem. Soc. 2020, 142, 15164–15171; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5g. Walton D. P., Dougherty D. A., J. Am. Chem. Soc. 2017, 139, 4655–4658; [DOI] [PubMed] [Google Scholar]
- 5h. Shell T. A., Shell J. R., Rodgers Z. L., Lawrence D. S., Angew. Chem. Int. Ed. 2014, 53, 875–878; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 894–897. [Google Scholar]
- 6. Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Angew. Chem. Int. Ed. 2016, 55, 10978–10999; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11140–11163. [Google Scholar]
- 7. Nihongaki Y., Suzuki H., Kawano F., Sato M., ACS Chem. Biol. 2014, 9, 617–621. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Engesaeter B. O., Bonsted A., Lillehammer T., Engebraaten O., Berg K., Maelandsmo G. M., Cancer Biol. Ther. 2006, 5, 1511–1520; [DOI] [PubMed] [Google Scholar]
- 8b. Dolmans D. E. J. G. J., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380–387; [DOI] [PubMed] [Google Scholar]
- 8c. Zhang Z., Dai R., BioMetals 2017, 30, 37–42; [DOI] [PubMed] [Google Scholar]
- 8d. Oleinick N. L., Morris R. L., Belichenko I., Photochem. Photobiol. Sci. 2002, 1, 1–21. [DOI] [PubMed] [Google Scholar]
- 9. Shamay Y., Adar L., Ashkenasy G., David A., Biomaterials 2011, 32, 1377–1386. [DOI] [PubMed] [Google Scholar]
- 10. Hentzen N. B., Mogaki R., Otake S., Okuro K., Aida T., J. Am. Chem. Soc. 2020, 142, 8080–8084. [DOI] [PubMed] [Google Scholar]
- 11. Mills E., Chen X., Pham E., Wong S., Truong K., ACS Synth. Biol. 2012, 1, 75–82. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Wysoczanski P., Mart R. J., Loveridge E. J., Williams C., Whittaker S. B., Crump M. P., Allemann R. K., J. Am. Chem. Soc. 2012, 134, 7644–7647; [DOI] [PubMed] [Google Scholar]
- 12b. Mart R. J., Errington R. J., Watkins C. L., Chappell S. C., Wiltshire M., Jones A. T., Smith P. J., Allemann R. K., Mol. BioSyst. 2013, 9, 2597–2603. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Borowiak M., Nahaboo W., Reynders M., Nekolla K., Jalinot P., Hasserodt J., Rehberg M., Delattre M., Zahler S., Vollmar A., Trauner D., Thorn-Seshold O., Cell 2015, 162, 403–411; [DOI] [PubMed] [Google Scholar]
- 13b. Sailer A., Ermer F., Kraus Y., Lutter F. H., Donau C., Bremerich M., Ahlfeld J., Thorn-Seshold O., ChemBioChem 2019, 20, 1305–1314. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Goto Y., Aoki K., bioRxiv 2020, 10.1101/2020.06.22.166264; [DOI] [Google Scholar]
- 14b. Zhang H., Aonbangkhen C., Tarasovetc E. V., Ballister E. R., Chenoweth D. M., Lampson M. A., Nat. Chem. Biol. 2017, 13, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Blanco B., Palasis K. A., Adwal A., Callen D. F., Abell A. D., Bioorg. Med. Chem. 2017, 25, 5050–5054; [DOI] [PubMed] [Google Scholar]
- 15b. Hansen M. J., Velema W. A., de Bruin G., Overkleeft H. S., Szymanski W., Feringa B. L., ChemBioChem 2014, 15, 2053–2057. [DOI] [PubMed] [Google Scholar]
- 16. Guo N., Peng Z., Asia Pac. J. Clin. Oncol. 2013, 9, 6–11. [DOI] [PubMed] [Google Scholar]
- 17. Tang Q., Wu P., Chen H., Li G., Life Sci. 2018, 207, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tsubuki S., Saito Y., Tomioka M., Ito H., Kawashima S., J. Biochem. 1996, 119, 572–576. [DOI] [PubMed] [Google Scholar]
- 19. Stein M. L., Cui H., Beck P., Dubiella C., Voss C., Krüger A., Schmidt B., Groll M., Angew. Chem. Int. Ed. 2014, 53, 1679–1683; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1705–1709. [Google Scholar]
- 20. Clute P., Pines J., Nat. Cell Biol. 1999, 1, 82–87. [DOI] [PubMed] [Google Scholar]
- 21. Dou Q. P., Zonder J. A., Curr. Cancer Drug Targets 2014, 14, 517–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Du Z. X., Yan Y., Zhang H. Y., Liu B. Q., Gao Y. Y., Niu X. F., Guan Y., Meng X., Wang H. Q., Endocr.-Relat. Cancer 2010, 17, 553–560; [DOI] [PubMed] [Google Scholar]
- 22b. Lee C. M., Kumar V., Riley R. I., Morgan E. T., Drug Metab. Dispos. 2010, 38, 2166–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Görner H., Photochem. Photobiol. Sci. 2005, 4, 822–828. [DOI] [PubMed] [Google Scholar]
- 24. Lage Robles J., Bochet C. G., Org. Lett. 2005, 7, 3545–3547. [DOI] [PubMed] [Google Scholar]
- 25. Krafft G. A., Sutton W. R., Cummings R. T., J. Am. Chem. Soc. 1988, 110, 301–303. [Google Scholar]
- 26. Hayward D., Alfonso-Perez T., Gruneberg U., FEBS Lett. 2019, 593, 2889–2907. [DOI] [PubMed] [Google Scholar]
- 27. Ciosk R., Zachariae W., Michaelis C., Shevchenko A., Mann M., Nasmyth K., Cell 1998, 93, 1067–1076. [DOI] [PubMed] [Google Scholar]
- 28. Huang X., Luan B., Wu J., Shi Y., Nat. Struct. Mol. Biol. 2016, 23, 778–785. [DOI] [PubMed] [Google Scholar]
- 29. Vlahakis S. R., Badley A. D., Curr. Opin. Clin. Nutr. 2006, 9, 42–47. [DOI] [PubMed] [Google Scholar]
- 30. Moreau P., Richardson P. G., Cavo M., Orlowski R. Z., San Miguel J. F., Palumbo A., Harousseau J. L., Blood 2012, 120, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O′Brien J., Wilson I., Orton T., Pognan F., Eur. J. Biochem. 2000, 267, 5421–5426. [DOI] [PubMed] [Google Scholar]
- 32. Han Y. H., Moon H. J., You B. R., Park W. H., Oncol. Rep. 2009, 22, 215–221. [PubMed] [Google Scholar]
- 33. Chaitanya G. V., Steven A. J., Babu P. P., Cell Commun. Signaling 2010, 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagata S., Annu. Rev. Immunol. 2018, 36, 489–517. [DOI] [PubMed] [Google Scholar]
- 35. van Engeland M., Nieland L. J. W., Ramaekers F. C. S., Schutte B., Reutelingsperger C. P. M., Cytometry 1998, 31, 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Supplementary