

Abstract
Alkynes are highly attractive motifs in organic synthesis due to their presence in natural products and bioactive molecules as well as their versatility in a plethora of subsequent transformations. A common procedure to insert alkynes into (hetero)arenes, such as the thiophenes studied herein, consists of a halogenation followed by a Sonogashira cross‐coupling. The regioselectivity of this approach depends entirely on the halogenation step. Similarly, direct alkynylations of thiophenes have been described that follow the same regioselectivity patterns. Herein we report the development of a palladium catalyzed C−H activation/alkynylation of thiophenes. The method is applicable to a broad range of thiophene substrates. For 3‐substituted substrates where controlling the regioselectivity between the C2 and C5 position is particularly challenging, two sets of reaction conditions enable a regiodivergent reaction, giving access to each regioisomer selectively. Both protocols use the thiophene as limiting reagent and show a broad scope, rendering our method suitable for late‐stage modification.
Keywords: alkynylation, C−H activation, regiodivergent, regioselectivity, thiophenes
A method for the direct C−H alkynylation of thiophenes has been developed. Complementary sets of reaction conditions enable a regiodivergence for 3‐substituted substrates, giving selective access to either the C2 or the C5 alkynylation products. The method works for various substitution patterns on the thiophene, features a broad scope, and uses the thiophene as the limiting reagent, rendering it suitable for late‐stage modification.
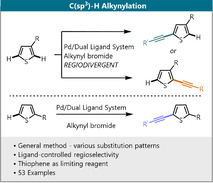
The direct functionalization of thiophenes, to access their valuable derivatives, is an important target in organic chemistry due to the broad use of thiophenes in material sciences and medicinal chemistry. [1] Alkynes are one of the key motifs for organic chemists and the most commonly used method for insertion of this motif into (hetero)arenes is the Sonogashira cross‐coupling, [2] where the regioselectivity of the product formation depends on (pseudo)halogenation step. Considering the importance of alkynylated thiophenes in pharmaceuticals and organic materials, [3] an alternative direct access to these products is highly desirable since this would not only make the method more atom‐ and step‐economic, but could also deliver complementary products in cases where there is a challenge in regioselectivity. In 2010, Waser and co‐workers reported a gold‐ and Brønsted‐acid‐catalyzed C5‐alkynylation of 2‐substituted thiophenes. [4] In this study, they also reported one example of an electron‐rich 3‐substituted thiophene, which was selectively alkynylated in the C2 position (Scheme 1). Furthermore, Su and co‐workers reported a Pd‐catalyzed oxidative cross‐coupling of 2‐substituted thiophenes and phenyl acetylenes, the latter being used as limiting reagent. [5] However, to the best of our knowledge no general method which enables the alkynylation of thiophenes irrespective of the substitution pattern has been reported to date. Especially for 3‐substituted thiophenes, the control over the regioselectivity between C5 and C2 position remains unaddressed.
Scheme 1.
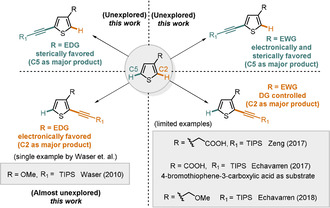
Explored and unexplored areas of regioselective C−H alkynylation of 3‐substituted thiophenes.
For such 3‐substituted thiophenes, the regioselectivity of the C−H activation is mainly governed by the steric and electronic properties of the substituents as well as the sensitivity of the catalyst system towards these effects.[ 6 , 7 , 8 ] For substrates bearing electron‐donating substituents (EDG) in the 3 position, the C2 product is electronically favored and hence an electrophilic reagent or catalyst is expected to induce the functionalization in this position. In contrast, a catalyst that is more sensitive to steric hindrance is expected to lead to C5‐substitution through a pathway in which the steric clash between the catalyst and the substituent in the 3 position is avoided. For electron‐withdrawing groups (EWG) in the 3 position another effect comes into play. Since many of these substituents are also Lewis‐basic, they can act as directing groups (DGs) thereby favoring the functionalization in the neighboring C2 position (and in principle also the often less reactive C4 position) through chelation control. In 2017, Zeng and co‐workers reported a directed Ir‐catalyzed ortho‐alkynylation of arenes, which included one example of such a carboxylate‐directed, C2‐selective alkynylation of a 3‐subsbtituted thiophene (Scheme 1). [9]
Also in 2017, Echavarren and co‐workers reported a Ru‐catalyzed ortho‐alkynylation of arenes. As part of this study they demonstrated the carboxylic acid‐directed alkynylation of 3‐substituted thiophenes to get di‐alkynylation at the C2 and C4 position. [10] One example of a C2‐selective mono‐alkynylation employing 4‐bromothiophene‐3‐carboxylic acid as substrate was also reported (Scheme 1). In 2018, the same group reported a Rh‐catalyzed ortho‐alkynylation of arenes, which included one example of a 3‐substituted thiophene with benzyl ether as weak DG to deliver the C2 product (Scheme 1). [11] As highlighted above, the direct C−H alkynylation of thiophenes remains a highly challenging yet attractive goal. For C2‐substituted substrates substantial limitations still exist with respect to the scope of substrates that can be addressed. The more challenging 3‐substituted substrates have to date only been addressed in isolated cases leading to C2‐selective functionalization.
Based on these observations and our recent experience in controlling the regioselectivity of C−H activations on heteroarenes, [6m] we hypothesized that through the design of suitable catalysts a regiodivergent reactivity enabling both a C2‐ and a C5‐selective alkynylation of thiophenes could be developed. [12] Additionally, we expected that one of these catalyst systems would likely display a broad scope with respect to thiophenes with simpler substitution patterns as well, thereby allowing us to develop a general method for the alkynylation of thiophenes.
We thus began our studies with 3‐hexyl thiophene 1 a as model compound. We expected that by applying our dual ligand‐enabled catalyst design, [13] which is known to deliver products under steric control, we would be able to induce an alkynylation in the C5 position. Although our initial experiments delivered poor regioselectivities, we observed a highly promising ligand control of the regioselectivity when we increased the steric demand of the substituent on the amino‐acid‐derived ligand (L1–L4, Scheme 2).
Scheme 2.
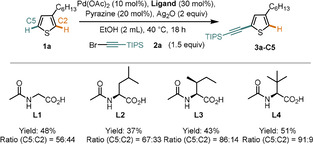
Effect of ligands on yield and regioselectivity. All reactions were conducted on a 0.1 mmol scale. Yields and ratios were determined by GC‐FID analysis using 1,3,5‐trimethoxybenzene as an internal standard.
Using L4, we proceeded to optimize the reaction conditions and identified the protocol shown in Entry 1 of Table 1. Under these conditions the target compound 3 a‐C5 was obtained in good yield (71 %) and regioselectivity (94:6). Importantly, our control experiments revealed that the reaction is indeed dual ligand‐enabled, since in the absence of either ligand, substantially worse reaction outcomes were observed (Entries 2–4).
Table 1.
Control Experiments (C5 selectivity).

|
Conditions[a] |
Conversion [%][b] |
Yield [%][b] |
Ratio (C5/C2)[b] |
|---|---|---|---|
|
As above |
97 |
71 |
94:6 |
|
No Pd, L4, pyrazine |
5 |
0 |
– |
|
No L4 |
26 |
19 |
47:53 |
|
No pyrazine |
22 |
9 |
67:33 |
[a] All reactions were conducted on a 0.1 mmol scale. [b] Conversions, yields, and ratios were determined by GC‐FID analysis using 1,3,5‐trimethoxybenzene as an internal standard.
With the optimized reaction conditions in hand, we proceeded to explore the scope of the reaction (Scheme 3).
Scheme 3.
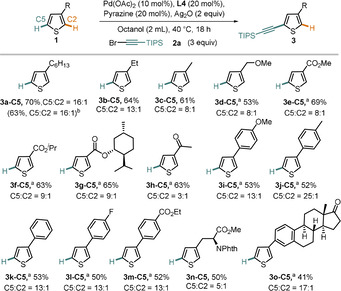
Scope of the C5‐selective alkynylation of thiophenes. All reactions were conducted on a 0.2 mmol scale. [a] For these reactions tAmyl‐OH was used as solvent. [b] Reaction conducted on a 5 mmol scale.
The sterically controlled nature of the catalyst system is visible if one compares the selectivity of the entries 3 a‐C5–3 c‐C5, where a decrease in the steric bulk of the alkyl substituent somewhat decreases the C5 selectivity, albeit still remaining good even for the small methyl group. Electron‐withdrawing ester and ketone substituents are also tolerated and give products with good C5 selectivity under both electronic and steric control (Scheme 3, 3 e‐C5–3 h‐C5). Our protocol works well for a series of 3‐aryl substituted thiophenes (Scheme 3, 3 i‐C5–3 m‐C5), which as well shows that a number of common functional groups are well tolerated under our reaction conditions, such as ethers, halides, and esters. Finally, our method can be applied to alkynylate a thiophene‐containing unnatural amino acid derivative (3 n‐C5) and an estrone derivative (3 o‐C5) with good C5 selectivity.
After realizing a broad scope in Scheme 3, we proceeded to attempt the development of a complementary, C2‐selective catalyst system. Although in our previous studies, dual ligand‐based catalysts always showed a preference for steric over electronic control, we reasoned that by increasing the electrophilicity of the catalyst, an electronically controlled reaction might be enabled. In our previous optimization studies, we noted that N‐acetyl‐β‐alanine (L5) and a COC6H11‐substituent on nitrogen of glycine as ligand (L6) both led to increased amounts of the C2‐alkynylated product 3 a‐C2 (Scheme 4).
Scheme 4.
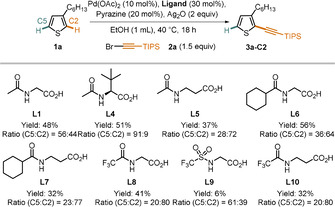
Ligand development for C2 selectivity. All reactions were conducted on a 0.1 mmol scale. Yields and ratios were determined using GC‐FID analysis using 1,3,5‐trimethoxybenzene as an internal standard.
Unfortunately, combining these effects in L7 did not deliver satisfactory results. We thus proceeded to test stronger electron‐withdrawing substituents on the ligand. We installed a COCF3 substituent (L8) and a SO2CF3 substituent (L9) on the nitrogen of glycine and, gratifyingly, L8 was able to deliver a 4:1 selectivity in favor of the C2 product. Lastly, we tested ligand L10, which constitutes a permutation of the positive effects seen in L5 and L8. However, this ligand gave a reduced yield and no improvement in the regioselectivity compared to L8, which was therefore chosen for further optimization studies. We proceeded to screen various other parameters which led us to identify reaction conditions under which a satisfactory yield and good regioselectivity are obtained (for details, see the Supporting Information).
These conditions were then used to explore the scope of the C2‐selective alkynylation (Scheme 5). The trend in C2 selectivity observed from hexyl to methyl substituent (Scheme 5, 3 a‐C2–3 c‐C2) shows that this catalyst system, while predominantly controlled by electronics, is sensitive to sterics as well, since the C2 selectivity is best when steric hindrance at this position is low. Halide substituents (3 p‐C2 and 3 q‐C2) as well as a strong electron‐donating methoxy (3 r‐C2) group give the C2 product exclusively. We also tested various aryl‐substituted thiophenes (Scheme 5, entries 3 i‐C2–3 m‐C2). In contrast to the C5‐selective protocol and as expected for an electronically controlled reaction, we observed a much stronger dependence of the reaction outcome on the electronic nature of the substituent. An electron‐donating methoxy substituent (3 i‐C2) on the phenyl ring gives substantially higher C2 selectivity than an electron‐poor ester substituent (3 m‐C2). Irrespective of these effects on the regioselectivity, the functional group compatibility was found to be good for the C2 selective protocol as well. Finally, this catalyst system can also be used to deliver alkynylation product from an amino‐acid‐derived thiophene derivative (3 n‐C2) and an estrone derivative (3 o‐C2) with good C2 selectivity.
Scheme 5.
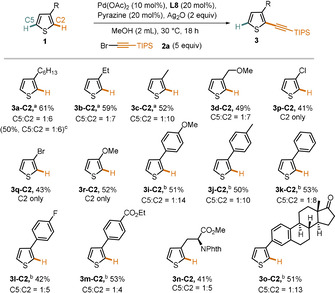
Reaction scope (C2 selectivity). All reactions were conducted on a 0.2 mmol scale. [a] Reaction conducted at 35 °C with 2 equivalents of reagent. [b] Reaction conducted at 50 °C. [c] Reaction conducted on a 1 mmol scale.
Having established the thiophene scope for our regiodivergent protocols, we were interested to evaluate the use of other bromoalkynes with both protocols (Scheme 6). [14] We found that a range of bromoalkynes could be used in both directions with satisfactory yields and selectivities. A TBS group was well tolerated (4‐C5 and 4‐C2), as well as various alkyl‐substituted bromoalkynes as reagent (5‐C5–12‐C5 and 5‐C2–12‐C2). Bromoalkynes bearing α‐non‐quaternary substituents, such as a cyclohexyl or n‐hexyl groups, as well as phenylacetylene‐derived bromoalkynes, gave no product formation under our reaction conditions. However, the regiodivergent reactivity developed herein can nevertheless be harnessed for such target compounds. To this end the TIPS‐group can be removed, followed by a Sonogashira cross‐coupling. We demonstrated the feasibility of this approach using our standard substrate (for details, see the Supporting Information).
Scheme 6.
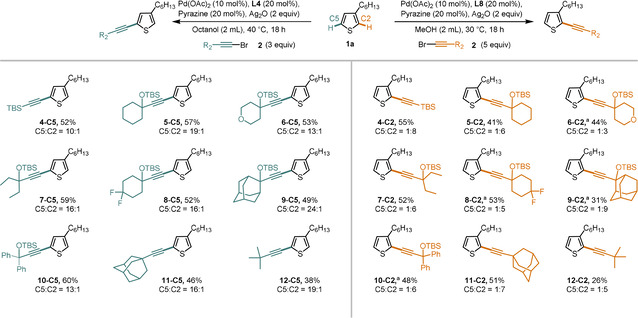
Alkyne scope. All reactions were conducted on a 0.2 mmol scale. [a] Reaction conducted at 35 °C with 2 equivalents of alkyne reagent.
As mentioned earlier, we expected that once the challenging regioselective alkynylation of 3‐substituted thiophenes would be addressed, the respective catalyst systems would likely also be able to functionalize regioselectivity‐wise less challenging thiophenes and thereby provide a general method for the alkynylation of all types of thiophenes.
We tried the conditions developed for the C5‐ and C2‐selective alkynylations on 2‐ethyl thiophene (13 a, Scheme 7) as model substrate and found that the latter delivered satisfactory results, giving Product 14 a in 68 % yield. An electron‐donating methoxy substituent (14 b) is well tolerated. Likewise, an aryl substituent in the 2 position led to a good reaction outcome (14 c). Finally, halide substituents (14 d,e) and 2,3‐disubstitution (14 f) were found to be well tolerated.
Scheme 7.
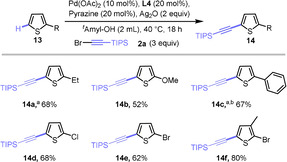
Scope of C2‐substituted thiophenes. All reactions were conducted on a 0.2 mmol scale. [a] Pd(OAc)2 (10 mol %), L8 (20 mol %), pyrazine (20 mol %), Ag2O (2 equiv), MeOH (2 mL), 2 a (5 equiv), 30 °C, 18 h. [b] Reaction conducted at 50 °C instead of 30 °C.
In summary, we have developed a pair of general catalyst systems for the Pd‐catalyzed non‐directed C−H activation/alkynylation of thiophenes that are suitable for all kinds of substitution patterns on the thiophene. For regioselectivity‐wise challenging 3‐substituted substrates the protocols are complementary, giving a regiodivergent access to the C5‐ and C2‐alkynylated products, respectively. Overall, a broad scope with respect to the thiophene and alkyne reaction partner can be addressed, including structurally complex examples. In all cases the thiophene substrate is used as the limiting reagent, which renders this protocol attractive in the context of late‐stage modification.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge financial support from the DFG (Emmy Noether Programme), Max Planck Society (Otto Hahn Award to M.v.G.), and WWU Münster. We thank the members of our NMR and MS departments for their excellent service, as well as M. Farizyan for providing starting materials and S. Bognar for confirming the reproducibility of the reaction. Also, thanks to A. Uttry, K. K. Ghosh and H. Chen for valuable discussions and proof reading of the manuscript. Furthermore, we are indebted to Prof. F. Glorius for his generous support. Open access funding enabled and organized by Projekt DEAL.
A. Mondal, M. van Gemmeren, Angew. Chem. Int. Ed. 2021, 60, 742.
Dedicated to Professor Bart Jan Ravoo on the occasion of his 50th birthday
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.12921362.v1).
References
- 1. The chemistry of Heterocyclic Compounds, Wiley-Interscience, New York, 1994. [Google Scholar]
- 2.
- 2a. Doucet H., Hierso J.-C., Angew. Chem. Int. Ed. 2007, 46, 834; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 850; [Google Scholar]
- 2b. Chinchilla R., Nájera C., Chem. Soc. Rev. 2011, 40, 5084. [DOI] [PubMed] [Google Scholar]
- 3. Diederich F., Stang P. J., Tykwinski R. R., Acetylene chemistry. Chemistry, biology, and material science, Wiley-VCH, Weinheim, 2005. [Google Scholar]
- 4. Brand J. P., Waser J., Angew. Chem. Int. Ed. 2010, 49, 7304; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7462. [Google Scholar]
- 5.
- 5a. Jie X., Shang Y., Hu P., Su W., Angew. Chem. Int. Ed. 2013, 52, 3630; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3718; [Google Scholar]
- 5b.additionally, a single example of a C5-selective alkynylation with a 2,3-disubstituted thiophene substrate has been described by Li and co-workers: Liu B., Ouyang W., Nie J., Gao Y., Feng K., Huo Y., Chen Q., Li X., Chem. Commun. 2020, 56, 11255. [DOI] [PubMed] [Google Scholar]
- 6.For C−C bond-forming reactions of 3-donor-substituted thiophenes, see:
- 6a. Forgione P., Brochu M.-C., St-Onge M., Thesen K. H., Bailey M. D., Bilodeau F., J. Am. Chem. Soc. 2006, 128, 11350; [DOI] [PubMed] [Google Scholar]
- 6b. Fournier Dit Chabert J., Marquez B., Neville L., Joucla L., Broussous S., Bouhours P., David E., Pellet-Rostaing S., Marquet B., Moreau N., et al., Bioorg. Med. Chem. 2007, 15, 4482; [DOI] [PubMed] [Google Scholar]
- 6c. Zhao J., Huang L., Cheng K., Zhang Y., Tetrahedron Lett. 2009, 50, 2758; [Google Scholar]
- 6d. Join B., Yamamoto T., Itami K., Angew. Chem. Int. Ed. 2009, 48, 3644; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3698; [Google Scholar]
- 6e. Liégault B., Petrov I., Gorelsky S. I., Fagnou K., J. Org. Chem. 2010, 75, 1047; [DOI] [PubMed] [Google Scholar]
- 6f. Zhang H., Liu D., Chen C., Liu C., Lei A., Chem. Eur. J. 2011, 17, 9581; [DOI] [PubMed] [Google Scholar]
- 6g. Zhang Y., Li Z., Liu Z.-Q., Org. Lett. 2012, 14, 226; [DOI] [PubMed] [Google Scholar]
- 6h. Jiang Z., Zhang L., Dong C., Cai Z., Tang W., Li H., Xu L., Xiao J., Adv. Synth. Catal. 2012, 354, 3225; [Google Scholar]
- 6i. Li Y., Wang J., Yan B., Huang M., Zhu Y., Wu Y., Wu Y., Tetrahedron 2015, 71, 2729; [Google Scholar]
- 6j. Gorsline B. J., Wang L., Ren P., Carrow B. P., J. Am. Chem. Soc. 2017, 139, 9605; [DOI] [PubMed] [Google Scholar]
- 6k. Wen Z.-K., Song T.-T., Liu Y.-F., Chao J.-B., Chem. Commun. 2018, 54, 3668; [DOI] [PubMed] [Google Scholar]
- 6l. Álvarez-Casao Y., Fernández-Ibáñez M. Á., Eur. J. Org. Chem. 2019, 1842; [Google Scholar]
- 6m. Chen H., Farizyan M., Ghiringhelli F., van Gemmeren M., Angew. Chem. Int. Ed. 2020, 59, 12213; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12311; [Google Scholar]
- 6n. Chen H., Farizyan M., van Gemmeren M., Eur. J. Org. Chem. 2020, 6318. [Google Scholar]
- 7.For C−C bond-forming reactions of 3-acceptor-substituted thiophenes, see:
- 7a. Lavenot L., Gozzi C., Ilg K., Orlova I., Penalva V., Lemaire M., J. Organomet. Chem. 1998, 567, 49; [Google Scholar]
- 7b. Glover B., Harvey K. A., Liu B., Sharp M. J., Tymoschenko M. F., Org. Lett. 2003, 5, 301; [DOI] [PubMed] [Google Scholar]
- 7c. Dong J. J., Doucet H., Eur. J. Org. Chem. 2010, 611; [Google Scholar]
- 7d. Park S. H., Kim J. Y., Chang S., Org. Lett. 2011, 13, 2372; [DOI] [PubMed] [Google Scholar]
- 7e. Bheeter C. B., Bera J. K., Doucet H., RSC Adv. 2012, 2, 7197; [Google Scholar]
- 7f. Graczyk K., Ma W., Ackermann L., Org. Lett. 2012, 14, 4110; [DOI] [PubMed] [Google Scholar]
- 7g. Padala K., Pimparkar S., Madasamy P., Jeganmohan M., Chem. Commun. 2012, 48, 7140; [DOI] [PubMed] [Google Scholar]
- 7h. Moon Y., Jeong Y., Kook D., Hong S., Org. Biomol. Chem. 2015, 13, 3918; [DOI] [PubMed] [Google Scholar]
- 7i. Gao S., Wu Z., Wu F., Lin A., Yao H., Adv. Synth. Catal. 2016, 358, 4129; and ref. [6l–n]. [Google Scholar]
- 8.For silylation and borylation reactions of 3-substituted thiophenes, see:
- 8a. Chotana G. A., Kallepalli V. A., Maleczka R. E., Smith M. R., Tetrahedron 2008, 64, 6103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Hatanaka T., Ohki Y., Tatsumi K., Chem. Asian J. 2010, 5, 1657; [DOI] [PubMed] [Google Scholar]
- 8c. Robbins D. W., Hartwig J. F., Org. Lett. 2012, 14, 4266; [DOI] [PubMed] [Google Scholar]
- 8d. Miller S. L., Chotana G. A., Fritz J. A., Chattopadhyay B., Maleczka R. E., Smith M. R., Org. Lett. 2019, 21, 6388; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Karmel C., Rubel C. Z., Kharitonova E. V., Hartwig J. F., Angew. Chem. Int. Ed. 2020, 59, 6074; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6130; [Google Scholar]
- 8f. Wright J. S., Scott P. J. H., Steel P. G., Angew. Chem. Int. Ed. 2020, 10.1002/anie.202001520; [DOI] [Google Scholar]; Angew. Chem. 2020, 10.1002/ange.202001520; and ref. [6b]. [DOI] [Google Scholar]
- 9. Chen C., Liu P., Tang J., Deng G., Zeng X., Org. Lett. 2017, 19, 2474. [DOI] [PubMed] [Google Scholar]
- 10. Tan E., Konovalov A. I., Fernández G. A., Dorel R., Echavarren A. M., Org. Lett. 2017, 19, 5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan E., Quinonero O., Elena de Orbe M., Echavarren A. M., ACS Catal. 2018, 8, 2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Beck E. M., Grimster N. P., Hatley R., Gaunt M. J., J. Am. Chem. Soc. 2006, 128, 2528; [DOI] [PubMed] [Google Scholar]
- 12b. Ueda K., Amaike K., Maceiczyk R. M., Itami K., Yamaguchi J., J. Am. Chem. Soc. 2014, 136, 13226; [DOI] [PubMed] [Google Scholar]
- 12c. Sasaki I., Taguchi J., Hiraki S., Ito H., Ishiyama T., Chem. Eur. J. 2015, 21, 9236; [DOI] [PubMed] [Google Scholar]
- 12d. Su Y., Gao S., Huang Y., Lin A., Yao H., Chem. Eur. J. 2015, 21, 15820; [DOI] [PubMed] [Google Scholar]
- 12e. Nájera C., Beletskaya I. P., Yus M., Chem. Soc. Rev. 2019, 48, 4515. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Chen H., Wedi P., Meyer T., Tavakoli G., van Gemmeren M., Angew. Chem. Int. Ed. 2018, 57, 2497; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2523; [Google Scholar]
- 13b. Zhao D., Xu P., Ritter T., Chem 2019, 5, 97; [Google Scholar]
- 13c. Chen H., Mondal A., Wedi P., van Gemmeren M., ACS Catal. 2019, 9, 1979; [Google Scholar]
- 13d. Mondal A., Chen H., Flämig L., Wedi P., van Gemmeren M., J. Am. Chem. Soc. 2019, 141, 18662. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Liu T., Qiao J. X., Poss M. A., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 10924; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11064; [Google Scholar]
- 14b. Porey S., Zhang X., Bhowmick S., Kumar Singh V., Guin S., Paton R. S., Maiti D., J. Am. Chem. Soc. 2020, 142, 3762. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary