

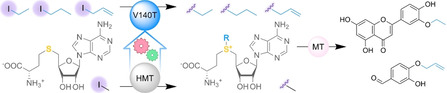
Abstract
Biocatalytic alkylations are important reactions to obtain chemo‐, regio‐ and stereoselectively alkylated compounds. This can be achieved using S‐adenosyl‐l‐methionine (SAM)‐dependent methyltransferases and SAM analogs. It was recently shown that a halide methyltransferase (HMT) from Chloracidobacterium thermophilum can synthesize SAM from SAH and methyl iodide. We developed an iodide‐based assay for the directed evolution of an HMT from Arabidopsis thaliana and used it to identify a V140T variant that can also accept ethyl‐, propyl‐, and allyl iodide to produce the corresponding SAM analogs (90, 50, and 70 % conversion of 15 mg SAH). The V140T AtHMT was used in one‐pot cascades with O‐methyltransferases (IeOMT or COMT) to achieve the regioselective ethylation of luteolin and allylation of 3,4‐dihydroxybenzaldehyde. While a cascade for the propylation of 3,4‐dihydroxybenzaldehyde gave low conversion, the propyl‐SAH intermediate could be confirmed by NMR spectroscopy.
Keywords: bioalkylation, halide methyltransferase, methylation, protein engineering, SAM analog
Biocatalytic alkylations are valuable for late‐stage functionalization but are limited by the availability of S‐adenosyl‐l‐methionine analogs. Directed evolution was used to create an engineered halide methyltransferase capable of converting cheap and readily available alkyl iodides into a number of SAM analogs. Used in cascades with methyltransferases, this enables chemo‐, regio‐ and stereoselective alkylations which are difficult to achieve by chemical means.
The “magic methyl effect” refers to the ability of an appropriately placed methyl group to dramatically alter the biological properties of a compound. [1] This makes selective alkylation of molecules highly desirable. [2] Bioalkylations are important because of the exquisite chemo‐, regio‐ and stereospecificity achievable using enzymes.[ 2 , 3 ] In nature, selective methylation is catalyzed by methyltransferases (MT, E.C. 2.1.1.X) that use S‐adenosyl‐l‐methionine (SAM, “nature's methyl iodide”) as methyl donor. [4] Interestingly, many methyltransferases are insensitive to the size of the alkyl substituent of SAM and could catalyze other alkylation reactions if the necessary SAM analogs were available. [5] This promiscuity can be harnessed to expand the structural and functional diversity of chemicals and enable various applications, such as site‐selective modification of molecules with fluorescent or “clickable” groups.[ 4a , 5a ]
SAM analogs enabling these diverse alkylation reactions are crucial not only for expanding the industrial relevance of biocatalytic alkylation but also for discovering promiscuous MTs.[ 1c , 6 ] However, very few naturally occurring SAM analogs[ 5a , 6 , 7 ] are known, making limited access to SAM analogs one of the most serious impediments to progress in the field. With the exception of S‐adenosyl‐l‐ethionine (SAE), SAM analogs are not readily available from commercial suppliers and have to be prepared on demand. Alkylation of S‐adenosyl‐l‐homocysteine (SAH) using alkyl halides results in low yields and contamination with the biologically inactive (R,S) diastereomers. [5b] These side products can be potent MT inhibitors and are not easily separated from the desired SAM analogs. [8] Enzymatic synthesis of SAM analogs is more specific and yields only the desired isomers. Methionine adenosyltransferases (MATs) or halogenases catalyze the production of SAM analogs by combining the adenosyl moiety of ATP or 5′‐chloro‐5′‐deoxyadenosine, respectively, to the sulfur atom of methionine analogs. [9] The major drawback of these approaches is that they require methionine analogs, which are expensive, if at all commercially available (Figure S1).
Liao and Seebeck recently reported the enzymatic synthesis of SAM from SAH and methyl iodide, using a halide methyltransferase (HMT) from Chloracidobacterium thermophilum (CtHMT). [10] We realized that, like many MTs, some HMTs would be promiscuous and that this would allow a diverse set of SAM analogs to be enzymatically synthesized using cheap and readily available alkyl iodides, without having to chemically synthesize methionine analogs.
We expressed and purified the HMT from Chloracidobacterium thermophilum (CtHMT) originally described by Liao and Seebeck. [10] We constructed an SAH nucleosidase‐deficient strain of E. coli BL21(DE3) to avoid contamination of recombinant enzymes with this SAH‐degrading enzyme (Method S1.3). We also prepared HMTs from Arabidopsis thaliana (AtHMT) and Raphanus sativus (RsHMT) as these have been well characterized and have high activities towards iodide. [11] We assessed the abilities of the three recombinant HMTs to alkylate SAH, using methyl iodide (MeI), ethyl iodide (EtI), propyl iodide (PrI), and butyl iodide (BuI) as alkyl donors. All three HMTs were active using methyl iodide, although CtHMT was significantly less active than AtHMT and RsHMT (Table S1). As expected, all three enzymes had promiscuous activity against ethyl iodide, although this was three orders of magnitude lower than methyltransferase activity for each of the enzymes (Table S1). However, none of the enzymes had significant activity towards propyl iodide or butyl iodide (less than 0.01 μmol min−1 mg−1). AtHMT had the highest ethyltransferase activity (2.69±0.15 μmol min−1 mg−1) and a solved crystal structure and was therefore chosen as the starting point for semi‐rational protein engineering.
We docked EtI, PrI, and BuI into the active site of the AtHMT crystal structure (PDB ID: 3LCC) with bound SAH (Figure 1 a). [12] The alkyl‐binding pocket is quite spacious and could accommodate even butyl iodide (in agreement with alkyl promiscuity of many methyltransferases). The iodine atoms of all docked substrates were superposed. We selected the residues forming the alkyl‐binding site (P20, V23, L27, W36, W47, Y139, V140, C143, Y172, and R214) for further investigation. For each of these positions, an NNK site‐saturation library was created using mutagenesis PCR. For each NNK library, 96 clones were screened by incubating crude lysate with 1 mm SAH and 5 mm ethyl iodide (Supporting Method S2). While the formation of SAE could be monitored by HPLC, this would require 20 hours per library, making it very tedious to investigate all randomized residues. Therefore, we needed a high‐throughput method for determining the amount of product formed in each assay. Because an iodide ion is released for each molecule of SAE formed (Figure 1 b), we initially considered using our recently published ultrasensitive halide assay. [13] However, we were concerned that background signals originating from chloride in crude cell lysates would complicate the interpretation of results. Therefore, we developed a modified assay that is insensitive to chloride, but highly sensitive to iodide.
Figure 1.
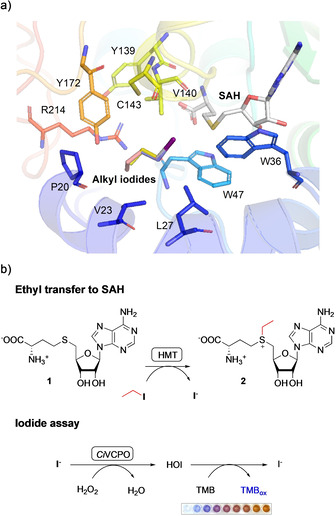
a) The crystal structure of AtHMT and the modelled alkyl iodides in the active site. The selected mutated sites, SAH, and the alkyl iodides, which include MeI, EtI, PrI, and BuI, are shown as sticks with elemental coloring. The carbons of SAH, MeI, EtI, PrI, and BuI are shown in white, grey, purple, yellow, and pink, respectively. b) Transfer of the ethyl group from ethyl iodide to SAH (1) and the production of SAE (2) and iodide. The iodide produced can be detected using hydrogen peroxide, a haloperoxidase, and TMB.
We used a recombinant Curvularia inaequalis vanadium‐dependent chloroperoxidase (CiVCPO) to oxidize iodide to hypoiodous acid (HOI). The HOI formed oxidizes the chromogen 3,3′,5,5′‐tetramethylbenzidine (TMB), resulting in the formation of blue color (Figure S2a). [14] In the process, iodide is released again, so that one iodide anion can catalyze the oxidation of multiple TMB molecules (Figure 1 b). This results in very high sensitivity, and only 1 μL of sample is required for the assay. The change in absorbance at 570 nm is directly proportional to iodide concentration in the range from 5 to 400 μm (Figure S3a). Chloride concentrations up to 1 m result in only slight color changes (Figure S2b). Most importantly, our assay is general and suitable for monitoring the release of iodide from a range of alkyl iodides. This enables both high‐throughput screening of mutant libraries and screening of HMT variants against various alkyl iodides. Activities of HMTs determined using the iodide assay were comparable to those determined using HPLC (Figure S3c), demonstrating the validity and reliability of the assay.
The 10 NNK libraries at the selected positions were screened and four hits (V23T, W36F, V140C, and V140T) with improved activities relative to the wild‐type AtHMT were identified (Figure S4). Based on these hits, we constructed NNK libraries at each other hit position, for a total of six additional libraries (Table S2). Hits from screening these libraries were double‐mutant combinations of V23K/L/T, W36F, and V140C/T. Finally, we constructed triple mutants based on the single and double mutants. We expressed and purified the wild‐type AtHMT and all these variants and determined their specific activities towards ethyl iodide (Figure 2). The specific activity of the most active V140T variant (14.02 μmol min−1 mg−1) towards ethyl iodide was fivefold higher than that of the wild‐type AtHMT (2.69 μmol min−1 mg−1).
Figure 2.
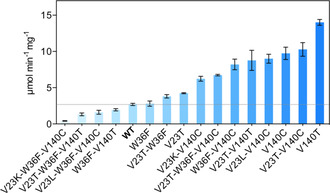
Specific activities of wild‐type AtHMT and several single, double, and triple mutants, determined using ethyl iodide as substrate. Data plotted are the means, with standard deviation, of three independent measurements. The data used for plotting this graph are shown in Table S3.
Kinetic analysis (Figures S5 and S6) showed that the activity improvement relied on an increased k cat and not decreased K m for ethyl iodide (Table 1). The k cat value for the V140T was 6‐fold higher than that for the wild type. The relative preference for ethyl transfer was 36‐fold higher for the V140T variant than for the wild‐type AtHMT.
Table 1.
Kinetic parameters of wild‐type AtHMT and the V140T variant.
|
Variant |
|
Methyltransferase activity |
|
Ethyltransferase activity |
|
Substrate preference [EtI]/[MeI][a] |
||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
k cat [min−1] |
K m, MeI [mm] |
k cat/K m, MeI [min−1/mm] |
k cat [min−1] |
K m, EtI [mm] |
k cat/K m, EtI [min−1/mm] |
|
|
|
|
WT |
|
364.4±12.7 |
1.43±0.19 |
254.5 |
|
0.36±0.03 |
9.36±1.66 |
0.038 |
|
1.5×10−4 |
|
V140T |
|
182.4±11.7 |
4.70±0.76 |
38.7 |
|
2.38±0.16 |
11.14±1.52 |
0.214 |
|
5.5×10−3 |
[a] Substrate preference for EtI relative to MeI is the ratio of k cat/K m values for the two substrates. This preference is ca. 36‐fold higher for the V140T variant.
In addition to increased ethyltransferase activity, the V140T variant was also more active towards propyl iodide and allyl iodide compared to the wild type (Figure S7). The V140T variant was used to convert SAH (15 mg) to SAE (2), S‐propyl‐l‐homocysteine (SAP (3)), and S‐allyl‐l‐homocysteine (SAA (4)) using ethyl iodide, propyl iodide, and allyl iodide. Conversions were 90, 50, and 70 % after 14, 24, and 14 h, respectively (Figure 3 a and Table S4).
Figure 3.
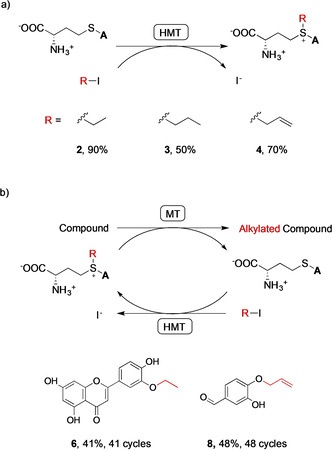
a) Preparative‐scale (15 mg) synthesis of SAE (2), SAP (3), and SAA (4) catalyzed by the V140T AtHMT. The “A” represents the adenosyl moiety. Conversions shown are from Table S4. b) Production (10–20 mg scale) of alkylated products using cyclic MT‐HMT cascades, employing 100 μm SAH and 80 mm alkyl iodide. The IeOMT variant T133M‐Y326L and the V140T AtHMT catalyzed the ethylation of luteolin and produced 4′‐O‐ethylluteolin (6) with 41 % conversion. Human COMT and V140T AtHMT catalyzed the allylation of 3,4‐dihydroxybenzaldehyde and produced 4‐allyloxy‐3‐hydroxybenzaldehyde (8) with 48 % conversion. Insignificant conversion took place if the HMT and MT were not added (Figures S9 and S11). Conversions are from Table S5 and numbers of SAH regeneration cycles were calculated as [product]/[SAH]t=0.
The V140T variant is therefore valuable for the synthesis of various SAM analogs. However, using stoichiometric amounts of these alkylating agents would be expensive due to poor atom economy and the high cost of SAH. Therefore, we used the V140T AtHMT for in vitro regeneration of SAE, SAP, and SAA, from catalytic amounts of SAH, in one‐pot biocatalytic alkylation cascades (Figure 3 b). The T133M–Y326L variant of isoeugenol O‐MT (IeOMT) and the V140T AtHMT catalyzed regioselective mono‐ethylation of luteolin to 3′‐O‐ethylluteolin (6) with 41 % conversion. [15] Similarly, human catechol O‐MT (COMT) in cascade with V140T AtHMT catalyzed regioselective mono‐allylation of 3,4‐dihydroxybenzaldehyde to produce 4‐allyloxy‐3‐hydroxybenzaldehyde (8) with 48 % conversion (Table S5). [16] The identities of the products were confirmed by 1H and 13C NMR spectroscopy (Figures S12–S17). The conversions achieved correspond to 41 and 48 SAH regeneration cycles. While a cascade of human COMT and V140T AtHMT catalyzed the propylation of 3,4‐dihydroxybenzaldehyde, the conversion was low (ca. 5 %) and the product could not be purified for NMR spectroscopy. Therefore, we isolated SAP synthesized using the V140T AtHMT (Figure S9b) and confirmed its structure by 1H and 13C NMR (Figures S18 and S19).
We have demonstrated that the scope of biocatalytic alkylations can be rapidly expanded by harnessing the promiscuity of an engineered halide methyltransferase. The HMT from Arabidopsis thaliana enabled us to produce three SAM analogs from cheap and readily available alkyl iodides. Importantly, we also demonstrated application of the engineered HMT in bioalkylation cascades. It could catalyze over 40 cycles of alkylation and SAH regeneration, allowing SAH to be used in catalytic rather than stoichiometric amounts (Figure 3 b). As noted by Liao and Seebeck, the use of toxic alkyl iodides as reagents might be considered a drawback of this approach. However, the same applies to chemical alkylation and the biocatalytic alternative functions under milder conditions and offers chemo‐, regio‐, and stereoselectivity. [17]
Our best variant (V140T) had the highest activity against all alkyl iodides tested (Figure S7), demonstrating that screening using ethyl iodide can increase activity against other alkyl iodides of interest. However, the ethyltransferase activity of V140T is significantly higher than its propyltransferase activity, as “you get what you screen for”. [18] We screened using ethyl iodide to maximize chances of finding improved variants, but the libraries and screening methodology reported here could be used to identify variants with improved activity towards other alkyl iodides. Exploring the intrinsic alkyl‐substituent promiscuity of naturally occurring HMTs presents a very promising avenue for future research. Not only might a highly promiscuous variant already exist in nature, but also chances of finding an optimal starting point for directed evolution would be improved by characterizing a larger number of extant proteins.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Q. T. thanks the China Scholarship Council for financial support of her PhD thesis project (File No.: 201606150073). A. S. A.‐Ü. thanks the European Union (722610 ES‐CAT). S. W. thanks the Alexander von Humboldt‐Stiftung for a Humboldt Research Fellowship. Open access funding enabled and organized by Projekt DEAL.
Q. Tang, C. W. Grathwol, A. S. Aslan-Üzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst, U. T. Bornscheuer, Angew. Chem. Int. Ed. 2021, 60, 1524.
This article is dedicated to Paul Kamer, an outstanding scientist
Contributor Information
Prof. Dr. Ioannis V. Pavlidis, Email: ipavlidis@uoc.gr.
Dr. Christoffel P. S. Badenhorst, Email: chris.badenhorst@uni-greifswald.de.
Prof. Dr. Uwe T. Bornscheuer, Email: uwe.bornscheuer@uni-greifswald.de.
References
- 1.
- 1a. Schönherr H., Cernak T., Angew. Chem. Int. Ed. 2013, 52, 12256–12267; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12480–12492; [Google Scholar]
- 1b. Barreiro E. J., Kümmerle A. E., Fraga C. A. M., Chem. Rev. 2011, 111, 5215–5246; [DOI] [PubMed] [Google Scholar]
- 1c. Andexer J. N., Rentmeister A., Nat. Chem. 2020, 12, 791–792. [DOI] [PubMed] [Google Scholar]
- 2. McKean I. J. W., Hoskisson P. A., Burley G. A., ChemBioChem 2020, 21, 2890–2897. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wu S., Snajdrova R., Moore J. C., Baldenius K., Bornscheuer U. T., Angew. Chem. Int. Ed. 2020, 10.1002/anie.202006648; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 10.1002/ange.202006648; [DOI] [Google Scholar]
- 3b. Hauer B., ACS Catal. 2020, 10, 8418–8427. [Google Scholar]
- 4.
- 4a. Struck A. W., Thompson M. L., Wong L. S., Micklefield J., ChemBioChem 2012, 13, 2642–2655; [DOI] [PubMed] [Google Scholar]
- 4b. Wessjohann L., Dippe M., Tengg M., Gruber-Khadjawi M., in Cascade Biocatalysis (Eds.: Riva S., Fessner W. D.), Wiley-VCH, Weinheim, 2014, pp. 393–426. [Google Scholar]
- 5.
- 5a. Huber T. D., Johnson B. R., Zhang J., Thorson J. S., Curr. Opin. Biotechnol. 2016, 42, 189–197; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Dalhoff C., Lukinavičius G., Klimas̆auskas S., Weinhold E., Nat. Chem. Biol. 2006, 2, 31–32; [DOI] [PubMed] [Google Scholar]
- 5c. Law B. J., Bennett M. R., Thompson M. L., Levy C., Shepherd S. A., Leys D., Micklefield J., Angew. Chem. Int. Ed. 2016, 55, 2683–2687; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2733–2737; [Google Scholar]
- 5d. Law B. J., Struck A.-W., Bennett M. R., Wilkinson B., Micklefield J., Chem. Sci. 2015, 6, 2885–2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herbert A. J., Shepherd S. A., Cronin V. A., Bennett M. R., Sung R., Micklefield J., Angew. Chem. Int. Ed. 2020, 59, 14950–14956; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15060–15066. [Google Scholar]
- 7.
- 7a. Kim J., Xiao H., Bonanno J. B., Kalyanaraman C., Brown S., Tang X., Al-Obaidi N. F., Patskovsky Y., Babbitt P. C., Jacobson M. P., Lee Y.-S., Almo S. C., Nature 2013, 498, 123–126; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. McKean I. J. W., Sadler J. C., Cuetos A., Frese A., Humphreys L. D., Grogan G., Hoskisson P. A., Burley G. A., Angew. Chem. Int. Ed. 2019, 58, 17583–17588; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17747–17752. [Google Scholar]
- 8.
- 8a. Borchardt R., Wu Y. S., J. Med. Chem. 1976, 19, 1099–1103; [DOI] [PubMed] [Google Scholar]
- 8b. Khani-Oskouee S., Jones J. P., Woodard R. W., Biochem. Biophys. Res. Commun. 1984, 121, 181–187. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Singh S., Zhang J., Huber T. D., Sunkara M., Hurley K., Goff R. D., Wang G., Zhang W., Liu C., Rohr J., Angew. Chem. Int. Ed. 2014, 53, 3965–3969; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4046–4050; [Google Scholar]
- 9b. Wang F., Singh S., Zhang J., Huber T. D., Helmich K. E., Sunkara M., Hurley K. A., Goff R. D., Bingman C. A., Morris A. J., FEBS J. 2014, 281, 4224–4239; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Thomsen M., Vogensen S. B., Buchardt J., Burkart M. D., Clausen R. P., Org. Biomol. Chem. 2013, 11, 7606–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao C., Seebeck F. P., Nat. Catal. 2019, 2, 696–701. [Google Scholar]
- 11.
- 11a. Nagatoshi Y., Nakamura T., Plant Biotechnol. 2007, 24, 503–506; [Google Scholar]
- 11b. Itoh N., Toda H., Matsuda M., Negishi T., Taniguchi T., Ohsawa N., BMC Plant Biol. 2009, 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidberger J. W., James A. B., Edwards R., Naismith J. H., O'Hagan D., Angew. Chem. Int. Ed. 2010, 49, 3646–3648; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3728–3730. [Google Scholar]
- 13. Aslan-Üzel A. S., Beier A., Kovář D., Cziegler C., Padhi S. K., Schuiten E. D., Dörr M., Böttcher D., Hollmann F., Rudroff F., Mihovilovic M. D., Buryška T., Damborský J., Prokop Z., Badenhorst C. P. S., Bornscheuer U. T., ChemCatChem 2020, 12, 2032–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bozeman P. M., Learn D. B., Thomas E. L., J. Immunol. Methods 1990, 126, 125–133. [DOI] [PubMed] [Google Scholar]
- 15. Tang Q., Vianney Y. M., Weisz K., Grathwol C. W., Link A., Bornscheuer U. T., Pavlidis I. V., ChemCatChem 2020, 12, 3721–3727. [Google Scholar]
- 16. Männistö P. T., Kaakkola S., Pharmacol. Rev. 1999, 51, 593–628. [PubMed] [Google Scholar]
- 17. Liao C., Seebeck F. P., Angew. Chem. Int. Ed. 2020, 59, 7184–7187; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7251–7254. [Google Scholar]
- 18. You L., Arnold F., Protein Eng. Des. Sel. 1996, 9, 77–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary