

Abstract
Chemical bonds are traditionally assigned as electron‐sharing or donor‐acceptor/dative. External criteria such as the nature of the dissociation process, energy partitioning schemes, or quantum chemical topology are invoked to assess the bonding situation. However, for systems with marked multi‐reference character, this binary categorization might not be precise enough to render the bonding properties. A third scenario can be foreseen: spin polarized bonds. To illustrate this, the case of a NaBH3 − cluster is presented. According to the analysis NaBH3 − exhibits a strong diradical character and cannot be classified as either electron‐sharing or a dative bond. Elaborated upon are the common problems of popular bonding descriptions. Additionally, a simple model, based on the bond order and local spin indicators, which discriminates between all three bonding situations, is provided.
Keywords: bond analysis, dative bonds, donor-acceptor systems, electronic structure, sodium
Conventional chemical bonding analysis distinguishes between two situations: electron sharing and donor‐acceptor. However, a third scenario is possible where the bond suffers spin polarization. Discussed here is the case of NaBH3 −, better classified as a spin‐polarized bond. A simple model to distinguish between these bonding types is provided.
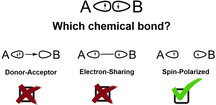
The chemical bond is a central paradigm for describing molecular structure and reactivity. [1] A fundamental approach towards understanding its properties consists in classifying the electron‐pair interactions between atoms or fragments. [2] There are two well‐established classes of bonding interactions, according to the origin of the electron‐pair. When each fragment contributes with one electron, the bonding is described as an electron‐sharing bond. When both electrons are contributed by one of the fragments, the interaction is interpreted as a dative or donor‐acceptor bond. [3]
The IUPAC recommends to analyse the nature of the chemical bond considering the minimum‐energy rupture in the gas phase or in inert solvents. [4] Following Haaland's guidelines, a bond is classified as dative if the minimum energy bond rupture proceeds heterolytically, while it is an electron‐sharing bond if this rupture proceeds homolytically. [5] Such a distinction oversees the electronic rearrangement happening during dissociation. Therefore, some systems can lead to heterolytic dissociation despite the fact that each fragment contributes with one electron. [6] Several methods based on valence bond theory, topological analysis, and molecular orbital theory have been used to assess the bonding interaction “without” the need of dissociation, but in most cases recurring to a seemingly unavoidable arbitrary fragmentation. [7]
In general, it is not trivial (maybe impossible) to distinguish between a dative and an electron‐sharing situation, without invoking an external criterion.[ 7 , 8 ] Arguably, the orbital‐based method energy decomposition analysis (EDA) and the quantum chemical topology (QCT) approaches are considered successful methods to solve such a task. [9] Within the EDA scheme, the bond is decomposed into an electrostatic interaction (ΔE elstat) between the frozen‐density fragments, the Pauli repulsion (ΔE Pauli) associated to the antisymmetrization of the wave function, and the stabilizing orbital term (ΔE orb), accounting for the final orbital relaxation. These terms depend on the specific reference electronic state of the fragments, thus there is no exclusive bond fragmentation. The lower the absolute values of the orbital term (ΔE orb), the better the representation of the chemical bond since this translates into a lower reorganization degree. [10] Aside from being a path function, this method depends on the correct representation of the ground state. [11] With QCT methods, specifically the atoms in molecules (QTAIM) approach, the value of different descriptors at the bond critical point are used to assess the nature of the chemical bond. [12] Since no reference states are needed, this method avoids the inherent problems carried by fragmentation schemes. Although physically well‐founded, it lacks predictive power and is prone to misinterpretations when it is connected with heuristic orbital models. [13]
Considering the AB system interacting through a dative bond, if A: is the donor and B is the acceptor, the contributions in terms of electron population of A: and B to the A−B bond would be 2‐δ and δ, respectively, where δ accounts for the donation of electron density upon bond formation. Instead, if the bonding interaction is electron‐sharing the atomic populations of A. and B. would be (assuming χ A>χ B) N A=1+p and N B=1−p, where p accounts for the bond polarization, induced by the different local electronegativity (χ) of A and B. Both pictures are naturally related by δ + p=1. When p → 0, the electron‐sharing fragmentation would likely lead to a smaller orbital interaction than the donor‐acceptor one. The contrary is expected as p → 1.
However, one can envisage a third scenario, where the bond suffers from spin polarization. In that case, the α and β atomic populations would be defined as N A α=1+p α, N B α=1−p α, N A β=1−p β and N B β=1+p β. The spin density on each fragment will be given by ps=|p α+p β|, while the overall bond polarization p=|p α−p β| would likely be small.
Spin polarization in bonds is a well‐documented phenomenon. For instance, high‐valent oxo‐iron species are key intermediates in the catalytic cycles of oxygen activating iron enzymes such as the cytochrome P450. The extent of spin polarization of the Fe=O unit stands behind debates over its electronic structure, namely oxo‐iron(IV) vs. oxyl‐iron(III) pictures. [14] In nitrosyl chemistry, spin polarization also plays a major role when it comes to assigning the oxidation states of the metal‐NO unit. [15] It also hinders the rationalization of metal‐metal multiple bonding. [16]
In the extreme case, spin‐polarization leads to a diradical species. Intermediate situations are usually referred as diradicaloids. Signatures of diradical character are a small singlet‐triplet gap and a spin‐polarized (broken‐symmetry, BS) solution below the closed‐shell (CS) description of single determinantal methods. In fact, incorporating static correlation is pivotal for the correct description of spin polarization.
In most EDA approaches, spin‐polarization in the fragments and the spin‐coupled intermediate state is not properly considered, with exceptions. [17] Importantly, the appearance of a BS solution below the CS one, increments the ΔE orb values for the donor‐acceptor and electron‐sharing patterns by the same amount. The other terms, namely ΔE elstat, ΔE Pauli and ΔE prep, keep the same magnitude. If the intermediate state, built up from A: + B, is higher in energy than that from A. + .B, irrespective of the nature of the ground‐state of AB, the lowest ΔEorb criterion would necessarily point towards an “electron‐sharing” situation, or better said, to a reference state with one electron per fragment. Hence, such a criterion appears to be useful merely to discriminate the dative picture from the other two. EDA is not designed to distinguish a classical electron‐sharing from a spin‐polarized interaction and, in the limiting case, from a diradical!
More suitable bonding indicators are bond orders and particularly the local spin. [18] In Mayer's local spin analysis (LSA), the expectation value of the spin‐squared operator is decomposed into atomic (local spins) and diatomic terms. The most relevant feature of LSA is that, even for pure singlet states, the method is able to differentiate a CS covalent molecule from an anti‐ferromagnetic system in which the local spins are coupled to a singlet, and intermediate situations. For the previously discussed A‐B interaction, in the limiting case of having a perfect singlet diradical, one would expect the local spins to be ⟨S2⟩A=⟨S2⟩B=3/4 and the diatomic term to amount to ⟨S2⟩AB=−3/4, indicating a perfect entanglement of the electrons. [10]
Considering a simple two‐electron single‐determinant minimal basis model for the AB system, the CS description leads to a Mayer's bond order of 1−p 2, that is, the covalent bond order decreases with the square of the bond polarization. The interaction can be considered as perfectly covalent as the local spin trivially vanishes. When spin polarization is allowed (via BS), the Mayer bond order varies as 1−p 2−p s 2, where ps indicates the spin polarization amount, that is, both bond polarization and spin polarization are responsible for the decrease of the bond order, at the same ratio. In the absence of bond polarization, the local spin amounts to ⟨S2⟩A=3/4 p s 2 (1−SAB 2), where SAB is the atomic overlap. That is, the increase of local spin is concomitant with the decrease of the covalent bond order due to spin polarization. Deviations from classical covalent bonding with increasing local spin have been observed for correlated wave functions. [19] Thus, the combined consideration of both bond order and local spin indicators affords the distinction between all three aforementioned bonding situations, as sketched in Table 1.
Table 1.
Chemical bonding analysis
|
Chemical Bond |
Bond order A‐B |
Local spin on A and B |
EDA A→B vs A–B |
|
Electron‐sharing |
Large |
Small/Null |
|ΔEorb(A. + .B)|<|ΔEorb(A: + B)| |
|
Donor‐acceptor |
Small |
Small/Null |
|ΔEorb(A. + .B)|>|ΔEorb(A: + B)| |
|
Spin‐Polarized |
Small |
Medium/Large |
|ΔEorb(A. + .B)|<|ΔEorb(A: + B)| |
Let us illustrate the issue with a controversial example. Liu et al. [20] have reported the realization of a NaBH3 − cluster featuring a Na−B bond. By combining anion photoelectron spectroscopy and bond dissociation energies (BDE), the authors claimed the bond as dative Na−→BH3. Later, Pan et al. [21] on the basis of EDA as sketched on Figure 1 reinterpreted the complex as a classical electron‐sharing covalent Na‐BH3 − bond. Only recently, and based on quantum chemical topological approaches, Foroutand‐Nejad classified Na‐B as an ionic enforced covalent bond, arguing that coulombic forces between the metal and the Hs direct the interaction. [22]
Figure 1.
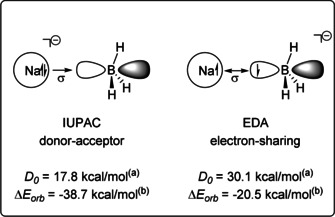
Orbital interactions (ΔE orb) and dissociation energies (D0) in NaBH3 −. aRef. [20b]. bRef. [21].
As already observed by Liu et al., [20] the NaBH3 − exhibits a close‐lying triplet state of C s symmetry. The singlet‐triplet gap obtained with the functionals used in the previous studies,[ 20a , 21 ] i.e., BP86 M06L, PBE0 are just 6.4, 1.5 and 1.7 kcal mol−1, respectively, in line with the CCSD(T) and CASPT2 results (5.2 and 4.3 kcal mol−1). Remarkably, as shown in Table 2 (Table S1), for HF, MP2 and some double‐hybrid functionals, the triplet state lies below the CS singlet state.
Table 2.
Triplet and open shell BS electronic energies (in kcal mol−1) relative to the CS state and Na‐B equilibrium distance (Re in Å) for NaBH3 −. ⟨S2⟩ and diradical character (n rad).[a]
|
Methodb |
CS (C 3v ) |
T (Cs) |
BS (C 3v ) |
|
|||
|---|---|---|---|---|---|---|---|
|
|
Re |
ΔE T |
Re |
ΔE BS |
Re |
⟨S2⟩ |
n rad [%] |
|
HF |
4.865 |
−7.4 |
2.557 |
−7.8 |
2.797 |
0.89 |
67 |
|
MP2 |
2.763 |
−3.9 |
2.581 |
−4.8 |
2.710 |
0.90 |
68[c] |
|
CCSD(T) |
2.719 |
5.2 |
2.580 |
|
|
|
0.14[d] |
|
CASPT2 |
2.666 |
4.3 |
2.552 |
|
|
|
56[e] |
|
BP86 |
2.707 |
6.4 |
2.579 |
−0.4 |
2.702 |
0.30 |
16 |
|
M06L |
2.699 |
1.5 |
2.482 |
−2.9 |
2.668 |
0.71 |
46 |
|
M06‐2X |
2.698 |
3.9 |
2.536 |
−3.8 |
2.701 |
0.55 |
33 |
|
PBE0 |
2.743 |
1.7 |
2.536 |
−2.8 |
2.681 |
0.61 |
37 |
|
B2PLYP |
2.753 |
−0.4 |
2.562 |
−4.1 |
2.732 |
0.70 |
45 |
[a] Computed from ⟨S2⟩ as described in Ref. [25]. [b] Combined with AVTZ, except for CASSCF (AVQZ). [c] From ⟨S2⟩ of the HF wavefunction. [d] Largest t 2 amplitude. [e] Derived from the CI coefficient of the doubly‐excited configuration.
Clearly unnoticed, the CS description of NaBH3 − is not a stable solution. The stability analysis [23] on the CS calculations revealed the presence of an unrestricted Broke Symmetry (BS) solution that leads to a lower electronic state by 0.4 to 8.2 kcal mol−1, depending on the functional. Noteworthy, the BS singlet solution lies below the triplet state in all cases (Table 2). In general, the BS equilibrium distances are also in better agreement with the high‐level CCSD(T) and CASPT2 results.
The BS description should come as no surprise due to the pronounced multi‐reference character of this system.[ 20 , 22 ] We have investigated the lowest singlet and triplet electronic states of NaBH3 − at the CASPT2 level. The CI coefficients for the CS and HOMO–LUMO double excited (22202000) configurations are c 0=0.9009 and cd=−0.3963, and Truhlar M diagnostic [24] amounts to 0.3, thus confirming the strong multi‐determinant nature of the system (Table S3). Noteworthy, the HOMO and LUMO consist of σ bonding and σ* anti‐bonding interactions between the Na 3s and BH3 A1 orbitals, which show fractional occupation numbers, as Figure 2 illustrates.
Figure 2.

CASSCF natural orbitals and occupations at the ground state CASPT2 equilibrium structure.
The ⟨S 2⟩ values of the BS states can be used as a global indicator of diradical character (n rad). [25] When a multi‐configurational wave function is used, n rad can be derived from the weights of appropriate configurations of the CI expansion. The values summarized in Table 2 suggest non‐negligible, although largely functional dependent, n rad. While for some GGA functionals n rad is about 16 % (BP86 or PBE), in the case of the double‐hybrid functionals n rad reaches 50 %, in line with the wave function methods.
We have also carried out EDA for both the CS and BS descriptions at PBE0/QZ4P (Table 3 and Tables S6,S7). For both the CS and BS solutions, the orbital term for the fragmentation Na(s 1) and BH3 −(A1 1) is lower than for Na−(s 2) and BH3(A1 0). Thus, the EDA interpretation remains unchanged, no matter the electronic state used. However, it is important to highlight that the CS results are linked to a misrepresentation of the electronic structure of the system, where two electrons are forced to occupy the σ‐bonding orbital, while the electronic structure of the BS description shows hints of deviation from a classical electron‐sharing bond.
Table 3.
EDA of NaBH3 − for CS and BS at PBE0/QZ4P//CCSD(T)/AVTZ. Energies in kcal mol−1.
|
|
CS |
BS |
||
|---|---|---|---|---|
|
|
Na−(s2); BH3(A1 0) |
Na(s1); BH3 −(A1 1) |
Na−(s2); BH3(A1 0) |
Na(s1); BH3 −(A1 1) |
|
ΔE int |
−18.5 |
−29.5 |
−21.1 |
−32.1 |
|
ΔE Pauli |
33.4 |
30.7 |
33.4 |
30.7 |
|
ΔE elstat |
−17.1 |
−43.6 |
−17.1 |
−43.6 |
|
ΔE orb |
−34.8 |
−16.6 |
−37.4 |
−19.2 |
|
ΔE prep |
1.4 |
12.3 |
1.4 |
12.3 |
|
De |
17.1 |
17.1 |
19.8 |
19.8 |
In the above minimal basis AB model, the three bonding scenarios translate into significant differences in the bond order and local spin electronic structure indicators. To illustrate this, we have considered the electronic structure of representative molecular systems exhibiting different bonding situations, that is, the NaBH3 −, BH4 − and NH3BH3. Relevant bond order, delocalization index, and local spin values (obtained in the framework of QTAIM) [26] are gathered in Table 4.
Table 4.
NBO and AIM charges (Q(E), E=H, NH3, Na), atomic spin density (ρ s), Wiberg Bond Order (WBONBO), AIM Delocalization Index (DIAIM), Local Spins (⟨S2⟩), EDA Orbital Interaction (ΔE orb in kcal mol−1) and electron density values at the E‐B (3,−1) point (ρ BCP) of BH4 −, NH3BH3, and CS and BS NaBH3 − at PBE0/AVTZ and CASSCF/AVTZ.[a,b]
|
|
BH4 − |
NH3BH3 |
NaBH3 − (CS) |
NaBH3 − (BS) |
|---|---|---|---|---|
|
PBE0/AVTZ |
|
|
|
|
|
Q(E)NBO Q(B)NBO |
−0.06 −0.76 |
+0.37 −0.23 |
−0.30 −0.45 |
−0.19 −0.56 |
|
Q(E)AIM Q(B)AIM |
−0.67 +1.70 |
+0.08 +1.84 |
−0.25 +1.52 |
−0.17 +1.43 |
|
WBONBO DIAIM |
1.00 0.55 |
0.65 0.34 |
0.91 0.43 |
0.52 0.29 |
|
ρ s(E)NBO ρ s(E)AIM |
– |
– |
– |
+0.66 +0.61 |
|
⟨S2⟩E/⟨S2⟩B |
– |
– |
– |
0.42/0.21 |
|
ρBCP |
0.15 |
0.11 |
0.013 |
0.014 |
|
ΔE orb(E. + .B) |
−106.3 |
−263.3 |
−16.6 |
−19.2 |
|
ΔE orb(E:+B) |
−154.2 |
−75.9 |
−34.8 |
−37.4 |
|
CASSCF/AVTZ |
|
|
|
|
|
Q(E)AIM Q(B)AIM |
−0.72 +1.91 |
+0.08 +2.04 |
−0.16 +1.61 |
|
|
DIAIM |
0.48 |
0.27 |
0.23 |
|
|
⟨S2⟩E/⟨S2⟩B |
0.02/0.02 |
0.03/0.03 |
0.33/0.26 |
|
|
ρ BCP |
0.15 |
0.10 |
0.015 |
|
|
c 0 [c] |
0.98 |
0.97 |
0.90 |
|
|
c d [c] |
−0.03 |
−0.05 |
−0.40 |
[a] On CCSD(T)/AVTZ structures. [b] See the Supporting Information for details. [c] CI coefficients.
Our minimal basis model explains the calculated Wiberg bond orders (WBO) in terms of the bond and spin polarization values, that can be easily derived from the NBO charges and spin populations (WBONBO). For instance, for BH4 − p=0.06, so the expected bond order is 1−0.062≈1. For NH3BH3, the donor NH3 unit has δ=0.37 and hence p=0.63, which would correspond to a bond order of 1−0.632=0.60, in line with the computed WBONBO=0.65. In the CS description of NaBH3 − p=0.30, leading to a bond order of 1−0.302=0.91, in perfect agreement with the WBONBO. In the BS case, the bond polarization is smaller (p=0.19) but there is significant spin polarization (ps=0.66), consistent with a bond order of 1−0.662−0.192=0.53, again in striking agreement with the exact WBONBO.
For BH4 −, the CASSCF(8,8) wave function displays a monodeterminantal character (c 0=0.98 and cd=−0.03). WBO is 1.00, while the local spin values on B and H are negligible. Within the KS‐DFT description, the DIAIM is somewhat smaller (0.55), driven by the large bond polarization produced by the QTAIM partitioning. EDA, QTAIM and NBO agree in an electron‐sharing picture as explained elsewhere. [21] NH3BH3 is also well‐represented by one single‐determinant at CASSCF(12,12) (c 0=0.97 and cd=−0.05). In this case, both the WBONBO and DIAIM are smaller, as compared to the electron‐sharing case (0.65 and 0.34, respectively), but the local spin is again negligible. EDA delivers a lower orbital term for the fragmentation NH3(A1 2) and BH3(A1 0), so all indicators point towards a dative picture. Remarkably, the marked multi‐configurational character of NaBH3 −, also captured by the BS solution, makes DIAIM to drop to just 0.29, while the local spins on Na (0.42) and B (0.21) are now significant. The DIAIM is significantly larger (0.43) for the CS solution, as the σ* contribution in the Na−B bond is absent. Note that a bonding analysis based on such a density would thus lead to inaccurately overestimated ionic interactions. [22] In fact, the −0.50 Na Mulliken charge calculated with monoconfigurational DFT [22] drops to −0.22 when switching to the CASSCF framework. The same trend is observed with the NBO and AIM charges for the CS and BS solutions. On the contrary, the BS solution mimics the CASSCF wave function, albeit with wrong spin symmetry (overall ⟨S2⟩=0.61). Both the DIAIM and the local spin values are in good agreement with the CASSCF results. EDA favors Na(s 1) and BH3 −(A1 1) fragmentation in both the CS and BS solutions. Thus, combining bond orders and local spins analysis suggests that the Na‐B interaction in NaBH3 − is better described as a spin‐polarized bond, revealing its σ diradicaloid character.
To conclude, the exotic case of NaBH3 − cluster underscores the fundamental limitations of the conventional chemical bond classification into electron‐sharing and dative bonds. This binary Scheme remains useful for molecules like BH4 − or NH3BH3, which are well‐represented by a single‐determinant, but fails for multiconfigurational systems such as NaBH3 −. Oversimplifying the wave function to a single CS configuration would essentially categorize a diradical as a conventional electron sharing bond. Within the KS‐DFT framework, the multi‐configurational character is partially recovered breaking the spin symmetry, allowing the localization of α and β electrons on distinct fragments. The assistance of other bonding indicators enables the identification of a third bonding category, namely a spin‐polarized bond, which captures the essence of the bonding in the NaBH3 − cluster.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
D.M.A. and P.S. thank ERC (EU805113) and MCIU (PGC2018‐098212‐B‐C22) projects. I.C. and E.V. thank Ramón y Cajal program, a FPU grant and the MCIU project PGC2018‐094644‐B‐C21. The CESGA Supercomputing Center and Centro de Computación Científica UAM (CCC‐UAM) are acknowledged. Open access funding enabled and organized by Projekt DEAL.
P. Salvador, E. Vos, I. Corral, D. M. Andrada, Angew. Chem. Int. Ed. 2021, 60, 1498.
Contributor Information
Dr. Pedro Salvador, Email: pedro.salvador@udg.edu.
Dr. Inés Corral, Email: ines.corral@uam.es.
Dr. Diego M. Andrada, Email: diego.andrada@uni-saarland.de.
References
- 1.
- 1a. Ruedenberg K., Schwarz W. H. E. in Pioneers of Quantum Chemistry, Vol. 1122, American Chemical Society, Washington, 2013, pp. 1–45; [Google Scholar]
- 1b. Maksic Z. B., Cremer D., Surjan P. R., The Concept of the Chemical Bond: Theoretical Models of Chemical Bonding Part 2, Springer Berlin Heidelberg, Berlin, 1990. [Google Scholar]
- 2. Pauling L., University C., Press C. U., The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, 1960. [Google Scholar]
- 3. Lewis G. N., Valence and the Structure of Atoms and Molecules, Chemical Catalog Company, Incorporated, 1923. [Google Scholar]
- 4. Minkin V. I., Pure Appl. Chem. 1999, 71, 1919. [Google Scholar]
- 5. Haaland A., Angew. Chem. Int. Ed. Engl. 1989, 28, 992–1007; [Google Scholar]; Angew. Chem. 1989, 101, 1017–1032. [Google Scholar]
- 6. Andrada D. M., Casals-Sainz J. L., Pendás A. M., Frenking G., Chem. Eur. J. 2018, 24, 9083–9089. [DOI] [PubMed] [Google Scholar]
- 7. Zhao L. L., Pan S., Holzmann N., Schwerdtfeger P., Frenking G., Chem. Rev. 2019, 119, 8781–8845. [DOI] [PubMed] [Google Scholar]
- 8. Frenking G., Krapp A., J. Comput. Chem. 2007, 28, 15–24. [DOI] [PubMed] [Google Scholar]
- 9. Zhao L. L., von Hopffgarten M., Andrada D. M., Frenking G., WIREs Comput. Mol. Sci. 2018, 8, e13450. [Google Scholar]
- 10. Li L. M., Parr R. G., J. Chem. Phys. 1986, 84, 1704–1711. [Google Scholar]
- 11. Andrada D. M., Foroutan-Nejad C., Phys. Chem. Chem. Phys. 2020, 22, 22459–22464. [DOI] [PubMed] [Google Scholar]
- 12. Cremer D., Kraka E., Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar]
- 13.
- 13a. Foroutan-Nejad C., Shahbazian S., Marek R., Chem. Eur. J. 2014, 20, 10140–10152; [DOI] [PubMed] [Google Scholar]
- 13b. Bader R. F. W., J. Phys. Chem. A 2009, 113, 10391–10396. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Ye S., Neese F., Proc. Natl. Acad. Sci. USA 2011, 108, 1228–1233; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Janardanan D., Wang Y., Schyman P., L. Que, Jr. , Shaik S., Angew. Chem. Int. Ed. 2010, 49, 3342–3345; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3414–3417; [Google Scholar]
- 14c. Dantignana V., Serrano-Plana J., Draksharapu A., Magallón C., Banerjee S., Fan R., Gamba I., Guo Y., Que L., Costas M., Company A., J. Am. Chem. Soc. 2019, 141, 15078–15091. [DOI] [PubMed] [Google Scholar]
- 15. Ampßler T., Monsch G., Popp J., Riggenmann T., Salvador P., Schröder D., Klüfers P., Angew. Chem. Int. Ed. 2020, 59, 12381–12386; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12480–12485. [Google Scholar]
- 16. Nakagaki M., Nakatani N., Sakaki S., Phys. Chem. Chem. Phys. 2019, 21, 22976–22989. [DOI] [PubMed] [Google Scholar]
- 17. Levine D. S., Horn P. R., Mao Y., Head-Gordon M., J. Chem. Theory Comput. 2016, 12, 4812–4820. [DOI] [PubMed] [Google Scholar]
- 18. Ramos-Cordoba E., Matito E., Mayer I., Salvador P., J. Chem. Theory Comput. 2012, 8, 1270–1279. [DOI] [PubMed] [Google Scholar]
- 19. Ramos-Cordoba E., Salvador P., Reiher M., Chem. Eur. J. 2013, 19, 15267–15275. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Liu G., Fedik N., Martinez-Martinez C., Ciborowski S. M., Zhang X., Boldyrev A. I., Bowen K. H., Angew. Chem. Int. Ed. 2019, 58, 13789–13793; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13927–13931; [Google Scholar]
- 20b. Liu G., Fedik N., Martinez-Martinez C., Ciborowski S. M., Zhang X., Boldyrev A. I., Bowen K. H., Angew. Chem. Int. Ed. 2020, 59, 8760–8764; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8840–8844. [Google Scholar]
- 21. Pan S., Frenking G., Angew. Chem. Int. Ed. 2020, 59, 8756–8759; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8836–8839. [Google Scholar]
- 22. Foroutan-Nejad C., Angew. Chem. Int. Ed. 2020, 10.1002/anie.202010024; [DOI] [Google Scholar]; Angew. Chem. 2020, 10.1002/ange.202010024. [DOI] [Google Scholar]
- 23.
- 23a. Seeger R., Pople J. A., J. Chem. Phys. 1977, 66, 3045–3050; [Google Scholar]
- 23b. Bauernschmitt R., Ahlrichs R., J. Chem. Phys. 1996, 104, 9047–9052. [Google Scholar]
- 24. Tishchenko O., Zheng J., Truhlar D. G., J. Chem. Theory Comput. 2008, 4, 1208–1219. [DOI] [PubMed] [Google Scholar]
- 25. Bachler V., Olbrich G., Neese F., Wieghardt K., Inorg. Chem. 2002, 41, 4179–4193. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Bader R. F. W., Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, 1990; [Google Scholar]
- 26b. Bader R. F. W., Stephens M. E., J. Am. Chem. Soc. 1975, 97, 7391–7399. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary