

Abstract
As one of the most demanded dicarboxylic acids, adipic acid can be directly produced from renewable sources. Hexoses from (hemi)cellulose are oxidized to aldaric acids and subsequently catalytically dehydroxylated. Hitherto performed homogeneously, we present the first heterogeneous catalytic process for converting an aldaric acid into muconic and adipic acid. The contribution of leached Re from the solid pre‐reduced catalyst was also investigated with hot‐filtration test and found to be inactive for dehydroxylation. Corrosive or hazardous (HBr/H2) reagents are avoided and simple alcohols and solid Re/C catalysts in an inert atmosphere are used. At 120 °C, the carboxylic groups are protected by esterification, which prevents lactonization in the absence of water or acidic sites. Dehydroxylation and partial hydrogenation yield monohexenoates (93 %). For complete hydrogenation to adipate, a 16 % higher activation barrier necessitates higher temperatures.
Keywords: aldaric acids, adipic acid, catalytic dihydroxylation, catalytic heterogenization, rhenium catalyst
In the present work, the conversion of bio‐based aldaric acids into muconic and adipic acid through a heterogeneous catalytic process, in the absence of corrosive reagents or gaseous H2 is reported. The mechanism of the selective removal of adjacent OH* groups on Re/C catalysts and the role of (m)ethanol in carboxylic group protection and hydrogen transfer in a slurry reactor are elucidated by DFT calculations and XPS, chemisorption, and HR‐TEM.

Introduction
Sustainable production of bio‐based platform chemicals, especially from underexploited lignocellulosic (LC) biomass, has attracted much attention in the last decade. [1] The selective reduction of oxygen‐containing groups (mainly ‐OH groups in cellulose‐based derivatives) has been a key target process to provide monomers for the polymer industry. Adipic acid, one of the most important dicarboxylic acids, is industrially produced from a mixture of cyclohexane and cyclohexanol in a multi‐step, exothermic oxidation process where nitric acid is used and NOx are emitted. Selective dehydroxylation of bio‐based feedstocks, such as aldaric acid from LC, into adipic acid (Figure 1) would eliminate the need for petrol‐based feedstocks and avoid the use of harmful chemicals and emissions of environmental pollutants. Homogeneous catalysts are well developed but suffer from purification difficulties and unwieldiness for implementation in a continuous process.
Figure 1.

The complete process of lignocellulosic feedstock to adipic acid. The challenging part is the selective reduction of aldaric acids, shown in the dotted box, which is the scope of this work.
Selective dehydroxylation of diols into olefins with homogeneous catalysts has been known for decades. Dehydration of diols into diolefins or alcohols over lithium pyrophosphate was published and patented by Weisang et al. in 1973. [2] Later on, rhenium appeared to be the key, several different homogeneous ReOx compounds were probed subsequently. [3] To avoid the formation of side products or the decomposition of sugar‐based compounds, [4] simple alcohols were tested as reducing agents at moderate temperatures (up to 180 °C). When using acids (TsOH or H2SO4), the required temperature is much lower. An efficient reductant is PPh3 (triphenylphosphine) in combination with cyclopentadienyl‐ and tris(pyrazolyl)borate)‐based oxo‐rhenium species.[ 5 , 6 ] Subsequent research was focused mostly on using different alcohols under moderate hydrogen pressure (1–20 bar).[ 7 , 8 , 9 ] Toste et al. obtained best results with 3‐pentanol and 3‐octanol. [10] Alcohols such as 1‐heptanol and cyclohexanol also showed some reactivity for deoxydehydration (DODH) over oxo‐rhenium catalysts. [11]
In all subsequent studies, homogeneous oxo‐rhenium complexes, mostly MeReO3,[ 3 , 12 , 13 , 14 ] were investigated. Gable and co‐workers[ 15 , 16 , 17 , 18 ] studied the kinetics of the reaction. The developed mechanism played a key role in further studies of selective dehydroxylation of vicinal diols. Vkuturi et al. [19] divided the mechanism of DODH by Gable and Brown [15] into three main steps: (i) condensation of the vicinal diol, (ii) reduction of rhenium(VII) to rhenium(V), and (iii) extrusion of the formed olefin from the rhenium(V) diolate, accompanied by reformation of the rhenium(VII) complex. It was found out that 1‐butanol is a better reductant than 2‐butanol, whereas 3‐pentanol performed better than 2‐pentanol. [20] The proposed DODH mechanism over oxo‐rhenium complexes was applied to numerous transformations of biobased compounds.[ 20 , 21 , 22 ]
Despite their good activity and selectivity, homogeneous Re catalysts are plagued by difficulties in separation from the reaction mixture and recycling. The first mentioned heterogenization of MeReO3 was developed by Denning, [23] who used carbon‐supported rhenium perrhenate under different reaction conditions and solvents on vicinal diols. However, a moderate loss in activity after four consecutive runs over recovered catalyst suggested that ReOx species dissolve and in fact catalyze the reaction homogeneously. To prevent leaching, lower reaction temperatures are desired. Using hot filtration in recyclability tests, Korstanje et al. [24] proved Re species leach in the liquid phase. Gebbink et al. performed comprehensive DFT calculations to elucidate the mechanism of MeReO3 catalyzed deoxydehydration. [25]
Thus, heterogeneous catalysts are intensely sought after. Tomishige et al. [26] used ceria‐supported Re and Pd to dehydroxylate and hydrogenate 1,4‐anhydroerythritol into tetrahydrofuran. Pd was used as a metal to activate H2, while the active site for dehydroxylation was suggested to be high valent ReOx (+6 or +7). It has been proposed that only isolated Re species are catalytically active. [27] Gold nanoparticles as a co‐catalyst showed good activity in a H2 atmosphere, while other tested co‐catalysts, (Ru, Rh, Pd, Pt, Ir, Ag and Au) performed poorly. It is proposed that Au has a promoting effect for the reduction of Re species. [28]
Lately, a few mechanistic studies of Re‐catalyzed DODH of vicinal diols were published. Shakeri et al. [29] studied two different pathways for DODH of phenyl‐1,2‐ethanediol to styrene over an oxo‐rhenium catalyst with PPh3. While previous studies focused on organometallic catalysts, the authors tested the process in the presence of perrhenate (ReO4 −). Xi et al. [30] performed a detailed mechanistic study of a ceria‐supported Re catalyst with Pd, which catalyzes the H2 spillover as an efficient hydrogenation catalyst. [31]
Among the studies on selective dehydroxylation, most focused on DODH of short‐chain vicinal alcohols with homogeneous Re catalysts. Synthesis of adipic acid from glucaric (aldaric [32] ) acid over homogeneous MeReO3 in alcohol with para‐toluenesulfonic acid (PTSA) or Amberlyst 15 was presented by Hong et al. [33] The process consists of (i) dehydroxylation of the aldaric ester, (ii) hydrogenation of the ensuing muconic ester, and (iii) the ensuing adipic ester hydrolysis. Similarly, Asikainen [34] patented selective deoxydehydration of aldaric acids into muconic and adipic acid over homogeneous MeReO3 in light alcohols without PTSA.
Although several studies report a selective dehydroxylation of aldaric acids into muconic acid, adipic acid and their esters over homogeneous oxorhenium catalysts with Pd as a co‐catalyst,[ 7 , 10 , 35 , 36 ] no successful application of heterogeneous Re catalysts for dehydroxylation beyond that of simple diols (for instance in sugar acids) has been reported. For instance, Kirilin et al. showed that for aqueous xylitol reforming, doping a Pt/TiO2 catalyst with Re improves yields, but still preferentially removes only the outer OH groups. [37]
In our study, we show how to remove hydroxyl groups from bio‐based aldaric acids (Figure 1, hatched square) in a highly selective heterogeneous process, producing muconic acid which has a high potential to be used in the polymer industry. [38] According to literature data, we first tested several homogeneous catalysts. As they cannot be easily recycled from the reaction mixture, we focused on Re‐based heterogeneous catalysts. To hydrogenate the double bonds in muconic acid and obtain adipic acid, a combination of Re/Pd or Re/Pt was further tested. To elucidate the mechanism of the reaction, density functional theory calculations were performed.
To the best of our knowledge, this is the first process of heterogeneous conversion of an aldaric acid to adipic acid. The use of a heterogeneous catalyst allows for an easy separation from the reaction mixture, while the efficiency of the process makes it suitable for use on bio‐based feedstocks. The prospect of simple catalyst recycling or fixation in a fixed‐bed flow reactor, facilitating the transfer from a batch to continuous process, makes the heterogeneous process desired by the chemical industry.
Results and Discussion
Carboxylic acid group protection with a solvent
In our previous study, [39] lactones readily formed from mucic acid even at room temperature in an inert atmosphere under aqueous conditions. To prevent the formation of lactones and to protect the carboxylic groups, methanol was used as a solvent. This also increased the solubility of the intermediates and products.
Upon dissolution in acidified methanol (1.25 M HCl), dimethyl esters rapidly began to form. At 323 K, esterification was complete in less than 30 minutes regardless of the short‐chained alcohol used (methanol, ethanol, 1‐propanol, 1‐butanol and 1‐pentanol), as schematically shown in Figure SI4. In non‐acidified alcohols, elevated temperatures were required for esterification. In all subsequent reactions, the temperature was high enough to form esters and to prevent lactone formation (they were never detected) even without the addition of an acid. However, low solubility and immediate esterification make it difficult to monitor the actual conversion of the reactant in the reaction mixture, therefore all the result shown below are based on the yields of dehydroxylated products quantified by GC calibrated with the external standards.
Homogeneous catalysis
Based on the literature data, we first tested three oxo‐rhenium complexes (MeReO3, KReO4, and HReO4) for dehydroxylation of mucic acid dissolved in MeOH, varying the temperature (100–140 °C), reaction time (up to 72 h), amount of catalyst (5–15 mol %), atmosphere (H2 and N2), and pressure (0.5–1.0 MPa). For a detailed list of the experiments performed, the reader is referred to the SI (Table SI1–SI3), while the results are listed in Table SI5 and summarized in Figure 2.
Figure 2.

Total yields of mucic acid dehydroxylation detected by GCMS after 72 h a) at various temperatures, atmospheres, loadings of CH3ReO3 and hydrogenation co‐catalysts; b) using different homogeneous Re‐based catalysts. Color code as in Figure 1, details in Table S1, experiment 6M represents the base conditions at 120 °C, 0.5 MPa H2 and 12 wt. % of CH3ReO3 catalyst.
As the reaction is rather slow having a yield of dehydroxylated products less than 5 % after 4 hours, most experiments were run for 72 hours with yields of up to 55 %, which is higher than in the study of Larson. [7] The effect of the solvent (EtOH, i‐PrOH, n‐BuOH) on the conversion and selectivity was low, warranting the use of readily available MeOH. The yields were higher at 120 °C than at 100 or 140 °C and comparable with Asikainen's patent. [34]
At elevated temperatures short‐chain carboxylic acids, partially deoxygenated products and furanics were formed. While H2 is required for hydrogenation of the dehydroxylated product (muconate), dehydroxylation itself occurs also in an inert atmosphere (N2). Yields of saturated adipate are strongly dependent on the hydrogen pressure. The increase of catalyst loading yields higher concentrations of products within the whole tested range (0–18 wt %). The yields of hydrogenated products increased when a heterogeneous hydrogenation catalyst was added (Pt/C or Pd/C). Pt/C improved the overall conversion, while Pd/C significantly improved the selectivity towards fully hydrogenated adipate, which was also demonstrated by Larson et al. [7] The other two homogeneous rhenium catalysts (HReO4 and KReO4) performed similarly. Among the test variations, the highest yields of dimethyl adipate (21.2 % and 19.2 %, respectively) were recorded with 10 mol % KReO4 + 15 wt % Pd/C at 120 °C and 0.5 MPa H2 and 10 mol % MeReO4 + 15 wt % Pd/C at 120 °C and 0.5 MPa H2. An increase in the hydrogen pressure would further increase the yield. Based on the experimental data, the following reaction pathway is postulated (dotted box in Figure 1 and Figure 7). Initially, a carboxylic acid forms a methyl ester, which undergoes catalytic transformations. First, either two adjacent or two outer hydroxyl groups are cleaved off, leading to the formation of a double bond. After the remaining OH groups are removed, yielding dimethyl muconate, the double bonds are quickly saturated and dimethyl adipate forms which can be easily hydrolyzed into adipic acid. The mechanism of homogeneous rhenium‐mediated dehydroxylation postulates an interconversion of Rh(+7) and Rh(+5) during the process, [10] while the heterogeneous mechanism is explained later on in this work.
Figure 7.

Mechanism of dehydroxylation of mucic acid (in a diester form) and hydrogenation of the ensuing muconate on Re(0001). The most probable pathway shown with bolded arrows with shaded compounds. The activation barriers, ΔG ≠ and reaction energies, ΔG, (in parenthesis) for each elementary step in eV (lateral interaction between co‐adsorbed products/reactants included). Free H* and OH*, involved in the reaction, not shown for clarity. For ZPE‐corrected energies, see Table S4 in the SI.
As there are 10 isomers of aldaric acids differing in the orientation of OH groups, [40] we also tested glucaric acid (Table SI3), which differs from mucic acid in the orientation on one chiral center. This significantly affects the reaction (Figure SI5). Despite extremely low conversions, the selectivity to adipate was nearly 100 %. A strong effect of the OH group orientation on yields has been identified before[ 5 , 21 , 41 ] and is even more pronounced with heterogeneous catalysts (vide infra).
Heterogeneous catalysis
Despite several studies extoling the high selectivity of homogeneous Re complexes for dehydroxylation of short chain polyalcohols and sugar based dicarboxylic acids,[ 4 , 21 , 25 , 35 ] a heterogeneous process is preferred. Thus, we tested myriad heterogeneous Re catalysts, varying the support, process gas, reaction conditions and co‐catalysts.
As yields on alumina‐supported and silica‐supported catalysts were negligible (Exp. 25M‐28M in Table SI5), only carbon‐supported catalysts were investigated further. Acidic supports (alumina) preferentially catalyzed decarboxylation and hydrodeoxygenation, which led to a loss of outer ester groups, cyclization and the formation of short‐chain products. In a reducing (H2) atmosphere, total yields at 120 °C were similar to yields at 140 °C in all cases. In fact, higher temperature accelerates the hydrogenation but not the dehydroxylation, merely shifting the selectivity towards more saturated hydrocarbons. Hydrogen pressure also increased the rate of hydrogenation due to higher hydrogen availability in the liquid phase and on the catalyst surface, [42] resulting in the formation of more dimethyl adipate.
Water has a deleterious effect on the reaction, as proved by a significant increase in total and adipate yields (from 62.2 % to 83.5 %, and 2.9 % to 19.0 %, respectively) when the catalyst had been dried before the reaction. Reducing the catalyst before the reaction had little effect on the formed product distribution (selectivity), but significantly increases the total yield, hence the reaction rate (Figure 3).
Figure 3.
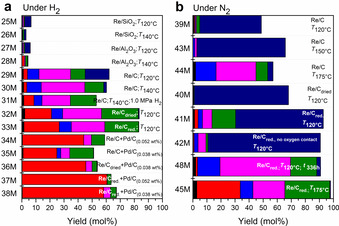
Total yields after dehydroxylation and hydrogenation of mucic acid over heterogeneous catalysts a) under H2 atmosphere; b) under N2 atmosphere. Reaction conditions (unless specified otherwise) were: the temperature of 120 °C, reaction time 72 h, total pressure of 0.5 MPa. Color code as in Figure 1. All yields of dehydroxylated products were obtained by GCMS, while NMR was used to determine the absence or presence of the reactant and/or secondary hydroxyl groups.
Selectivity toward adipate can be further improved with Pd as a co‐catalyst as it was reported to provide sufficient hydrogen affinity, hence promoting hydrogenation. [43] Its addition as separate Pd/C particles also significantly increased the yield of dimethyl adipate without affecting the conversion. The highest yield was obtained with a reduced 0.42 wt % Re/C+0.052 wt % Pd/C catalyst at 120 °C and 0.5 MPa H2, reaching 60.5 % for adipate with the yield of all dehydroxylated products of 63.6 %. Pd improved the yields of saturated products to the point of almost perfect selectivity without having to increase hydrogen pressure, a huge advantage from the industrial point of view. However, solely Pt/C, Pd/C or C did not show any dehydroxylation activity.
In an inert atmosphere, mostly dimethyl muconate forms on Re/C. The yield is markedly increased if the catalyst is dried and/or reduced prior to the reaction (Figure 5). However, small amounts of (partially) saturated products are also detected, especially when using the pre‐reduced catalyst. In the gaseous phase, high amounts of hydrogen were detected when using a pre‐reduced catalyst (see Figure SI6) at high temperature (175 °C), resulting in dimethyl adipate, which started forming after the first double bond (either on second or third place) was saturated (see Figure 4 b for concentrations and Table SI6 for yields). The hydrogen source is methanol, which is readily oxidized into formaldehyde on Re. Subsequently, it undergoes C−C coupling and etherification with the solvent (see Figure SI7).
Figure 5.

Catalyst recycling tests done with Re/C catalyst, when catalyst was untreated before experiment, reduced only before first experiment and reduced before each experiment.RKC conditions same as in the exp. 39M, except the prereduction of the catalys, which was specified for each run. Colour code as in Figure 1.
Figure 4.

Time profile of the products with standard deviation during the a) long‐term experiment at 120 °C (reaction conditions as in the exp. 48 M) and b) 72 h experiment at 175 °C (reaction conditions as in the exp. 45M). Color code as in Figure 7. Muconic acid methyl ester (blue; all diastereomers), 2‐Hexenedioic acid methyl ester (green; Z and E), 3‐Hexenedioic acid methyl ester (violet; Z and E) and dimethyl adipate (red).
The total yields increased at higher temperatures (Table SI6, Exp. 45M), reaching 98 % at 175 °C, which is to the best of our knowledge the highest published yield of any bio‐based nylon precursor. NMR analyses (see the SI for details) of the final product confirmed that the dehydroxylation of mucic acid ester was complete (Figure SI6), while the presence of secondary hydroxyl groups in other products was still determined (Figure SI9, experiment 39M).
Analogously to the homogeneous process, other short chain alcohols can be used. In an inert atmosphere, 2‐propanol and 1‐butanol showed a high conversion to esters and similar total yields (Table SI6, Exp. 46M‐47M).
In order to validate the long‐term dehydroxylation and hydrogenation performance of the catalyst at low temperature in an inert atmosphere, a 336‐hour test was performed at 120 °C over a pre‐reduced Re/C catalyst. The highest yield of dimethyl muconate (non‐saturated product) was measured after 60 hours. Over the course of the reaction, it completely converted into partially hydrogenated (one double bond) products (3–6), as shown in the transient profile in Figure 4 a. At this temperature, hexenedioic acid methyl esters with only one double bond (predominantly located on the third carbon in Z configuration) were ultimately yielded.
Further hydrogenation to saturated product (adipate) had negligible reaction rate, which is in accordance with our DFT study (Figure 7) that also predicted higher energy barrier for hydrogenation of hexenedioate compared to muconate. The long‐term experiment at 120 °C demonstrated that even without external H2 yields of dimethyl muconate can exceed 80 % (at 60 h), while hexenedioic acid methyl esters can be yielded with nearly 93 % selectivity (at 270 h). In case that a complete saturation of double bonds is required, the temperature should be elevated, as demonstrated in Figure 4 b.
Recycling tests, performed by four consecutive experiments after separating the catalyst from the mixture, washing, and drying it, showed that the catalyst activity starts to decrease. When the catalyst was unreduced, the yield of dehydroxylated products dropped by 60 % in the next experimental run, to less than 10 % after the fourth run. The hydrogenation of double bonds dropped to zero after the first run. Although the yield of dehydroxylated products remains the same for two consecutive runs, the distribution of the products shows a significant decrease in catalyst activity for hydrogenation. The activity of the catalyst started to decrease significantly after the second run, regardless of the catalyst reduction after each experiment (Figure 5).
This is explained by a slow oxidation of the catalyst, which makes it more soluble in the liquid phase. In addition to increasing catalyst activity, the reduction also increases its stability. The ICP‐MS (Inductively coupled plasma mass spectrometry) analysis (detailed explanation in SI) showed that the loading of Re in the pre‐reduced catalysts dropped by 8–14 % after one experiment. The leaching of active metal from solid surface was also observed by Sharkey and Jentoff. [44] The reduced form of the Re (+3, 0) is less soluble compared to the oxidized (+6 and +7), thus the leaching was more significant when unreduced catalyst was used. The optimisation of support selection and Re‐impregnation protocol should be a subject of further research, alongside with the stability tests.
Re/C catalyst characterization
Activity of a catalyst is determined by several factors, such as surface area, presence of the impurities on the surface, active metal oxidation state, exposure of certain facets due to particle size and other factors. Rhenium particles are much larger after reduction in H2 at 400 °C for 3 h (d Re‐avg=4.7 nm) in comparison to the fresh sample (d Re‐avg<1 nm) as observed from Figures 6 a–c. In fact, the rhenium in the fresh Re/C sample is in the form of finely dispersed rhenium oxide in the range of the size of single molecule. From the XPS measurements we observe on average an almost 6‐times increase of the rhenium content in the reduced sample due to an oxygen coverage on the oxidized rhenium (Table 1). We can observe that for the fresh sample and Re/C sample after reduction and oxidation, that rhenium on the surface (≈3 nm in depth) is in the form of ReO3, while for the reduced sample it is mainly in the form of elementary Re and Re2O3. Additional information about the XPS Re 4f spectra deconvolution can be found in SI. The H2 consumption from H2 TPR is displayed in Table 1 along the BET surface area. There was no consumption of H2 observed in the reduced sample, therefore the presence of Re3+ in XPS could be due to slight exposure to air during the insertion into the vacuum chamber prior to the XPS analysis. Re reduction peak area for the Re/C red&ox (369 μmol g−1) is immensely smaller than for the fresh sample (1311 μmol g−1) as observed in Table 1. With the combination of the XPS results, this indicates Re‐ReO3 core–shell structure of the red&ox sample. H2 consumption quantification for rhenium reduction is problematic due to a simultaneous formation of CH4 and CO because of the decomposition of the carbon support. The reader is referred to the SI about the H2 TPR details. BET surface area of the fresh catalyst is equal to 649 m2 g−1. Using XRD analyses (in SI) we could not find presence of rhenium species; however, we detected the presence of SiO2 and graphitic phase.
Figure 6.

Comparison between a) fresh (dark field mode) and b) reduced sample (bright field mode). The bright spots on the fresh samples and the dark spots on the reduced catalyst represent rhenium particles. c) Particle size distribution determined using TEM. d) The Re is reduced to elementary state and the sample exposed to air for several hours is in the same oxidation as the fresh catalyst.
Table 1.
Catalyst properties under different treatments.
|
|
Re/C fresh |
Re/C reduced |
Re/C red&ox |
|---|---|---|---|
|
Re content XPS (4f7/2) [wt %] |
0.34 |
2.1 |
0.41 |
|
H2 consumption up to 400 °C [μmol gcat −1] |
1311 |
0 |
369 |
|
CRe [μmol gcat −1]‐CO TPD/ CO pulse ads. |
[a] |
14.7/ 24.7 |
[a] |
[a] Only applicable for Re in metallic state.
It is observed that MeOH can indeed reduce oxidized rhenium at 175 °C as determined by oxidation in O2 followed by a treatment in MeOH vapor and H2 TPR described in SI.
From the experimental observations (Exp. 45M), the pre‐reduced Re/C catalyst is the most active catalyst due to rhenium in reduced state or/and increased Re particles size and therefore increased surfaces of regular facets of Re metal. Additionally, DFT observations confirm that the Re(0001) facet is active for mucic acid (in diester form) dehydroxylation and methanol dehydrogenation. For this reason, the metallic site density had been measured for Re using CO‐TPD and confirmed CO pulse adsorption (Table 1). As seen from CO‐TPD (Figure SI15) there is one minor peak between 40 °C and 250 °C which corresponds to the desorption from rhenium,[ 45 , 46 ] while the larger peak above 400 °C corresponds to the decomposition of functional groups on carbon support.[ 47 , 48 ]
The concentration of surface sites obtained from CO‐TPD represents only the highly coordinated rhenium atoms (i.e. Re(0001)) and not atoms which are part of steps where CO desorbs at higher temperature (250 °C–700 °C). [45] Re surface site density from CO‐TPD is also lower than the concentration from pulse CO adsorption due possible adsorption on carbon support.
The catalyst, which did not come into contact with air after reduction (Exp. 42M), showed very similar performance to the catalyst being exposed to air during the transfer from reduction oven (Exp. 41M), with dehydroxylation degree of 90.6 % and 92.8 % respectively. However, we can observe an around 3‐times higher yield of hydrogenated products in the case of partially oxidized catalyst (30.4 %) in comparison with non‐oxidized sample (yield 10.7 %). As observed using DFT, adsorbed OH is able to promote methanol decomposition causing a larger coverage of H species on the catalyst surface. Owing to the present H species, oxygen from ReOx is hydrogenated to form OH species which additionally promote hydrogen formation and subsequently hydrogenation of double bonds on dehydroxylated products. The catalyst does not oxidize during the transfer, however, extensive purging of the catalyst with air (in a TPR apparatus) leads to an increase in the Re oxidation state and a subsequent decrease of the catalyst's activity.
Activity tests also revealed that initial Re/C pre‐reduction is crucial to improve the stability, while further reduction and air exposure cycles are disadvantageous. Further studies are required (beyond the aim of this work) to tune the catalyst synthesis and activation protocols for this group of catalysts for their long‐term activity, stability against deactivation and to further increase their performance. The catalyst stability could be improved by controlling the metal‐support interactions as proposed by Deelen et al. [49]
Dehydroxylation mechanism and DFT calculations
DFT calculations on a four‐layer Ru(0001) surface to shed more light on the mechanism were performed using vasp. For detailed information on the model used and how the zero‐point energy (ZPE) corrections, entropic contributions and the Gibbs free energies (at 120 °C) were calculated, the reader is referred to the SI. As shown in Figure 7, dimethyl ester of mucic acid (1 a) dehydroxylates by cleaving the C−O bonds of the hydroxyl groups. The reactions are fast (ΔG ≠ around 0.4–0.6 eV) and exothermic. After the OHα group is cleaved off first, it is followed by the removal of the OHβ and not by immediate hydrogenation of the CαH radical, which has a higher barrier and is endothermic. A direct consequence of the stereochemistry, only two adjacent OH* groups cleave off as the other two are not in contact with the catalyst (see Figure 8 a). For further dehydroxylation, the molecule (1 b) must desorb and re‐adsorb with the remaining OH groups coming into contact with the catalyst. In the last two steps, the γ‐ and δ‐OH groups are removed and dimethyl muconate is formed (2). Note that due to the limited accuracy of DFT calculations, geometric isomerism (cis, trans) was not taken into account.
Figure 8.

Due to chirality, mucic acid can only bind with two hydroxyl groups in contact with the catalyst structure (a). Methanol molecule (b) is a better hydrogen donor than H* on Re(0001); c) Gibbs free energy surface for methanol decomposition on Re(0001) at 120 °C from DFT calculations.
Unsaturated muconic acid ester is hydrogenated step‐wise. When reacting with the adsorbed H*, most reaction steps are endothermic and with high barriers (>1 eV). First, Cα and then Cβ are hydrogenated, yielding (3) and (4) rather than (5) or (6) (again, we do not distinguish geometric isomers), which is consistent with experimental observations (see the yields in Table SI6). Later on, Cδ and Cγ hydrogenate, yielding an adipate (7). Rather higher activation barriers and endothermicity of the hydrogenation are mirrored by low yields of the fully saturated product (7) on heterogeneous Re catalysts. However, hydrogenation of multiple bonds on Pd(111) is known to be exothermic and have lower barriers, [50] explaining much higher yields of (7) when adding Pd/C.
The overall yield of unsaturated products under comparable conditions is slightly lower in an inert atmosphere because nitrogen actually binds quite strongly to the catalyst (from DFT calculations: E ads=−0.63 eV), decreasing the number of accessible active sites. DFT calculations confirm that CH3OH acts as a hydrogen source on metallic Re (see Figure 8 c and Table 2). Upon a moderately strong adsorption (E ads=−0.37 eV), methanol readily gives off the hydroxylic hydrogen (ΔG ≠=0.25 eV) in an exothermic reaction (Figure 8 c). The ensuing H* can recombine to form H2 but this endothermic reaction has a high Gibbs barrier of 0.99 eV. Therefore, H* will hydrogenate the double bonds of muconic acid whenever possible. CH3O* can lose an aliphatic hydrogen in a moderately endothermic reaction (ΔG=+0.09 eV) upon overcoming a Gibbs barrier of 0.71 eV, yielding CH2O*. A potential energy surface diagram of the entire catalytic cycle is shown in Figure S3.
Table 2.
Calculated Gibbs free reaction energies and activation barriers for methanol decomposition and hydrogen adsorption at 120 °C. For ZPE‐corrected energies, see Table S4 in the SI.
|
# |
Reaction |
ΔG [eV] |
ΔG ≠ [eV] |
|---|---|---|---|
|
1 |
CH3OH → CH3OH* |
−0.26 |
n/a |
|
2 |
CH2O → CH2O* |
−0.69 |
n/a |
|
3a |
CH3OH* → CH3O* + H* |
−0.70 |
0.25 |
|
3b |
CH3OH* + OH* → CH3O* + H2O* |
−0.01 |
0.05 |
|
4a |
CH3O* → CH2O* + H* |
+0.09 |
0.71 |
|
4b |
CH3O* + OH* → CH2O* + H2O* |
−0.20 |
1.07 |
|
5 |
CH3O* + CH3O* → CH2O* + CH3OH* |
+0.00 |
0.95 |
|
6 |
OH* + H* → H2O* |
+0.87 |
1.20 |
|
7 |
H* + H* → H2* |
+0.75 |
0.99 |
|
8 |
H2 → H2* |
−0.01 |
n/a |
|
9 |
N2 → N2* |
−0.07 |
n/a |
|
10 |
H2O → H2O* |
+0.35 |
n/a |
After dehydroxylation, the OH* species is also present on the surface. It facilitates the oxidation of methanol, lowering the barrier for the decomposition of CH3OH* to a meagre 0.05 eV. Also, the OH‐assisted decomposition of CH3O* to CH2O* and H2O* becomes exothermic (ΔG=−0.20 eV) thanks to a strong attractive interaction between the co‐adsorbed products, although the barrier is higher (ΔG ≠=1.07 eV vs. 0.71 for the unassisted route). Lastly, it is also possible for two co‐adsorbed CH3O* to disproportionate into CH2O* and CH3OH*(ΔG ≠=0.95 eV, ΔG=0.00 eV).
CH2O* readily undergoes further aldol condensation reactions because it is strongly bound to the surface (E ads=−1.41 eV) and unlikely to desorb (ΔG ads 120 °C=−0.69 eV). Experimentally, this manifests as a number of long‐chain ethers and alcohols as the products of the aldol condensation and subsequent hydrogenation. For strongly adsorbed species, adsorbate‐adsorbate interactions are usually considerable, decreasing the adsorption energies as the coverage increases and preventing total saturation of the surface. For instance, the lateral interaction for two co‐adsorbed CH2O molecules is calculated to be +0.47 eV. Despite occupying multiple active sites, C6 species analogously have large adsorption energies and strong lateral interactions.
Furthermore, it is possible for CH3OH* to directly hydrogenate double bonds, as shown in Figure 8 b. For instance, the calculated barrier for the first hydrogenation of muconic acid is lowered from 1.05 eV (with H*, Figure SI3a) to 0.66 eV when CH3OH directly donates its hydroxylic hydrogen. Moreover, such reaction is exothermic (ΔG=−0.25 eV), as shown in Figure SI3b. When CH3O is the hydrogen donor for muconic acid, the barrier is increased to 1.15 eV and the reaction is endothermic (ΔG=+0.39 eV). This explains why the reaction proceeds without Pd/C and even in an inert atmosphere, albeit slower and to a lesser extent.
In short, while the elementary step of dehydroxylation is a straight‐forward C−O bond scission, there are multiple competing hydrogen sources. Surface H* forms from the dissociative adsorption of H2 or, under inert atmosphere, from the dissociation of CH3OH. The latter can also directly hydrogenate double bonds. Both cases are shown in Figure S3. In reality, a strict delineation is not sensible because the pathways compete and cross over. A full microkinetic study would be required to ascertain the contribution of individual reaction pathway under the given conditions on Re(0001). That being said, Re(0001) is the most stable reduced rhenium surface but it is conceivable that there are also other facets and defects present. On undercoordinated surface terminations and defects, reaction barriers are even lower, making the catalyst more active [51] and changing the contributions of different reaction pathways. Such modelling is beyond the scope of this work.
While it is known that DFT values, especially when using the inexpensive generalized gradient approximation, must be always taken cum grano salis, the trends can be trusted for metallic surfaces. However, the solvent effect is another factor which is often omitted in theoretical heterogeneous catalysis because of its computational cost. While a full quantum sampling of hundreds of solvent molecules remains intractable, most intermediate approaches focus on aqueous phases. For the C−H bond cleavage, where hydrogen bonding does not play an important role, the effect is negligible (ΔΔG ≠=−0.16 eV for ethylene glycol on Pt(111)). With strong hydrogen bonding (such as for proton migration reactions), the effect is up to −0.50 eV. [52] In this work, the reaction proceeds in methanol, which can form half as many hydrogen bonds as water and has a dielectric constant of 33.3 (as opposed to 78.4 for water), meaning that the effect will be smaller.
Conclusion
This study presents successful selective dehydroxylation of an aldaric acid (mucic acid) over rhenium species (via homogeneous and heterogeneous catalysis) to bio‐based nylon precursor and similar industrially desirable polymer products. Using homogeneous catalysis, we were able to achieve up to 55 mol % yield of the desired products, which was in line with previously published literature and patents. However, a significant breakthrough was achieved with the use of heterogeneous catalysis (Re/C), as a 98 mol % product(s) yield was achieved with complete conversion (based on NMR analysis).
Reducing the catalyst (Re/C) before the reaction and avoiding contact with air (oxidizing atmosphere) have proven to be crucial contributions to achieving higher yields of desired products. Extensive characterization of the catalyst revealed that the most active catalyst form (reduced one) has a lower oxidation state (0 or +3), while a higher oxidation state (+6; oxidized form) is associated with a lower catalyst (dehydroxylation) activity and higher solubility of the rhenium species in the liquid phase. Reducing the catalyst increases its activity and extends its stability for several consecutive experimental tests.
The study showed that the selective production of dimethyl adipate (adipic acid precursor) does not require an external source of hydrogen, normally used for hydrogenation of double bonds produced by dehydroxylation. Methanol used as solvent acts as a hydrogen donor. Dehydroxylated product with two double bonds is thus selectively obtained at lower temperature (120 °C), while dimethyl adipate is selectively obtained at higher temperature (175 °C). At higher temperatures, the oxidation of methanol to formaldehyde is more considerable, with hydrogen being released (detected in the gas phase).
The mechanism of hydrogen generation from the solvent was supported by extensive theoretical calculations (DFT). On Re(0001), methanol readily gives off its hydroxylic hydrogen in a low‐barrier (0.25 eV) exothermic reaction (−0.70 eV), which serves as the hydrogen source. In the second mildly endothermic step, the ensuing CH3O* gives off the aliphatic hydrogen and is ultimately oxidized to formaldehyde, which can undergo further polymerization reactions. In the reaction with a spectator OH* species, this oxidation to formaldehyde becomes exothermic.
Esters of mucic acid undergo dehydroxylation by cleaving the C−O bonds in fast and exothermic reactions, which explains the fast formation of hexadienoates. Hydrogenation of the newly formed double bonds is a slower endothermic reaction. Due to stereochemistry of mucic acid, only two adjacent hydroxyl groups can be cleaved off in a heterogeneous surface process at a time.
It is noteworthy that the process is green, because a solid, reusable catalyst is used, and the solvent (methanol or other short‐chain alcohols, also the internal source of hydrogen) can also be recovered and reused. However, catalyst recycling is still an area that needs improvements to achieve higher stability and, consequently, life‐cycle performance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the Slovenian Research Agency (ARRS) under core funding P2‐0152 and postdoctoral project Z2‐9200. B. H. acknowledges funding from the Young Researchers Programme from ARRS. Dr. Janez Kovač is acknowledged for XPS measurement. Brett Pomeroy and Florian Harth are acknowledged for English proofreading. Ab initio calculations were performed on the computational resources provided by the National Institute of Chemistry (Ljubljana).
B. Hočevar, A. Prašnikar, M. Huš, M. Grilc, B. Likozar, Angew. Chem. Int. Ed. 2021, 60, 1244.
Contributor Information
Matej Huš, Email: matej.hus@ki.si.
Prof. Miha Grilc, Email: miha.grilc@ki.si.
Prof. Blaž Likozar, Email: blaz.likozar@ki.si.
References
- 1. Kawaguchi H., Hasunuma T., Ogino C., Kondo A., Curr. Opin. Biotechnol. 2016, 42, 30–39. [DOI] [PubMed] [Google Scholar]
- 2.“Dehydration Catalysts, Particularly for the Dehydration of Diols”: Weisang J., Szabo G., Maurin J., US3862964A, 1973.
- 3. Boucher-Jacobs C., Nicholas K. M. in Selective Catalysis for Renewable Feedstocks and Chemicals (Ed.: Nicholas K. M.), Springer International Publishing, Cham, 2014, pp. 163–184. [Google Scholar]
- 4. Arceo E., Ellman J. A., Bergman R. G., J. Am. Chem. Soc. 2010, 132, 11408–11409. [DOI] [PubMed] [Google Scholar]
- 5. Raju S., Moret M.-E., Klein Gebbink R. J. M., ACS Catal. 2015, 5, 281–300. [Google Scholar]
- 6. Herrmann W. A., Herdtweck E., Flöel M., Kulpe J., Küsthardt U., Okuda J., Polyhedron 1987, 6, 1165–1182. [Google Scholar]
- 7. Larson R. T., Samant A., Chen J., Lee W., Bohn M. A., Ohlmann D. M., Zuend S. J., Toste F. D., J. Am. Chem. Soc. 2017, 139, 14001–14004. [DOI] [PubMed] [Google Scholar]
- 8. Yanagi T., Suzuki H., Oishi M., Chem. Lett. 2013, 42, 1403–1405. [Google Scholar]
- 9. Ziegler J. E., Zdilla M. J., Evans A. J., Abu-Omar M. M., Inorg. Chem. 2009, 48, 9998–10000. [DOI] [PubMed] [Google Scholar]
- 10. Shiramizu M., Toste F. D., Angew. Chem. Int. Ed. 2012, 51, 8082–8086; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8206–8210. [Google Scholar]
- 11. Yi J., Liu S., Abu-Omar M. M., ChemSusChem 2012, 5, 1401–1404. [DOI] [PubMed] [Google Scholar]
- 12. Metzger J. O., ChemCatChem 2013, 5, 680–682. [Google Scholar]
- 13. Dutta S., ChemSusChem 2012, 5, 2125–2127. [DOI] [PubMed] [Google Scholar]
- 14. Makshina E. V., Dusselier M., Janssens W., Degrève J., Jacobs P. A., Sels B. F., Chem. Soc. Rev. 2014, 43, 7917–7953. [DOI] [PubMed] [Google Scholar]
- 15. Gable K. P., Brown E. C., J. Am. Chem. Soc. 2003, 125, 11018–11026. [DOI] [PubMed] [Google Scholar]
- 16. Gable K. P., Phan T. N., J. Am. Chem. Soc. 1994, 116, 833–839. [Google Scholar]
- 17. Gable K. P., Juliette J. J. J., J. Am. Chem. Soc. 1995, 117, 955–962. [Google Scholar]
- 18. Gable K. P., Ross B., Feedstocks for the Future, American Chemical Society, Washington, 2006, pp. 143–155. [Google Scholar]
- 19. Vkuturi S., Chapman G., Ahmad I., Nicholas K. M., Inorg. Chem. 2010, 49, 4744–4746. [DOI] [PubMed] [Google Scholar]
- 20. Shiramizu M., Toste F. D., Angew. Chem. Int. Ed. 2013, 52, 12905–12909; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13143–13147. [Google Scholar]
- 21. Dethlefsen J. R., Fristrup P., ChemSusChem 2015, 8, 767–775. [DOI] [PubMed] [Google Scholar]
- 22. Gossett J., Srivastava R., Tetrahedron Lett. 2017, 58, 3760–3763. [Google Scholar]
- 23. Denning A. L., Dang H., Liu Z., Nicholas K. M., Jentoft F. C., ChemCatChem 2013, 5, 3567–3570. [Google Scholar]
- 24. Korstanje T. J., de Waard E. F., Jastrzebski J. T. B. H., Klein Gebbink R. J. M., ACS Catal. 2012, 2, 2173–2181. [Google Scholar]
- 25. Korstanje T. J., Jastrzebski J. T. B. H., Klein Gebbink R. J. M., Chem. Eur. J. 2013, 19, 13224–13234. [DOI] [PubMed] [Google Scholar]
- 26. Yoshinao N., Sibao L., Masazumi T., Keiichi T., ChemSusChem 2015, 8, 1114–1132.25711481 [Google Scholar]
- 27. Ota N., Tamura M., Nakagawa Y., Okumura K., Tomishige K., Angew. Chem. Int. Ed. 2015, 54, 1897–1900; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1917–1920. [Google Scholar]
- 28. Tazawa S., Ota N., Tamura M., Nakagawa Y., Okumura K., Tomishige K., ACS Catal. 2016, 6, 6393–6397. [Google Scholar]
- 29. Shakeri J., Hadadzadeh H., Farrokhpour H., Joshaghani M., Weil M., J. Phys. Chem. A 2017, 121, 8688–8696. [DOI] [PubMed] [Google Scholar]
- 30. Xi Y., Yang W., Ammal S. C., Lauterbach J., Pagan-Torres Y., Heyden A., Catal. Sci. Technol. 2018, 8, 5750–5762. [Google Scholar]
- 31. Vilé G., Albani D., Nachtegaal M., Chen Z., Dontsova D., Antonietti M., López N., Pérez-Ramírez J., Angew. Chem. Int. Ed. 2015, 54, 11265–11269; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11417–11422. [Google Scholar]
- 32. de Lederkremer R., Marino C., Adv. Carbohydr. Chem. Biochem. 2003, 58, 199–306. [DOI] [PubMed] [Google Scholar]
- 33.“Synthetic Process of Adipic Acid”: Hong C. H., Kim Y. G., Shin N., US9447012B2, 2016.
- 34. Asikainen M., Thomas D., Harlin A., United States Pat. Appl. US Patent No. US20170137363A1, 2017.
- 35. Li X., Wu D., Lu T., Yi G., Su H., Zhang Y., Angew. Chem. Int. Ed. 2014, 53, 4200–4204; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4284–4288. [Google Scholar]
- 36. Shin N., Kwon S., Moon S., Hong C. H., Kim Y. G., Tetrahedron 2017, 73, 4758–4765. [Google Scholar]
- 37. Kirilin A. V., Tokarev A. V., Manyar H., Hardacre C., Salmi T., Mikkola J.-P., Murzin D. Y., Catal. Today 2014, 223, 97–107. [Google Scholar]
- 38. Khalil I., Quintens G., Junkers T., Dusselier M., Green Chem. 2020, 22, 1517–1541. [Google Scholar]
- 39. Hočevar B., Grilc M., Likozar B., Catalysts 2019, 9, 286. [Google Scholar]
- 40. de Lederkremer R. M., Marino C., Advances in Carbohydrate Chemistry and Biochemistry, Academic Press, New York, 2003, pp. 199–306. [DOI] [PubMed] [Google Scholar]
- 41. Ota N., Tamura M., Nakagawa Y., Okumura K., Tomishige K., ACS Catal. 2016, 6, 3213–3226. [Google Scholar]
- 42. Nishimura S., Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis, Wiley, New York, 2001. [Google Scholar]
- 43. Rylander P. N., ULLMANN'S Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- 44. Sharkey B. E., Jentoft F. C., ACS Catal. 2019, 9, 11317–11328. [Google Scholar]
- 45. Ducros R., Fusy J., Jupille J., Pareja P., Tatarenko S., Appl. Surf. Sci. 1987, 29, 179–193. [Google Scholar]
- 46. Ducros R., Housley M., Piquard G., Alnot M., Surf. Sci. 1981, 108, 235–252. [Google Scholar]
- 47. Guerrero-Ruiz A., Badenes P., Rodríguez-Ramos I., Appl. Catal. A 1998, 173, 313–321. [Google Scholar]
- 48. Kunkes E. L., Simonetti D. A., Dumesic J. A., Pyrz W. D., Murillo L. E., Chen J. G., Buttrey D. J., J. Catal. 2008, 260, 164–177. [Google Scholar]
- 49. van Deelen T. W., Hernández Mejía C., de Jong K. P., Nat. Catal. 2019, 2, 955–970. [Google Scholar]
- 50. Ziegler T., Chem. Rev. 1991, 91, 651–667. [Google Scholar]
- 51. Kopač D., Likozar B., Huš M., Appl. Surf. Sci. 2019, 497, 143783. [Google Scholar]
- 52. Saleheen M., Heyden A., ACS Catal. 2018, 8, 2188–2194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary