

Abstract
Hydrosilylation reactions, which allow the addition of Si−H to C=C/C≡C bonds, are typically catalyzed by homogeneous noble metal catalysts (Pt, Rh, Ir, and Ru). Although excellent activity and selectivity can be obtained, the price, purification, and metal residues of these precious catalysts are problems in the silicone industry. Thus, a strong interest in more sustainable catalysts and for more economic processes exists. In this respect, recently disclosed hydrosilylations using catalysts based on earth‐abundant transition metals, for example, Fe, Co, Ni, and Mn, and heterogeneous catalysts (supported nanoparticles and single‐atom sites) are noteworthy. This minireview describes the recent advances in this field.
Keywords: alkenes, alkynes, hydrosilylation, silanes
This minireview describes recent advancements in catalysts for the hydrosilylation of olefins and alkynes. Emphasis is given on developments of alternative catalyst systems such as non‐noble metal complexes and heterogeneous catalysts.
1. Introduction
Considering silicon as the second most abundant element on earth and the considerable number of organosilicon compounds used in our daily life, organosilicon chemistry is of significant importance for the development of more sustainable and greener chemistry. In general, organosilicon compounds are characterized by their stable and inert carbon–silicon bonds.[ 1 , 2 , 3 ] In particular, organosilanes, organosilyl halides, and the corresponding ethers are readily available and offer straightforward possibilities for versatile functionalizations. Compared to their ordinary pure carbon analogues, organosilicons have complementary physical properties, which make them attractive for a variety of industrial applications. Hence, organosilicon compounds are widely found in adhesives and coatings and used as oils, rubbers, and resins. [4]
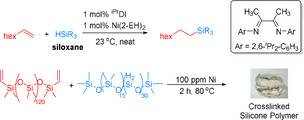
Among the different methods available for the creation of C−Si bonds, catalytic hydrosilylations allow the straightforward addition of silanes (Si−H) to multiple bonds, for example, olefins and alkynes.[ 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 ] These reactions are not only used on the laboratory scale, but have been also implemented in the chemical industry for the production of functional organosilicon compounds. In fact, they have been proven to be one of the most efficient reactions in the silicone industry. Theoretically, hydrosilylations are 100 % atomic economic without generating other products or wastes. Performing hydrosilylations with functionalized silanes (Figure 1 a) offers direct prospects to modify the properties of polymers or inorganic materials. For example, poly(dimethylsiloxane), an important kind of silicone rubber, can be functionalized by crosslinking with another vinyl silicon reagent (Figure 1 b). [16] Moreover, organosilicon compounds offer versatile properties as bonding or bridging agents in the preparation of composites from organic polymers and inorganic materials, for example, glass, minerals, and metal oxides. [17] Thus, apart from their importance for applications in the silicone industry, hydrosilyations are increasingly attractive for basic material sciences.[ 9 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 ]
Figure 1.
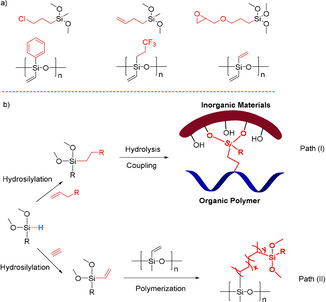
a) Selected functionalized silanes and silicone polymers; b) utilization of silane reagent: path (I): interfacial bonding by a silane coupling agent; path (II): crosslinking reaction between vinyl‐containing silane and silicone polymers.
Since the first hydrosilylation reaction appeared in the academic literature in 1947, platinum‐based catalysts dominated this area. [26] Originally, the introduction of Speier's catalyst (H2PtCl6) was a major breakthrough. Later on, Karstedt made an important contribution to this area by developing a platinum(0) complex containing vinyl‐siloxane ligands. [27] Today, this lipophilic complex represents an efficient benchmark catalyst in industrial hydrosilylation processes. Despite the efficiency of this system, obviously there are certain disadvantages of homogeneous Pt‐based catalysts in some applications. For example, platinum complexes can be easily trapped in the product and it is difficult to recover them due to the viscous properties of the resulting products.[ 28 , 29 , 30 , 31 , 32 ] Notably, it was estimated that consumption of platinum accounts for up to 30 % of the cost of silicones. [33] Hence, from an industrial point of view, the high price of platinum strongly motivates researchers to develop recyclable and less expensive catalysts to reduce the precious metal consumption.
The development and importance of hydrosilylation reactions has been discussed intensively in the scientific literature, and a number of comprehensive reviews, articles, and books were published mainly before 2015.[ 3 , 4 , 34 ] Since then, a variety of robust homogeneous catalysts with well‐defined ligands have been developed. [35] More recently, also heterogeneous catalysts evolved for this process. [36] Interestingly, heterogeneous single‐atom catalysts (SACs), which are considered to combine the advantages of molecular‐defined and heterogeneous catalysis, were disclosed and displayed comparable activities and selectivities to their homogeneous counterparts. [37]
This minireview covers the most important catalyst developments from the past five years (Figure 2). To make it easy for the reader, it is organized according to catalyst developments into two sections: a) the use homogeneous non‐noble metal complexes specifically Co, Fe, and Ni derivatives; b) the usage of recyclable heterogeneous catalysts focusing on supported noble/non‐noble nanoparticles (NP) and single‐atom catalysts. Finally, we will give indications for future progress in this field.
Figure 2.
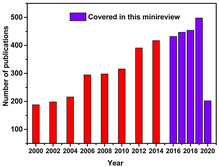
Number of publications using the term “hydrosilylation” from 2000 to May of 2020 according to the ISI Web of Science.
2. The Development of Homogeneous Non‐noble Metal Catalysts
The replacement of traditional platinum‐based catalysts in alkene hydrosilylations by more sustainable and economic metals is a long‐standing goal of the silicone industry.[ 31 , 38 , 39 , 40 , 41 ] Following this goal, several molecularly defined catalysts based on earth‐abundant metals (Fe, Co, Ni, et. al.) have been developed in the past decade.[ 5 , 6 , 7 , 9 , 13 , 42 , 43 , 44 , 45 , 46 ] Moreover, the regioselectivity can be controlled by modification of ligands leading to anti‐Markovnikov or Markovnikov selective products. The resulting silane products are widely used as silicon fluids and silicon curing agents. Furthermore, the anti‐Markovnikov silanes are also important moieties for life science applications.[ 47 , 48 , 49 ]
2.1. Catalysts with Monodentate Ligands
In 2017, Deng et al. reported cobalt N‐heterocyclic carbene (NHC)‐catalyzed anti‐Markovnikov hydrosilylations of aliphatic alkenes with tertiary silanes (Scheme 1). [50] Specifically, they developed a cobalt(II) amide/NHC catalyst system, which facilitated the selective hydrosilylation of monosubstituted aliphatic alkenes with HSi(OEt)3 to linear products in moderate to very high yields (42–98 %). However, when highly reactive Ph3SiH was used with 1‐octene, a lower yield of only 28 % was observed. According to mechanistic studies Co‐silyl intermediates are involved in the catalytic cycle with cobalt(I) species proposed as the active intermediates.
Scheme 1.
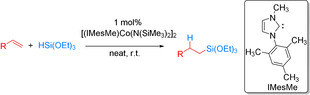
Hydrosilylation of aliphatic alkenes with tertiary silanes using a cobalt‐NHC catalyst. [50]
One year later, the same group disclosed that related CoI‐NHC complexes such as [(IAd)(PPh3)‐CoCl] and [(IMes)2CoCl] displayed distinct performance in catalyzing the reaction of diverse alkenes with Ph2SiH2 (Scheme 2). [51] Interestingly, [(IAd)(PPh3)CoCl] proved to be efficient in catalyzing anti‐Markovnikov hydrosilylation of Ph2SiH2 with both alkyl‐ and aryl‐substituted alkenes, while [(IMes)2CoCl] promoted Markovnikov hydrosilylation of Ph2SiH2 with aryl‐substituted alkenes. The authors explain the changed catalytic performance by the different steric nature of the carbene ligands IAd vs. IMes.
Scheme 2.
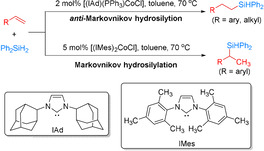
Catalytic behavior of cobalt(I)‐NHC complexes in the hydrosilylation of alkenes with diphenylsilane. [51]
Here, mechanistic studies suggested that cobalt(I) silyl species are the key active species for the hydrosilylation process. On the basis of their results, a preliminary mechanism of this transformation was proposed (Scheme 3): Initial reaction of cobalt(I)‐NHC chloride with Ph2SiH2 gives a cobalt(I) hydride intermediate, which interacts with Ph2SiH2 to form the corresponding cobalt(I) silyl intermediate and H2. Subsequent reaction of the cobalt(I) silyl intermediate with an alkene via migratory insertion forms a cobalt alkyl complex that further reacts with Ph2SiH2 to give the desired hydrosilylation products and regenerates the active cobalt(I) silyl species for the next catalytic cycle.
Scheme 3.
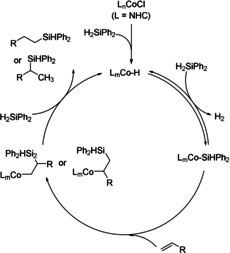
Proposed catalytic cycle for the Co‐NHC‐catalyzed hydrosilylation. [51]
In 2016, Petit and colleagues described the HCo(PMe3)4‐catalyzed highly regio‐ and stereoselective hydrosilylation of internal alkynes (Scheme 4). [52] The reaction was applied to a variety of hydrosilanes and symmetrical as well as unsymmetrical alkynes, giving in many cases a single hydrosilylation isomer in varying yields (19–96 %). The authors suggested that the regio‐ and stereocontrol of the reaction is predominantly governed by steric features of the substrates.
Scheme 4.
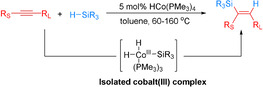
HCo(PMe3)4‐catalyzed highly regio‐ and stereoselective hydrosilylation of internal alkynes. [52]
According to mechanistic studies, dihydridocobalt species are most likely involved in this catalytic process (Scheme 5). Oxidative addition of the silane to a CoI hydride center forms the dihydridocobalt(III) intermediate, which undergoes alkyne insertion into one of the Co−H bonds after coordination of the alkyne. Direct reductive elimination releases the vinylsilane as the major product with the observed regioselectivity and regenerates the catalytically active hydridocobalt(I) species.
Scheme 5.
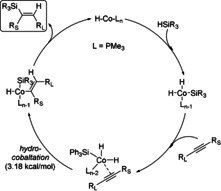
Proposed mechanism for HCo(PMe3)4‐catalyzed highly regio‐ and stereoselective hydrosilylation of internal alkynes. [52]
2.2. Catalysts with Bidentate Ligands
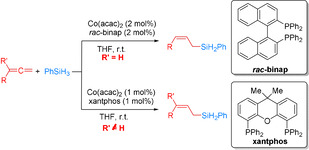
In 2017, Wang et al. developed a regiodivergent cobalt‐catalyzed hydrosilylation of alkenes by careful choice of ligands and hydrosilane substrates (Scheme 6). [53] More specifically, a Co(acac)2/Xantphos catalyst system showed excellent activity for Markovnikov hydrosilylation of styrene derivatives with PhSiH3, whereas the Co(acac)2/dppf catalyst system facilitated the anti‐Markovnikov hydrosilylation of styrene derivatives with Ph2SiH2. In contrast, utilizing aliphatic alkenes with PhSiH3, in the presence of Co(acac)2/mesPDI as the catalyst, produced branched organosilanes in high yields (50 %–98 %) with high to excellent regioselectivities (b/l ratio: 91:9 to >99:1), while the corresponding linear products were obtained with Co(acac)2 and Xantphos. Deuterium‐labeling studies support the classic Chalk–Harrod mechanism with a Co–H intermediate resulting from oxidative addition of the silane to the metal center for the cobalt/bisphosphine system. Interestingly, a modified mechanistic proposal with Co–Si intermediates has been suggested for the cobalt/pyridine‐2,6‐diimine system.
Scheme 6.
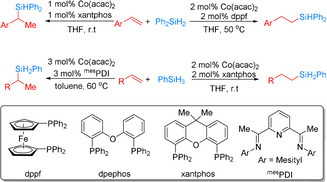
Ligand‐ and silane‐dependent cobalt‐catalyzed regiodivergent hydrosilylation of vinylarenes and aliphatic alkenes. [53] acac=acetylacetonate.
Furthermore, β‐diketiminate‐based catalysts, such as the dimeric β‐diketiminate manganese hydride (Mn‐1) and the dimeric β‐diketiminatomagnesium hydride (Mg‐1) in Figure 3, were also developed for anti‐Markovnikov alkene hydrosilylation.[ 54 , 55 ] Using Mn‐1 as catalyst, aliphatic alkenes underwent anti‐Markovnikov hydrosilylation to afford (E)‐β‐vinylsilanes with 37–99 % conversion, while the hydrosilylation of styrenes afforded α‐vinylsilanes with 19–99 % conversion through Markovnikov hydrosilylation.
Figure 3.
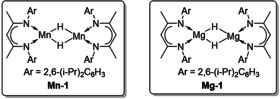
The structures of dimeric β‐diketiminate manganese and magnesium hydride.[ 54 , 55 ]
In 2018, Zhu et al. described selective hydrosilylations of alkenes with Fe‐based catalysts in the presence of 1,10‐phenanthroline ligands (Scheme 7). [56] In particular, the combination of FeCl2 with 2,9‐diaryl‐1,10‐phenanthroline ligands exhibited good reactivity and selectivity for hydrosilylation of both styrenes and aliphatic alkenes.
Scheme 7.
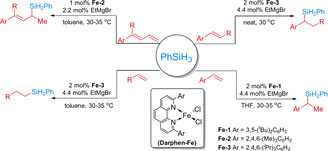
Iron‐catalyzed hydrosilylation of alkenes with phenylsilane. [56]
As shown in Scheme 7, Fe‐1 and Fe‐2 showed very high Markovnikov selectivity (≥98 %) in the hydrosilylation of terminal styrenes, 1‐substituted, and 1,1‐disubstituted buta‐1,3‐dienes and led to the corresponding products in high yields (88–95 %). However, in the presence of Fe‐3, anti‐Markovnikov products were obtained in the hydrosilylation of 1‐alkyl ethylene derivatives in 72–98 % yields. Kinetic isotope effect experiments and density functional theory (DFT) calculations suggest direct Si migration as the rate‐ determining step. Unfortunately, these iron catalyst systems seem to be limited to hydrosilylations with PhSiH3 and required Grignard reagents (EtMgBr) for catalyst activation.
Compared to the hydrosilylations of simple alkenes with primary or secondary silanes (vide supra), hydrosilylation of alkenes or vinylsilanes with tertiary alkoxy‐ or siloxyhydrosilanes for silicone synthesis are considered more challenging from an industrial perspective. The resulting silicones represent industrially relevant fluids and curing materials. However, competitive hydrogenation of alkenes and alkynes can easily occur during the hydrosilylation process when the ligands are changed slightly. In this respect the work of Chirik and co‐workers is notable. Here a nickel(II) bis(carboxylate) catalyst was used, which displayed high activity in the hydrosilylation of alkenes with a variety of industrially relevant tertiary alkoxy‐ and siloxy‐substituted silanes. Under optimal conditions, selective anti‐Markovnikov hydrosilylation of aliphatic alkenes with commercially relevant silanes and siloxanes was achieved in a practical manner (Scheme 8). [57]
Scheme 8.
Alkene hydrosilylation with α‐diimine nickel catalysts. 2‐EH=2‐ethylhexanoate. [57]
In 2017, Lee and co‐workers synthesized more than 25 cobalt‐(aminomethyl)pyridine complexes and explored their catalytic performance for anti‐Markovnikov hydrosilylations (Scheme 9). [58] With Co‐3 as the optimal catalyst system, various alkoxy(vinyl)silanes including mono‐, di‐, and triethoxy(vinyl)silanes and their corresponding methoxy derivatives reacted with alkoxy‐ or siloxyhydrosilanes to afford the desired anti‐Markovnikov products in 70–99 % yield with >98 % anti‐Markovnikov selectivity.
Scheme 9.
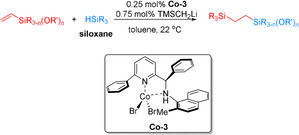
Cobalt‐catalyzed anti‐Markovnikov hydrosilylation of alkoxy‐ or siloxy(vinyl)silanes with alkoxy‐ or siloxyhydrosilanes. [58] TMS=trimethylsilyl.
Shortly thereafter, Ge and co‐workers developed the highly selective cobalt‐catalyzed stereoconvergent Markovnikov 1,2‐hydrosilylation of conjugated dienes (Scheme 10 a). [59] In the presence of 1 mol % of Co(acac)2 and xantphos at room temperature, a wide range of conjugated trans‐dienes encompassed aryl‐substituted, alkyl‐substituted, and multiple‐substituted dienes reacted smoothly with PhSiH3 and Ph2SiH2 affording the corresponding (E)‐allylsilanes in 64–92 % yield with excellent regioselectivities (b/l>99:1). Furthermore, asymmetric Markovnikov 1,2‐hydrosilylation of (E)‐1‐aryl‐1,3‐dienes with PhSiH3 in the presence of Co(acac)2/(R)‐difluorphos proceeded smoothly to afford the desired products in 61–80 % yield with good enantioselectivities (88:12–90:10 e.r.) (Scheme 10 b). Mechanistic studies revealed that this stereoconvergence resulted from a σ–π–σ isomerization of an allylcobalt species generated by the 1,4‐hydrometalation of (Z)‐dienes. A Chalk–Harrod mechanism involving the 2,1‐insertion of the terminal double bond of the diene into the Co−H bond was proposed.
Scheme 10.
Markovnikov 1,2‐hydrosilylation of conjugated dienes: a) regioselective stereoconvergent and b) asymmetric variant. [59]
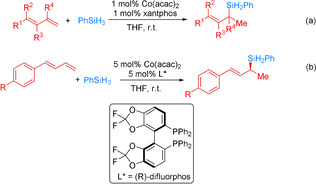
More recently, the Huang group developed a quinoline‐oxazoline‐based cobalt complex for the asymmetric 1,2‐Markovnikov hydrosilylation of conjugated dienes with primary silanes (Scheme 11). [60] The tBu‐substituted analogue (QuinOxtBu)CoCl2 (Co‐4) was identified as an effective catalyst for the highly regio‐ and enantioselective 1,2‐Markovnikov hydrosilylation of various conjugate dienes bearing aryl/alkyl substituents with PhSiH3, furnishing chiral allyl silanes in high yields with high regioselectivity (up to >99:1) and enantioselectivity (up to 96 % ee) in the presence of NaBEt3H. Ph2SiH2 was less reactive than Ph2SiH3, while tertiary silanes (Et3SiH and (EtO)2MeSiH) are unreactive. A modified Chalk–Harrod mechanism involving the 1,2‐insertion of the terminal double bond of the diene into the Co−Si bond is assumed.
Scheme 11.
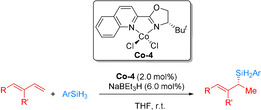
The asymmetric 1,2‐Markovnikov hydrosilylation of conjugated dienes with primary silanes using pyridine/quinoline‐oxazoline chiral CoII dichloride complexes. [60]
Apart from olefins and dienes, the hydrosilylation of alkynes has been investigated with non‐noble metal complexes bearing bidentate ligands. [61] In this context, Ge et al. reported a Co‐catalyzed anti‐Markovnikov hydrosilylation of terminal alkynes with both primary and secondary hydrosilanes PhR′SiH2 (Scheme 12). [62] With Co(acac)2 and dpephos or xantphos as ligands, a broad range of alkynes containing either aromatic or aliphatic substituents underwent this hydrosilylation reaction smoothly at room temperature to afford (E)‐vinylsilanes in 59–91 % yield with high regioselectivity ((E)‐β:α 89:11 to 99:1).
Scheme 12.

The anti‐Markovnikov hydrosilylation of terminal alkynes using Co(acac)2 and bisphosphine ligands. [62]
Complementary to that work, Jin and co‐workers developed an efficient cobalt‐catalyzed Markovnikov‐selective hydrosilylation of alkynes using bidentate CImPy ligands such as L1 in 2019 (Scheme 13). [63] The hydrosilylation of aromatic and aliphatic alkynes with primary and secondary silanes proceeded well to form the corresponding products in 36–98 % yield with moderate to high regioselectivities (α/β 63:37 to 99:1). The comparably high catalytic activity enabled by the CImPy ligand is ascribed to the high stereoelectronic tunability and rigid environment, which suppresses the deactivation of the catalyst.
Scheme 13.
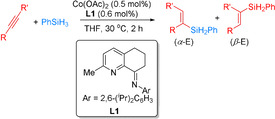
Cobalt‐catalyzed Markovnikov‐selective hydrosilylation of alkynes using bidentate CImPy ligands such as L1. [63]
Recently, Zhu and co‐workers described also an Fe‐catalyzed dihydrosilylation of aliphatic terminal alkynes and primary silanes for the synthesis of geminal bis(silanes) (Scheme 14). [64] Reactions of PhSiH3 and n‐C12H25SiH3 with a range of aliphatic terminal alkynes generated the corresponding geminal bis(silanes) as the sole hydrosilylation products in 85–95 % yield. This method allowed the efficient synthesis of previously unreported geminal bis(silanes) with secondary silyl groups. Mechanistic studies demonstrated that the reaction proceeds via two iron‐catalyzed hydrosilylation reactions, the first generating β‐(E)‐vinylsilanes and the second producing geminal bis(silanes).
Scheme 14.
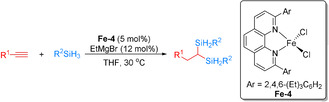
Iron‐catalyzed dihydrosilylation of alkynes for the synthesis of geminal bis(silanes). [64]
Based on their work on alkyne hydrosilylation, Ge and co‐workers also disclosed a highly regio‐ and stereoselective hydrosilylation of allenes by employing a bench‐stable catalyst system consisting of Co(acac)2 and binap or xantphos ligands (Scheme 15). [65] Allyl‐ or vinylsilanes can be easily prepared by selective hydrosilylation of allenes with hydrosilanes in the presence of transition metal catalysts. The major difficulty in these reactions is to control the regio‐ and stereoselectivity preventing the formation of different vinyl‐ and allylsilane products. Here, a variety of mono‐ and disubstituted terminal allenes reacted with primary and secondary hydrosilanes to produce the desired disubstituted (Z)‐allylsilanes in high yields (57–95 %) with excellent stereoselectivity (Z:E=99:1). Unfortunately, hydrosilylation of allenes did not occur when tertiary hydrosilanes were used. The regio‐ and stereocontrol of the reaction is explained by the steric repulsion between the substituent on the allyl group and the ligand of the cobalt catalyst.
Scheme 15.
Cobalt‐catalyzed (Z)‐selective allene hydrosilylation. [65]
Another elegant example of asymmetric hydrosilylation was presented by Buchwald and co‐workers in 2017. They demonstrated an enantioselective Cu–H catalyzed Markovnikov hydrosilylation of vinylarenes and associated vinyl heterocycles with Ph2SiH2 (Scheme 16). [66] These reactions gave bench‐stable silanes and chiral alcohol derivatives in 61–89 % yield with good to excellent enantioselectivity (70–98 % ee).
Scheme 16.
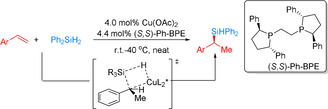
Copper hydride‐catalyzed asymmetric Markovnikov hydrosilylation of vinylarenes and vinyl heterocycles. [66]
2.3. Catalysts with Pincer Ligands
In the past two decades, catalysts with so‐called pincer ligands have been extensively exploited for all kind of catalytic reactions, including hydrosilylations. In general, the corresponding complexes contain tridentate ligands, which bind to the metal center with three adjacent coplanar sites in a meridional configuration. Often pincer ligands have a central, σ‐donating moiety that contains two side donor groups, typically either amino‐ or phosphinomethyl groups in ortho‐position. Advantages of pincer complexes are their high thermal stability and well‐defined reactivity with remaining coordination sites. In this section, recent studies on hydrosilylation using pincer catalysts will be discussed.
As an early example of alkene hydrosilylations, Chirik and co‐workers used a nickel complex bearing a bis(amino)amide NNN‐pincer ligand. Their work was inspired by a related iron pincer catalyst, which, however, tolerated no carbonyl groups in the reaction. [67] Hydrosilylation of 1‐octene with Ph2SiH2 using 0.025 mol % of Ni‐1 achieved a 98 % conversion in only 3 minutes, thus resulting in an impressive TOF of 83 000 h−1. Ni‐1 was capable of efficiently converting differently substituted alkenes and cyclic alkenes with up to 94 % yield. Interestingly, selective hydrosilylations of alkenes containing ketones and aldehydes groups were successfully conducted with yields from 71–74 % (Scheme 17). In these reactions Ni hydride species are assumed to be potential intermediates. [6]
Scheme 17.
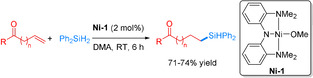
Ni‐catalyzed hydrosilylation of olefins containing carbonyl groups. [67] DMA=dimethylacetamide.
In general, low‐valent Fe pincer complexes are known to be more unstable and difficult than iron(II/III) complexes. With this in mind, Thomas and co‐workers prepared a series of FeII NNN‐pincer complexes bearing Cl, Br, and OTf as counterions. Activation of such higher valent complexes was possible using tertiary amines at room temperature. Thus, in the presence of (iPr)2NEt the Fe‐pincer catalyst bearing OTf exhibited the best activity for the hydrosilylation of 1‐octene with PhSiH3, achieving 95 % yield for the anti‐Markovnikov product. This behavior explained by the fact that the ion/counterion bond strength of Fe‐halides is stronger than that of Fe‐OTf. With exception of nitro and nitrile groups, a broad scope of substituents was tolerated in the presence of this catalyst system, including carbonyl groups. [69]
An interesting one‐pot cascade‐like approach for alkane dehydrogenation/isomerization/hydrosilylation yielding terminal silanes was developed using a dual iridium/iron pincer catalytic system. Here, initially 1‐octane is dehydrogenated in the presence of 1 mol % iridium catalyst and 1.2 mol % of NaOtBu at 200 °C. For the isomerization/hydrosilylation reactions, anti‐Markovnikov products were obtained (67 % yield) in the presence of 10 mol % of Fe‐NNN pincer catalyst and 20 mol % of NaHBEt3 (Scheme 18). Control experiments revealed that the iridium catalyst played no role in the tandem isomerization/hydrosilylation reaction. Notably, the iron‐catalyzed alkene hydrosilylation occurred with a high reaction rate, thus inhibiting reversible isomerization of the alkene. [68]
Scheme 18.
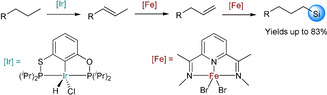
One‐pot dehydrogenation, isomerization and hydrosilylation catalyzed by iridium and iron pincer catalysts. [68]
The application of pincer ligands with less hindered imine groups favored hydrosilylation instead of dehydrogenative hydrosilylation of olefins. [7] In this context, Chirik and co‐workers developed a Co‐NNN pincer catalyst derived from Co‐5 a for hydrosilylations. The catalyst bearing a CH2SiMe3 group showed high activity for 1‐octene hydrosilylation with HSi(OEt)3 at room temperature. Unfortunately, this catalyst is not bench‐stable. Hence, more stable complexes Co‐5 b and Co‐6 b were prepared as catalysts by ligand modification. These stable catalysts exhibited excellent activity for hydrosilylations with alkoxysilanes. As an example, the hydrosilylation of sensitive allyl glycidyl ether was successfully conducted in the presence of Co‐6 b on a 10 g scale with 98 % yield for the trialkoxysilane product, which finds widespread applications in industry (Scheme 19). [70]
Scheme 19.
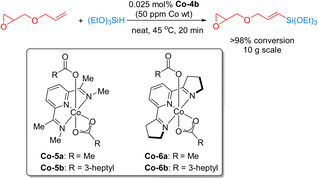
Hydrosilylation of allyl glycidyl ether using a Co pincer catalyst. [70]
Apart from olefins and alkynes, the hydrosilylation of 1,3‐ and 1,4‐dienes was studied in the presence of Co‐5. Thus, an anti‐Markovnikov hydrosilylation of (E)‐1,3‐dodecadiene with phenylsilane occurred selectively. The following activity trend was observed for substituents on the pincer framework: 2,4,6‐tri‐Me <2,6‐di‐Et <2,6‐di‐iPr. With this catalytic system, primary and secondary silanes (PhSiH3, PhMeSiH2, and Ph2SiH2) were successfully converted into the desired anti‐Markovnikov products in up to 92 % yield, while tertiary silanes showed no activity for hydrosilylation, but instead led to hydrogenation of the corresponding dienes. [72]
Recently, manganese complexes including pincer ligands have become relatively popular in homogeneous catalysis. [73] Nevertheless, the use of such complexes for hydrosilylations of alkenes is still quite rare. As an exception several Mn‐NNN pincer complexes (Mn‐2 to Mn‐4) were used for hydrosilylation of terminal alkenes. These catalysts displayed excellent regioselectivity for a broad scope of alkenes and silanes, yielding the corresponding products up to 95 % with >99 % regioselectivity in the presence of NaOtBu. Apparently, the increased steric bulk of the ligand is favorable for this transformation. Hence, Mn‐4 led to higher hydrosilylation yields, which was also proven for the hydrosilylation of 1‐octene with HSi(OEt)3 in a gram‐scale reaction (Scheme 20). [71]
Scheme 20.
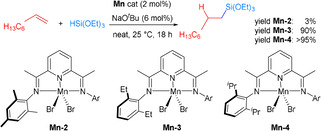
Manganese‐catalyzed alkene hydrosilylation. [71]
In 2019, a series of quinoline‐derived Fe‐PNN pincer complexes (Fe‐5 to Fe‐7) were prepared, aiming for a bench‐stable, activator‐ and solvent‐free non‐noble metal catalyst system (Scheme 21). In the presence of stoichiometric amounts of LiCH2SiMe3 and KOPiv, Fe‐5 can be transformed to Fe‐6 and Fe‐7, which are active catalysts for the hydrosilylation under base‐free conditions. Using 1‐octene and PhSiH3 as starting materials, Fe‐6 and Fe‐7 displayed excellent catalytic performance affording TONs of 480 000 and 100 000, respectively. Interestingly, Fe‐7 proved to be stable under air exposure, while Fe‐6 immediately decomposed upon air exposition. It was confirmed that Fe–H species, which are believed to be the active intermediates in the catalytic cycle, were produced in the presence of Fe‐6 and PhSiH3. [74]
Scheme 21.
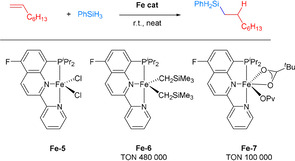
Iron pincer‐catalyzed 1‐octene hydrosilylation. [74]
While pyridyl‐derived NNN and quinoline‐derived PNN pincer complexes favored the formation of the hydrosilylation products from aliphatic olefins (vide supra),[ 75 , 76 ] Lu and co‐workers demonstrated a Markovnikov (enantio)selective hydrosilylation of terminal alkenes using different Fe‐PNN pincer catalysts (Fe‐8). In their elegant studies, they investigated a series of pyridine‐based ligands with different oxazoline and imino substituents. An increased steric bulk of the imino group not only increased the selectivity of the hydrosilylation, but also improved the regio‐ and enantioselectivity. Comparing diverse oxazoline moieties, the iPr‐substituted one exhibited better activity than that with Me and tBu groups. Under optimal conditions, alkenes bearing bioactive molecules, such as naproxen, ibuprofen, and desloratadine, gave the desired products in a highly enantioselective manner in 91 %, 97 % and 88 % yield, respectively. Moreover, an experiment with 1,5‐hexadiene with 2.4 equiv of PhSiH3 provided the disilylated product in 90 % yield with 96/4 branched/linear ratio and 97 % ee (Scheme 22). With respect to asymmetric catalysis this development is noteworthy because it allows highly enantioselective functionalization of plain aliphatic olefins without any additional coordination sites. [77]
Scheme 22.
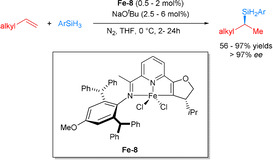
Iron‐catalyzed enantioselective Markovnikov hydrosilylation of alkenes. [77]
Compared to most known pincer ligands, phosphinite‐iminopyridine ligands can be easily decomposed due to P−O bond cleavage. [9] Obviously, related phosphino‐iminopyridine (PNN) ligands are more stable and have been explored with respect to their steric hindrance (Scheme 23). More specifically, Fe‐9 and Co‐7 pincer catalysts were prepared for regioselective Markovnikov alkene hydrosilylations. Interestingly, in the hydrosilylation of 1‐octene with PhSiH3, the Markovnikov product was mainly obtained using Fe‐9, while the anti‐Markovnikov was the major one in the presence of Co‐7. The different product formation with the use of Co‐7 was ascribed to a different silyl migration mechanism involving a CoI–silyl intermediate. Ligand screening experiments revealed increased activity with the bulkier ligand for Fe‐9, whereas the less bulky ligand was more active for Co‐7. [78]
Scheme 23.
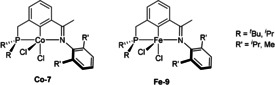
Cobalt and iron PNN pincer catalysts used in alkene hydrosilylations. [78]
The pincer catalysts also exhibited high activities in the selective hydrosilylation of alkynes.[ 79 , 80 ] In 2016, Lu and co‐workers described an improved sequential hydrosilylation/asymmetric hydrogenation of terminal phenylalkynes to obtain chiral silanes using chiral Co‐NNN pincer catalysts. After detailed investigations of the steric bulk of substitutents on the pincer ligand, Co‐8 was identified as the optimal complex, which exhibited excellent catalytic performance for hydrosilylations of phenylacetylenes with Ph2SiH2 with up to 91 % yield and >99 % ee. Mechanistic studies revealed that cobalt–hydride species are intermediates in this hydrosilylation reaction. [81] Notably, the hydrosilylations of phenylacetylenes proceeded extremely fast. In fact, a reaction with Ph2SiH2 using 1 mol % of Co‐8 was completed in 5 seconds, which corresponds formally to a TOF of 65 000 h−1 (Scheme 24 a). When the hydrosilylation of 4‐vinylphenylacetylene was performed, a chemoselective reaction for the alkyne moiety was observed, although in lower yield (Scheme 24 b). [81] Later on, sequential double hydrosilylation of aliphatic alkynes to yield highly enantioenriched gem‐bis(silyl)alkanes was achieved by the same group. Both experimental results and DFT calculations were analyzed to understand the reaction mechanism. It was shown that the highly enantioselective synthesis of gem‐bis‐(silyl)alkanes is a result of the sequential asymmetric double 1,1‐hydrosilylation of aliphatic alkynes in the presence of CoBr2⋅Xantphos, and CoBr2⋅OIP (OIP=oxazoline‐iminopyridine). [82]
Scheme 24.
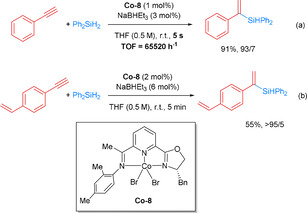
Cobalt pincer complex‐catalyzed alkyne hydrosilylation. [81]
Moreover, asymmetric hydrosilylation of unsymmetric alkynes with dihydrosilanes producing silicon‐stereogenic vinylhydrosilanes was realized with high regio‐ and enantioselectivity by Co‐NNN catalysts. More specifically, Huang and co‐workers reported the Markovnikov hydrosilylation of terminal alkynes with diphenylsilane in the presence of pyridine‐bis(oxazoline) Co catalysts. Later on, the same group described alkyne hydrosilylations with prochiral dihydrosilanes in the presence of the same kind of cobalt/pyridine‐bis(oxazoline)) complex producing Markovnikov silanes with up to 99 % selectivity.[ 83 , 84 ]
In 2017, a series of cobalt pyridine‐2,6‐diimine complexes were prepared and tested for the Z‐selective hydrosilylation of terminal alkynes with PhSiH3. In this work the less sterically hindered ligands favored the formation of α‐vinylsilanes, while (Z)‐β‐vinylsilanes were preferred using mes PDI and ipr PDI. This behavior was attributed to the accessibility of the respective cobalt center. A broad range of alkynes including phenylacetylene and aliphatic alkynes were successfully converted to the desired products with Z/E ratios higher than 91:9 (Scheme 25). However, highly reactive nitro and aldehyde substituents were not tolerated. Moreover, aliphatic alcohol and carboxylic acid moieties inhibited the reaction. [85]
Scheme 25.
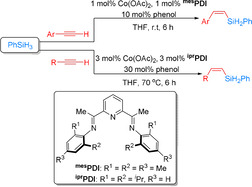
Cobalt‐catalyzed Z‐selective hydrosilylation of terminal alkynes. [85]
3. Recyclable Heterogeneous Catalysts
In classical liquid‐phase reactions, heterogeneous catalysts are more easily recycled than homogeneous ones. Although this advantage is also also holds for many catalytic hydrosilylations,[ 31 , 86 ] in a number of (industrial) cases, the corresponding products are either highly viscous or even polymers. In such cases, the practical benefit of a heterogeneous material is less obvious. Here, instead, the avoidance of (costly) ligands might be the main driver to search for heterogeneous catalysts. For this reason, an increasing number of heterogeneous catalysts have been explored in recent years. In this section, heterogeneous catalysts based on supported nanoparticles, including so‐called supported single‐atom catalysts, fabricated by different processes and their applications in hydrosilylation will be emphasized.
3.1. Precious Metal Catalysts
3.1.1. Pt Nanoparticle Catalysts
For a long time, the molecularly defined Karstedt complex has been regarded as the state‐of‐the‐art catalyst for hydrosilylation reactions. Thus, also several attempts were made to immobilize this complex or derivatives on different supports. For example, a silica‐supported Karstedt complex, which displayed initial high activity, was first prepared in 2003. However, for this material poor catalytic performance was observed after several reuses due to its low stability. Since then, other heterogeneous Pt catalysts have been broadly investigated. [87] In 2017, a mesostructured silica framework was used as a support where Pt nanoparticles (NPs) were embedded into walls of the silica matrix. The resulting material exhibited excellent catalyst turnover numbers of TON=105 for the hydrosilylation of 1‐octene with polymethylhydrosiloxane (PMHS). Due to the physical trapping of the Pt NPs in the silica framework, no Pt leaching was observed after recycling. [88]
Pt0 NPs were also embedded in a modified silica xerogel (SiliaCatPt(0)), which is obtained by sol–gel polycondensation of organosilanes. The resulting catalyst has a uniform spherical morphology and proved to be highly active and selective for a broad scope of olefins. However, the activity deceased sharply to 65 % after the fourth cycle, probably caused by the slight size increase of Pt NPs. [89] To obtain effective catalyst separation from liquid silicone products, magnetic silica particles (Fe3O4@SiO2) were used as support. After the material was modified by addition of ethylenediaminetetraacetic acid (EDTA) or diethylenetriaminepentaacetic acid (DTPA) and immobilization of Pt, the resulting catalysts displayed good activity even for the isomerization–hydrosilylation of internal alkenes. [90]
Recently, a graphene‐supported platinum catalyst was synthesized using electrostatic adsorption techniques with solventless microwave irradiation. Using this special method, additional defects or holes in graphene are formed and simultaneously small Pt nanoparticles are stabilized with an average diameter of 6.8 nm. The resulting catalyst material displayed a superior efficiency in the hydrosilylation of 1,1,1,3,5,5,5‐heptamethyltrisiloxane (MDM) and 1‐octene, leading to a TON of 9.4×106 which was tenfold higher than that obtained by the parent Karstedt catalyst (0.9×106). [36]
3.1.2. Pt Single‐Atom Catalysts (Pt SACs)
Although several supported Pt nanoparticles were developed for catalytic hydrosilylations (vide supra), in some cases poor recyclability caused by leaching was observed. This was attributed to the weak binding of Pt particles to the support. Complementary to materials based on supported nanoparticles, single‐atom catalysis provides a new concept to prepare materials with isolated metal centers, which are stabilized by neighboring sites of the support. In 2017, a Pt single‐atom catalyst was synthesized by impregnation of platinum salts on aluminum oxide nanorods. The resulting Pt‐SAC was applied for the selective hydrosilylation of all kinds of terminal olefins and exhibited excellent activity comparable to the original Karstedt system (Scheme 26). Interestingly, this Pt‐SAC displayed also high stability, which is explained by the strong binding of the individual Pt atoms to their neighboring oxygen atoms. [37] Later on, superparamagnetic Fe3O4–SiO2 core–shell nanoparticles (NPs) were used as the support in Pt single‐atom catalysts. The material was easily separated from high‐viscosity products by applying a magnetic field (Scheme 27). Notably, the Pt loading decreased from 1.5 % to 1.26 % after four cycles. [91] Furthermore, a partially charged Pt single‐atom catalyst was fabricated on the surface of anatase TiO2 (Pt1 δ+/TiO2) by an electrostatic‐induction ion exchange. DFT calculations explained the excellent catalytic performance of this material by the intrinsic nature of partially charged Pt(δ+) atoms on TiO2. The authors also concluded that the lower oxidation state of Pt is favorable for the desired transformation compared to platinum in higher oxidation states (II or IV). [92]
Scheme 26.
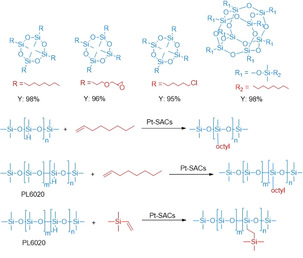
Pt SACs used for the selective hydrosilylation of various alkenes. [37]
Scheme 27.
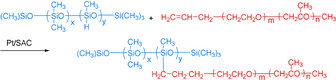
Pt‐SAC for hydrosilylation. [91]
In order to prevent metal agglomeration in SACs, the metal loading is often (very) low. Recently, an interesting material was disclosed which contains isolated Pt single atoms in a dense distribution. It was successfully synthesized by the NaCO3‐assisted one‐pot pyrolysis of an EDTA‐Pt complex on N‐doped graphene (Pt‐ISA/NG in Figure 4). Here, the Pt centers are coordinated to N species instead of O. The Pt‐ISA/NG exhibited microstructure and morphology features typical for atomically thin 2D graphene‐like analogues, with a specific surface area of 1892 m2 g−1 and 5.3 wt % Pt loading. Pt‐ISA/NG displayed high selectivity, activity, and stability for anti‐Markovnikov hydrosilylation of different terminal alkenes with silanes under mild conditions. [93]
Figure 4.

Characterization of the Pt‐ISA/NG catalyst: a) Typical transmission electron microscopy (TEM) image. b) High‐resolution TEM (HRTEM) image. c) Energy‐dispersive X‐ray (EDX) mapping. d,e) Aberration‐corrected high‐angle annular dark‐field scanning transmission electron microscopy (AC‐HAADFSTEM) image and corresponding enlarged view. Reproduced with permission. [93] Copyright 2018, American Chemical Society.
In the case of Pt NPs and SACs, the leaching of the metal is an important aspect, which has to be avoided. Although the average loss of Pt was calculated to be several ppm in each round of reaction, the involvement of the leached Pt could not be excluded because the hydrosilylation easily initiated even by a ppm amounts of Pt. Moreover, the leaching problem leads to difficulties in mechanistic studies and contamination in the final silicon products.
3.1.3. Other Precious Metal Catalysts
Apart from platinum, other precious metal catalysts based on Au, Rh, and Ru have been reported for hydrosilylation reactions of olefins and related substrates. For example, highly regioselective alkene hydrosilylation is possible in the presence of gold nanoparticles on TiO2 or Al2O3. In this case, ionic gold(I) species at the interface between the nanoparticles and the support were suggested as the active sites. [86]
Recently, RhI complexes were co‐immobilized with tertiary amines on the surface of SiO2 (referred to as SiO2/Rh‐NEt2). SiO2/Rh‐NEt2 exhibited excellent turnover numbers for the hydrosilylation of olefins with a wide range of substrates. The good catalytic performance was ascribed to the electron donation from amine groups to the Rh complex, which promotes both the oxidative addition and insertion steps during the hydrosilylation cycle. [94]
Despite significant progress with heterogeneous catalysts using supported noble metals, the hydrosilylation of internal alkenes is still challenging. In this context, Prieto and co‐workers demonstrated an interesting isomerization–hydrosilylation of internal alkenes. Very recently, they successfully developed a process involving tandem catalysis by Rh and Ru single‐atom catalysts on CeO2. When these two SACs were combined in a single reaction, a synergetic effect was observed and high selectivity was obtained for the hydrosilylation of different internal alkenes with Et3SiH. DFT calculations ascribed the observed selectivity to differences in the binding strength of the alkene substrate where the single Ru atoms bind more strongly than the Rh counterparts. [95]
3.2. Non‐Precious Metal Catalysts
3.2.1. Nickel‐Based Heterogeneous Catalysts
In 2016, Hu and co‐workers developed the first nickel nanoparticle catalyst, which is able to catalyze alkene hydrosilylation with tertiary silanes. In this case, nickel nanoparticles with an average size of 3.5 nm were formed in situ using a particular nickel alkoxide potassium salt (Ni(OtBu)2⋅x KCl). Interestingly, the resulting Ni nanoparticles displayed high activity not only in the hydrosilylation of terminal alkenes with high anti‐Markovnikov selectivity, but also in the tandem isomerization–hydrosilylation of cis and trans internal alkenes in high yields (Scheme 28). Consequently, this non‐precious metal catalyst is able to synthesize a single terminal alkyl silane from a mixture of different internal and terminal olefin isomers. [96] Later on, other isolated Ni nanoparticles stabilized by n‐octylsilane were prepared by decomposition of Ni(COD)2 (COD=cycloocta‐1,5‐diene) in toluene under 4 bar of H2. The resulting Ni colloid (Ni3Si2) consists of very small nanoparticles 1.2 nm in diameter. However, with this system only moderate conversion (70 %) and low selectivity (30 %) were achieved for the model hydrosilylation of triethoxyvinylsilane with triethoxysilane. [97]
Scheme 28.
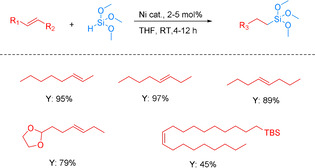
Ni‐catalyzed tandem isomerization–hydrosilylation of internal alkenes. [96]
In 2019, a novel supported nickel catalyst was prepared whereby isolated nickel centers were anchored on a metal—organic framework (MOF) carrier. The single metal sites are considered to be stabilized by the hydroxyl groups from the MOF forming the active centers. Using this catalyst, the hydrosilylation of n‐octene and diphenylsilane was carried out on a 150 mmol scale under mild conditions with high conversion and selectivity. [98] Furthermore, isolated Ni centers stabilized by heterogeneous ligands have been applied for hydrosilylation reactions very recently. In this latter case, a porous organic polymer containing Xantphos (POP‐Xantphos) moieties was used to stabilize the isolated nickel centers. The prepared Ni‐POP‐Xantphos catalyst demonstrated high regio‐ and stereoselectivity in the hydrosilylation of alkynes. The obtained selectivity was considered to be controlled by the microporous structure of POP‐Xantphos. [99]
3.2.2. Cobalt‐Based Heterogeneous Catalysts
Comparable to the Ni‐POP‐Xantphos catalyst (vide supra), isolated Co sites were coordinated with a POP‐PPh3 ligand (Co‐POP‐PPh3). The resultant Co‐POP‐PPh3 material catalyzed the hydrosilylation of alkynes with PhSiH3 with high regio‐ and stereoselectivity. Moreover, the reusability of Co‐POP‐PPh3 was tested in a continuous‐flow system. Even after several rounds of recycling only little loss of activity and selectivity was observed. [100] Recently, also Co/TiO2 was synthesized where the cobalt ions were doped onto the TiO2 surface. After hydrogen treatment, CoTiO3 species were formed, which exhibited excellent catalytic performance for the hydrosilylation of various alkenes under neat conditions. Importantly, the CoTiO3 species were not leached after recycling due to the strong interaction between Co and TiO2, leading to high stability and reusability. [101]
3.2.3. Iron‐Based Heterogeneous Catalysts
In 2016, Lin and co‐workers reported an iron‐based catalyst for several hydrosilylation reactions of terminal alkenes using a two‐dimensional (2D) metal–organic layer (MOL) carrier. The synthesized MOL was composed of [Hf6O4(OH)4(HCO2)6] as secondary building units (SBUs) and benzene‐1,3,5‐tribenzoate (BTB) as bridging ligands. After connecting the MOL with 4′‐(4‐benzoate)‐(2,2′,2′′‐terpyridine)‐5,5′′‐dicarboxylate (TPY), the resulting MOL‐TPY was used to immobilize iron centers, affording single‐site solid catalysts (Fe‐MOL‐TPY, Figure 5). A variety of terminal alkenes were converted to the corresponding alkyl silanes with PhSiH3 in high yields. Fe‐MOL‐TPY is free from diffusional constraints, leading to high activity and reusability. [102]
Figure 5.
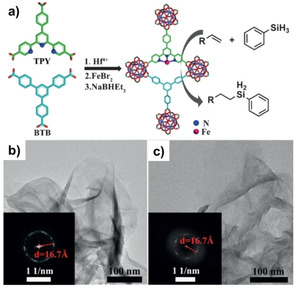
a) Preparation of the MOL catalyst Fe‐TPY‐MOL. b,c) HRTEM and FFT images of Fe‐TPY‐MOL before (b) and after catalysis (c). Reproduced with permission. [102] Copyright 2016, Wiley‐VCH.
In addition to Fe‐SACs, iron oxide nanoparticles (Fe2O3) were synthesized in a practical way using iron(III) acetylacetonate as the precursor and N,N‐dimethylformamide (DMF) as both the reducing and protecting agent. The DMF‐stabilized Fe2O3 nanoparticles were monodispersed in the solvent and showed high catalytic activity for the hydrosilylation of alkenes in the absence of any additives. Furthermore, the colloidal catalyst can be recycled by simple extraction with a hexane/DMF system for fifth run. [103]
3.2.4. Bimetallic Heterogeneous Catalysts
Bimetallic nanoparticles can exhibit exclusive catalytic performances, distinct from those of monometallic NPS, due to their unique electronic states and structures. As an example, Li and co‐workers developed a bimetallic catalyst by immobilizing Pt1Ni1 nanoparticles on the surface of nitrogen‐doped carbon (NC). The Pt1Ni1/NC‐1000 catalyst, which was obtained by pyrolysis of metal–organic frameworks at 1000 °C, displayed the highest catalytic performance for the hydrosilylation benchmark reaction of 1‐octene with HSi(OEt)3. Characterizations revealed a high graphitization degree, which should favor a stronger charge transfer between the NC support and Pt1Ni1, forming more positively charged Pt centers. These charged Pt species possibly lead to the higher catalytic activity. [105]
In 2017, Cai and co‐workers developed an efficient and recyclable Pd1Cu2 bimetallic catalyst. The Pd1Cu2 NPs were supported on SiO2 and exhibited superior activity and selectivity toward the hydrosilylation of internal and terminal alkynes. The catalytic performance is improved by the ultrasmall size (2.8 nm) and the high dispersion of the Pd–Cu nanoparticles as well as the enrichment of Pd on the catalyst surface (Figure 6). [104]
Figure 6.
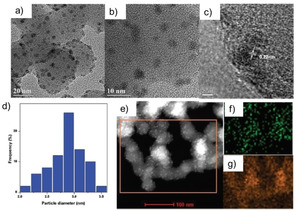
Characterization of the Pd1Cu2/SiO2 catalyst: a,b) TEM images. c) High‐resolution TEM (HRTEM) image of the Pd1Cu2/SiO2 catalyst. d) Size distribution of Pd1Cu2 bimetallic nanoparticles. e) High‐angle annular dark field (HAADF) STEM image of Pd1Cu2/SiO2. f,g) EDS elemental maps for Pd (f) and Cu (g). Reproduced with permission. [104] Copyright 2017, The Royal Society of Chemistry.
Finally, heterogeneous catalysts using bimetallic materials have been used for hydrosilylations of alkynes. Here, Shishido and co‐workers reported the use of Pd‐Au bimetallic NPs at ambient temperature. After careful screening of supports and metal ratios, Pd1Au5/Nb2O5 catalyst was identified as the most active catalyst for the hydrosilylation of alkynes with complete trans‐configured products. High activity was observed for the catalyst with a relatively low Pd/Au ratio of 1:5, which is in accord with the isolated single Pd atoms characterized in Pd1Au5/Nb2O5. [106]
4. Challenges and Perspectives
The catalytic addition of silanes to olefins and alkynes (hydrosilylation reaction) continues to attract significant interest among academic and industrial chemists. In fact, this methodology remains a key technology for innovations in the silicone industry. For the advancement of this field, the development of new and improved catalysts is a prerequisite. In this respect, the present report discusses briefly actual developments of nonclassical homogeneous and heterogeneous catalysts for hydrosilylation reactions. In addition to molecularly defined non‐noble metal catalysts, also recently discovered heterogeneous catalysts such as supported noble/non‐noble NPs and single‐atom catalysts are discussed. Considering all these achievements, what are the major challenges in research on catalytic hydrosilylation reactions in the coming decade?
Apart from few examples, most of the academic developments—which are by no means scientifically very interesting—are far from being relevant for industry and real‐world applications. This is because of the model reactions used, the reaction conditions applied, and the substrates investigated. We believe there is an enormous innovation potential, if the gap between academic and industrial research can be bridged more efficiently.
In the development of new powerful homogeneous catalysts, the complexity of the ligand part is often underestimated, sometimes fully neglected. Obviously, an ideal catalytic system not only constitutes an available, less toxic metal center, for example, iron or manganese, but also inexpensive, stable, and modular ligands. Looking at recently disclosed ligand scaffolds, a number can be synthesized only on small scale under special conditions, for example, in a glovebox. Especially, for non‐noble metal catalysts it is still a challenge to develop practical ligands.
The use of heterogeneous catalysts for hydrosilylation reactions is a fascinating, growing scientific area. Here, the detailed understanding of the structural features that control the regio‐ and stereoselectivity of a given catalyst is still missing. Clearly, such knowledge is the basis for any rational catalyst development. Here, the use of heterogeneous SACs with a limited and defined number of atomic species, where the metal centers are spatially isolated from each other, might give new impetus. Furthermore, the combination of supported (multi)metallic NPs in the presence or absence of ligands will offer alternative solutions for directing regio‐ and stereoselectivities of hydrosilylations.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Leandro Duarte de Almeida, born in Jundiaí (Brazil) in 1992, obtained his bachelor's and master's degrees at the Universidade Federal de Minas Gerais (Brazil). In 2018, he started his PhD at the same university under the direction of Prof. Patricia Alejandra Robles‐Azocar with a research stay in Rostock at the Leibniz Institute of for Catalysis (Germany) supervised by Prof. Matthias Beller. His work is focused on valorization of terpenes and their derivatives using catalytic systems..

Biographical Information
Hongli Wang was born in Shaanxi Province (China) in 1984. He obtained his PhD degree in 2013 at Lanzhou University, under the supervision of Prof. Yong‐Min Liang and Prof. Shang‐Dong Yang. From 2013–2015 he worked as a postdoctoral research fellow with Prof. Jin‐Quan YuHe at Shanghai Institute of Organic Chemistry, CAS. He joined the faculty of Lanzhou Institute of Chemical Physics as an assistant research fellow in 2015 and was promoted to associate professor in 2019. His research focuses on catalytic synthesis of high value‐added chemicals with C1 molecules.

Biographical Information
Kathrin Junge, born in 1967 in northern Germany, received her PhD degree in Chemistry from the University of Rostock in 1997 working with Prof. E. Popowski. After a postdoctoral position in the Max‐Planck group of Uwe Rosenthal, she joined the group led by Matthias Beller in 2000. Since 2008 she has been the group leader for sustainable redox reactions at LIKAT. Her current main research interest is the development of environmentally benign and efficient homogeneous and heterogeneous catalytic reactions based on inexpensive, non‐precious metals.

Biographical Information
Xinjiang Cui, born in China in 1984, obtained his PhD in 2013 supervised by Prof. Youquan Deng and Prof. Feng Shi at Lanzhou Institute of Chemical Physics (LICP), CAS. Then he worked one and half years at LICP as an assistant researcher. He conducted postdoctoral research at the Leibniz Institute for Catalysis with Prof. Matthias Beller (2014–2017) and at École Polytechnique Fédérale de Lausanne (EPFL) with Prof. Paul J. Dyson (2018–2019). In 2020, he returned to LICP and started his academic career independently as a full professor. His interests focus on the transformation of light chain hydrocarbons and the synthesis of fine chemicals with olefins by heterogeneous catalysis.

Biographical Information
Matthias Beller, born in Gudensberg (Germany) in 1962, obtained his PhD in 1989 working with Lutz F. Tietze at the University of Göttingen. After one year of postdoctoral research with Barry Sharpless at MIT (USA), he worked at Hoechst AG in Frankfurt (1991–1995) before he started his academic career at TU Munich. In 1998, he relocated to Rostock to head the Leibniz Institute for Catalysis. The research of his group focuses on applying homogeneous and heterogeneous catalysis for the synthesis of fine/bulk chemicals as well as energy technologies.

Acknowledgements
We acknowledge financial support by National Key Research and Development Program of China (2017YFA0403103), Chinese Academy of Sciences, Key Research Program of Frontier Sciences of CAS (QYZDJ‐SSW‐SLH051), Light of West China, and Youth Innovation Promotion Association (2019409). We also acknowledge the State of Mecklenburg‐Western Pomerania, the Federal State of Germany (BMBF), and the EU (ERC Advanced Grant NoNaCat) for financial support. Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001. Open access funding enabled and organized by Projekt DEAL.
L. D. de Almeida, H. Wang, K. Junge, X. Cui, M. Beller, Angew. Chem. Int. Ed. 2021, 60, 550.
Contributor Information
Prof. Xinjiang Cui, Email: XinjiangCui@licp.cas.cn.
Prof. Matthias Beller, Email: Matthias.Beller@catalysis.de.
References
- 1. Rochow E. G., Silicon and Silicones: About Stone-age Tools, Antique Pottery, Modern Ceramics, Computers, Space Materials and How They All Got That Way, Springer, Berlin, 1987. [Google Scholar]
- 2. Murugavel R., Voigt A., Walawalkar M. G., Roesky H. W., Chem. Rev. 1996, 96, 2205–2236. [DOI] [PubMed] [Google Scholar]
- 3. Fleming I., Barbero A., Walter D., Chem. Rev. 1997, 97, 2063–2192. [DOI] [PubMed] [Google Scholar]
- 4. Marciniec B., Coord. Chem. Rev. 2005, 249, 2374–2390. [Google Scholar]
- 5. Chen C., Hecht M. B., Kavara A., Brennessel W. W., Mercado B. Q., Weix D. J., Holland P. L., J. Am. Chem. Soc. 2015, 137, 13244–13247. [DOI] [PubMed] [Google Scholar]
- 6. Buslov I., Becouse J., Mazza S., Montandon-Clerc M., Hu X., Angew. Chem. Int. Ed. 2015, 54, 14523–14526; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14731–14734. [Google Scholar]
- 7. Atienza C. C. H., Diao T. N., Weller K. J., Nye S. A., Lewis K. M., Delis J. G. P., Boyer J. L., Roy A. K., Chirik P. J., J. Am. Chem. Soc. 2014, 136, 12108–12118. [DOI] [PubMed] [Google Scholar]
- 8. Simonneau A., Oestreich M., Angew. Chem. Int. Ed. 2013, 52, 11905–11907; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12121–12124. [Google Scholar]
- 9. Peng D. J., Zhang Y. L., Du X. Y., Zhang L., Leng X. B., Walter M. D., Huang Z., J. Am. Chem. Soc. 2013, 135, 19154–19166. [DOI] [PubMed] [Google Scholar]
- 10. Fasulo M. E., Lipke M. C., Tilley T. D., Chem. Sci. 2013, 4, 3882–3887. [Google Scholar]
- 11. Tondreau A. M., Atienza C. C. H., Weller K. J., Nye S. A., Lewis K. M., Delis J. G. P., Chirik P. J., Science 2012, 335, 567–570. [DOI] [PubMed] [Google Scholar]
- 12. Abinet E., Spaniol T. P., Okuda J., Chem. Asian J. 2011, 6, 389–391. [DOI] [PubMed] [Google Scholar]
- 13. Wu J. Y., Stanzl B. N., Ritter T., J. Am. Chem. Soc. 2010, 132, 13214–13216. [DOI] [PubMed] [Google Scholar]
- 14. Díez-González S., Nolan S. P., Acc. Chem. Res. 2008, 41, 349–358. [DOI] [PubMed] [Google Scholar]
- 15. Buch F., Brettar H., Harder S., Angew. Chem. Int. Ed. 2006, 45, 2741–2745; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2807–2811. [Google Scholar]
- 16. Roy A. K., Adv. Organomet. Chem. 2008, 55, 1–59. [Google Scholar]
- 17. Plueddemann P. E., Silane Coupling Agents, Plenum Press, New York, 1991. [Google Scholar]
- 18. Zhao Z. Y., Nie Y. X., Tang R. H., Yin G. W., Cao J., Xu Z., Cui Y. M., Zheng Z. J., Xu L. W., ACS Catal. 2019, 9, 9110–9116. [Google Scholar]
- 19. Zhang Y. Q., Funken N., Winterscheid P., Gansauer A., Angew. Chem. Int. Ed. 2015, 54, 6931–6934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7035–7038. [Google Scholar]
- 20. Liberman-Martin A. L., Bergman R. G., Tilley T. D., J. Am. Chem. Soc. 2015, 137, 5328–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bleith T., Wadepohl H., Gade L. H., J. Am. Chem. Soc. 2015, 137, 2456–2459. [DOI] [PubMed] [Google Scholar]
- 22. Yang Z. Y., Iqbal M., Dobbie A. R., Veinot J. G. C., J. Am. Chem. Soc. 2013, 135, 17595–17601. [DOI] [PubMed] [Google Scholar]
- 23. Königs C. D. F., Klare H. F. T., Oestreich M., Angew. Chem. Int. Ed. 2013, 52, 10076–10079; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10260–10263. [Google Scholar]
- 24. Stoelzel M., Prasang C., Inoue S., Enthaler S., Driess M., Angew. Chem. Int. Ed. 2012, 51, 399–403; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 411–415. [Google Scholar]
- 25. Osakada K., Angew. Chem. Int. Ed. 2011, 50, 3845–3846; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 3929–3930. [Google Scholar]
- 26. Sommer L. H., Pietrusza E. W., Whitmore F. C., J. Am. Chem. Soc. 1947, 69, 188.20291058 [Google Scholar]
- 27. Karstedt B. D., General Electric Company, US3775452A, 1973.
- 28. Markó I. E., Stérin S., Buisine O., Mignani G., Branlard P., Tinant B., Declercq J. P., Science 2002, 298, 204–206. [DOI] [PubMed] [Google Scholar]
- 29. Markó I. E., Stérin S., Buisine O., Berthon G., Michaud G., Tinant B., Declercq J. P., Adv. Synth. Catal. 2004, 346, 1429–1434. [Google Scholar]
- 30. Gigler P., Drees M., Riener K., Bechlars B., Herrmann W. A., Kuhn F. E., J. Catal. 2012, 295, 1–14. [Google Scholar]
- 31. Marciniec B., Posala K., Kownacki I., Kubicki M., Taylor R., ChemCatChem 2012, 4, 1935–1937. [Google Scholar]
- 32. Igarashi M., Kobayashi T., Sato K., Ando W., Matsumoto T., Shimada S., Hara M., Uchida H., J. Organomet. Chem. 2013, 725, 54–59. [Google Scholar]
- 33. Hu M. Y., He P., Qiao T. Z., Sun W., Li W. T., Lian J., Li J. H., Zhu S. F., J. Am. Chem. Soc. 2020, 142, 16894–16902. [DOI] [PubMed] [Google Scholar]
- 34. Troegel D., Stohrer J., Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar]
- 35. Obligacion J. V., Chirik P. J., Nat. Rev. Chem. 2018, 2, 15–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong C. J., Gilliland S. E., Clark B. R., Gupton B. F., Chem. Commun. 2018, 54, 13343–13346. [DOI] [PubMed] [Google Scholar]
- 37. Cui X., Junge K., Dai X., Kreyenschulte C., Pohl M. M., Wohlrab S., Shi F., Bruckner A., Beller M., ACS Cent. Sci. 2017, 3, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buisine O., Berthon-Gelloz G., Briere J. F., Sterin S., Mignani G., Branlard P., Tinant B., Declercq J. P., Marko I. E., Chem. Commun. 2005, 3856–3858. [DOI] [PubMed] [Google Scholar]
- 39. Downing C. M., Kung H. H., Catal. Commun. 2011, 12, 1166–1169. [Google Scholar]
- 40. Dunsford J. J., Cavell K. J., Kariuki B., J. Organomet. Chem. 2011, 696, 188–194. [Google Scholar]
- 41. Ortega-Moreno L., Peloso R., Maya C., Suarez A., Carmona E., Chem. Commun. 2015, 51, 17008–17011. [DOI] [PubMed] [Google Scholar]
- 42. Benítez Junquera L., Puerta M. C., Valerga P., Organometallics 2012, 31, 2175–2183. [Google Scholar]
- 43. Kamata K., Suzuki A., Nakai Y., Nakazawa H., Organometallics 2012, 31, 3825–3828. [Google Scholar]
- 44. Lipschutz M. I., Tilley T. D., Chem. Commun. 2012, 48, 7146–7148. [DOI] [PubMed] [Google Scholar]
- 45. Greenhalgh M. D., Frank D. J., Thomas S. P., Adv. Synth. Catal. 2014, 356, 584–590. [Google Scholar]
- 46. Srinivas V., Nakajima Y., Ando W., Sato K., Shimada S., Catal. Sci. Technol. 2015, 5, 2081–2084. [Google Scholar]
- 47. Rémond E., Martin C., Martinez J., Cavelier F., Chem. Rev. 2016, 116, 11654–11684. [DOI] [PubMed] [Google Scholar]
- 48. Langkopf E., Schinzer D., Chem. Rev. 1995, 95, 1375–1408. [Google Scholar]
- 49. Blumenkopf T. A., Overman L. E., Chem. Rev. 1986, 86, 857–873. [Google Scholar]
- 50. Liu Y., Deng L., J. Am. Chem. Soc. 2017, 139, 1798–1801. [DOI] [PubMed] [Google Scholar]
- 51. Gao Y. F., Wang L. J., Deng L., ACS Catal. 2018, 8, 9637–9646. [Google Scholar]
- 52. Rivera-Hernández A., Fallon B. J., Ventre S., Simon C., Tremblay M. H., Gontard G., Derat E., Amatore M., Aubert C., Petit M., Org. Lett. 2016, 18, 4242–4245. [DOI] [PubMed] [Google Scholar]
- 53. Wang C., Teo W. J., Ge S., ACS Catal. 2017, 7, 855–863. [Google Scholar]
- 54. Garcia L., Dinoi C., Mahon M. F., Maron L., Hill M. S., Chem. Sci. 2019, 10, 8108–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mukhopadhyay T. K., Flores M., Groy T. L., Trovitch R. J., Chem. Sci. 2018, 9, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hu M. Y., He Q., Fan S. J., Wang Z. C., Liu L. Y., Mu Y. J., Peng Q., Zhu S. F., Nat. Commun. 2018, 9, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pappas I., Treacy S., Chirik P. J., ACS Catal. 2016, 6, 4105–4109. [Google Scholar]
- 58. Lee K. L., Angew. Chem. Int. Ed. 2017, 56, 3665–3669; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3719–3723. [Google Scholar]
- 59. Sang H. L., Yu S., Ge S., Chem. Sci. 2018, 9, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wen H., Wang K., Zhang Y., Liu G., Huang Z., ACS Catal. 2019, 9, 1612–1618. [Google Scholar]
- 61. Wu G. J., Chakraborty U., Jacobi Von Wangelin A., Chem. Commun. 2018, 54, 12322–12325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu C., Teo W. J., Ge S., ACS Catal. 2018, 8, 5896–5900. [Google Scholar]
- 63. Zong Z., Yu Q., Sun N., Hu B., Shen Z., Hu X., Jin L., Org. Lett. 2019, 21, 5767–5772. [DOI] [PubMed] [Google Scholar]
- 64. Hu M. Y., Lian J., Sun W., Qiao T. Z., Zhu S. F., J. Am. Chem. Soc. 2019, 141, 4579–4583. [DOI] [PubMed] [Google Scholar]
- 65. Wang C., Teo W. J., Ge S., Nat. Commun. 2017, 8, 2258–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gribble M. W., Pirnot M. T., Bandar J. S., Liu R. Y., Buchwald S. L., J. Am. Chem. Soc. 2017, 139, 2192–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tondreau A. M., Atienza C. C. H., Darmon J. M., Milsmann C., Hoyt H. M., Weller K. J., Nye S. A., Lewis K. M., Boyer J., Delis J. G. P., Lobkovsky E., Chirik P. J., Organometallics 2012, 31, 4886–4893. [Google Scholar]
- 68. Jia X. Q., Huang Z., Nat. Chem. 2016, 8, 157–161. [DOI] [PubMed] [Google Scholar]
- 69. Challinor A. J., Calin M., Nichol G. S., Carter N. B., Thomas S. P., Adv. Synth. Catal. 2016, 358, 2404–2409. [Google Scholar]
- 70. Schuster C. H., Diao T., Pappas I., Chirik P. J., ACS Catal. 2016, 6, 2632–2636. [Google Scholar]
- 71. Carney J. R., Dillon B. R., Campbell L., Thomas S. P., Angew. Chem. Int. Ed. 2018, 57, 10620–10624; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10780–10784. [Google Scholar]
- 72. Raya B., Jing S., RajanBabu T. V., ACS Catal. 2017, 7, 2275–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang X. X., Wang C. Y., Angew. Chem. Int. Ed. 2018, 57, 923–928; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 935–940. [Google Scholar]
- 74. Kamitani M., Kusaka H., Yuge H., Chem. Lett. 2019, 48, 1196–1198. [Google Scholar]
- 75. Cheng B., Lu P., Zhang H. Y., Cheng X. P., Lu Z., J. Am. Chem. Soc. 2017, 139, 9439–9442. [DOI] [PubMed] [Google Scholar]
- 76. Guo J., Shen X. Z., Lu Z., Angew. Chem. Int. Ed. 2017, 56, 615–618; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 630–633. [Google Scholar]
- 77. Cheng B., Liu W. B., Lu Z., J. Am. Chem. Soc. 2018, 140, 5014–5017. [DOI] [PubMed] [Google Scholar]
- 78. Du X., Zhang Y., Peng D., Huang Z., Angew. Chem. Int. Ed. 2016, 55, 6671–6675; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6783–6787. [Google Scholar]
- 79. Kong D. G., Hu B. W., Chen D. F., Chem. Asian J. 2019, 14, 2694–2703. [DOI] [PubMed] [Google Scholar]
- 80. Zhang S. C., Ibrahim J. J., Yang Y., Org. Lett. 2018, 20, 6265–6269. [DOI] [PubMed] [Google Scholar]
- 81. Guo J., Lu Z., Angew. Chem. Int. Ed. 2016, 55, 10835–10838; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10993–10996. [Google Scholar]
- 82. Guo J., Wang H. L., Xing S. P., Hong X., Lu Z., Chem-Us 2019, 5, 881–895. [Google Scholar]
- 83. Wen H. A., Wan X. L., Huang Z., Angew. Chem. Int. Ed. 2018, 57, 6319–6323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6427–6431. [Google Scholar]
- 84. Zuo Z. Q., Yang J., Huang Z., Angew. Chem. Int. Ed. 2016, 55, 10839–10843; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10997–11001. [Google Scholar]
- 85. Teo W. J., Wang C., Tan Y. W., Ge S., Angew. Chem. Int. Ed. 2017, 56, 4328–4332; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4392–4396. [Google Scholar]
- 86. Kidonakis M., Stratakis M., Org. Lett. 2015, 17, 4538–4541. [DOI] [PubMed] [Google Scholar]
- 87. Shao D., Li Y., RSC Adv. 2018, 8, 20379–20393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Galeandro-Diamant T., Sayah R., Zanota M. L., Marrot S., Veyre L., Thieuleux C., Meille V., Chem. Commun. 2017, 53, 2962–2965. [DOI] [PubMed] [Google Scholar]
- 89. Pandarus V., Ciriminna R., Gingras G., Béland F., Kaliaguine S., Pagliaro M., Green Chem. 2019, 21, 129–140. [Google Scholar]
- 90. Li L. M., Li Y. X., Yan J. C., Cao H., Shao D. Y., Bao J. J., Rsc Adv. 2019, 9, 12696–12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zai H. C., Zhao Y. Z., Chen S. Y., Ge L., Chen C. F., Chen Q., Li Y. J., Nano Res. 2018, 11, 2544–2552. [Google Scholar]
- 92. Chen Y., Ji S., Sun W., Chen W., Dong J., Wen J., Zhang J., Li Z., Zheng L., Chen C., Peng Q., Wang D., Li Y., J. Am. Chem. Soc. 2018, 140, 7407–7410. [DOI] [PubMed] [Google Scholar]
- 93. Zhu Y., Cao T., Cao C., Luo J., Chen W., Zheng L., Dong J., Zhang J., Han Y., Li Z., Chen C., Peng Q., Wang D., Li Y., ACS Catal. 2018, 8, 10004–10011. [Google Scholar]
- 94. Motokura K., Maeda K., Chun W.-J., ACS Catal. 2017, 7, 4637–4641. [Google Scholar]
- 95. Sarma B. B., Kim J., Amsler J., Agostini G., Weidenthaler C., Pfander N., Arenal R., Concepcion P., Plessow P., Studt F., Prieto G., Angew. Chem. Int. Ed. 2020, 59, 5806–5815; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5855–5864. [Google Scholar]
- 96. Buslov I., Song F., Hu X., Angew. Chem. Int. Ed. 2016, 55, 12295–12299; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12483–12487. [Google Scholar]
- 97. Galeandro-Diamant T., Suleimanov I., Veyre L., Bousquie M., Meille V., Thieuleux C., Catal. Sci. Technol. 2019, 9, 1555–1558. [Google Scholar]
- 98. Zhang Z. K., Bai L. C., Hu X. L., Chem. Sci. 2019, 10, 3791–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhou Y. B., Liu Z. K., Fan X. Y., Li R. H., Zhang G. L., Chen L., Pan Y. M., Tang H. T., Zeng J. H., Zhan Z. P., Org. Lett. 2018, 20, 7748–7752. [DOI] [PubMed] [Google Scholar]
- 100. Li R. H., An X. M., Yang Y., Li D. C., Hu Z. L., Zhan Z. P., Org. Lett. 2018, 20, 5023–5026. [DOI] [PubMed] [Google Scholar]
- 101. Mitsudome T., Fujita S., Sheng M., Yamasaki J., Kobayashi K., Yoshida T., Maeno Z., Mizugaki T., Jitsukawa K., Kaneda K., Green Chem. 2019, 21, 4566–4570. [Google Scholar]
- 102. Cao L., Lin Z., Peng F., Wang W., Huang R., Wang C., Yan J., Liang J., Zhang Z., Zhang T., Long L., Sun J., Lin W., Angew. Chem. Int. Ed. 2016, 55, 4962–4966; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5046–5050. [Google Scholar]
- 103. Azuma R., Nakamichi S., Kimura J., Yano H., Kawasaki H., Suzuki T., Kondo R., Kanda Y., Shimizu K.-i., Kato K., Obora Y., ChemCatChem 2018, 10, 2378–2382. [Google Scholar]
- 104. Zhang J. W., Lu G. P., Cai C., Green Chem. 2017, 19, 2535–2540. [Google Scholar]
- 105. Wen J., Chen Y., Ji S., Zhang J., Wang D., Li Y., Nano Res. 2019, 12, 2584–2588. [Google Scholar]
- 106. Miura H., Endo K., Ogawa R., Shishido T., ACS Catal. 2017, 7, 1543–1553. [Google Scholar]