

Abstract
The application of arynes as radical acceptors is described. The stable radical TEMPO (2,2,6,6‐tetramethyl piperidine 1‐oxyl) is shown to add to various ortho‐substituted benzynes generating the corresponding aryl radicals which engage in 5‐exo or 6‐endo cyclizations. The cyclized radicals are eventually trapped by TEMPO. The introduced method provides ready access to various dihydrobenzofurans, oxindoles, and sultones by a conceptually novel approach.
Keywords: arynes, cyclization, heterocycles, radicals, TEMPO
Arynes generated in situ are shown to be good acceptors for the persistent TEMPO radical. The adduct aryl radicals engage in typical radical reactions such as direct TEMPO trapping, cyclization, or hydrogen atom transfer. Final TEMPO trapping provides bisalkoxyamines. Cyclizations, dihydrobenzofurans, oxindoles, and sultones can be prepared by this conceptually novel aryne chemistry, nicely complementing existing aryne methodology.
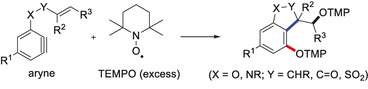
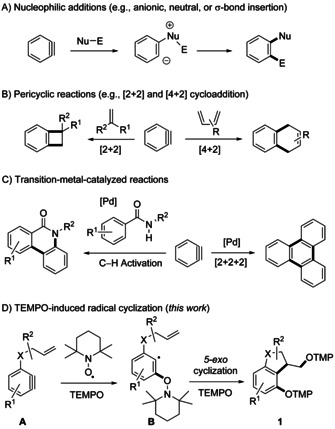
Arynes are an interesting class of reactive intermediates which can be used as versatile building blocks in organic chemistry, as convincingly documented by their application in natural product synthesis. [1] Structurally, arynes exhibit a strained triple bond with a large singlet–triplet gap in the ground state (37.7 kcal mol−1 for benzyne). [2] A direct consequence of this unusual bent alkyne structure is their low‐lying LUMO which renders arynes highly reactive.[ 1c , 3 ] The electrophilic character of arynes has been intensively studied and multi‐component, [4] aryne relay, [5] and σ‐bond insertion [6] reactions have been developed (Scheme 1 A). Furthermore, the aryne triple bond engages in pericyclic reactions, which has been exploited in [2+2] and [4+2] cycloadditions (Scheme 1 B). [7] Arynes have also found use as intermediates in transition‐metal‐catalyzed [2+2+2] reactions, [8] σ‐bond insertions, [9] C−H activations, [10] and multicomponent reactions [11] (Scheme 1 C). For example, in the presence of a Pd catalyst, benzyne undergoes a cyclotrimerization to give triphenylene. [8a]
Scheme 1.
Arynes as reactive intermediates in synthesis (TEMPO=2,2,6,6‐tetramethyl piperidine 1‐oxyl, TMP=(2,2,6,6‐tetramethyl)‐piperidin‐1‐yl).
The low‐lying LUMO of arynes should also make them ideal radical acceptors; however, cascades comprising arynes as acceptors are nearly unexplored. [12] The main challenge in such transformations lies in the low concentrations of both aryne and radical, as both reaction components are highly reactive intermediates. [13] Therefore, coupling of a radical with an aryne in a chain reaction is highly challenging and, not surprisingly, only very few aryne radical reactions have been reported, mostly discovered as unexpected processes. For example, Shioji and co‐workers found that benzyne reacts with sterically highly congested thiones not via the targeted [2+2] cycloaddition but as a biradical adding to the C−S double bond. [12c] Wang and co‐workers [12h] isolated dibenzoselenophenes by reacting tetraynes generated in a hexadehydro Diels–Alder process with diphenyldiselenide. They suggested a mechanism based on a free‐radical reaction. Murphy and Tuttle showed that benzyne can act as a radical initiator in base‐promoted homolytic aromatic substitutions. [12g] However, to the best of our knowledge, preparative valuable radical cascades comprising arynes as acceptors exploiting their biradical character have not been reported to date.
We envisioned to address the critical “concentration problem” of aryne radical chemistry by using nitroxides that are stable and persistent radicals as the aryne reaction partners.[ 13b , 14 ] To our knowledge, reactions of arynes with nitroxides have not been reported to date. Our strategy is depicted in Scheme 1 D. An in situ generated aryne of type A bearing a pendant second radical acceptor should react with TEMPO, added as a stable reagent, to the aryl radical B. This in turn will undergo a fast 5‐exo‐cyclization to the corresponding cyclized alkyl radical, which will be finally trapped by a second equivalent of TEMPO to give the trapping product 1. This unique transformation, comprising three consecutive σ‐bond formations, deserves further comments: Since TEMPO addition to an unactivated alkene is not an efficient reaction, [15] we expect a highly chemoselective initial addition of TEMPO to the highly reactive aryne functionality in A. Moreover, due to the bulkiness of TEMPO, addition onto the aryne should occur with high regioselectivity. [16] The subsequent 5‐exo‐cyclization should be faster than direct intermolecular TEMPO trapping of B and the terminating radical/radical cross coupling of the cyclized radical with the second TEMPO should be selective due to the high relative concentration of the persistent TEMPO radical. [13b]
To evaluate the feasibility of such an unprecedented cascade, we first investigated the reactivity of benzyne towards TEMPO. TEMPO is a cheap, commercially available and bench‐stable nitroxyl radical which has been used as a radical trapping reagent, in living radical polymerizations and in oxidations of alcohols in combination with stoichiometric amounts of a cooxidant. [17] The Kobayashi method was selected for aryne generation.[ 18 , 19 , 20 ] Pleasingly, we found that the reaction of the triflate 2 with CsF (3.0 equiv) and 18‐crown‐6 ether (3.0 equiv) in the presence of TEMPO (5.0 equiv) in n‐hexane provided the TEMPO‐bisadduct 3 in 48 % yield, clearly documenting that TEMPO addition onto an aryne is occurring (Scheme 2). Product 3 is formed by the trapping of the intermediately generated TEMPO adduct aryl radical with a second equivalent of TEMPO. Notably, bisalkoxyamine 3 was found to be moderately stable and its structure was unambiguously confirmed by X‐ray crystallography. [21]
Scheme 2.
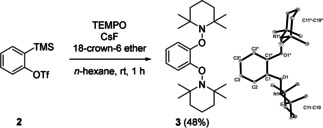
Radical reaction of benzyne with TEMPO to give the bisalkoxyamine 3 and crystal structure of 3 (H‐atoms are omitted).
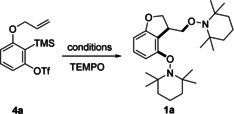
Encouraged by this result, we next investigated the radical cascade suggested in Scheme 1 D. To this end, triflate 4 a was prepared as the model substrate (Table 1). We were very pleased to find that upon reacting 4 a with CsF (3.0 equiv) and 18‐crown‐6 ether (3.0 equiv) in the presence of TEMPO (5.0 equiv) in n‐hexane at room temperature the targeted dihydrobenzofuran 1 a was obtained in 59 % yield (Table 1, entry 1). [22] Decreasing the amount of TEMPO to 2 equivalents led to a lower yield of 4 a, whereas increasing the TEMPO amount provided a similar yield (35 % and 57 %, Table 1, entries 2 and 3). Solvent screening revealed n‐hexane, toluene, and acetonitrile to be good solvents for this cascade, but a worse result was achieved in 1,2‐dichloroethane (Table 1, entry 9). The good solvents show small differences in the amount of product formed (Table 1, entries 4–6). For reactions carried out in n‐hexane, 18‐crown‐6 ether was required in order to increase the solubility of the fluoride source. Variation of the temperature (−20 °C to rt) showed only little impact on the yield (Table 1, entries 6–8). CsF can be replaced by TBAT or K2CO3 at 70 °C for aryne generation, although lower yields were noted in these cases (30 %, 53 %, Table 1, entries 10 and 11).
Table 1.
Reaction optimization.[a]
|
Entry |
TEMPO (equiv) |
Solvent |
T |
Yield 1 a [%] |
|---|---|---|---|---|
|
1[b] |
5 |
n ‐hexane |
rt |
59 |
|
2[c] |
2 |
n‐hexane |
rt |
35 |
|
3[b] |
10 |
n‐hexane |
rt |
57 |
|
4[b] |
5 |
n‐hexane |
0 °C |
59 |
|
5[b] |
5 |
PhMe |
0 °C |
60 |
|
6 |
5 |
MeCN |
rt |
58 |
|
7 |
5 |
MeCN |
0 °C |
50 |
|
8 |
5 |
MeCN |
−20 °C |
56 |
|
9 |
5 |
1,2‐DCE |
rt |
34 |
|
10[d] |
5 |
MeCN |
70 °C |
30 |
|
11[e] |
5 |
MeCN |
rt |
53 |
[a] Reaction conditions: 4 a (0.1 mmol, 1.0 equiv), TEMPO (0.5 mmol, 5.0 equiv), CsF (0.3 mmol, 3.0 equiv), and solvent (1 mL, 0.1 m). Yields represent isolated yields. [b] 18‐crown‐6 ether (0.3 mmol, 3.0 equiv) was added. [c] CsF (0.2 mmol, 2.0 equiv) and 18‐crown‐6 ether (0.2 mmol, 2.0 equiv) were used. [d] K2CO3 (0.40 mmol, 4.0 equiv) and 18‐crown‐6 ether (0.40 mmol, 4.0 equiv) were used to prepare the aryne. [e] Tetrabutylammonium difluorotriphenylsilicate (TBAT, 0.30 mmol, 3.0 equiv) was used to prepare the aryne. rt=room temperature.
With optimized conditions in hand (Table 1, entry 1), we tested different triflates 4 b–n in this novel cascade (Scheme 3). For the preparation of the starting materials, we refer to the Supporting Information (SI). A substituent at the 5‐position of the intermediate 3‐allyoxy aryne is tolerated: Electron‐donating groups such as phenyl (1 b) and methoxy (1 c) led to lower yields, whereas the electron‐withdrawing chloro‐substituent showed little effect on the yield (1 d). In the NMR spectra of products 1 b to 1 d an inseparable side product was identified in each case (3–9 %, see SI). These side products derive from a 1,6‐HAT from the TEMPO‐methyl group of the intermediate aryl radical B with subsequent TEMPO trapping (see analogous compound 6 e in Scheme 4 below). Moreover, the generally moderate yields observed are also caused by the instability of the products (labile N−O bond in aryl‐TEMPO alkoxyamines).
Scheme 3.
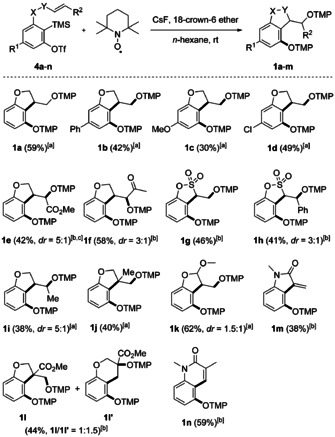
Radical reaction of o‐substituted arynes with TEMPO and cyclization to bisalkoxyamines. Yields represent isolated yields. Reaction time t=1–18 hours. Conditions: [a] Method A: 4 (0.20 mmol, 1.0 equiv), TEMPO (1.0 mmol, 5.0 equiv), CsF (0.60 mmol, 3.0 equiv), 18‐crown‐6 ether (0.60 mmol, 3.0 equiv), and n‐hexane (2.0 mL). [b] Method B: 4 (0.20 mmol, 1.0 equiv), TEMPO (1.0 mmol, 5.0 equiv), CsF (0.60 mmol, 3.0 equiv), and MeCN (2.0 mL). [c] Reaction was performed on 0.25 mmol scale.
Scheme 4.
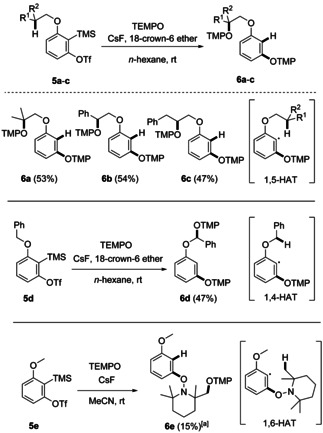
Reactions of arynes with TEMPO involving 1,5‐ or 1,4‐HAT to give bisalkoxyamines 6 a–d. Reaction conditions: 5 (0.20 mmol, 1.0 equiv), TEMPO (1.0 mmol, 5.0 equiv), CsF (0.60 mmol, 3.0 equiv), 18‐crown‐6 ether (0.60 mmol, 3.0 equiv), and n‐hexane (2.0 mL). [a] Reaction condition: 5 e (1.0 mmol, 1.0 equiv), TEMPO (3.0 mmol, 3.0 equiv), CsF (3.0 mmol, 3.0 equiv), and MeCN (10 mL). Yields represent isolated yields.
The allyloxy group can be further substituted (R2 group) by an ester (1 e), acetyl (1 f) or methyl group (1 i) providing the corresponding dihydrobenzofurans in 38–58 % yields [22] with moderate diastereoselectivities for the TEMPO‐trapping step (3:1 to 5:1). The structure of the major isomer of ester 1 e was confirmed by X‐ray crystallography (Figure 1). [21] α,β‐Unsaturated sulfonate esters 4 g and 4 h gave the targeted sultones 1 g and 1 h in 46 % and 41 % yield, respectively. The sultone 1 h was formed as a diastereomeric mixture (dr=3:1) and the structure of the major isomer was unambiguously assigned by X‐ray crystallography (Figure 1). [21]
Figure 1.
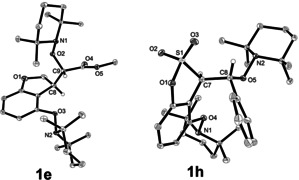
X‐ray crystal structures of the major diastereomers of ester 1 e and sultone 1 h. H‐atoms are only shown at the stereocenters.
By installing an α‐substituent at the allyloxy group we also addressed the diastereoselectivity of the fast radical 5‐exo‐cyclization. For a tested system (4 k), only a moderate selectivity was noted and the 2,3‐disubstituted dihydrobenzofuran 1 k was obtained in 62 % yield (dr=1.5:1). As expected, for β‐substituted 3‐allyloxyarynes, the regioselectivity of the radical cyclization was not complete. Hence, with the activating ester moiety, a significant amount of the 6‐endo product 1 l′ was formed (5‐exo/6‐endo=1:1.5). However, the β‐methyl congener afforded exclusively the 5‐exo product 1 j. In the NMR spectra of 1 j we identified an inseparable side product that is derived from a 1,6‐HAT from the TEMPO moiety of intermediate B with subsequent TEMPO trapping. To our surprise, reaction of the acryl amide 4 m with TEMPO under optimized conditions did not afford the expected bisalkoxyamine, but the oxindole 1 m was isolated in 38 % yield. Methacrylamide 4 n furnished the quinolinone 1 n in 59 % yield via a 6‐endo cyclization. In these two cases (1 m, 1 n), facile TEMPOH elimination is occurring from the targeted products via an ionic or a radical pathway. [23]
Finally, we investigated the reactivity of the intermediate aryl radical generated by TEMPO addition to an aryne towards intramolecular hydrogen atom transfer (HAT), further leveraging the potential of radical aryne chemistry (Scheme 4). Of note, radical translocation via 1,5‐HAT to aryl radicals has been successfully used in organic synthesis. [24] The ortho‐alkoxyl substituted aryne precursors 5 a–c bearing a C−H bond at the β‐position of the alkoxyl group engaged in the cascade and the bisalkoxyamines 6 a, 6 b, and 6 c were obtained with similar yields (47–54 %). These reactions proceed via TEMPO addition to the intermediate aryne to give the corresponding adduct aryl radical that further reacts via a 1,5‐HAT. The thus generated translocated alkyl radical is eventually trapped by the second equivalent of TEMPO to give compounds of type 6. Considering triflate 5 c, the bisalkoxyamine derived from a 1,6‐HAT was not identified. In analogy, triflate 5 d reacted via a 1,4‐HAT to the mixed acetal 6 d (47 %). Surprisingly, subjecting triflate 5 e to the standard reaction conditions did not provide the expected 1,4‐HAT‐derived acetal; instead, we isolated the 1,6‐HAT/TEMPO trapping product 6 e (15 %).
In summary, we have shown that arynes react as in situ generated radical acceptors with the persistent TEMPO radical. The adduct aryl radical thus generated can then engage in different typical radical reactions such as direct TEMPO trapping, cyclization and intramolecular hydrogen atom transfer. The rearranged radicals generated in the latter two cases can finally be trapped by the persistent TEMPO radical in a highly selective radical/radical cross coupling. In all cases, bisalkoxyamines result in rather good yields considering the complexity of these cascades. Aryne radical chemistry nicely complements existing ionic or transition‐metal based reactions of arynes opening new doors in that timely research area.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for supporting this project.
M. Scherübl, C. G. Daniliuc, A. Studer, Angew. Chem. Int. Ed. 2021, 60, 711.
Dedicated to Professor Ilhyong Ryu on the occasion of bis 70th birthday
References
- 1.For reviews see:
- 1a. Tadross P. M., Stoltz B. M., Chem. Rev. 2012, 112, 3550–3577; [DOI] [PubMed] [Google Scholar]
- 1b. Wenk H. H., Winkler M., Sander W., Angew. Chem. Int. Ed. 2003, 42, 502–528; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 518–546; [Google Scholar]
- 1c. Sanz R., Org. Prep. Proced. Int. 2008, 40, 215–291; [Google Scholar]
- 1d. Gampe C. M., Carreira E. M., Angew. Chem. Int. Ed. 2012, 51, 3766–3778; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3829–3842; [Google Scholar]
- 1e. Takikawa H., Nishii A., Sakai T., Suzuki K., Chem. Soc. Rev. 2018, 47, 8030–8056; [DOI] [PubMed] [Google Scholar]
- 1f. Wentrup C., Aust. J. Chem. 2010, 63, 979–986; [Google Scholar]
- 1g. Jiang X., Feng M., Synthesis 2017, 49, 4414–4433; [Google Scholar]
- 1h. Bhunia A., Reddy Yetra S., Biju A. T., Chem. Soc. Rev. 2012, 41, 3140–3152. [DOI] [PubMed] [Google Scholar]
- 2. Leopold D. G., Miller A. E. S., Lineberger W. C., J. Am. Chem. Soc. 1986, 108, 1379–1384. [Google Scholar]
- 3. Cahill K. J., Ajaz A., Johnson R. P., Aust. J. Chem. 2010, 63, 1007–1012. [Google Scholar]
- 4.
- 4a.Review: Bhojgude S. S., Bhunia A., Biju A. T., Acc. Chem. Res. 2016, 49, 1658–1670; [DOI] [PubMed] [Google Scholar]
- 4b. Sha F., Huang X., Angew. Chem. Int. Ed. 2009, 48, 3458–3461; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3510–3513; [Google Scholar]
- 4c. Bhojgude S. S., Biju A. T., Angew. Chem. Int. Ed. 2012, 51, 1520–1522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1550–1552; [Google Scholar]
- 4d. Bhunia A., Roy T., Pachfule P., Rajamohanan P. R., Biju A. T., Angew. Chem. Int. Ed. 2013, 52, 10040–10043; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10224–10227; [Google Scholar]
- 4e. Bhunia A., Porwal D., Gonnade R. G., Biju A. T., Org. Lett. 2013, 15, 4620–4623; [DOI] [PubMed] [Google Scholar]
- 4f. Allan K. M., Gilmore C. D., Stoltz B. M., Angew. Chem. Int. Ed. 2011, 50, 4488–4491; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4580–4583. [Google Scholar]
- 5.
- 5a. Yoshida S., Nakamura Y., Uchida K., Hazama Y., Hosoya T., Org. Lett. 2016, 18, 6212–6215; [DOI] [PubMed] [Google Scholar]
- 5b. Yoshida S., Shimizu K., Uchida K., Hazama Y., Igawa K., Tomooka K., Hosoya T., Chem. Eur. J. 2017, 23, 15332–15335; [DOI] [PubMed] [Google Scholar]
- 5c. Xiao X., Hoye T. R., J. Am. Chem. Soc. 2019, 141, 9813–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Tambar U. K., Stoltz B. M., J. Am. Chem. Soc. 2005, 127, 5340–5341; [DOI] [PubMed] [Google Scholar]
- 6b. Liu Z., Larock R. C., J. Am. Chem. Soc. 2005, 127, 13112–13113; [DOI] [PubMed] [Google Scholar]
- 6c. Peña D., Pérez D., Guitián E., Angew. Chem. Int. Ed. 2006, 45, 3579–3581; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 3659–3661; [Google Scholar]
- 6d. Hoye T. R., Baire B., Niu D., Willoughby P. H., Woods B. P., Nature 2012, 490, 208–212; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Yoshida H., Yoshida R., Takaki K., Angew. Chem. Int. Ed. 2013, 52, 8629–8632; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8791–8794; [Google Scholar]
- 6f. Li Y., Chakrabarty S., Mück-Lichtenfeld C., Studer A., Angew. Chem. Int. Ed. 2016, 55, 802–806; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 813–817; [Google Scholar]
- 6g. Mesgar M., Nguyen-Le J., Daugulis O., J. Am. Chem. Soc. 2018, 140, 13703–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Mariet N., Ibrahim-Ouali M., Santelli M., Tetrahedron Lett. 2002, 43, 5789–5791; [Google Scholar]
- 7b. Maurin P., Ibrahim-Ouali M., Parrain J.-L., Santelli M., J. Mol. Struct. Theochem 2003, 637, 91–100; [Google Scholar]
- 7c. Buszek K. R., Brown N., Luo D., Org. Lett. 2009, 11, 201–204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7d. Yoshida S., Morita T., Hosoya T., Chem. Lett. 2016, 45, 726–728; [Google Scholar]
- 7e. Li Y., Mück-Lichtenfeld C., Studer A., Angew. Chem. Int. Ed. 2016, 55, 14435–14438; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14649–14653; [Google Scholar]
- 7f. Umezu S., dos Passos Gomes G., Yoshinaga T., Sakae M., Matsumoto K., Iwata T., Alabugin I., Shindo M., Angew. Chem. Int. Ed. 2017, 56, 1298–1302; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1318–1322. [Google Scholar]
- 8.
- 8a. Peña D., Escudero S., Pérez D., Guitián E., Castedo L., Angew. Chem. Int. Ed. 1998, 37, 2659–2661; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2804–2806; [Google Scholar]
- 8b. Peña D., Pérez D., Guitián E., Castedo L., Org. Lett. 1999, 1, 1555–1557; [Google Scholar]
- 8c. Sato Y., Tamura T., Mori M., Angew. Chem. Int. Ed. 2004, 43, 2436–2440; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2490–2494; [Google Scholar]
- 8d. Caeiro J., Peña D., Cobas A., Pérez D., Guitián E., Adv. Synth. Catal. 2006, 348, 2466–2474; [Google Scholar]
- 8e. Qiu Z., Xie Z., Angew. Chem. Int. Ed. 2009, 48, 5729–5732; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5839–5842. [Google Scholar]
- 9.
- 9a. Yoshida H., Ikadai J., Shudo M., Ohshita J., Kunai A., J. Am. Chem. Soc. 2003, 125, 6638–6639; [DOI] [PubMed] [Google Scholar]
- 9b. Yoshida H., Tanino K., Ohshita J., Kunai A., Angew. Chem. Int. Ed. 2004, 43, 5052–5055; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 5162–5165; [Google Scholar]
- 9c. Yoshida H., Kawashima S., Takemoto Y., Okada K., Ohshita J., Takaki K., Angew. Chem. Int. Ed. 2012, 51, 235–238; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 239–242. [Google Scholar]
- 10.
- 10a. Liu Z., Zhang X., Larock R. C., J. Am. Chem. Soc. 2005, 127, 15716–15717; [DOI] [PubMed] [Google Scholar]
- 10b. Gerfaud T., Neuville L., Zhu J., Angew. Chem. Int. Ed. 2009, 48, 572–577; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 580–585; [Google Scholar]
- 10c. Peng X., Wang W., Jiang C., Sun D., Xu Z., Tung C.-H., Org. Lett. 2014, 16, 5354–5357. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Jayanth T. T., Jeganmohan M., Cheng C.-H., Org. Lett. 2005, 7, 2921–2924; [DOI] [PubMed] [Google Scholar]
- 11b. Henderson J. L., Edwards A. S., Greaney M. F., J. Am. Chem. Soc. 2006, 128, 7426–7427; [DOI] [PubMed] [Google Scholar]
- 11c. Yoo W.-J., Nguyen T. V. Q., Kobayashi S., Angew. Chem. Int. Ed. 2014, 53, 10213–10217; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10377–10381; [Google Scholar]
- 11d. Garve L. K. B., Werz D. B., Org. Lett. 2015, 17, 596–599. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Gassman P. G., Richmond G. D., J. Am. Chem. Soc. 1970, 92, 2090–2096; [Google Scholar]
- 12b. Usieli V., Sarel S., J. Org. Chem. 1973, 38, 1703–1708; [Google Scholar]
- 12c. Okuma K., Sonoda S., Koga Y., Shioji K., J. Chem. Soc. Perkin Trans. 1 1999, 2997–3000; [Google Scholar]
- 12d. Rao U. N., Biehl E., J. Org. Chem. 2002, 67, 3409–3411; [DOI] [PubMed] [Google Scholar]
- 12e. Rao U. N., Sathunuru R., Maguire J. A., Biehl E., J. Heterocycl. Chem. 2004, 41, 13–21; [Google Scholar]
- 12f. Yamabe S., Minato T., Ishiwata A., Irinamihira O., Machiguchi T., J. Org. Chem. 2007, 72, 2832–2841; [DOI] [PubMed] [Google Scholar]
- 12g. Zhou S., Anderson G. A., Mondal B., Doni E., Ironmonger V., Kranz M., Tuttle T., Murphy J. A., Chem. Sci. 2014, 5, 476–481; [Google Scholar]
- 12h. Hu Y., Ma J., Li L., Hu Q., Lv S., Liu B., Wang S., Chem. Commun. 2017, 53, 1542–1545; [DOI] [PubMed] [Google Scholar]
- 12i. Yang X., Chit Tsui G., Chem. Sci. 2018, 9, 8871–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Berry R. S., Clardy J., Schafer M. E., Tetrahedron Lett. 1965, 6, 1011–1017; [Google Scholar]
- 13b. Leifert D., Studer A., Angew. Chem. Int. Ed. 2020, 59, 74–108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 74–110. [Google Scholar]
- 14.
- 14a. Karoui H., Le Moigne F., Ouari O., Tordo P. in Stable Radicals: fundamentals and applied aspects of odd-electron compounds (Ed.: Hicks R. G.), Wiley, Chichester, 2010, pp. 173–230; [Google Scholar]
- 14b. Tebben L., Studer A., Angew. Chem. Int. Ed. 2011, 50, 5034–5068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5138–5174. [Google Scholar]
- 15. Coseri S., Ingold K. U., Org. Lett. 2004, 6, 1641–1643. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Cheong P. H.-Y., Paton R. S., Bronner S. M., Im G.-Y. J., Garg N. K., Houk K. N., J. Am. Chem. Soc. 2010, 132, 1267–1269; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Medina J. M., Mackey J. L., Garg N. K., Houk K. N., J. Am. Chem. Soc. 2014, 136, 15798–15805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Studer A., Schulte T., Chem. Rec. 2005, 5, 27–35; [DOI] [PubMed] [Google Scholar]
- 17b. Vogler T., Studer A., Synthesis 2008, 1979–1993. [Google Scholar]
- 18. Himeshima Y., Sonoda T., Kobayashi H., Chem. Lett. 1983, 12, 1211–1214. [Google Scholar]
- 19.The Knochel method operating with Grignard reagents does not work since such organometallic species are known to react efficiently with TEMPO, see ref. [20]. Sapountzis I., Lin W., Fischer M., Knochel P., Angew. Chem. Int. Ed. 2004, 43, 4364–4366; [Google Scholar]; Angew. Chem. 2004, 116, 4464–4466. [Google Scholar]
- 20.
- 20a. Whitesides G. M., Newirth T. L., J. Org. Chem. 1975, 40, 3448–3450; [Google Scholar]
- 20b. Maji M. S., Pfeifer T., Studer A., Angew. Chem. Int. Ed. 2008, 47, 9547–9550; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9690–9692; [Google Scholar]
- 20c. Studer A., Maji M., Synthesis 2009, 2467–2470. [Google Scholar]
- 21.Deposition numbers 2030811 (3), 2030809 (1e), and 2030810 (1 h) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 22.The lower yield of 1 i as compared to the parent 1 a may be caused by a competing intramolecular ene reaction, as observed by Lautens and co-workers on related systems. Unfortunately, we were not able to isolate the suggested side products. Candito D. A., Dobrovolsky D., Lautens M., J. Am. Chem. Soc. 2012, 134, 15572–15580. [DOI] [PubMed] [Google Scholar]
- 23. Knoop C. A., Studer A., J. Am. Chem. Soc. 2003, 125, 16327–16333. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Curran D. P., Kim D., Liu H. T., Shen W., J. Am. Chem. Soc. 1988, 110, 5900–5902; [Google Scholar]
- 24b. Snieckus V., Cuevas J.-C., Sloan C. P., Liu H., Curran D. P., J. Am. Chem. Soc. 1990, 112, 896–898. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary