

Abstract
Characterizing ecological relationships between viruses, bacteria and phytoplankton in the ocean is critical to understanding the ecosystem; however, these relationships are infrequently investigated together. To understand the dynamics of microbial communities and environmental factors in harmful algal blooms (HABs), we examined the environmental factors and microbial communities during Akashiwo sanguinea HABs in the Jangmok coastal waters of South Korea by metagenomics. Specific bacterial species showed complex synergistic and antagonistic relationships with the A. sanguinea bloom. The endoparasitic dinoflagellate Amoebophrya sp. 1 controlled the bloom dynamics and correlated with HAB decline. Among nucleocytoplasmic large DNA viruses (NCLDVs), two Pandoraviruses and six Phycodnaviruses were strongly and positively correlated with the HABs. Operational taxonomic units of microbial communities and environmental factors associated with A. sanguinea were visualized by network analysis: A. sanguinea–Amoebophrya sp. 1 (r = .59, time lag: 2 days) and A. sanguinea–Ectocarpus siliculosus virus 1 in Phycodnaviridae (0.50, 4 days) relationships showed close associations. The relationship between A. sanguinea and dissolved inorganic phosphorus relationship also showed a very close correlation (0.74, 0 day). Microbial communities and the environment changed dynamically during the A. sanguinea bloom, and the rapid turnover of microorganisms responded to ecological interactions. A. sanguinea bloom dramatically changes the environments by exuding dissolved carbohydrates via autotrophic processes, followed by changes in microbial communities involving host‐specific viruses, bacteria and parasitoids. Thus, the microbial communities in HAB are composed of various organisms that interact in a complex manner.
Keywords: Akashiwo sanguinea, endoparasitic Amoebophrya sp., environmental change, harmful algal bloom, microbial community nucleocytoplasmic large DNA viruses
1. INTRODUCTION
In ecology, phytoplankton are considered as a double‐edged sword (Zhou et al., 2018). Although phytoplankton is an essential component of the marine ecosystem because of its multiple roles in matter cycling (Arrigo, 2005), a few phytoplankton taxa form harmful algal blooms (HABs), which can adversely impact marine ecosystems and human health (Anderson, 1997). Most known HABs are dinoflagellates (Smayda, 1997), among which, Akashiwo sanguinea causes frequent blooms worldwide (Du et al., 2011; Yang et al., 2012). A. sanguinea produces surfactants that saturate the feathers of marine birds with water and cause severe hypothermia (Jessup et al., 2009) and have also been associated with fish kills and marine mammal strandings (Amorim Reis‐Filho et al., 2012). However, the environmental changes caused by these strategies in dissolved organic matter and specific nutrient sources of this bloom are poorly understood.
Marine microbial communities are diverse (viruses, bacteria, fungi and some parasitic algae) and support other marine organisms; particularly, they have the potential to impact population dynamics of HAB organisms (Chen et al., 2018). Viruses are the most common biological entities in the marine environment and greatly contribute to the flux of energy and matter as well as influence biogeochemical cycling (Fuhrman, 1999). Nucleocytoplasmic large DNA viruses (NCLDVs) infect both animals and unicellular eukaryotes (Colson et al., 2013). Members of the Phycodnaviridae family are large icosahedral NCLDVs that are mostly known to infect eukaryotic algae (Van Etten et al., 2002). However, not all viral families have not yet been assigned a species‐specific host group. For example, Mimiviridae infect Acanthamoeba and other protists which serve as natural hosts (Claverie & Abergel, 2018), but members of this group were recently shown to infect various phytoplankton species. Thus, the role of each group of NCLDVs in host‐specific infection remains unclear (Claverie & Abergel, 2018; Schulz et al., 2017). Particularly, the relationship between NCLDVs and hosts in ecological systems has not been studied.
Interactions between phytoplankton and bacteria are important in shaping their environment, and consequently, the biogeochemical cycles (Azam & Malfatti, 2007). Phytoplankton rely on bacteria to remineralize organic matter back to its inorganic substituents (Worden et al., 2015). Recently, specific bacterial phylotypes were shown to be associated with different microalgae. Yang et al. (2016) reported species‐specific relationships between bacterial communities and A. sanguinea bloom. In eukaryotic parasitoids, Amoebophrya sp. kills its host and controls the dinoflagellate bloom (Mazzillo et al., 2011). Amoebophrya sp. has a relatively short generation time and high prevalence in nature (Coats et al., 1996; Coats & Park, 2002). Studies on the interaction between Amoebophrya sp. and A. sanguinea bloom as host are performed in the laboratory (Coats & Park, 2002); however, ecological species‐specific host–parasitoid interactions remain unclear.
Interactions among microbial communities in an ecosystem are very complex. Therefore, assessing changes in environmental characteristics and their interactions with microorganisms in A. sanguinea bloom can increase the understanding of microbial communities. With the advancement of metagenomic next‐generation sequencing (mNGS) technology, a large volume of sequencing data have been analysed and baseline information regarding genetic traits has been determined. In addition, many ecological studies have used mNGS to estimate changes in population dynamics and communities (Kim et al., 2016). New technologies for studying aquatic microbial diversity require smaller volumes and masses of DNA (Flaviani et al., 2017). To explore changes in environmental characteristics and microbial communities in the phycosphere of A. sanguinea bloom and estimate the potential control mechanisms for A. sanguinea bloom, we investigated the ecological phenomena when A. sanguinea bloomed in the Jangmok Bay Time‐series Monitoring Site (JBTMS). Specifically, we used an intensive monitoring plan (i.e. daily sampling) to understand the dynamics of microbial communities.
2. MATERIALS AND METHODS
2.1. Sample collection
The sampling site (Jangmok Bay Time‐series Monitoring Site (JBTMS): 34°59′37″N and 128°40′27″E) is a semi‐closed bay on the southern coast of South Korea (Kim et al., 2016; Figure S1). The JBTMS is a eutrophic system subjected to strong mixing between the surface and bottom layers. Its maximum tidal range is approximately 2.2 m, and the mean water depth at the sampling station is approximately 8.5 m. A total of 90 subsamples during June 2016 and June 2017 were obtained from the surface water (sampling depth: 1 m under sea surface). In particular, when A. sanguinea bloomed (11 November–26 December 2016), we conducted intensive collections (daily sampling). We also explored the differences between A. sanguinea bloom and no‐bloom conditions daily between 14 November and 26 December 2017. We drew 10 L samples of seawater from the surface layer and stored them in an ice cooler (approximately 4°C) until arrival (5 min) at the laboratory of the South Sea Institute of Korea Institute of Ocean Science & Technology (KIOST, Geoje, South Korea), where the seawater was prepared immediately.
2.2. Monitoring of environmental factors
Temperature, salinity, pH and dissolved oxygen (DO) were evaluated using a portable YSI environmental multiparameter (YSI 6920 Inc.). A 100 ml aliquot of each subsample was filtered through a 47‐mm glass fibre filter (GF/F, Whatman), and the filtered seawater was added to an acid‐cleaned polyethylene bottle and stored at −80°C until nutritional analysis. Subsequently, the concentrations of dissolved inorganic nutrients, such as dissolved inorganic nitrogen (DIN; NO2 − + NO3 − + NH4 +), dissolved inorganic phosphorus (DIP) and dissolved silica (DSi), were determined in each sample using an automatic nutrient analyser (QuAAtro39; Seal Analytical Instrument). To analyse the dissolved organic carbon (DOC) concentration, a 10 ml aliquot of each water sample was filtered through a GF/F filter (precombusted at 450°C overnight) under gravity pressure, and the DOC concentration was determined using a high‐temperature catalytic combustion instrument (TOC‐VCPH; Shimadzu). To determine the chlorophyll a concentration, 500 ml of each sample was filtered through a GF/F filter under low vacuum pressure. Each filter was soaked in 15 ml of cold 90% acetone‐distilled water (v/v) and sonicated to break the cell walls. Chlorophyll a was extracted for 24 hr at 4°C in the dark, and its concentration was measured with a 10‐AU Fluorometer (Turner Designs, Inc.).
2.3. Microscopic observation
To count total heterotrophic bacteria, a 10 ml aliquot was collected from each subsample in a 15‐ml sterilized polyethylene bottle and fixed immediately with a final concentration 2% glutaraldehyde. The sample was stored in the dark at 4°C prior to analysis. The fixed bacterial cells were filtered through a black isopore membrane filter (GTBP 02500; Millipore) and stained with 1 μg/ml of 4′,6‐diamidino‐2‐phenylindole solution (Porter & Feig, 1980). At least 600 stained bacterial cells per sample were counted at a magnification of 1,000× using an epifluorescence microscope (Axioplan, Zeiss). To count and identify phytoplankton communities, a 900 ml subsample was collected from into a 1‐L sterilized polyethylene bottle and fixed immediately with 2% Lugol's iodine solution (final concentration). The fixed water samples were left undisturbed for 1 day, after which the supernatant was removed to concentrate the phytoplankton. In the concentrated sample, at least 500 phytoplankton cells per subsample were identified and counted using a phytoplankton (Sedgwick–Rafter) counting chamber under a light microscope (Axioplan) at a magnification of 400–1,000×
2.4. Preparation for DNA extraction of microbial communities
The microbial communities are multiphylotype communities, ranging from numerically dominant viruses to phylogenetically diverse eukaryotic plankton. For mNGS, Flaviani et al. (2017) concluded that 250 ml of seawater is sufficient to analyse microbial diversity (from double‐stranded DNA virome to eukaryotic plankton). Therefore, we analysed NCLDVs, bacteria and eukaryotic planktonic organisms (including the endoparasitic dinoflagellate Amoebophrya spp.) from 1 L surface seawater. Moreover, to analyse various microbial communities, we harvested the microbes in three steps according to their size fraction: first, a 10 μm polycarbonate filter (TCTP04700, Millipore) was used for >10 μm eukaryotic plankton and dinospores of Amoebophrya sp. in A. sanguinea cells. The filters were washed three times with approximately 50 ml distilled water at approximately 50–60°C (Jung et al., 2018) to remove organisms <10 μm in size and particles attached to A. sanguinea cell surfaces. Second, a 2‐μm polycarbonate filter (TTTP04700) was used for free‐living Amoebophrya spp. and nano‐sized phytoplankton at cell size of 10–2 μm. Finally, a 0.2‐μm polycarbonate filter (GTTP04700) was used for bacteria and NCLDVs at 0.2–2 μm. The filters were stored at −80°C until DNA extraction.
2.5. mNGS analyses of bacteria and eukaryotic plankton
The filters at each size fraction were cut into several pieces with sterilized scissors before genomic DNA (gDNA) extraction. To analyse the bacterial community in the filter sample, gDNA was extracted using the DNeasy Powersoil Kit (Qiagen) from the 0.2–2 μm size fraction; the gDNA was diluted to a final concentration of 20 ng/μl. The quantity and quality of the total gDNA were determined using NanoDrop (Nano‐MD‐NS, SCINCO Ltd.). The V3‐V4 hypervariable regions of bacterial 16S rDNA genes were amplified using the universal Illumina‐tagged forward (341F) and reverse (800R) primers (Table S1). To analyse eukaryotic plankton, gDNA was extracted from the 10 μm (>10 μm eukaryotic plankton) and 2 μm (2–10 μm size fractions) samples using a DNeasy PowerSoil Kit; gDNA was diluted to each final concentration of 30 and 20 ng/μl, respectively. The V4‐V5 region of the 18S rDNA gene was targeted using the Illumina‐tagged forward (TAReuk454FWD1) and reverse (TAReukREV3) primers (Table S2). Although we did not perform replicate experiments, we attempted to overcome the experimental bias and obtain more accurate results by intensive daily continuous monitoring in JBTMS, performing three PCRs in distinct tubes and mixing the PCR products to obtain more accurate mNGS results (Jung et al., 2018). The amplified products from the first PCR were individually purified using a QIAquick PCR Purification Kit (Qiagen). The second PCR was performed for 10 cycles using tags of Nextera XT 96 Index Kit v2 (Illumina). DNA concentration was measured with a Bioanalyzer 2100 (Agilent Technologies). Equal concentrations of the PCR products for each sample were pooled, and the merged samples were analysed using a MiSeq platform (Illumina).
After each sequencing procedure, the data were preprocessed using miseq control Software v2.4.1. Raw sequences were first analysed using FastQC (Andrew, 2010) to check the basic statistics, such as the GC%. Furthermore, the quality score distribution per base and poor‐quality sequences were flagged. Ambiguous and chimeric reads were removed, and noised sequences (denoising), which involved operational taxonomic units (OTUs) with 1, 2 and 3 reads, were removed at a cut‐off of 97%. The processed pair‐end reads were then merged using the fast length adjustment of short reads (flash) software tool (Magoč & Salzberg, 2011). After each sequencing procedure, a quality check was performed to remove short sequence reads (<150 bp), low‐quality sequences (score < 25 in analysis of 16s rDNA; score < 33 in analysis of 18s rDNA), singletons and nontarget sequences. Using the Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1990), all sequence reads were compared with those from the National Center for Biotechnology Information (NCBI) database. Sequence reads with an E‐value (<0.01) were considered for further analysis. Pairwise global alignment was performed on the selected candidate hits to identify the most similar sequences. The taxonomy of the sequence with the highest similarity was assigned to the sequence read (species level with >97% similarity). To analyse the otus, cd‐hit‐otu software (Li & Chang, 2016) was used for clustering and metagenomic functional information. To calculate alpha diversity, including Shannon–Weaver diversity, Chao richness and Simpson evenness, we used the closed‐reference protocol published by Mothur (Schloss et al., 2009) and QIIME (Caporaso et al., 2010) based on the OTU table.
2.6. mNGS analysis of NCLDVs
To analyse NCLDVs, gDNA extracted for analysis of bacterial metagenomics was used, and a sequencing library of NCLDVs was generated using NEBNext Ultra DNA Library Prep Kit (Illumina) following the manufacturer's instructions. The library was prepared by random fragmentation of the DNA sample, followed by 5′ and 3′ adapter ligation. “Tagmentation,” which combines the fragmentation and ligation reactions into a single step and greatly increases the efficiency of library preparation, was used. Adapter‐ligated fragments were amplified in 12 PCR cycles and purified by gel electrophoresis and a gel extraction kit (Qiagen). Libraries were analysed for their size distribution by using a Bioanalyser 2100 model (Agilent Technologies), which indicated that the final library contained inserts of 35–1,000 bp (Hwang et al., 2018). The index‐coded samples were clustered on a cBot Cluster Generation System according to the manufacturer's instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq 2500 platform (Illumina).
FASTAQ files were imported into the CLC Genomics Workbench v. 20.0.3 (Qiagen). Reads below the 0.05 quality score cut‐off and adapter trimming were removed from subsequent analyses. The remaining reads were trimmed of any ambiguous and low‐quality 5′ bases, and reads approximately 100 bp in length were retained for assembly. Quality‐controlled reads were then assembled using the SPAdes 3.13.0 assembler (Bankevich et al., 2012) with four iterative k‐mer assemblies (21, 33, 55 and 77). Assembled fasta files were imported into CLC workbench for contig assessment and analysis. Quality‐controlled contigs (E‐value < 10−5 and minimum > 300 bp) were subjected to a BLASTN search against viral reference genome sequences, using the NCBI virus genome database (http://www.ncbi.nlm.nih.gov/genome/viruses/). Blasted contigs were performed by taxonomic assignment using the coding of r program.
2.7. Statistical interpretation of the data
Pearson's correlation analysis was performed to examine the relationships between the measured parameters using SPSS v.12 (SPSS, Inc.). Cluster analysis was performed using group average clustering by the Bray–Curtis similarity method on the most abundant OTUs of bacteria and NCLDVs (each displaying a relative abundance > 1% in at least one sample). To test the null hypothesis (no significant difference between the groups discriminated according to the agglomerative clustering analysis), similarities were analysed with ANOSIM using PRIMER version 6.1.13 (Clarke, 1993). Using the ranked similarity matrix, an ordination plot was produced by nonmetric multidimensional scaling (nMDS) using the primer 6 program. Hierarchical agglomerative clustering using the group average method was carried out on the most abundant OTUs based on groups selected from nMDS analysis. Alpha diversity (including Chao1 richness, Shannon diversity and Simpson evenness metrics) was plotted using a combination of custom R using R Studio (v. 1.2.5042) with the vegan (Oksanen et al., 2019), ape (Paradis et al., 2019) and ggplot2 packages (Wickham et al., 2020).
Extended local similarity analysis (eLSA) (Xia et al., 2011) was used for the data from 2016 and 2017 with 33 and 29 days (a time interval of 2 days), respectively, to analyse covariation between the most abundant OTUs (over 1% in at least one sample), resulting in 110 and 77 OTUs of microbial communities and nine environmental parameters each in 2016 and 2017, respectively. The p‐value was determined by statistical approximation followed by permutation testing to reduce the computing time and ensure accuracy, and the Q‐value (false discovery rate) was calculated to estimate the likelihood of false positives (Xia et al., 2013). The eLSA network of delay‐shifted Spearman correlation coefficients between variables was visualized using Cytoscape v3.7.2 (Shannon et al., 2003) with p < .01 and Q < 0.05. Because the sampling was evenly spaced at two‐day intervals, the maximum time lags were considered to be 10 days. The networks were selected by A. sanguinea (2016) and Bathycoccus prasinos (2017) identifications or edge types (or example, correlations between specific OTUs). Random undirected networks of equal size by number of nodes and edges were calculated by the Erdös–Rényi model using the Random Network plugin in Cytoscape. Network statistics were calculated with the network analyser as undirected networks using the defaults (Assenov et al., 2008).
3. RESULTS
3.1. Environmental characteristics during Akashiwo sanguinea bloom
Variations were observed in the environmental characteristics of JBTMS from June 2016 to June 2017 (Figure S2). A. sanguinea bloom was sustained for 44 days; it developed on October 31 and declined on 13 December 2016 (Figure 1). During this blooming period, the mean abundance of A. sanguinea was 542 cells/ml, with a maximum abundance of 2,935 cells/ml on 18 November; the water temperature gradually decreased, and A. sanguinea bloom rapidly declined below 16°C (after November 21). DSi concentrations remained between 21.18 and 30.67 μM and showed no significant correlation with the abundance of A. sanguinea. DIN concentrations rapidly decreased at the beginning of A. sanguinea bloom and remained between 1.19 and 2.87 μM. DIP, DOC and chlorophyll a concentrations showed similar changes following A. sanguinea bloom and were significantly correlated with changes in the A. sanguinea abundance. Daily monitoring of the JBTMS from 14 November to 26 December 2017 showed that the dominant phytoplankton was B. prasinos (Chlorophyta) (Figure S3). We also observed that the water temperature was lower in 2017 than in 2016, rising over 16°C for only 2 days in 2017. DIN, DIP and DOC concentrations did not change with B. prasinos abundance. Changes in the DIP concentration showed no significant correlation with B. prasinos.
FIGURE 1.
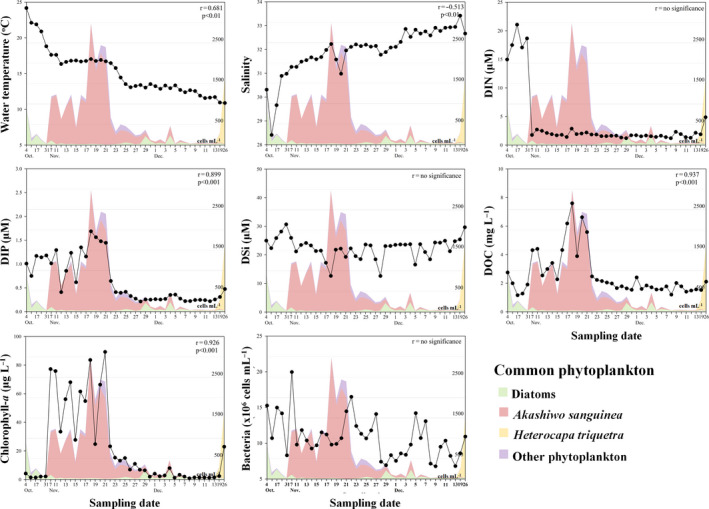
Daily changes in environmental factors before, during and afterAkashiwo sanguineabloom periods in 2016. Coloured areas in the figure correspond to the common phytoplankton groups in 2016. r value in each figure (upper right) indicates Pearson's correlation coefficient between each environmental factor andA. sanguineaabundance
3.2. Species‐specific bacterial community during A. sanguinea bloom
The mNGS results for the bacterial community in JBTMS are summarized in Table S3. In 2016, the bacterial community was classified into four groups at 73% similarity by nMDS analysis (Figure 2a). Group I was associated with “before A. sanguinea bloom” (4 and 31 October). This group comprised communities of Alphaproteobacteria (73%), Flavobacteriia (13%), Gammaproteobacteria (8%) and other bacteria. Groups II and III were associated with “during A. sanguinea bloom” (7–28 November), wherein Flavobacteriia increased rapidly to 36% and 57%, respectively. Group IV was associated with “after A. sanguinea bloom” (29 November to 26 December). In this group, the abundance of Gammaproteobacteria (13%) increased from that of group III. In 2017, the bacterial community was divided into two groups at 70% similarity by nMDS analysis (Figure 2b). Group I was associated with “dominance of B. prasinos” (14 November to 13 December), and the group also comprised Alphaproteobacteria (44%), Flavobacteriia (20%), Gammaproteobacteria (7%) and others (28%). In group II (after decrease in B. prasinos abundance, 19 and 26 December), Alphaproteobacteria rapidly increased to a proportion of 80%.
FIGURE 2.
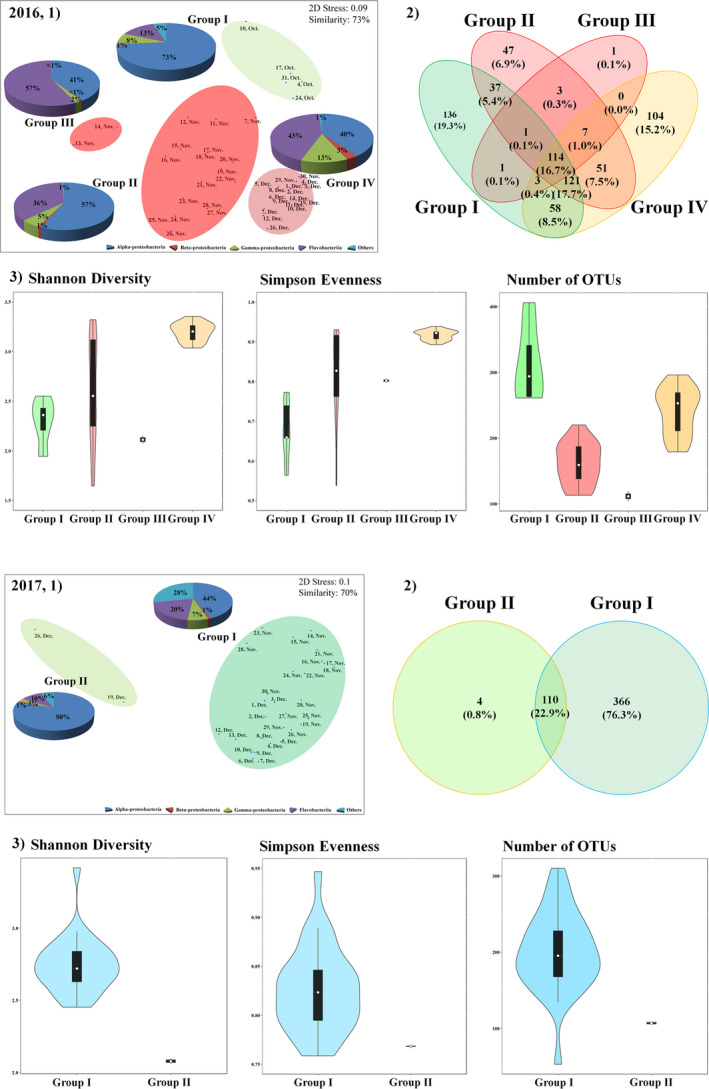
Distribution of OTUs among bacterial communities in 2016 and 2017. (1), Nonmetric multidimensional scaling (nMDS) plot by the Bray–Curtis dissimilarity method. In a nMDS plot, the pie chart plots indicate high‐ranking taxonomy distribution of the class level of bacteria community. (2), Venn diagram showing the shared and unique bacterial operational taxonomic units (OTUs; 97% identification). (3), Violin plots, which includes the box plot (median, min and max) showing alpha diversity (Shannon diversity, Simpson evenness and number of OTUs)
The number of OTUs and alpha diversity showed a similar trend to that in the read counts and varied according to the period; a Venn diagram was drawn to show OTUs shared between groups (I–IV in 2016 and I–II in 2017) (Figure 2, Table S3). The most abundant bacterial OTUs in 2016 belonged to Alphaproteobacteria (9 OTUs), Gammaproteobacteria (7), Flavobacteriia (11) and other bacterial species (2) (Figure 3). Before A. sanguinea bloom (group I), eight bacterial OTUs were common species, and Cribrihabitans marinus (Alphaproteobacteria) was dominant at 53.91%. During A. sanguinea bloom (groups II and III), 18 bacterial OTUs were common species, and uncultured Alphaproteobacterium (OTU #2) and C. marinus (Alphaproteobacteria; 21.01% and 13.83%, respectively), and Tenacibaculum aiptasiae and Polaribacter marinivivus (Flavobacteriia; 10.95% and 10.41%, respectively) were dominant with an accumulated proportion of 56.2%. Particularly, the changes in uncultured bacterium (OTU #2) and A. sanguinea cells were significantly correlated (r = .90, p < .001). After A. sanguinea bloom (group IV), 22 bacterial OTUs were common species, including C. marinus, Amylibacter ulvae (Alphaproteobacteria; 7.98% and 15.94%, respectively), Mesonia algae (Flavobacteriia; 16.61%) and Methylophilus methylotrophus (Betaproteobacteria; 3.14%). In 2017, the most abundant bacterial OTUs belonged mainly to Alphaproteobacteria (7), Gammaproteobacteria (6), Flavobacteriia (11) and other bacteria (2) (Figure 3). During the dominance of B. prasinos (group I), C. marinus (20.77%), A. ulvae (11.71%) and Euzebya tangerine (25.83%, Actinobacteria) were the predominant species in 16 common bacterial OTUs. After the decrease in abundance of B. prasinos (group II), A. ulvae, Lentibacter algarum and Planktomarina temperata (Alphaproteobacteria; 40.71%, 11.49% and 21.37%, respectively), and E. tangerine (5.52%) were the dominant species in the nine common bacterial OTUs.
FIGURE 3.
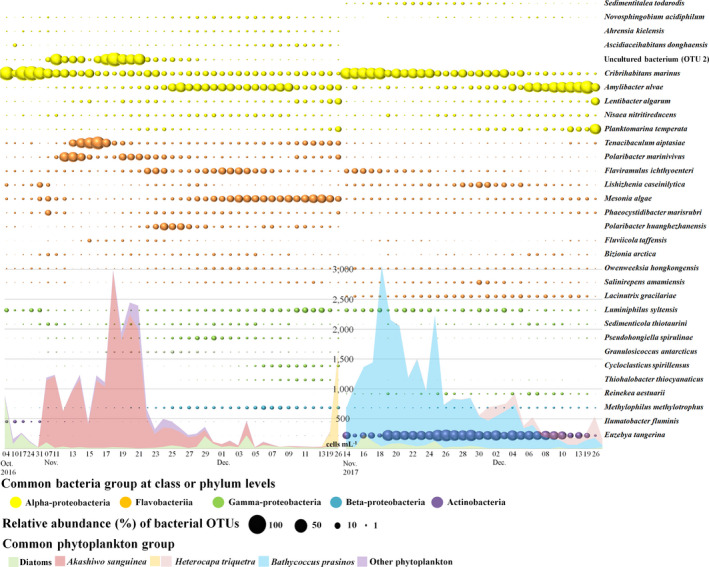
Time‐series circle plot showing the most abundant bacterial operational taxonomic units (OTUs) (each displaying a relative abundance > 1% in at least one sample) in 2016 and 2017. The colours in the circle plots correspond to the common bacterial groups. The coloured areas correspond to the common phytoplankton groups in 2016 and 2017. To show the differences in relative abundance for the displayed OTUs, the circle is on a 0–100 scale representing relative abundance (%)
3.3. Potential NCLDV infection of A. sanguinea bloom
The mNGS results for NCLDVs of JBTMS (2016 and 2017) are summarized in Table S4. The number of OTUs and alpha diversity showed a similar trend to that in the read counts and varied according to the period; and the Venn diagram shows the OTUs shared between groups (I–IV in 2016 and I–II in 2017) (Figure 4). In 2016, the most abundant NCLDV OTUs belonged mainly to Phycodnaviridae (49.74% and 29 OTUs) > Mimiviridae (24.53% and 6 OTUs) > Pandoraviridae (8.23% and 6 OTUs) > Iridoviridae (5.57% and 12 OTUs) > Poxviridae (3.93% and 12 OTUs) > other NCLDVs involving Ascoviridae, Pithoviridae and unassigned classified Mollivirus sibericum (2.08% and 4 OTUs). According to nMDS analysis of the relative abundances of NCLDVs in 2016, NCLDVs were clustered in four groups at 73% similarity (Figure 4a). Before A. sanguinea bloom (group I), Phycodnaviridae (39.01%), Iridoviridae (20.10%) and Pandoraviridae (11.08%) were the most abundant NCLDVs (>10%). During early A. sanguinea bloom (groups II and III), Phycodnaviridae, Pandoraviridae and Mimiviridae increased rapidly, whereas other NCLDVs (at family levels) decreased during early HABs periods. In periods of gradually declining HABs (group IV), Phycodnaviridae (53.04%) and Pandoraviridae (6.61%) were decreased, while other family levels (including Ascoviridae, Iridoviridae, Mimiviridae, Pandoraviridae, Phycodnaviridae, Pithoviridae and Poxviridae) increased from that of groups II and III. Specifically, Pandoravirus macleodensis and Pandoravirus salinus in Pandoraviridae and Acanthocystis turfacea chlorella virus 1, Heterosigma akashiwo virus 01, Ostreococcus mediterraneus virus 1, Paramecium bursaria chlorella virus CVA‐1, Yellowstone lake phycodnavirus 1 and Ectocarpus siliculosus virus 1 in Phycodnaviridae were positively correlated with A. sanguinea abundance (r > .5 and p < .01) (Figure 5a).
FIGURE 4.
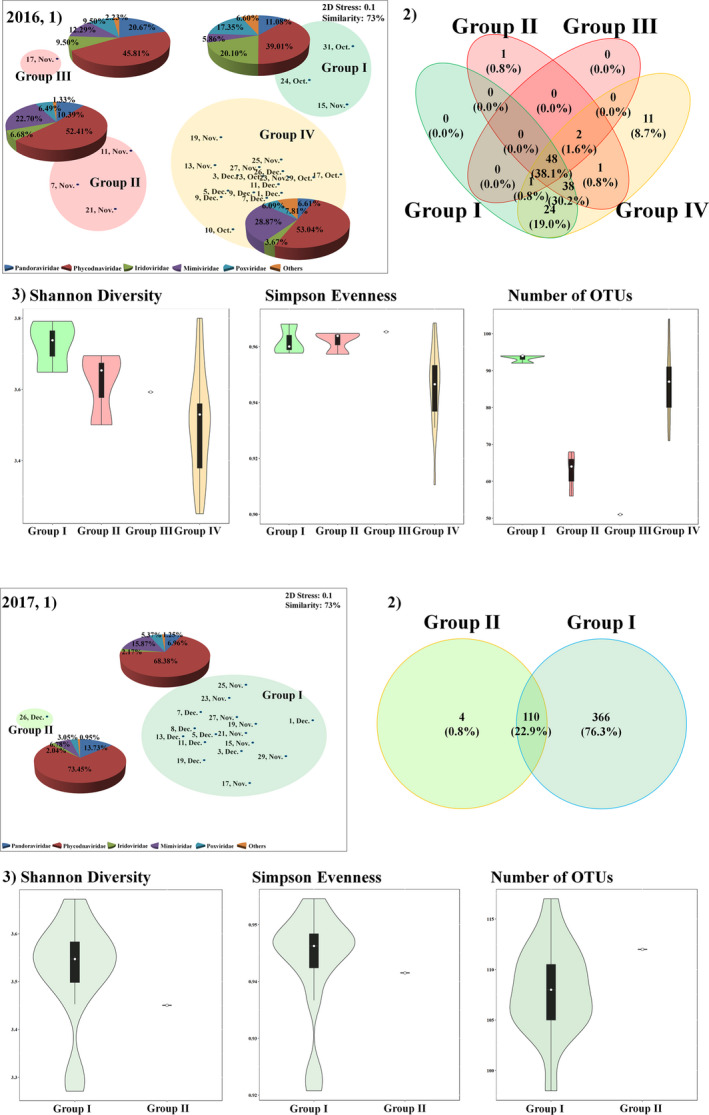
Distribution of OTUs among the nucleocytoplasmic large DNA virus (NCLDV) communities in 2016 and 2017. (1), Nonmetric multidimensional scaling (nMDS) plot by the Bray–Curtis dissimilarity method. In a nMDS plot, the pie chart plots indicate high‐ranking taxonomy distribution of the family level of NCLDV community. (2), Venn diagram showing the shared and unique NCLDV operational taxonomic units (OTUs). (3), Violin plots, which includes the box plot (median, min and max) showing alpha diversity (Shannon diversity, Simpson evenness and number of OTUs)
FIGURE 5.
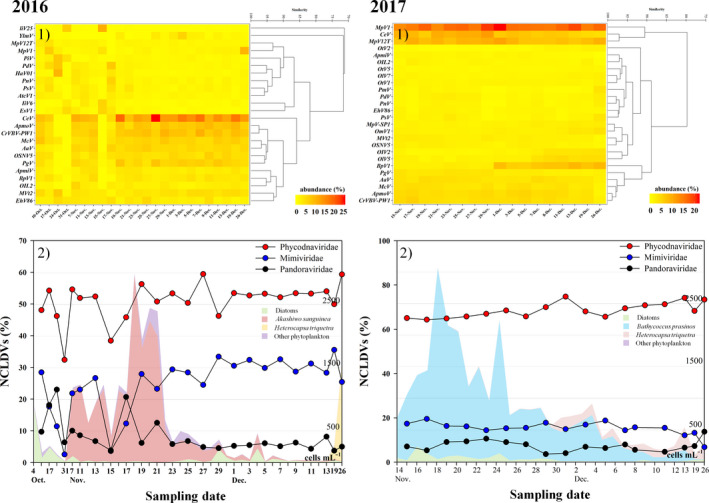
Heat map analysis of the nucleocytoplasmic large DNA virus (NCLDV) communities at the species level showing the most abundant bacterial operational taxonomic units (OTUs) (each displaying a relative abundance > 1%) in 2016 and 2017 (1). (2), The abundant NCLDVs at family level and in a figure, each coloured area in 2016 and 2017 corresponds to the common phytoplankton groups
In 2017, the most abundant NCLDV OTUs mainly belonged to Phycodnaviridae (21 OTUs), Pandoraviridae (6), Mimiviridae (6) and 2 other NCLDVs. According to nMDS analysis of the relative abundances of NCLDVs in 2017, NCLDVs were clustered into two groups at 85% similarity (Figure 4b). During the development and termination periods of B. prasinos (group I), Phycodnaviridae (64.93%) and Mimiviridae (15.87%) were dominant in the most abundant NCLDVs, comprising 80.80% of the total relative abundance. In group II (only 1 day on 26 December), Phycodnaviridae were dominant at 70.03% and Pandoraviridae were relatively increased to 13.73%. Only Syngen Nebraska virus 5 and Phaeocystis globosa virus (Phycodnaviridae) and Acanthamoeba polyphaga mimivirus (Mimiviridae) were strongly and positively significant correlated with the B. prasinos abundance (r > .6 and p < .01), whereas Bathycoccus sp. RCC1105 virus BpV1, Ostreococcus lucimarinus virus 2, O. lucimarinus virus 7 and Ostreococcus tauri virus 2 (Phycodnaviridae) were strongly negative correlated with the B. prasinos abundance (Figure 5b).
3.4. Endoparasitic dinoflagellate dynamics during A. sanguinea bloom
To explore co‐occurrence patterns, focusing primarily on potential parasitic interactions between endoparasitic dinoflagellate Amoebophrya sp. ex. A. sanguinea, we assessed the relationship between Amoebophrya sp. (Syndiniales) and A. sanguinea in JBTMS. The mNGS results of eukaryotic (18S rDNA) communities in JBTMS are summarized in Figure 6 and Table S5. In 2016, Amoebophrya sp. 1 trends were strongly associated with those of A. sanguinea bloom in the JBTMS (Figure 6a). Moreover, dinospores of Amoebophrya sp. 1 changed similarly to A. sanguinea cells. After A. sanguinea disappeared, Heterocapsa triquetra emerged, and another OTU (Amoebophrya sp. 2) was detected (26 December, Figure 5a). Other Syndiniales seldom appeared during A. sanguinea bloom. In 2017, low levels of Amoebophrya sp. 1 were detected in seawater, but Amoebophrya sp. 2 increased rapidly when H. triquetra emerged (Figure 6b).
FIGURE 6.
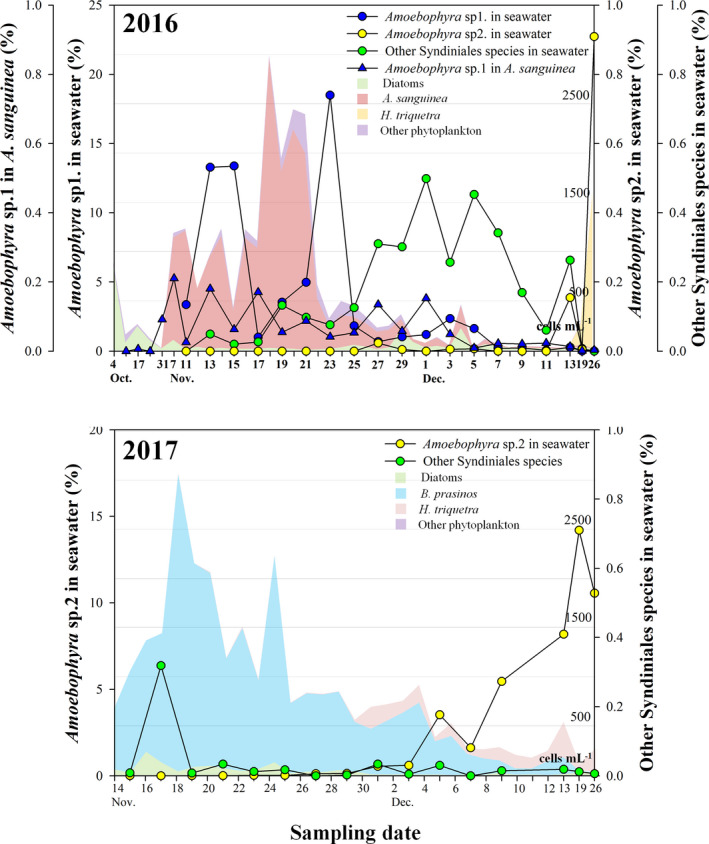
Daily changes in operational taxonomic units (OTUs) of endoparasitic dinoflagellate,Amoebophryaspp. (displaying a relative abundance), in 2016 (a) and 2017 (b).Amoebophryaspp. are mostly divided intoAmoebophryasp. 1, sp. 2 and other Syndiniales species. Each coloured area in 2016 and 2017 corresponds to the common phytoplankton groups
3.5. Network analysis during A. sanguinea bloom in 2016 and no bloom in 2017
Network analyses of microbial communities focused on A. sanguinea in 2016 and B. prasinos in 2017 and revealed a distinct associated interaction with specific microbial communities and environmental factors (Figure 7, Tables S6–S8). The 2016 network showed significantly correlated biological and environmental factors with 78 nodes and 514 edges (Figure 7a). In the network, A. sanguinea association networks identified factors that were highly correlated with specific OTUs, such as bacteria (10 OTUs), NCLDVs (1), parasitic dinoflagellates (2) and environmental factors (4). Our association network supports the paradigm that A. sanguinea bloom is regulated by both Amoebophrya sp. 1 (r = .59 and time lag: −2 days) and E. siliculosus virus 1 (.50, −4 days). In networks with bacterial communities, uncultured Alphaproteobacterium (OTU #2), Fluviicola taffensis and P. marinivivus, were strongly and positively correlated, whereas other specific bacterial species (i.e. A. ulvae) were negatively linked. In the network with environmental factors, strong positive connectivity of DIP (0.74, 2 days) may reflect A. sanguinea‐selective interactions.
FIGURE 7.
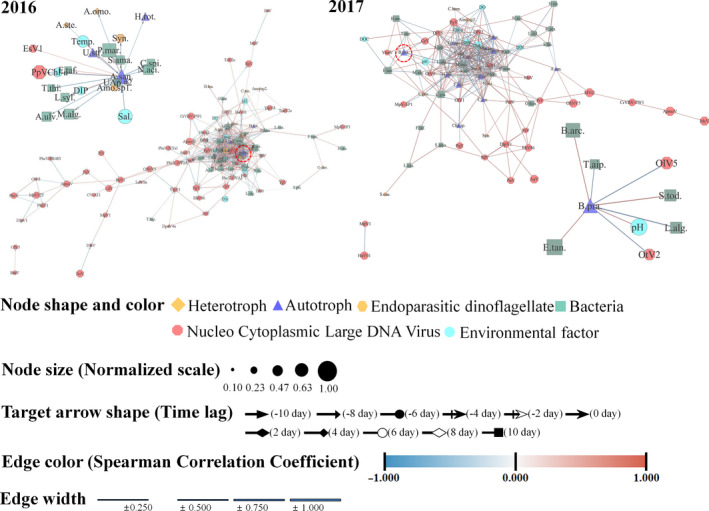
Network analysis derived from the most abundant operational taxonomic units (OTUs) of microbial communities and environmental factors showing significant correlations (p < .01; false discoveryQ < 0.05) in 2016 and 2017. Zoom in the subnetwork in figure is associated withAkashiwo sanguineaand associated withBathycoccus prasinos
The 2017 network showed biological and environmental factors with 74 nodes and 345 correlations (Figure 7b). Association networks of B. prasinos were significantly correlated with eight specific OTUs (five bacteria and two NCLDVs) and one environmental factor. In networks with bacterial communities, Sedimentitalea todarodis (0.86, 2 days), Bizionia arctica (0.74, −2 days) and E. tangerine (0.70, 0 day) were positively correlated at different time lags with B. prasinos, while L. algarum (−0.89, 8 days) and T. aiptasiae (−0.66, 0 day) were negatively correlated with B. prasinos. The network of B. prasinos was mildly negatively correlated with O. lucimarinus virus OIV5 (−0.82, 0 day) and O. tauri virus 2 (−0.74, 2 days), but was not correlated with Amoebophrya sp. 1. This interaction may reflect B. prasinos‐selective infection with specific bacteria and NCLDVs. In the network with environmental factors, B. prasinos showed no correlation with DIP as compared with the A. sanguinea network in 2016.
4. DISCUSSION
In our study of the dynamics of microbiomes focused on HABs, a major strength was that we used a high‐resolution sampling approach. Day‐to‐day sampling of an A. sanguinea bloom spanning is useful for understanding the fine‐scale dynamics of the bloom life cycle. We showed that the A. sanguinea abundance initially increased by taking up DIN from the surrounding waters, and DIP and DOC concentrations strongly and immediately increased with the development of A. sanguinea bloom. This may be because of the dissolved carbohydrate (DCHO) being released by A. sanguinea cells. In marine systems, evidence for strong correlations between DCHO concentrations and phytoplankton biomass was found in oceanic surface waters (Børsheim et al., 1999; Fajon et al., 1999; Pakulski & Benner, 1994). DCHO production by marine phytoplankton depends on the species, growth stage and environmental conditions (Chen & Wangersky, 1996; Myklestad, 1995; Penna et al., 1999). Urbani et al. (2005) reported the biodegradability of DCHO released by Thalassiosira pseudonana and Skeletonema costatum in centric diatoms. Thus, A. sanguinea bloom markedly increases biological carbon export into the surrounding waters. A. sanguinea bloom is dominant worldwide in cold seasons (Du et al., 2011; Yang et al., 2012).
According to Yang et al. (2012), bacterial abundance greatly increased in the A. sanguinea bloom area during a bloom in the Xiamen sea, and DO concentration dropped as a result of bacterial decomposition of A. sanguinea. However, bacterial abundance was not significantly associated with A. sanguinea bloom and did not affect DO levels in our study. Nevertheless, certain bacterial communities were closely related to A. sanguinea bloom. Specific bacteria, such as P. marinivivus and uncultured Alphaproteobacterium (OTU #2), may have a symbiotic association in A. sanguinea bloom, whereas A. ulvae, M. algae and L. syltensis may be inhibited in the HABs. Thus, it is important to elucidate the ecological role of specific bacteria associated with HABs (Croft et al., 2005; Naviner et al., 1999). Our results agree with those of previous studies (Croft et al., 2005) showing the state that phytoplankton harbour (habitat “phycosphere”) specific bacterial communities. We found that the bacterial species composition varied at different growth stages of A. sanguinea (Figure 3). Antibacterial metabolites are produced by some phytoplankton (Naviner et al., 1999), which may inhibit certain bacterial species. These antibacterial substances are specifically associated with DCHO excreted from a phytoplankton cell (Myklestad, 1995); thus, the physiological flexibility of bacteria may support their colonization. In previous studies, algicidal bacteria were shown to suddenly increase in the presence of HABs (Jung et al., 2008). Mayali and Azam (2004) suggest that algicidal bacteria affect HAB dynamics, as their abundance increases with the decline of algal blooms. In our study, the most common algicidal bacteria belonged to Gammaproteobacteria and Bacteroidetes (Table S9), and no sign of HAB control by algicidal bacteria was observed: Alteromonas and Pseudoalteromonas in Gammaproteobacteria as well as Saprospira and Cytophaga in Bacteroidetes (i.e. common algicidal bacteria) exhibited low abundance (or not detected), which did not increase after the decline of A. sanguinea bloom. Therefore, no specific bacteria in this study showed the potential to control the A. sanguinea bloom.
Our results revealed that two Pandoraviruses and six Phycodnaviruses were positively correlated with A. sanguinea bloom, indicating that these NCLDVs can infect A. sanguinea. In further studies, similar interactions between species‐specific NCLDVs and A. sanguinea blooms should be investigated. In most common NCLDVs, P. macleodensis and P. salinus mainly infect amoebas (Philippe et al., 2013) and hosts of O. mediterraneus virus 1 and H. akashiwo virus 01 are eukaryotic phytoplankton such as O. mediterraneus and H. akashiwo (Bellec et al., 2014; Nagasaki & Yamaguchi, 1998). However, additional studies are needed to evaluate these different hosts, and there is no substantial evidence to determine whether Pandoraviruses and Phycodnaviruses can infect A. sanguinea or whether this behaviour is normal for the viruses in lower water temperature periods in any host (Yutin & Koonin, 2013). Moreover, Pandoraviruses are highly evolved from Phycodnaviridae (Legendre et al., 2018). The dynamics of Pandoraviruses and Phycodnaviruses are key to understanding the termination of A. sanguinea bloom because the distribution of Pandoraviruses and Phycodnaviruses is closely related to the distribution of A. sanguinea bloom; however, it is very difficult to determine the ecological relationship between the virus and host based on the “killing the winner hypothesis” (Winter et al., 2010) or “piggyback‐winner hypothesis” (Silveira & Rohwer, 2016). Therefore, further studies are required to identify specific infection mechanisms of specific Pandoraviruses and Phycodnaviruses against A. sanguinea and estimate the ecological role of these viruses in nature.
An endoparasitic dinoflagellate, Amoebophrya sp., can efficiently control populations of their dinoflagellate hosts, and infection, as this parasitoid spreads rapidly through dense dinoflagellate populations, facilitating the decline of the dinoflagellate bloom (Chambouvet et al., 2008). Many marine psychologists (Chen et al., 2018; Coats et al., 1996; Coats & Park, 2002) have reported that Amoebophrya sp. can easily infect A. sanguinea. mNGS has revealed the enormous genetic diversity of Amoebophrya‐like organisms within the marine Alveolata group II (Lima‐Mendez et al., 2015). In this study, there were genetic divergences among several Amoebophrya spp. [i.e. Amoebophrya sp. 1 (OTU #24) and Amoebophrya sp. 2 (OTU #14)]. The OTUs of Amoebophrya sp. 1 in seawater (free‐living Amoebophrya sp. 1) in A. sanguinea (infected dinospores of Amoebophrya sp.) in the JBTMS verified the control of A. sanguinea blooms by Amoebophrya sp. 1 (Figure 5). This can be explained by the short generation time of Amoebophrya (Coats et al., 1996). The ecological role of host‐specific Amoebophrya infection may have a greater impact on the population dynamics of toxic bloom‐forming dinoflagellates than microzooplankton grazing (Figure S4); Amoebophrya can eliminate an entire host population within a few days (Montagnes et al., 2008). This study revealed a natural phenomenon wherein an endoparasitic dinoflagellate controls its host. However, we did not consider the mechanisms of killing by Amoebophrya sp. Thus, further studies of host–parasitoid interactions are needed to estimate the ecological role of Amoebophrya and determine how various Amoebophrya species coexist in nature.
Network analysis revealed that the A. sanguinea bloom in 2016 was associated with specific microbial communities and an environmental factor and showed differentiation of network results of 2017. In 2016, connected partners with A. sanguinea blooms included taxa (OTUs) from all trophic positions (i.e. infections, parasites, phototrophs and heterotrophs). DIP can respond to A. sanguinea bloom and showed changes in this environmental factor derived from DCHO released by A. sanguinea bloom. Moreover, specific Phycodnavirus and Pandoravirus, and endoparasitic Amoebophrya sp. 1 with positive associations with A. sanguinea have been found and regulated to A. sanguinea bloom. The high connectivity displayed by some microbes with negative association with competition, niche partitioning, grazing (Eiler et al., 2012; Fuhrman & Steele, 2008), and environmental factors with A. sanguinea bloom suggests similarity in their ecological properties (symbiosis and inhibition as well as infection). Our study revealed the value of frequent sampling to evaluate community responses and microbial interactions among protists by reinforcing recent predictions on the rapid dynamics and the importance of parasites.
We propose three stages of interactions between environmental characteristics and microbial communities in A. sanguinea bloom: (a) “before A. sanguinea bloom”: diatoms and Alphaproteobacteria were common phytoplankton and bacterial groups, respectively. A low abundance of Amoebophrya sp. 1 was observed in this stage. The relatively high NCLDV groups were Iridoviridae and Poxviridae. For environmental characteristics, most parameters (particularly DIN) were detected to be higher than those in the other stages; (b) “during A. sanguinea bloom”: A. sanguinea bloom showed marked changes in environmental characteristics (which were exported into the surrounding waters), followed by changes involving species‐specific viruses in Pandoraviridae and Phycodnaviridae, bacteria and parasitoids. A. sanguinea abundance initially increased when they took up DIN and DIP from the surrounding waters, but changes in DIP and DOC concentrations were strongly positive correlated with changes in HABs. A. sanguinea bloom harboured and promoted specific bacterial populations. Particularly, the host‐specific bacterial group (Flavobacteriia increased rapidly) that remineralizes extracellular products from A. sanguinea participates in micro‐environments and plays an important role in microbial community dynamics. Specific NCLDVs in Phycodnaviridae and Pandoraviridae, increased following an increase in the A. sanguinea abundance, specifically during bloom peaks. The endoparasitic dinoflagellate Amoebophrya sp. 1 has attracted attention regarding its roles in trophic interactions; (c) “after A. sanguinea bloom”: when A. sanguinea bloom was terminated, the water temperature was below 16°C, and most environmental characteristics showed minor changes. Succession of common phytoplankton groups occurred from A. sanguinea to diatoms (Pseudo‐nitzschia delicatissima and Skeletonema marinoi‐dorhnii complex species) and H. triquetra (dinoflagellate). Daily changes in the bacteria and NCLDV groups were observed, such as a relative increase in Gammaproteobacteria and Mimiviridae, respectively. Amoebophrya sp. 1 rapidly decreased with the termination of A. sanguinea bloom. Consequently, microbial communities and the environment dynamically and changed in a complex manner in A. sanguinea bloom, and the rapid turnover of microorganisms could respond to ecological interactions. Microbial communities in HAB ecology are composed of various organisms which interact in a complex way. Therefore, to interpret their ecosystem, the complex reactions among various microorganisms should be studied rather than evaluated a simple 1:1 reaction, such as a prey–predator interaction.
AUTHOR CONTRIBUTIONS
The research plan was designed by S.W.J. and T.‐K.L. The experiments were performed by J.K., J.S.P., S.W.J., H.‐J.K., H.M.J., H.S. and S.K., and data were analysed by M.C.J., K.L., S.J.O., S.L. and T.‐K.L. The results were discussed by J.K., J.S.P., S.W.J., H.M.J., S.K., M.C.J., K.L., S.J.O. and S.L. The manuscript was written by J.K., J.S.P., S.W.J. and T.‐K.L.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The stored gDNA samples and fixed phytoplankton samples were obtained from the Library of Marine Samples of Korea Institute of Ocean Science & Technology (KIOST), South Korea. This research was supported by the National Research Foundation (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF‐2020R1A2C2005970 and NRF‐2017M3A9E4072753).
Kang J, Park J, Jung SW, et al. Zooming on dynamics of marine microbial communities in the phycosphere of Akashiwo sanguinea (Dinophyta) blooms. Mol Ecol. 2021;30:207–221. 10.1111/mec.15714
Contributor Information
Seung Won Jung, Email: diatoms@kiost.ac.kr.
Taek‐Kyun Lee, Email: tklee@kiost.ac.kr.
DATA AVAILABILITY STATEMENT
The raw sequencing data (FASTAQ files) of 18s rDNA and 16s rDNA genes obtained from the MiSeq platform were deposited in the Sequence Read Archive database at NCBI under Accession Numbers: SRR11123089–SRR 11123147 (PRJNA 607609), SRR11136919–SRR11136959 (PRJNA 607810) and SRR11131500–SRR11131531 (PRJNA 607814). The raw sequencing files of NCLDVs obtained from the HiSeq platform were deposited in the Sequence Read Archive database at NCBI under Accession Number SRR11172603–SRR11172644 (PRJNA 608210).
REFERENCES
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215(3), 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Amorim Reis‐Filho, J. , da Silva, E. M. , de Anchieta, C. , da Costa Nunes, J. , & Barros, F. (2012). Effects of a red tide on the structure of estuarine fish assemblages in northeastern Brazil. International Review of Hydrobiology, 97(5), 389–404. 10.1002/iroh.201101457 [DOI] [Google Scholar]
- Anderson, D. M. (1997). Turning back the harmful red tide. Nature, 388(6642), 513–514. [Google Scholar]
- Andrew, S. (2010). FastQC: A quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Arrigo, K. R. (2005). Marine microorganisms and global nutrient cycles. Nature, 437(7057), 349–355. [DOI] [PubMed] [Google Scholar]
- Assenov, Y. , Ramírez, F. , Schelhorn, S.‐E. , Lengauer, T. , & Albrecht, M. (2008). Computing topological parameters of biological networks. Bioinformatics, 24(2), 282–284. 10.1093/bioinformatics/btm554 [DOI] [PubMed] [Google Scholar]
- Azam, F. , & Malfatti, F. (2007). Microbial structuring of marine ecosystems. Nature Reviews Microbiology, 5(10), 782–791. 10.1038/nrmicro1747 [DOI] [PubMed] [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellec, L. , Clerissi, C. , Edern, R. , Foulon, E. , Simon, N. , Grimsley, N. , & Desdevises, Y. (2014). Cophylogenetic interactions between marine viruses and eukaryotic picophytoplankton. BMC Evolutionary Biology, 14(1), 59 10.1186/1471-2148-14-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Børsheim, K. Y. , Myklestad, S. M. , & Sneli, J.‐A. (1999). Monthly profiles of DOC, mono‐and polysaccharides at two locations in the Trondheimsfjord (Norway) during two years. Marine Chemistry, 63(3–4), 255–272. 10.1016/S0304-4203(98)00066-8 [DOI] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , Fierer, N. , Peña, A. G. , Goodrich, J. K. , Gordon, J. I. , Huttley, G. A. , Kelley, S. T. , Knights, D. , Koenig, J. E. , Ley, R. E. , Lozupone, C. A. , McDonald, D. , Muegge, B. D. , Pirrung, M. , … Knight, R. (2010). QIIME allows analysis of high‐throughput community sequencing data. Nature Methods, 7(5), 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambouvet, A. , Morin, P. , Marie, D. , & Guillou, L. (2008). Control of toxic marine dinoflagellate blooms by serial parasitic killers. Science, 322(5905), 1254–1257. [DOI] [PubMed] [Google Scholar]
- Chen, T. , Liu, Y. , Song, S. , & Li, C. (2018). Characterization of the parasitic dinoflagellate Amoebophrya sp. infecting Akashiwo sanguinea in coastal waters of China. Journal of Eukaryotic Microbiology, 65(4), 448–457. [DOI] [PubMed] [Google Scholar]
- Chen, W. , & Wangersky, P. J. (1996). Rates of microbial degradation of dissolved organic carbon from phytoplankton cultures. Journal of Plankton Research, 18(9), 1521–1533. [Google Scholar]
- Clarke, K. R. (1993). Non‐parametric multivariate analyses of changes in community structure. Australian Journal of Ecology, 18(1), 117–143. [Google Scholar]
- Claverie, J.‐M. , & Abergel, C. (2018). Mimiviridae: An expanding family of highly diverse large dsDNA viruses infecting a wide phylogenetic range of aquatic eukaryotes. Viruses, 10(9), 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats, D. W. , Adam, E. , Gallegos, C. L. , & Hedrick, S. (1996). Parasitism of photosynthetic dinoflagellates in a shallow subestuary of Chesapeake Bay, USA. Aquatic Microbial Ecology, 11(1), 1–9. [Google Scholar]
- Coats, D. W. , & Park, M. G. (2002). Parasitism of photosynthetic dinoflagellates by three strains of Amoebophyra (Dinophyta): Parasite survival, infectivity, generation time, and host specificity. Journal of Phycology, 38(3), 520–528. [Google Scholar]
- Colson, P. , De Lamballerie, X. , Yutin, N. , Asgari, S. , Bigot, Y. , Bideshi, D. K. , Cheng, X.‐W. , Federici, B. A. , Van Etten, J. L. , Koonin, E. V. , La Scola, B. , & Koonin, E. V. (2013). “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Archives of Virology, 158(12), 2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft, M. T. , Lawrence, A. D. , Raux‐Deery, E. , Warren, M. J. , & Smith, A. G. (2005). Algae acquire vitamin B 12 through a symbiotic relationship with bacteria. Nature, 438(7064), 90–93. [DOI] [PubMed] [Google Scholar]
- Du, X. , Peterson, W. , McCulloch, A. , & Liu, G. (2011). An unusual bloom of the dinoflagellate Akashiwo sanguinea off the central Oregon, USA, coast in autumn 2009. Harmful Algae, 10(6), 784–793. 10.1016/j.hal.2011.06.011 [DOI] [Google Scholar]
- Eiler, A. , Heinrich, F. , & Bertilsson, S. (2012). Coherent dynamics and association networks among lake bacterioplankton taxa. The ISME Journal, 6(2), 330–342. 10.1038/ismej.2011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajon, C. Ã. , Cauwet, G. , Lebaron, P. , Terzic, S. , Ahel, M. , Malej, A. , Mozetic, P. , & Turk, V. (1999). The accumulation and release of polysaccharides by planktonic cells and the subsequent bacterial response during a controlled experiment. FEMS Microbiology Ecology, 29(4), 351–363. 10.1111/j.1574-6941.1999.tb00626.x [DOI] [Google Scholar]
- Flaviani, F. , Schroeder, D. , Balestreri, C. , Schroeder, J. , Moore, K. , Paszkiewicz, K. , Pfaff, M. , & Rybicki, E. (2017). A pelagic microbiome (viruses to protists) from a small cup of seawater. Viruses, 9(3), 47 10.3390/v9030047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman, J. A. (1999). Marine viruses and their biogeochemical and ecological effects. Nature, 399(6736), 541–548. [DOI] [PubMed] [Google Scholar]
- Fuhrman, J. A. , & Steele, J. A. (2008). Community structure of marine bacterioplankton: Patterns, networks, and relationships to function. Aquatic Microbial Ecology, 53(1), 69–81. 10.3354/ame01222 [DOI] [Google Scholar]
- Hwang, J. , Park, S. Y. , Lee, S. , & Lee, T.‐K. (2018). High diversity and potential translocation of DNA viruses in ballast water. Marine Pollution Bulletin, 137, 449–455. 10.1016/j.marpolbul.2018.10.053 [DOI] [PubMed] [Google Scholar]
- Jessup, D. A. , Miller, M. A. , Ryan, J. P. , Nevins, H. M. , Kerkering, H. A. , Mekebri, A. , Crane, D. B. , Johnson, T. A. , & Kudela, R. M. (2009). Mass stranding of marine birds caused by a surfactant‐producing red tide. PLoS One, 4(2), e4550 10.1371/journal.pone.0004550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, S. W. , Kang, D. , Kim, H.‐J. , Shin, H. H. , Park, J. S. , Park, S. Y. , & Lee, T.‐K. (2018). Mapping distribution of cysts of recent dinoflagellate and Cochlodinium polykrikoides using next‐generation sequencing and morphological approaches in South Sea. Korea. Scientific Reports, 8(1), 7011 10.1038/s41598-018-25345-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, S. W. , Kim, B.‐H. , Katano, T. , Kong, D.‐S. , & Han, M.‐S. (2008). Pseudomonas fluorescens HYK0210‐SK09 offers species‐specific biological control of winter algal blooms caused by freshwater diatom Stephanodiscus hantzschii . Journal of Applied Microbiology, 105(1), 186–195. [DOI] [PubMed] [Google Scholar]
- Kim, H. J. , Jung, S. W. , Lim, D.‐I. , Jang, M.‐C. , Lee, T.‐K. , Shin, K. , & Ki, J.‐S. (2016). Effects of temperature and nutrients on changes in genetic diversity of bacterioplankton communities in a semi‐closed bay. South Korea. Marine Pollution Bulletin, 106(1–2), 139–148. 10.1016/j.marpolbul.2016.03.015 [DOI] [PubMed] [Google Scholar]
- Legendre, M. , Fabre, E. , Poirot, O. , Jeudy, S. , Lartigue, A. , Alempic, J.‐M. , Beucher, L. , Philippe, N. , Bertaux, L. , Christo‐Foroux, E. , Labadie, K. , Couté, Y. , Abergel, C. , & Claverie, J.‐M. (2018). Diversity and evolution of the emerging Pandoraviridae family. Nature Communications, 9, 2285 10.1038/s41467-018-04698-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , & Chang, Y. (2016). CD‐HIT‐OTU‐MiSeq, an improved approach for clustering and analyzing paired end MiSeq 16S rRNA sequences. BioRxiv [Preprint]. Retrieved from https://www.biorxiv.org/content/early/2017/06/22/153783 [Google Scholar]
- Lima‐Mendez, G. , Faust, K. , Henry, N. , Decelle, J. , Colin, S. , Carcillo, F. , Chaffron, S. , Ignacio‐Espinosa, J. C. , Roux, S. , Vincent, F. , Bittner, L. , Darzi, Y. , Wang, J. , Audic, S. , Berline, L. , Bontempi, G. , Cabello, A. M. , Coppola, L. , Cornejo‐Castillo, F. M. , … Raes, J. (2015). Determinants of community structure in the global plankton interactome. Science, 348(6237), 1262073 10.1126/science.1262073 [DOI] [PubMed] [Google Scholar]
- Magoč, T. , & Salzberg, S. L. (2011). FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27(21), 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayali, X. , & Azam, F. (2004). Algicidal bacteria in the sea and their impact on algal blooms. Journal of Eukaryotic Microbiology, 51(2), 139–144. [DOI] [PubMed] [Google Scholar]
- Mazzillo, F. F. , Ryan, J. P. , & Silver, M. W. (2011). Parasitism as a biological control agent of dinoflagellate blooms in the California Current System. Harmful Algae, 10(6), 763–773. 10.1016/j.hal.2011.06.009 [DOI] [Google Scholar]
- Montagnes, D. J. , Chambouvet, A. , Guillou, L. , & Fenton, A. (2008). Responsibility of microzooplankton and parasite pressure for the demise of toxic dinoflagellate blooms. Aquatic Microbial Ecology, 53(2), 211–225. 10.3354/ame01245 [DOI] [Google Scholar]
- Myklestad, S. M. (1995). Release of extracellular products by phytoplankton with special emphasis on polysaccharides. Science of the Total Environment, 165(1–3), 155–164. 10.1016/0048-9697(95)04549-G [DOI] [Google Scholar]
- Nagasaki, K. , & Yamaguchi, M. (1998). Intra‐species host specificity of HaV (Heterosigma akashiwo virus) clones. Aquatic Microbial Ecology, 14(1), 109–112. 10.3354/ame014109 [DOI] [Google Scholar]
- Naviner, M. , Berge, J. P. , Durand, P. , & Le Bris, H. (1999). Antibacterial activity of the marine diatom Skeletonema costatum against aquacultural pathogens. Aquaculture, 174(1–2), 15–24. 10.1016/S0044-8486(98)00513-4 [DOI] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Minchin, P. R. , O'Hara, R. B. , Simpson, G. L. , Solymos, P. , Henry, M. , Stevens, H. , Szoecs, E. , & Wagner, H. (2019). Package ‘vegan’. Community ecology package. Retrieved from https://github.com/vegandevs/vegan [Google Scholar]
- Pakulski, J. D. , & Benner, R. (1994). Abundance and distribution of carbohydrates in the ocean. Limnology and Oceanography, 39(4), 930–940. 10.4319/lo.1994.39.4.0930 [DOI] [Google Scholar]
- Paradis, E. , Blomberg, S. , Bolker, B. , Brown, J. , Claude, J. , Cuong, H. S. , Desper, R. , Didier, G. , Durand, B. , Dutheil, J. , Ewing, R. J. , Gascuel, O. , Guillerme, T. , Heibl, C. , Ives, A. , Jones, B. , Krah, F. , Lawson, D. , Lefort, V. , … de Vienne, D. (2019). Package ‘ape’. Analyses of phylogenetics and evolution. Retrieved from http://ape-package.ird.fr/ [Google Scholar]
- Penna, A. , Berluti, S. , Penna, N. , & Magnani, M. (1999). Influence of nutrient ratios on the in vitro extracellular polysaccharide production by marine diatoms from the Adriatic Sea. Journal of Plankton Research, 21(9), 1681–1690. 10.1093/plankt/21.9.1681 [DOI] [Google Scholar]
- Philippe, N. , Legendre, M. , Doutre, G. , Couté, Y. , Poirot, O. , Lescot, M. , Arslan, D. , Seltzer, V. , Bertaux, L. , Bruley, C. , Garin, J. , Claverie, J.‐M. , & Bruley, C. (2013). Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science, 341(6143), 281–286. [DOI] [PubMed] [Google Scholar]
- Porter, K. G. , & Feig, Y. S. (1980). The use of DAPI for identifying and counting aquatic microflora. Limnology and Oceanography, 25(5), 943–948. [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , Lesniewski, R. A. , Oakley, B. B. , Parks, D. H. , Robinson, C. J. , Sahl, J. W. , Stres, B. , Thallinger, G. G. , Van Horn, D. J. , & Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75(23), 7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz, F. , Yutin, N. , Ivanova, N. N. , Ortega, D. R. , Lee, T. K. , Vierheilig, J. , Daims, H. , Horn, M. , Wagner, M. , Jensen, G. J. , Kyrpides, N. C. , Koonin, E. V. , Woyke, T. , & Jensen, G. J. (2017). Giant viruses with an expanded complement of translation system components. Science, 356(6333), 82–85. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , Amin, N. , Schwikowski, B. , & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira, C. B. , & Rohwer, F. L. (2016). Piggyback‐the‐Winner in host‐associated microbial communities. NPJ Biofilms and Microbiomes, 2(1), 1–5. 10.1038/npjbiofilms.2016.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smayda, T. J. (1997). Harmful algal blooms: Their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnology and Oceanography, 42(5), 1137–1153. 10.4319/lo.1997.42.5_part_2.1137 [DOI] [Google Scholar]
- Urbani, R. , Magaletti, E. , Sist, P. , & Cicero, A. M. (2005). Extracellular carbohydrates released by the marine diatoms Cylindrotheca closterium, Thalassiosira pseudonana and Skeletonema costatum: Effect of P‐depletion and growth status. Science of the Total Environment, 353(1–3), 300–306. 10.1016/j.scitotenv.2005.09.026 [DOI] [PubMed] [Google Scholar]
- Van Etten, J. L. , Graves, M. , Müller, D. , Boland, W. , & Delaroque, N. (2002). Phycodnaviridae–large DNA algal viruses. Archives of Virology, 147(8), 1479–1516. 10.1007/s00705-002-0822-6 [DOI] [PubMed] [Google Scholar]
- Wickham, H. , Chang, W. , Henry, L. , Pedersen, T. L. , Takahashi, K. , Wilke, C. , Woo, K. , Yutani, H. , & Dunnington, D. (2020). . Package ‘ggplot2’. Create elegant data visualisations using the grammar of graphics. Retrieved from https://github.com/tidyverse/ggplot2 [Google Scholar]
- Winter, C. , Bouvier, T. , Weinbauer, M. G. , & Thingstad, T. F. (2010). Trade‐offs between competition and defense specialists among unicellular planktonic organisms: The “killing the winner” hypothesis revisited. Microbiology and Molecular Biology Reviews, 74(1), 42–57. 10.1128/MMBR.00034-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden, A. Z. , Follows, M. J. , Giovannoni, S. J. , Wilken, S. , Zimmerman, A. E. , & Keeling, P. J. (2015). Rethinking the marine carbon cycle: Factoring in the multifarious lifestyles of microbes. Science, 347(6223), 1257594 10.1126/science.1257594 [DOI] [PubMed] [Google Scholar]
- Xia, L. C. , Ai, D. , Cram, J. , Fuhrman, J. A. , & Sun, F. (2013). Efficient statistical significance approximation for local similarity analysis of high‐throughput time series data. Bioinformatics, 29(2), 230–237. 10.1093/bioinformatics/bts668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, L. C. , Steele, J. A. , Cram, J. A. , Cardon, Z. G. , Simmons, S. L. , Vallino, J. J. , Fuhrman, J. A. , & Sun, F. (2011). Extended local similarity analysis (eLSA) of microbial community and other time series data with replicates. BMC Systems Biology, 5, S15 10.1186/1752-0509-5-S2-S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Li, Y. I. , Zhou, Y. , Lei, X. , Zheng, W. , Tian, Y. , Van Nostrand, J. D. , He, Z. , Wu, L. , Zhou, J. , & Zheng, T. (2016). A comprehensive insight into functional profiles of free‐living microbial community responses to a toxic Akashiwo sanguinea bloom. Scientific Reports, 6, 34645 10.1038/srep34645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. , Li, Y. , Zhou, Y. , Zheng, W. , Tian, Y. , & Zheng, T. (2012). Bacterial community dynamics during a bloom caused by Akashiwo sanguinea in the Xiamen sea area, China. Harmful Algae, 20, 132–141. 10.1016/j.hal.2012.09.002 [DOI] [Google Scholar]
- Yutin, N. , & Koonin, E. V. (2013). Pandoraviruses are highly derived phycodnaviruses. Biology Direct, 8(1), 25 10.1186/1745-6150-8-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Richlen, M. L. , Sehein, T. R. , Kulis, D. M. , Anderson, D. M. , & Cai, Z. (2018). Microbial community structure and associations during a marine dinoflagellate bloom. Frontiers in Microbiology, 9, 1201 10.3389/fmicb.2018.01201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The raw sequencing data (FASTAQ files) of 18s rDNA and 16s rDNA genes obtained from the MiSeq platform were deposited in the Sequence Read Archive database at NCBI under Accession Numbers: SRR11123089–SRR 11123147 (PRJNA 607609), SRR11136919–SRR11136959 (PRJNA 607810) and SRR11131500–SRR11131531 (PRJNA 607814). The raw sequencing files of NCLDVs obtained from the HiSeq platform were deposited in the Sequence Read Archive database at NCBI under Accession Number SRR11172603–SRR11172644 (PRJNA 608210).