

Abstract
Protein-xenobiotic adducts are a byproduct of xenobiotic metabolism. While there is a correlation between protein adduction and target organ toxicity, a cause and effect relationship is not often clear. Naphthoquinone (NQ) and monocrotaline pyrrole (MCTP) are two pneumotoxic electrophiles that form covalent adducts with a similar select group of proteins rich in reactive thiols. In this study, we treated human pulmonary artery endothelial cells (HPAEC) with NQ, MCTP, or preformed NQ or MCTP adducts to the protein galectin-1 (gal-1) and examined indicators of reactive oxygen species (ROS) oxidative injury, markers of apoptosis (caspase-3 and annexin V) and gene responses of cellular stress. ROS production was assayed fluorescently using CM-H2DCFDA. NQ adducts to gal-1 (NQ-gal) produced 183% more intracellular ROS than gal-1 alone (p<.0001). Caspase-3 activity and annexin V staining of phosphatidylserine were used to assess apoptotic activity in treated cells. HPAEC exposed to MCTP-gal had increases in both caspase-3 activation and membrane translocation of annexin V relative to gal-1 alone (p<.0001). Direct application of NQ produced significantly more ROS and induced significant caspase-3 activation, whereas MCTP did not. Human bronchial epithelial cells were also exposed to MCTP-gal and found to have significant increases in both caspase-3 activation and annexin V staining in comparison to gal-1 (p<.05). Western blot analysis showed that both NQ and MCTP significantly induced the Nrf2 mediated stress response pathway despite differences in ROS generation. ER stress was not induced by either adducts or parent compounds as seen by quantitative RT-PCR, but HOX-1 expression was significantly induced by NQ-gal and MCTP alone. Electrophile adduction to gal-1 produces different cytotoxic effects specific to each reactive intermediate.
Introduction
Metabolism of xenobiotics via Phase I and/or Phase II enzymes generates hydrophilic metabolites that are more easily eliminated by excretion. Many phase I metabolites are more reactive than the parent compound and readily form covalent bonds with cellular macromolecules such as nucleic acids and proteins. There is a correlation between protein-drug adduct formation and target organ toxicity, but whether this represents a primary mechanism of cellular injury, a detoxification mechanism, or epiphenomenon remains uncertain (1-2). Most cell toxicology research focuses on identification of (1) toxic electrophilic metabolites of xenobiotics, (2) specifically bound proteins in target cells, and (3) cellular responses to injury (3). Few specific mechanisms directly linking protein-xenobiotic adducts to altered cell function have been described. One such is acetaminophen hepatotoxicity where protein binding correlates with altered phosphatases and downstream perturbations in protein phosphorylation states (4).
Protein adduction is hypothesized to initiate cellular toxicity by several mechanisms. The first suggests that covalent adduction of a target protein disrupts normal protein function by altering folding characteristics or blocking a functional site thus preventing enzymatic processes in signal transduction or regulatory pathways. The second mechanism involves secondary activation of the immune response against the now modified protein (5). A third, more general, mechanism postulates that adduction of protein thiols and glutathione depletes cellular antioxidant capacities thus compromising cellular redox homeostasis.
Phase I metabolism of monocrotaline (a pyrrolizidine alkaloid and experimental lung toxicant) and naphthalene (a common component of cigarette smoke and automobile exhaust) generates electrophilic metabolites. Monocrotaline is oxidized to the reactive metabolite monocrotaline pyrrole (MCTP) which can be hydrolyzed to dehydroretronecine and monocrotalic acid (6-7). Naphthalene is metabolized into 1,2-epoxide, 1,2-naphthoquinone, or 1,4-naphthoquinone (NQ) (8-9). Both precursor compounds are activated by cytochrome P450 related enzymes in the liver and, for naphthalene, in pulmonary Clara cells. MCTP and NQ are both pneumotoxic electrophiles that form adducts to proteins and cellular macromolecules in pulmonary arterial endothelial cells (PAECs) and nonciliated epithelial cells (Clara cells) (10-11). Phase II metabolism leading to formation of glutathione conjugates of MCTP and NQ has been characterized using bovine PAEC, isolated hepatic perfusions, and liver extracts (12-14). NQ causes depletion of glutathione and results in increases in reactive oxygen species (ROS) and subsequent cytotoxicity (15-16). While MCTP also diminishes GSH levels, it does not do so to an extent sufficient to cause oncosis (17). Instead, MCTP treatment in vitro characteristically induces apoptosis (18).
Ex vivo studies demonstrate that quinone arylation of thiol rich enzymes inhibits enzymatic function through conformational change. NQ related redox cycling also generates reactive oxygen species (ROS) that oxidize thiol and methionine groups on proteins. The relative contribution of arylation versus ROS mediated sulfhydryl oxidation appears to be dependent upon time and xenobiotic concentration (19-20).
The NE-F2 related factor - Kelch-like ECH-associated protein 1 (Nrf2-Keap1) pathway is a key detection and signaling transcriptional activator responding to cellular stressors such as ROS and reactive electrophilic metabolites. Cellular stressors ultimately result in the dissociation of the Nrf2-Keap1 complex and the activation of Nrf2 as a transcription factor (21). Induction of the Nrf2-Keap1 signaling pathway can occur through either thiolphilic binding of the cysteines on Keap1 allowing release of Nrf2, or by kinase activity (PI 3-kinase, MAP kinase, and protein kinase C) that directly phosphorylates Nrf2, which undergoes conformational change and dissociates from Keap1 (22). Gene targets transcribed in response to Nrf2 signaling include chaperone and proteasome proteins in addition to Phase II detoxifying enzymes, antioxidant proteins and GSH generating enzymes (23).
We previously identified and compared the binding of MCTP and NQ to proteins in pulmonary cells and found them to prefer targets rich in reactive thiols (24-25). One such thiol rich protein was galectin-1 (gal-1), a mammalian lectin found both intracellularly and associated with the surface of a variety of cells, including tumor cells and activated endothelium (26). Generally, gal-1 function is attributed based on its cellular localization. On the cell surface, gal-1 has lectin dependent functionality with roles in cell-cell regulation, cell-matrix interactions, immune response, apoptosis, and neoplastic transformation (27). Intracellular gal-1 has biological roles in cell cycle regulation, RNA splicing, and transcriptional regulation (28). Since this protein is abundantly expressed in both cytosol and at the cell surface, we chose to clone and express it to compare the functional consequences of adduct formation by NQ and MCTP.
Experiments in the present study examined the effect of adding preformed NQ or MCTP-galectin adducts to human pulmonary artery endothelial cells. ROS production, apoptosis initiation, Nrf2-Keap1 activation and cellular stress gene responses were evaluated.
Materials and Methods
Cell Culture
HPAEC from a 28 year old female were obtained from Cascade Biologics (Portland, OR) and were cultured in EGM-2 supplemented media (Lonza Walkersville; Walkersville, MD). Normal human bronchial epithelial cells (HBE) cultured with retinoic acid from a 29 year old female were obtained from Lonza Walkersville (Lonza) and were cultured in BEGM supplemented media (Lonza). Passages 6 and 7 were used for these experiments. Prior to experimental methods or treatments, cells were pre-equilibrated in new medium for one hour.
Synthesis of NQ and MCTP
NQ was synthesized from naphthalene as previously described (24, 29). NQ was stored in ethanol at -80 °C until just prior to use. Monocrotaline (MCT) (Sigma-Aldrich, St. Louis, MO) was converted to MCTP according to the method of Mattocks et al. (30) and re-crystallized from anhydrous ethyl ether as previously described (31-33). MCTP was stored in N,N-dimethylformamide (DMF) at −80 °C until just prior to use. Electrospray ionization mass spectra confirmed the stability of MCTP during storage.
Synthesis of adducted Galectin-1
Escherichia coli strain M15 (Qiagen, Valencia, CA) containing human galectin-1 cDNA incorporated into pQAE9 (Qiagen) was used to express the protein. This expression system was the generous gift of Richard D. Cummings (University of Oklahoma Health Sciences Center, Dept. of Biochemistry and Molecular Biology, Oklahoma Center for Molecular Medicine, Oklahoma City, Oklahoma). Purification of the product is detailed in a previous publication (24). The protein (2 mg/500 μL) was dialyzed overnight against PBS pH 7.4 using a Slide-A-Lyzer cassette (10,000 MWCO) and subsequently reduced with a 7-fold molar excess of tributylphosphine (Sigma-Aldrich). The reaction was allowed to progress under argon in the absence of light for 1.5 h. The solution of reduced protein was extracted 6 × with hexane and the residual hexane removed on a SpeedVac. To form protein adducts, a 6-fold molar excess of MCTP dissolved in DMF was added to a stirring solution of gal-1 and the reaction terminated after 1 h by passing the reaction mix over a column of Sephadex G-10 resin previously equilibrated in distilled water. Fractions were analyzed for protein using the BCA assay (Pierce, Rockford, IL).
Similarly adducts to NQ were generated by adding 6-fold molar excess to the stirring protein. The reaction was terminated after 7 min. The presence of adducts of pyrrole or quinone on gal-1 was determined using electrospray MS, as described below. In all cases, cells were exposed to 10 μg of adducted protein/mL of media. For control reactions gal-1 was processed identically to that for adduct formation in the presence of vehicle but without electrophiles.
Mass Spectrometry
Electrospray ionization mass spectra were generated in positive ion mode using a Waters (Milford, MA) LCT Premier mass spectrometer interfaced to a Shimadzu LC-20AD HPLC. The mass scale was calibrated using cluster ions from a 0.1% H3PO4 solution. Proteins were analyzed using on-line desalting that employed a 1 × 10 mm BetaBasic cyanopropyl guard column (Thermo Scientific, Waltham, MA) and a gradient of solvent A = 0.15% aqueous formic acid and solvent B = acetonitrile from 5%B to 70%B over 6 minutes. Mass spectra were transformed to zero charge state spectra using MaxEnt1 algorithm of the Waters MassLynx software.
Synthesis and uptake determination of Fluorescein conjugated Galectin-1
Galectin-1 (133 nmoles) was dissolved in 1 mL of PBS and reduced with a 6-fold molar excess of TCEP HCL (Pierce) for 30 min. The reduced product was subsequently reacted with a 2-fold molar excess of Fluorescein-5-Maleimide (Pierce) delivered in 50 μL of DMF for 1 h under a nitrogen atmosphere in the dark. The reaction was quenched with excess glutathione. Material was dialyzed (10,000 MW cutoff) extensively against PBS at 4°C. The fluorescein tagged galectin-1 (Gal-FITC) was used to estimate the total uptake of the protein over a 3.5 h interval. Briefly HPAEC were grown to confluency in 96-well, black, clear bottom Costar 3603 plates (Corning-Costar, Corning, NY). Cells were exposed to Gal-FITC (10 μg/mL) with and without ProJect (Pierce) in unsupplemented phenol red free EBM-2 (Lonza) medium for 3.5 h. ProJect is a cationic lipid protein transfection reagent that transports biologically active molecules into target cells by endocytosis and releases them into cytosol. Subsequently total well fluorescence (cells and media) and fluorescence after removal of the initial media and washing cells one time with PBS, was recorded using a Synergy HT multi-mode microplate reader (BioTek Instruments, Winooski, VT).
Treatments
Gal-1 adducts were transfected into cell cytoplasm using Pro-Ject. In this study, gal-1 alone or adducted to the compounds NQ or MCTP were transfected into HPAECs and HBEs according to manufacturer’s protocol. Briefly, cells grown to confluency on fibronectin coated glass coverslips, 8-well chamber slides or 6-well culture plates were rinsed three times with basal media prior to loading of gal-1 transfection complexes at a final concentration of 10 μg/mL. Preliminary experiments with Gal-FITC demonstrated an optimal uptake time of 3.5 h so the effect of protein adducts was evaluated at that time point. HPAEC transfected with either NQ or MCTP adducts to gal-1 were assayed for ROS production and apoptosis induction.
The effects of unadducted NQ and MCTP on HPAEC were also characterized. NQ in ethanol was premixed in medium prior to addition to culture wells or flasks at a final concentration of 4 μM. MCTP in DMF was directly pipetted into the overlaying media in culture wells or delivered by positive displacement syringes into media in culture flasks at a final concentration of 31 μM or 185 μM. Nrf2 translocation was evaluated at time points of 1 and 2 h. All other experiments were evaluated at 3.5 h. Control treatments for NQ and MCTP experiments were their vehicles, ethanol (0.5 μL/mL) and DMF (1 μL/mL), respectively.
ROS detection
CM-H2DCFDA (Molecular Probes, Eugene, OR) is a nonpolar compound that readily diffuses into cells where it is hydrolyzed to the nonfluorescent derivative dichlorodihydrofluorescein (H2DCF) and trapped within the cytoplasm. In the presence of an appropriate oxidant, H2DCF is oxidized to the highly fluorescent 2,7-dichlorofluorescein (DCF). The dye was dissolved in dimethyl sulfoxide (DMSO) and diluted in Krebs Henseleit buffer. HPAECs were grown on fibronectin coated coverslips placed in 24-well Costar medical grade polystyrene plates. Cells were rinsed with Krebs Henseleit buffer and incubated with CM-H2DCFDA in Krebs Henseleit buffer (0.578 μg/mL) for 45 min in the dark. After CM-H2DCFDA uptake, monolayers were rinsed and allowed a 1 h recovery in complete medium prior to treatments as described above. Cells were transfected with the gal-1 adducted compounds (NQ-gal, MCTP-gal) or treated with NQ (4 μM), and MCTP (31 μM or 185 μM) as described above. At the conclusion of treatment times, cells were rinsed twice in pre-warmed PBS and fixed in 1% PFA in PBS for 10 min. Coverslips were mounted on microscope slides (Corning, Lowell, MA) using ProLong Gold antifade reagent (Molecular Probes). Slides were examined with a Provis AX70 fluorescent microscope (Olympus, Central Valley, PA) and digital images captured with AxioCam software (Carl Zeiss Inc, Thornwood, NY). Digital images were analyzed using the modified methods of Lehr et al with Adobe Photoshop CS to detect increases in ROS induced pixel intensity (34). Briefly, under the select tab, the color range tool was used to select bright areas of pixels which represent ROS production. This initial selection was saved, and loaded for all subsequent images to be analyzed. The number of selected pixels was recorded. The total number of pixels for the cell monolayer was determined by selecting the background and using the inverse command to select the imaged cells. The average percentage of bright pixels per cellular monolayer was calculated for a sample size of at least six.
Western Blot Analysis
HPAECs were cultured in 175 cm2 tissue culture flasks (Corning). NQ and MCTP were pipetted directly into the culture flask. After treatment, flasks were placed on ice for 5 min, and then cells were scraped from the bottom of the flask into the treatment media. The cell suspension was transferred to a 50 mL centrifuge tube and pelleted for 5 min at 2000 rpm. Supernatant was removed and pellets were transferred to 1.5 mL microcentrifuge tubes. Pellets were rinsed with ice cold PBS three times followed by centrifugation after each subsequent rinse (3000 rpm for 3 min). Pellets were lysed using the NE-PER Extraction Kit (Pierce) following the manufacturer’s protocol with modifications. For nuclear extraction, pellets were sonicated using a Branson digital sonifier at 30% power for six – one second bursts with one second rests, before incubation with intermittent vortexing, all performed on ice. Protein determination was performed using the BCA assay (Pierce). Total protein (75 μg) from each treatment group was separated on an 11%T, 2.75%C SDS-PAGE gel and transferred onto PVDF membranes (Bio-Rad, Hercules, CA). PVDF membranes were blocked with 5% non-fat milk (Bio-Rad) in Tris buffered saline (TBS) and subsequently incubated at 4°C, overnight with anti-Nrf2 (polyclonal rabbit, 1:200, Santa Cruz Biotechnology, Santa Cruz, CA), anti-p53 (monoclonal mouse, 1:1000, Cell Signaling Technology, Danvers, MA), or β-actin (monoclonal mouse, 1:10,000, Sigma-Aldrich). Membranes were developed using HRP conjugated donkey anti-rabbit IgG or sheep anti-mouse IgG, respectively (GE Healthcare, Pittsburgh, PA). Blots were developed with the ECL+ detection system according to manufacturer’s specifications (GE Healthcare). Films were digitally scanned using an Epson Expression 1680 flatbed scanner with transparency adaptor. Densitometric analysis of major gel bands were analyzed using Image Quant. A paired Student’s t-test was used to compare densities between treatment groups from two separate experiments.
Apoptosis Assay
The Dual Apoptosis Assay Kit (Biotium, Hayward, CA) was used for fluorescent probes of two markers of apoptosis in HPAEC and HBE: caspase-3 activity and phosphatidylserine (PS) translocation. Cells were grown on fibronectin coated chamber slides or coverslips were transfected and treated as described above. Treated cells were assessed for apoptosis following the manufacturer’s protocol. NucView 488 caspase-3 substrate was added to treated cells. Upon cleavage by intracellular caspase-3, the substrate migrates to the nucleus and fluoresces green. Fluorescently labeled annexin V (Texas Red) is a phospholipid binding protein with high affinity for PS on the outer leaflet of the plasma membrane. DAPI was used to stain nuclei to determine total cell counts. Slides were examined with an Olympus AX70 fluorescent microscope and digital images captured with AxioCam software. Five microscopic fields were randomly selected from peripheral and central portions of each coverslip. Images of each field and emission spectrum were layered using Adobe Photoshop CS to demonstrate enhanced colocalization of FITC labeled caspase-3 nuclei with the DAPI stain. Cells positive for caspase-3 activity appeared teal whereas nonapoptotic cells stained only for DAPI and were royal blue. Cells with membrane translocation of annexin V had peripheral membrane associated red fluorescence.
Real time RT-PCR
Total RNA from HPAEC was isolated using the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. 1.5 μg of total RNA was reverse-transcribed to obtain cDNA in a final volume of 20 μL reaction buffer consisting of random hexamer primers, dNTPs, DTT, RNaseOUT, and Superscript III reverse transcriptase (Invitrogen). Real time RT-PCR (ABI PRISM 7700 Sequence detection system; PE Applied Biosystems, Foster City, CA) using SYBR green (Applied Biosystems) as fluorescent reporter was used to quantify the expression of BIP, HSP40, HSP70, SEPX1, MSRB3, SOD-1, catalase (CAT) and HOX-1. All the gene specific primers (Table 1) were designed either with PrimerQuest mFold Oligo Analyzer 3.0 or free software from Integrated DNA Technologies, INC (www.idtdna.com). Triplicate reactions for each sample were carried out in 384-well optical well plate containing 18.75 ng cDNA/well. The RNA quantity was further normalized by amplifying cDNA samples simultaneously with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) specific primers. The 2-ΔΔCT method (35) was used to calculate relative changes in gene expression determined from real-time RT-PCR experiments (Applied Biosystems User Bulletin No.2 (P/N4303859). The threshold cycle, CT, which correlates inversely with the target mRNA levels, was measured as the cycle number at which the SYBR Green emission increases above a threshold level. Specific mRNA transcripts were expressed as fold difference in the expression relative to their corresponding vehicle control.
Table 1.
Primer sequences for real-time RT-PCR
| Gene | Alternate Names | Primer sequence (5’-3’) |
|---|---|---|
| GAPDH | Sense-CACCAACTGCTTAG | |
| Antisense-TGGTCATGAGTCCT | ||
| BIP | HSPA5, GRP78 | Sense-TTGAATGGCTGGAAAGCCACCAAG |
| Antisense-AGGGCCTGCACTTCCATAGAGTTT | ||
| HSP40 | DNAJB1 | Sense-AGTTCTTCGGTGGCAGAAATCCCT |
| Antisense-CCAAAGTTCACGTTGGTGAAGCCA | ||
| HSP70 | HSPA4 | Sense-AAGATGCAAGTGGACCAGGAGGAA |
| Antisense-TGATTCTCGATTGGCAGGTCCACA | ||
| SEPX1 | MSRB1 | Sense-TCTGAAGCCTTGAAGGTGTCCTGT |
| Antisense-ACCCTGGGAGGCAGAAGTTTCTTT | ||
| MSRB3 | Sense-AATTTGACTCCGGTTCAGGTTGGC | |
| Antisense-TGTTTCCACCCTGTGCATCCCATA | ||
| SOD-1 | Sense-GTCGTAGTCTCCTGCAGCGTC | |
| Antisense-CTGGTTCCGAGGACTGCAA | ||
| CAT | Sense-CGGAGATTCAACACTGCCAA | |
| Antisense-GAATGCCCGCACCTGAGTAA | ||
| HOX-1 | Sense-CAGCCCTCTCACTGTGTCCC | |
| Antisense-GCCATAGGCTCCTTCCTCCTT |
Statistics
Student’s t-test or ANOVA were used, as appropriate, for all experiments except for analysis of ROS production in NQ-gal transfected HPAEC and Western blot analysis. These exceptions were analyzed using a paired Student’s t-test. Bonferroni post hoc tests were used to determine individual interactions between treatment groups. Statistical analyses were performed using Microsoft Excel Analysis Toolpak software (Microsoft Corp., Redmond, WA) and SPSS software (SPSS, Inc., Chicago, IL). p<0.05 was considered statistically significant.
Results
Electrophile adduct formation with Galectin-1
Transformed electrospray ionization mass spectra verified the formation of NQ and MCTP adducts with gal-1 (Figure 2). Two findings are prominent in the spectra generated from reactions with NQ. First, the sequential addition of structure III (Figure 1) giving uniform mass increase of 158 from 15479 to 16584 corresponds to the addition of as many as seven adducts within this mass span. The mass value consistent with unadducted Gal-1 was below the level of detection. The second prominent feature is the appearance of additional peaks with mass increases of one or more multiples of 16 Da from the base adduct peak. For example the cluster of masses, 15952, 15968, 15983 and 15999 denote as much as three additions of oxygen to the adducted protein. Therefore, the reaction of the quinone with gal-1 not only resulted in the formation of adducts but also stimulated the generation of ROS and the extensive oxidation of the protein. NQ adducts can exist in various redox states and are capable of redox cycling, and the nature of the redox states cannot be discerned from ESI spectra.
Figure 2. Mass spectra of Gal-1, MCTP-gal, and NQ-gal.
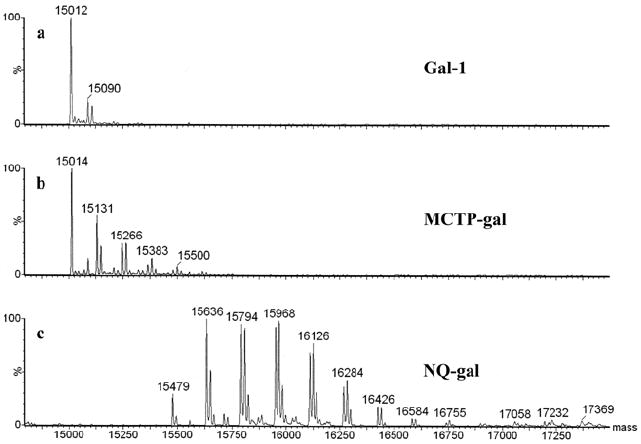
Unadducted Gal-1 is represented by a mass peak at ~15012 Da (a). Peaks in MCTP adducted Gal-1 represent combinations of structures I, IIa, and IIb (b). Mass increased of 158 Da in NQ spectra represent sequential addition of structure III with additional 16 Da peaks representing addition of oxygen. Lack of a 15012 peak demonstrates adduction of all Gal-1 starting material (c). (See Figure 1 and text for details).
Figure 1. Structures and Schemes.
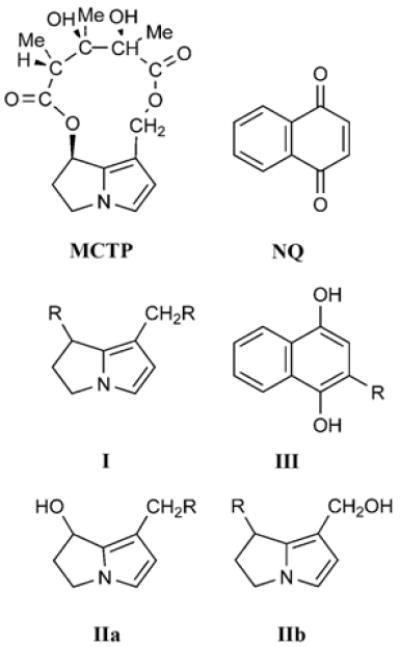
The structures of monocrotaline pyrrole (MCTP) and 1,4-naphthoquinone (NQ). Scheme I, IIa, and IIb show possible sites where pyrrole can form adducts with gal-1 (R). Scheme III shows the possible site where NQ can form adducts with gal-1 (R).
The reaction of MCTP with gal-1 produced mass increases at 15131 and 15148 Da which are generated from the addition of structure I (117 Da) and structure II (a or b) (135 Da) (Figure 1) respectively. The di-adducted peaks at 15248 and 15266 are adducted with 2 × I and I + II respectively. Tri-adducted gal-1 appears at mass 15383 and tetra-adducted gal-1 appears at mass 15500 containing (2 × I) + II and (3 × I) + II, respectively. The largest peak corresponds to non-adducted protein. Peak area calculations indicate, however, that approximately 68% of the gal-1 was adducted. Table 2 contains the data relative to the percent of total ion current (TIC) attributed to galectin and peaks composed of single and multiple adductions with NQ or MCTP to the protein.
Table 2.
Percent of total ion current (TIC) for Gal-1, mono and multiple adductions with NQ and MCTP
| NQ-gal adductsa | MCTP-gal adductsb | ||
|---|---|---|---|
| # of adducts | % TIC | # of adducts | % TIC |
| 0 | 0 | 0 | 31.8 |
| 3 | 2.7 | 1 | 32.2 |
| 4 | 13.7 | 2 | 21.5 |
| 5 | 23.3 | 3 | 10.8 |
| 6 | 27.6 | 4 | 2.8 |
| 7 | 19.7 | 5 | 0.8 |
| 8 | 9.7 | ||
| 9 | 1.7 | ||
| 10 | 0.6 | ||
| 11 | 0.5 | ||
| 12 | 0.4 | ||
Overall adduction is 5.9 nmoles adduct/nmol protein; only trace amounts of unmodified gal-1 was detected
Overall adduction is 1.5 nmoles adduct/nmole protein; 31.8% unmodified gal-1
Uptake determination of Gal-FITC
Gal-FITC experiments, utilizing a 96 well format, each well containing 1.39 μg of material (10 μg/mL) resulted in the uptake of 5% of the fluorescently conjugated material in 3.5 h. Transfected cells, cultured on fibronectin coated coverslips, when examined by fluorescent microscopy revealed a uniform carpet of labeled cells devoid of non-transfected elements. Assuming that NQ and MCTP adducted galectin-1 would approximate the behavior of Gal-FITC, the following calculations were made. Relative to MCTP, we have 1.5 nmoles of adduct/15 μg of gal-1 (Table 2). For a 24-well plate, at 5 μg of gal-1 per well, only 5% of the adducted material gains access to the cell or 0.25 μg. This translates into 0.025 nmoles of adduct in a confluent layer that has ~ 12 μg of total cellular protein. This is equivalent to 2 ng of adduct/mg of protein for MCTP adducted gal-1 and 8 ng of adduct/mg of protein for NQ (5.9 nmoles/15 μg gal-1, Table 2). The exposure levels for the present series of experiments are within what has been observed by other investigators. For instance Hoffmann et al (1985) studying covalent adducts with acetaminophen found levels of covalent binding approaching 1 nmol/mg of cytosolic protein 4 hours after a 200 mg/kg dose, with hepatic microsomal incubations generating 3 nmol/mg (36). However in the present study the label is localized to one source protein and not disseminated among numerous cellular macromolecules.
Detection of Reactive Oxygen Species
After 3.5 h, HPAECs transfected with NQ-gal produced an average 183% more intracellular ROS than gal-1 controls (paired t-test P<.0001) (Figure 3a and 3b). In comparison to gal-1, the MCTP-gal treatment resulted in a large amount of highly fluorescent debris left on the coverslips. This interfered with ROS assays in the remaining cells. The results of this particular experiment led to testing for markers of apoptosis.
Figure 3. ROS generation in NQ-gal transfected cells.
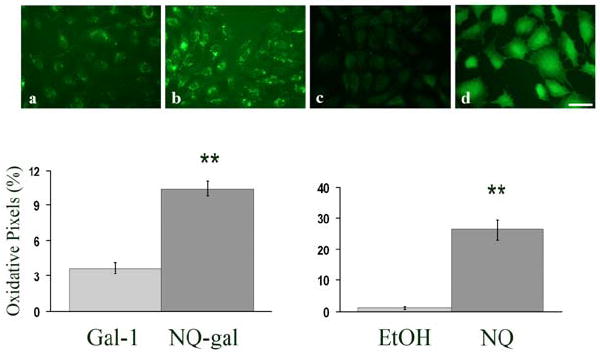
(a-d) Representative images of ROS generation in HPAEC, preloaded with CM-H2DCFDA, transfected with (a) galectin-1 (gal-1), (b) NQ adducted to galectin-1 (NQ-gal), (c) ethanol (EtOH), or (d) NQ. Graphs represent the percent of oxidative pixels within the whole cell population. Each bar represents the mean and the standard error of the results of four experiments. NQ-gal transfected cells and NQ treated cells had a significantly higher percentage of oxidative pixels in comparison to gal-1 and ethanol control cells after 3.5 h, respectively (**p<.0001). Bar= 10 μm.
In the NQ experiments, significant differences in ROS production were found between test and control where p<0.0001 (Figure 3c and 3d). There was an average 470-fold increase in DCF fluorescence over the control. For MCTP experiments, two concentrations (31 μM or 185 μM) were used for treatment. In both MCTP treatments, ROS production was too low to be distinguished from controls.
Western blot analysis of nuclear extracts for Nrf2 translocation
For NQ experiments, Nrf2 translocation was significantly more in NQ treated HPAEC than ethanol controls, at both 1 h (p<.0006) and 2 h (p<.0002) (Figure 4a). The average density of the Nrf2 band for NQ treated cells at two hours was 53% greater than ethanol treated cells.
Figure 4. Western blot analysis of Nrf2 in nuclear extracts.
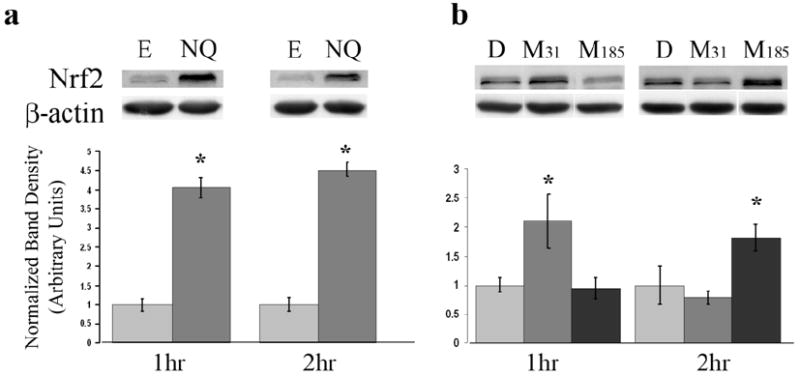
Western blot analysis was performed on nuclear lysates isolated from HPAEC treated with NQ (a) or MCTP (b). Membranes were probed with Nrf2 and β-actin. Graphs represent band density of treatments as normalized to respective vehicle controls. Each bar represents the mean and the standard error of the results of three experiments. Ethanol (E), DMF (D), MCTP 31 μM (M31), MCTP 185 μM (M185). *p<.05
In MCTP experiments, Nrf2 translocation was dependent upon concentration and time (Figure 4b). At 1 h, significant differences were found between HPAEC treated with the lower concentration of MCTP (31 μM) and DMF control (p<.011). The Nrf2 band for MCTP 31 μM treated cells at 1 h had an average 111% increase in density compared to DMF controls. At 2 h significant differences were found between cells treated with MCTP (185 μM) and DMF control (p<.017). Nrf2 band intensity for MCTP 185 μM treated cells at 2 h had an 82% increase in density relative to DMF controls.
Apoptosis Assay
HPAEC and HBE treated with gal-1 adducted compounds were fluorescently stained for two markers of apoptosis (caspase-3 or PS translocation via Annexin V stain). Apoptotic cells positive for caspase-3 and PS translocation had FITC labeled nuclei, and Texas Red labeled membrane, respectively. Figure 5(a-c) shows MCTP-gal adducts induced increased apoptosis in HPAEC as compared to the gal-1 control. Caspase-3 activity and annexin V staining were significantly higher with MCTP-gal test cells than both the NQ-gal test and the gal-1 control (p<.0001) after 3.5 h. Cells treated with MCTP-gal had 15% caspase-3 activation and 10% stained positive for annexin V. In comparison, NQ-gal treated cells had 9% caspase-3 activation and 5% annexin V staining. Gal-1 control cells had 6% caspase-3 activity and 3% annexin V staining.
Figure 5. Apoptosis increased in MCTP-gal treated HPAEC.
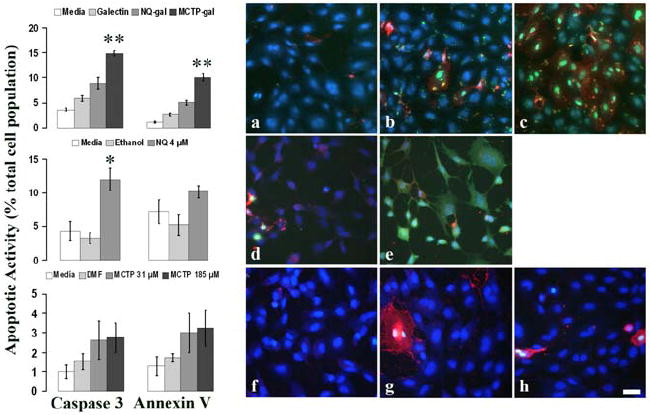
Caspase-3 and annexin V were used to detect apoptosis in treated HPAEC. (a-h) Representative images of HPAEC transfected with (a) galectin-1, (b) NQ-gal, (c) MCTP-gal, (d) ethanol, (e) NQ, (f) DMF, (g) MCTP 31 μM, and (h) MCTP 185 μM. FITC labeled nuclei are positive for caspase-3 activity. Texas-Red labeling indicates annexin V staining of phosphatidylserine (PS) found on the outer leaflet of the plasma membrane. Cell nuclei were counterstained with DAPI (blue) to determine total cell counts. Graphs represent the percent of the total cell population positive for caspase-3 activity and annexin V staining of PS. Each graph is representative for each treatment row to its right. Each bar represents the mean and the standard error of the results of four experiments. Bar=10 μm. *p<.05, **p<.0001.
In addition to the gal-1 adducted compounds, HPAEC were also treated with NQ and MCTP alone. The direct application of unadducted NQ resulted in a significant increase in caspase-3 activation in comparison to ethanol controls (p<.002) (Figure 5d, e). There were no significant changes in annexin V staining between test and control. The direct application of MCTP did not show any significant differences in caspase-3 activity or PS translocation in comparison to DMF (Figure 5f-h).
Figure 6 shows MCTP-gal adducts induced increased apoptosis in HBE as compared to the gal-1 control. Caspase-3 activity and annexin V staining were significantly higher with MCTP-gal test cells than both the NQ-gal test and the gal-1 control (p<.05) after 3.5 h. HBE treated with MCTP-gal had 7% caspase-3 activation and 12% stained positive for annexin V. In comparison, NQ-gal treated cells had 3% caspase-3 activation and 5% annexin V staining. Gal-1 control cells had 3% caspase-3 activity and 7% annexin V staining.
Figure 6. Apoptosis increased in MCTP-gal treated HBE.
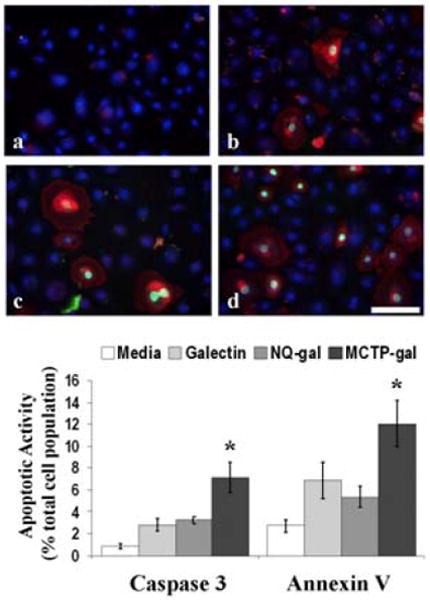
Caspase-3 and annexin V were used to detect apoptosis in treated HBE. (a-d) Representative images of HBE transfected with (a) media, (b) galectin-1, (c) NQ-gal, and (d) MCTP-gal. The graph represents the percent of the total cell population positive for caspase-3 activity and annexin V staining of PS. Each bar represents the mean and the standard error of the results of four experiments. Bar=10 μm. *p<.05.
Western blot analysis of p53
Our previous findings with bovine PAEC demonstrated that MCTP induced apoptosis most likely as a consequence of DNA binding that result in activation of the p53 pathway. Since MCTP did not induce caspase-3 activity at early time points, we compared the effects of un-adducted NQ and MCTP on p53 induction after 24 h of treatment. SDS-PAGE separation of whole cell lysates showed that MCTP (185 μM) was the only treatment to show banding for p53, in comparison to its vehicle control and other treatments (MCTP 31 μM and NQ 4 μM) (Figure 7).
Figure 7. Western blot analysis of p53.
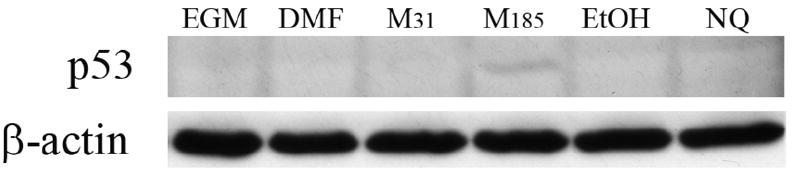
Western blot analysis was performed on cellular lysates isolated from HPAEC treated with medium (EGM), DMF, MCTP 30.9 μM (M31), MCTP 185 μM (M185), ethanol (EtOH), or NQ 4 μM (NQ). Membranes were probed with p53 and β-actin. This blot is representative of four experiments.
Real time RT-PCR expression of cellular stress genes
In order to better specify cellular responses to our treatments, we evaluated the gene response to ER stress (BIP), cytosolic stressors such as heat shock (HSP40 and HSP70), and antioxidant regulation including antioxidant repair and induction (methionine sulfoxide reductases (SEPX1 and MSRB3), SOD-1, CAT and HOX-1). After 3.5 h, HPAEC transfected with gal-1 adducted compounds (Figure 8, top) had modest changes in gene expression of all the genes except for BIP and SEPX-1. NQ-gal transfected cells had a significant decrease in transcript levels of HSP70, MSRB3 and CAT by 11, 14 and 16%, respectively, compared to gal-1. NQ-gal induced a significant increase of HOX-1 by 23% against gal-1 controls. Conversely, MCTP-gal had a significant decrease of HOX-1 by 22%. Significant decreases by MCTP-gal were also found in MSRB3 and SOD-1 by 4% and 8%, respectively. The only significant increase was by HSP40 by 17%.
Figure 8. Expression of cellular stress genes.
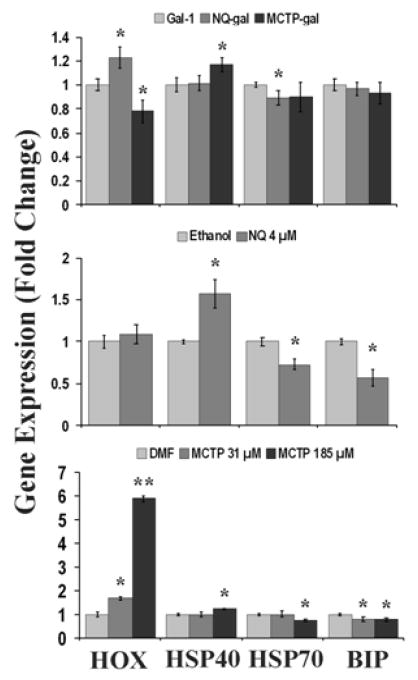
Real-time quantitative RT-PCR was used to determine gene expression in treated HPAEC. The graphs show the gene expression results for HOX-1 (HOX), HSP40, HSP70, and BIP. The top graph represents HPAEC transfected with gal-adduct. The middle and bottom graphs represent HPAEC treated with NQ and MCTP, respectively. (n≤3; *p<.05, **p<.0001).
NQ treatment after 3.5 h produced significant decreases in the expression of BIP, HSP70, SEPX1 and MSRB3 in comparison to ethanol controls by 43, 27, 19, and 28%, respectively. HSP40 expression was significantly increased by 57% in NQ treatments (Figure 8, middle).
Within the MCTP treatments, MCTP 185 μM had the most gene expression changes (Figure 8, bottom). Both doses of MCTP (31 and 185 μM) significantly decreased the expression of BIP by 20 and 21%, respectively. HSP70, SEPX1, MSRB3 and CAT were significantly decreased by the high dose MCTP 185 μM alone by 24, 42, 36, and 22%, respectively. Increases in gene expression were found in HSP40 and HOX-1. MCTP 185 μM alone increased HSP40 expression by 24%. Both doses of MCTP increased HOX-1 expression by 69 and 487% in comparison to DMF controls.
Discussion
This study demonstrated that two different reactive intermediates trigger distinct responses of cytotoxicity when adducted to the same protein (galectin-1), indicating that reactive intermediates are not completely inactivated after covalent binding to gal-1. NQ-gal produced significant amounts of intracellular ROS thus inducing cellular oxidative stress. In comparison to unbound NQ (4 μM), NQ-gal induced less cytotoxicity. Direct application of NQ caused production of ROS and activation of caspase-3. In contrast, adduction of MCTP to galectin-1 was found to be more cytotoxic than the unbound counterpart (MCTP) in HPAEC. MCTP-gal activated caspase-3 and induced phosphatidylserine translocation to the outer membrane, indicating apoptosis in both HPAEC and HBE. MCTP (31 μM and 185 μM) treated HPAEC did not produce ROS or induce apoptotic markers at 3.5 h.
Mass spectra of NQ adducted gal-1 indicated that the reaction progressed to completion and no un-adducted gal-1 remained. Calculation of molar equivalency gives an estimate that treating cells with 10 μg of protein/mL corresponds to an equivalent dose of ~ 4 μM of quinone moieties. The appearance of more than 6 adducts per gal-1 molecule suggests that quinones react with gal-1 at non-thiol nucleophilic sites. Adduct formation was also seen to coincide with the generation of mass increases of 16 Da, resulting in distinctive clusters suggesting the addition of one or more oxygens. The extensive oxidation of galectin is consistent with adduct driven generation of ROS and redox cycling. This generation of ROS is suggestive of at least two prominent mechanisms by which quinone adducted proteins could affect cellular insult. Adducted proteins could act as a focus of the generation of ROS resulting in extensive oxidation of both self and neighboring molecules. Additionally, cycling between quinone and hydroquinone could allow cross-linking other macromolecules to the adducted protein.
The cellular toxicity of quinones and quinones adducted to galectin-1 would be expected to be difficult to dissect mechanistically, more so than cellular insult derived from exposure to MCTP. This stems from the complex behavior of electrophiles similar to 1,4-naphthoquinone and alterations in the quinone following its interaction with nucleophiles. Introduction of 1,4-naphthoquinone into the cells could undergo one electron reduction to a semiquinone radical with a subsequent electron transfer to an acceptor molecule like oxygen, regenerating the original quinone (redox cycling) (37). In the Haber-Weiss reaction, superoxide is converted to hydrogen peroxide which in turn reduces Fe+3 to Fe+2. Hydrogen peroxide can react with Fe+2 resulting in the formation of a macromolecule damaging hydroxyl radical (38). Hydrogen peroxide could also react with catalase and be converted to water and oxygen or with glutathione peroxidase resulting in the conversion of glutathione to the disulfide. Reaction with glutathione, protein thiols or D,T-diaphorase would produce a hydroquinone that is less reactive than the semiquinone. The hydroquinone, free or adducted to glutathione or protein thiols, has potential to undergo auto-oxidation producing more superoxide. The hydroquinone can be conjugated with sulfate or glucuronic acid and eliminated from the cell, blocking further redox cycling. Phenolic conjugates with glutathione are superior to the original hydroquinone in the reduction of oxygen and as such are more toxic. Thiol conjugates on galectin would be expected to exhibit reactivity similar to glutathione conjugates, but degradation and elimination from cells would be prolonged, providing for longer-lived catalysis for redox cycling. The original complex nature of quinones would be retained on adducted galectin with celluar damage being precipitated through redox cycling, simple inactivation of adducted galectin through intra cross-linking with self or inter cross-linking with self or other proteins. Adduction of quinone to galectin could also alter its tertiary structure and its ability to interact with other proteins, or more seriously compete with productive protein associations.
The ability of quinone-thiol Michael adducts to retain redox cycling function has been previously demonstrated in N2A cells treated with arylated gamma tocopherol quinones (16). Similarly, previous ex vivo studies have shown that the adduction of NQ with urease under aerobic conditions resulted in the inhibition of the enzyme by a combination of two mechanisms, the direct arylation of thiol groups and ROS generation (19-20). Glutathione depletion due to extensive conjugation or oxidation driven by NQ, leads to alterations of the cell’s redox homeostasis/environment and subsequent increase in ROS levels (15). ROS triggers Nrf2 activation presumably to increase the expression of glutathione and antioxidants such as NAD(P)H:quinone oxidase (NQO1) (39). In this study, direct application of NQ significantly increased Nrf2 translocation to the nucleus.
Mass spectra of gal-1 treated with MCTP found approximately 68% of the material was adducted indicating that the formation of gal-1 adducts was less efficient than for NQ. Estimates of molar equivalents suggest that the relative available concentration of pyrrole adducts in media of treated cells is below the NQ estimate of 4 μM. The lower efficiency of MCTP adduct formation is consistent with hydrolytic side reactions that convert MCTP to products including dehydroretronecine (DHR) that are less reactive than MCTP. MCTP is a bifunctional alkylating reagent, and ESI spectra of adducted protein document intramolecular pyrrole cross-links. As depicted in Figure 1, adduction can occur on C7 or C9 or both linking two macromolecules together or linking internal sites within the same molecule.
The finding that MCTP-gal adducts retain the ability to induce apoptosis was an unexpected result, but was found to be a consistent finding in two different cell lines. Our previous studies in bovine PAEC demonstrated MCTP induced apoptosis in conjunction with DNA binding of MCTP metabolites (18). These findings, in conjunction with additional experiments demonstrating alterations in cell cycle checkpoint activities, suggested the apoptosis was in response to DNA cross-linking by the bifunctional necine base characteristic of pyrrolizidine alkaloids (40). We used two doses of MCTP previously found to have significant biological effect (18, 41), but in this study, treatment of HPAEC with MCTP did not initiate apoptosis at 3.5 h. Since the response to MCTP appears to depend on a delayed response to DNA cross-linking, we demonstrated p53 activation of DNA damage at 24 h by the higher concentration of MCTP (185 μM), but not in NQ treated cells.
Previous work (42) has attempted to explain the mechanism of delivery of aqueous labile MCTP from its hepatic site of production to remote pulmonary sites. Red blood cells (RBC) appeared to be likely carriers since tracer studies showed extensively labeled RBCs. The lipophilic membranes of RBC were suspected to shield the parent pyrrole (MCTP) from the aqueous environment with the close association between the RBC membrane and the membranes of pulmonary endothelial cells resulting in adduct formation on the surface of endothelial cells (25) or interior by diffusion of MCTP and its less reactive hydrolysis product DHR. The latter has been shown to be ineffective in vivo compared to MCTP due to reduced alkylating potential and ease of excretion (43). Our finding that adducts to gal-1 are toxic is intriguing considering that the major protein component of RBCs, globins, were found to be adducted after exposing animals to MCTP. This suggests an alternative mechanism of reactive intermediate transport since RBC globins have previously been suggested to adsorb to the surface of endothelial cells and, due to their hydrophobic nature, enter the cell by pinocytosis (44).
While continuing redox cycling of adducted NQ provides a plausible explanation of continuing cytotoxicity, the mechanism of enhanced toxicity by galectin adducts of MCTP seems less clear. Since the spectra of pyrrole adducted gal-1 contains additions of 135 Da to the protein the electrophile still retains the ability to alkylate other nucleophilic sites present on glutathione, or macromolecules like DNA, RNA and proteins. Adduction to gal-1 could facilitate nuclear transport of pyrrole where binding to DNA would initiate apoptosis. An alternative explanation is consistent with adduct-induced interference in gal-1 mediated transcriptional processes. Gal-1, along with galectin-3, is critical in the splicing of pre-mRNA and cells depleted in these two galectins lose splicing activity (45). Pyrrole adducts to gal-1 and other proteins would also be expected to prolong cellular exposure to the electrophile since it would not be subjected to rapid cellular excretion as anticipated for glutathione adducts (46). Loss of an active alkylating group in cases of both C7 and C9 bound to nucleophilic sites either intra or inter-molecular does not necessarily nullify toxicity. Adducted macromolecules could still compete with functional macromolecules and disrupt normal homeostasis in a manner more far reaching than the loss of protein. This competition might be expanded in time if the cell fails to detect xenobiotic modifications of proteins or target them for destruction and removal.
Direct application of MCTP did not induce apoptosis or produce detectable amounts of ROS after 3.5 h, but significant Nrf2 movement to the nucleus was observed by Western analysis. In previous experiments from our laboratory, glutathione levels in HPAEC exposed to MCTP (100 μM) decreased to 40% of normal after 15 min of treatment, but recovered within 4-6 h (17). As in the case of NQ, MCTP induced depletion of glutathione could initiate Nrf2 signaling by altering cellular redox homeostasis. Alternatively, previous studies from our laboratory demonstrate an affinity of MCTP for sulfhydryl groups (25) and Nrf2 activation could occur in response to MCTP binding to the SH residues on Keap1. Finally, MCTP has been shown to modify proteins in both the ER and Golgi (11, 47-48). In addition, previous investigators have shown that ER stress (49) and the covalent modification of intramitochondrial proteins (HSP60, HSP70, aspartate aminotransferase, aconitase, and α-ketoglutarate dehydrogenase) by compounds such as tetrafluoroethylcysteine (a metabolite of tetrafluorethylene) can trigger Nrf2 movement (50). Both intermediates (NQ and MCTP) activated the Nrf2-Keap1 pathway, indicating a response to cellular stress, though likely through differing mechanisms.
We used quantitative RT-PCR to evaluate potential cellular response mechanisms for all of the treatments in this study. There was no induction of ER stress or heat shock response genes BIP (GRP78) and HSP70. BiP (GRP78) is a molecular chaperone located in the ER that is selectively up-regulated during unfolded protein response (UPR), and stress conditions such as glucose deprivation, hypoxia and in the presence of toxic agents (51). HSP70, an inducible heat shock protein, works as a chaperone to repair denatured proteins (52). In this study, HOX-1 (heme oxygenase 1) was significantly induced by NQ-gal. HOX-1 is an enzyme that increases reduced glutathione levels and breaks down heme into the antioxidants biliverdin and bilirubin (53-54). HOX-1 transcription is Nrf2 dependent inferring that NQ-gal transfection would induce Nrf2 to the nucleus within our 3.5 h interval. Conversely, NQ alone does not induce HOX-1, despite the Nrf2 translocation shown at 1 and 2 h. HSP40 (heat shock protein 40), which was also induced, is a cytosolic molecular chaperone that works with HSP70 in repair of stress damaged proteins (52). In addition to protein repair, HSP70 has roles in anti-apoptotic signaling by directly blocking the release of pro-apoptotic factors such as cytochrome c in mitochondria (51). This anti-apoptotic signaling can be inhibited through the Akt/FOXO3a signaling pathway. FOXO3a is a transcription factor that suppresses HSP70 expression in endothelial cells, resulting in caspase-9 activation and subsequent apoptosis (55). In this study, NQ treated cells had decreased HSP70 transcripts which may correlate with the increased caspase-3 activity.
In the MCTP-gal adduct experiments, HOX-1 was not induced along with HSP70, though the latter was not significantly decreased in comparison to gal-1 adducts. Again, HSP40 was found to be significantly increased, suggesting an attempt at cytoprotection despite HSP70 non-induction. With the significant induction of apoptotic markers, caspase-3 and annexin V, it’s not surprising that most of the genes assayed had decreased transcript levels. Within the MCTP experiments, high dose (MCTP 185 μM) had the most change in comparison to DMF controls. MCTP (185 μM) had the largest induction of HOX-1 of all the treatments, including the gal-adducts, which correlates with the significant translocation of Nrf2 found at 2 h.
In this study, we showed that electrophile adduction to galectin-1 produced different cytotoxic effects specific to NQ and MCTP. In the case of NQ, neat or adducted to galectin-1, resulted in significant cellular ROS, but only free NQ was sufficient in inducing caspase activity. In the case of MCTP, the adduction to galectin-1 was seen to accentuate the electrophile’s apoptotic properties over a shorter time frame in HPAEC. Previously MCTP was shown to induce apoptosis under conditions of prolonged exposure. Future experiments showing the effects of adduction to different proteins would be an interesting and important study.
Acknowledgments
This research has been funded in part by NIH grant #R01HL48411 from NHLBI by the United States Environmental Protection Agency through STAR grant RD832414 to the University of California at Davis. It has not been subjected to the Agency’s required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred.
Footnotes
The authors declare that there are no conflicts of interest.
References
- 1.Yang XX, Hu ZP, Chan SY, Zhou SF. Monitoring drug-protein interaction. Clin Chim Acta. 2006;365:9–29. doi: 10.1016/j.cca.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 2.Zhou S, Chan E, Duan W, Huang M, Chen YZ. Drug bioactivation, covalent binding to target proteins and toxicity relevance. Drug Metab Rev. 2005;37:41–213. doi: 10.1081/dmr-200028812. [DOI] [PubMed] [Google Scholar]
- 3.Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicol Appl Pharmacol. 1997;143:1–12. doi: 10.1006/taap.1996.8074. [DOI] [PubMed] [Google Scholar]
- 4.Bruno MK, Khairallah EA, Cohen SD. Inhibition of protein phosphatase activity and changes in protein phosphorylation following acetaminophen exposure in cultured mouse hepatocytes. Toxicol Appl Pharmacol. 1998;153:119–132. doi: 10.1006/taap.1998.8512. [DOI] [PubMed] [Google Scholar]
- 5.Pumford NR, Halmes NC. Protein targets of xenobiotic reactive intermediates. Annu Rev Pharmacol Toxicol. 1997;37:91–117. doi: 10.1146/annurev.pharmtox.37.1.91. [DOI] [PubMed] [Google Scholar]
- 6.Mattocks AR. Toxicity of pyrrolizidine alkaloids. Nature. 1968;217:723–728. doi: 10.1038/217723a0. [DOI] [PubMed] [Google Scholar]
- 7.Mattocks AR. Dihydropyrrolizine derivatives from unsaturated pyrrolizidine alkaloids. J Chem Soc Perkin 1. 1969;8:1155–1162. doi: 10.1039/j39690001155. [DOI] [PubMed] [Google Scholar]
- 8.Tingle MD, Pirmohamed M, Templeton E, Wilson AS, Madden S, Kitteringham NR, Park BK. An investigation of the formation of cytotoxic, genotoxic, protein-reactive and stable metabolites from naphthalene by human liver microsomes. Biochem Pharmacol. 1993;46:1529–1538. doi: 10.1016/0006-2952(93)90319-r. [DOI] [PubMed] [Google Scholar]
- 9.Wilson AS, Davis CD, Williams DP, Buckpitt AR, Pirmohamed M, Park BK. Characterisation of the toxic metabolite(s) of naphthalene. Toxicology. 1996;114:233–242. doi: 10.1016/s0300-483x(96)03515-9. [DOI] [PubMed] [Google Scholar]
- 10.Chichester CH, Buckpitt AR, Chang A, Plopper CG. Metabolism and cytotoxicity of naphthalene and its metabolites in isolated murine Clara cells. Mol Pharmacol. 1994;45:664–672. [PubMed] [Google Scholar]
- 11.Lame MW, Jones AD, Wilson DW, Dunston SK, Segall HJ. Protein targets of monocrotaline pyrrole in pulmonary artery endothelial cells. J Biol Chem. 2000;275:29091–29099. doi: 10.1074/jbc.M001372200. [DOI] [PubMed] [Google Scholar]
- 12.Lame MW, Jones AD, Morin D, Segall HJ, Wilson DW. Biliary excretion of pyrrolic metabolites of [14C]monocrotaline in the rat. Drug Metab Dispos. 1995;23:422–429. [PubMed] [Google Scholar]
- 13.Lame MW, Morin D, Jones AD, Segall HJ, Wilson DW. Isolation and identification of a pyrrolic glutathione conjugate metabolite of the pyrrolizidine alkaloid monocrotaline. Toxicol Lett. 1990;51:321–329. doi: 10.1016/0378-4274(90)90075-w. [DOI] [PubMed] [Google Scholar]
- 14.Tsuruda LS, Lame MW, Jones AD. Formation of epoxide and quinone protein adducts in B6C3F1 mice treated with naphthalene, sulfate conjugate of 1,4-dihydroxynaphthalene and 1,4-naphthoquinone. Arch Toxicol. 1995;69:362–367. doi: 10.1007/s002040050185. [DOI] [PubMed] [Google Scholar]
- 15.Miller MG, Rodgers A, Cohen GM. Mechanisms of toxicity of naphthoquinones to isolated hepatocytes. Biochem Pharmacol. 1986;35:1177–1184. doi: 10.1016/0006-2952(86)90157-7. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Thomas B, Sachdeva R, Arterburn L, Frye L, Hatcher PG, Cornwell DG, Ma J. Mechanism of arylating quinone toxicity involving Michael adduct formation and induction of endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2006;103:3604–3609. doi: 10.1073/pnas.0510962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reid MJ, Dunston SK, Lame MW, Wilson DW, Morin D, Segall HJ. Effect of monocrotaline metabolites on glutathione levels in human and bovine pulmonary artery endothelial cells. Res Commun Mol Pathol Pharmacol. 1998;99:53–68. [PubMed] [Google Scholar]
- 18.Thomas HC, Lame MW, Dunston SK, Segall HJ, Wilson DW. Monocrotaline pyrrole induces apoptosis in pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1998;151:236–244. doi: 10.1006/taap.1998.8458. [DOI] [PubMed] [Google Scholar]
- 19.Krajewska B, Zaborska W. Double mode of inhibition-inducing interactions of 1,4-naphthoquinone with urease: arylation versus oxidation of enzyme thiols. Bioorg Med Chem. 2007;15:4144–4151. doi: 10.1016/j.bmc.2007.03.071. [DOI] [PubMed] [Google Scholar]
- 20.Zaborska W, Krajewska B, Kot M, Karcz W. Quinone-induced inhibition of urease: elucidation of its mechanisms by probing thiol groups of the enzyme. Bioorg Chem. 2007;35:233–242. doi: 10.1016/j.bioorg.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Giudice A, Montella M. Activation of the Nrf2-ARE signaling pathway: a promising strategy in cancer prevention. Bioessays. 2006;28:169–181. doi: 10.1002/bies.20359. [DOI] [PubMed] [Google Scholar]
- 22.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol. 2006;38:317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 23.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1-Nrf2 pathway. Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 24.Lame MW, Jones AD, Wilson DW, Segall HJ. Protein targets of 1,4-benzoquinone and 1,4-naphthoquinone in human bronchial epithelial cells. Proteomics. 2003;3:479–495. doi: 10.1002/pmic.200390062. [DOI] [PubMed] [Google Scholar]
- 25.Lame MW, Jones AD, Wilson DW, Segall HJ. Monocrotaline pyrrole targets proteins with and without cysteine residues in the cytosol and membranes of human pulmonary artery endothelial cells. Proteomics. 2005;5:4398–4413. doi: 10.1002/pmic.200402022. [DOI] [PubMed] [Google Scholar]
- 26.Thijssen VL, Postel R, Brandwijk RJ, Dings RP, Nesmelova I, Satijn S, Verhofstad N, Nakabeppu Y, Baum LG, Bakkers J, Mayo KH, Poirier F, Griffioen AW. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci U S A. 2006;103:15975–15980. doi: 10.1073/pnas.0603883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Case D, Irwin D, Ivester C, Harral J, Morris K, Imamura M, Roedersheimer M, Patterson A, Carr M, Hagen M, Saavedra M, Crossno J, Jr, Young KA, Dempsey EC, Poirier F, West J, Majka S. Mice deficient in galectin-1 exhibit attenuated physiological responses to chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;292:L154–164. doi: 10.1152/ajplung.00192.2006. [DOI] [PubMed] [Google Scholar]
- 28.Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–157R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- 29.Periasamy M. A Convenient Method for the Oxidation of Polycyclic Aromatic Hydrocarbons to Quinones. Synthesis 1977 [Google Scholar]
- 30.Mattocks AR, Jukes R, Brown J. Simple procedures for preparing putative toxic metabolites of pyrrolizidine alkaloids. Toxicon. 1989;27:561–567. doi: 10.1016/0041-0101(89)90117-7. [DOI] [PubMed] [Google Scholar]
- 31.Lame MW, Jones AD, Morin D, Wilson DW, Segall HJ. Association of dehydromonocrotaline with rat red blood cells. Chem Res Toxicol. 1997;10:694–701. doi: 10.1021/tx960173v. [DOI] [PubMed] [Google Scholar]
- 32.Pan LC, Wilson DW, Lame MW, Jones AD, Segall HJ. COR pulmonale is caused by monocrotaline and dehydromonocrotaline, but not by glutathione or cysteine conjugates of dihydropyrrolizine. Toxicol Appl Pharmacol. 1993;118:87–97. doi: 10.1006/taap.1993.1013. [DOI] [PubMed] [Google Scholar]
- 33.Taylor DW, Wilson DW, Lame MW, Dunston SD, Jones AD, Segall HJ. Comparative cytotoxicity of monocrotaline and its metabolites in cultured pulmonary artery endothelial cells. Toxicol Appl Pharmacol. 1997;143:196–204. doi: 10.1006/taap.1996.8083. [DOI] [PubMed] [Google Scholar]
- 34.Lehr HA, Mankoff DA, Corwin D, Santeusanio G, Gown AM. Application of photoshop-based image analysis to quantification of hormone receptor expression in breast cancer. J Histochem Cytochem. 1997;45:1559–1565. doi: 10.1177/002215549704501112. [DOI] [PubMed] [Google Scholar]
- 35.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 36.Hoffmann KJ, Streeter AJ, Axworthy DB, Baillie TA. Identification of the major covalent adduct formed in vitro and in vivo between acetaminophen and mouse liver proteins. Mol Pharmacol. 1985;27:566–573. [PubMed] [Google Scholar]
- 37.Koster AS. Bioreductive activation of quinones: a mixed blessing. Pharm Weekbl Sci. 1991;13:123–126. doi: 10.1007/BF01981528. [DOI] [PubMed] [Google Scholar]
- 38.Monks TJ, Lau SS. The pharmacology and toxicology of polyphenolic-glutathione conjugates. Annu Rev Pharmacol Toxicol. 1998;38:229–255. doi: 10.1146/annurev.pharmtox.38.1.229. [DOI] [PubMed] [Google Scholar]
- 39.Nioi P, Hayes JD. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat Res. 2004;555:149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 40.Thomas HC, Lame MW, Morin D, Wilson DW, Segall HJ. Prolonged cell-cycle arrest associated with altered cdc2 kinase in monocrotaline pyrrole-treated pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1998;19:129–142. doi: 10.1165/ajrcmb.19.1.2895. [DOI] [PubMed] [Google Scholar]
- 41.Ramos M, Lame MW, Segall HJ, Wilson DW. Monocrotaline pyrrole induces Smad nuclear accumulation and altered signaling expression in human pulmonary arterial endothelial cells. Vascul Pharmacol. 2007;46:439–448. doi: 10.1016/j.vph.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan LC, Lame MW, Morin D, Wilson DW, Segall HJ. Red blood cells augment transport of reactive metabolites of monocrotaline from liver to lung in isolated and tandem liver and lung preparations. Toxicol Appl Pharmacol. 1991;110:336–346. doi: 10.1016/s0041-008x(05)80016-x. [DOI] [PubMed] [Google Scholar]
- 43.Mattocks AR. Role of the acid moieties in the toxic actions of pyrrolizidine alkaloids on liver and lung. Nature. 1970;228:174–175. doi: 10.1038/228174a0. [DOI] [PubMed] [Google Scholar]
- 44.Tsemakhovich VA, Bamm VV, Shaklai M, Shaklai N. Vascular damage by unstable hemoglobins: the role of heme-depleted globin. Arch Biochem Biophys. 2005;436:307–315. doi: 10.1016/j.abb.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Patterson RJ, Wang W, Wang JL. Understanding the biochemical activities of galectin-1 and galectin-3 in the nucleus. Glycoconj J. 2004;19:499–506. doi: 10.1023/B:GLYC.0000014079.87862.c7. [DOI] [PubMed] [Google Scholar]
- 46.Cole SP, Deeley RG. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci. 2006;27:438–446. doi: 10.1016/j.tips.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 47.Mukhopadhyay S, Sehgal PB. Discordant regulatory changes in monocrotaline-induced megalocytosis of lung arterial endothelial and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1216–1226. doi: 10.1152/ajplung.00535.2005. [DOI] [PubMed] [Google Scholar]
- 48.Mukhopadhyay S, Shah M, Patel K, Sehgal PB. Monocrotaline pyrrole-induced megalocytosis of lung and breast epithelial cells: Disruption of plasma membrane and Golgi dynamics and an enhanced unfolded protein response. Toxicol Appl Pharmacol. 2006;211:209–220. doi: 10.1016/j.taap.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho HK, White CC, Fernandez C, Fausto N, Kavanagh TJ, Nelson SD, Bruschi SA. Nrf2 activation involves an oxidative-stress independent pathway in tetrafluoroethylcysteine-induced cytotoxicity. Toxicol Sci. 2005;86:354–364. doi: 10.1093/toxsci/kfi205. [DOI] [PubMed] [Google Scholar]
- 51.Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calderwood SK, Murshid A, Prince T. The shock of aging: molecular chaperones and the heat shock response in longevity and aging--a mini-review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stocker R, McDonagh AF, Glazer AN, Ames BN. Antioxidant activities of bile pigments: biliverdin and bilirubin. Methods Enzymol. 1990;186:301–309. doi: 10.1016/0076-6879(90)86123-d. [DOI] [PubMed] [Google Scholar]
- 54.Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, Ikehara S, Wolin MS, Abraham NG. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol. 2005;289:H701–707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 55.Kim HS, Skurk C, Maatz H, Shiojima I, Ivashchenko Y, Yoon SW, Park YB, Walsh K. Akt/FOXO3a signaling modulates the endothelial stress response through regulation of heat shock protein 70 expression. FASEB J. 2005;19:1042–1044. doi: 10.1096/fj.04-2841fje. [DOI] [PubMed] [Google Scholar]