

ABSTRACT
The free hormone hypothesis postulates that only the nonbound fraction (the free fraction) of hormones that otherwise circulate in blood bound to their carrier proteins is able to enter cells and exert biologic effects. In this review, I will examine four hormone groups—vitamin D metabolites (especially 25OHD), thyroid hormones (especially thyroxine [T4]), sex steroids (especially testosterone), and glucocorticoids (especially cortisol)—that are bound to various degrees to their respective binding proteins—vitamin D‐binding protein (DBP), thyroid‐binding globulin (TBG), sex hormone‐binding globulin (SHBG), and cortisol‐binding globulin (CBG)—for which a strong case can be made that measurement of the free hormone level provides a better assessment of hormonal status than the measurement of total hormonal levels under conditions in which the binding proteins are affected in levels or affinities for the hormones to which they bind. I will discuss the rationale for this argument based on the free hormone hypothesis, discuss potential exceptions to the free hormone hypothesis, and review functions of the binding proteins that may be independent of their transport role. I will then review the complications involved with measuring the free hormone levels and the efforts to calculate those levels based on estimates of binding constants and levels of both total hormone and total binding protein. In this review, the major focus will be on DBP and free 25OHD, but the parallels and differences with the other binding proteins and hormones will be highlighted. Vitamin D and its metabolites, thyroid hormones, sex steroids, and glucocorticoids are transported in blood bound to serum proteins. The tightness of binding varies depending on the hormone and the binding protein such that the percent free varies from 0.03% for T4 and 25OHD to 4% for cortisol with testosterone at 2%. Although the major function of the primary carrier proteins (DBP, TBG, SHBG, and CBG) may be to transport their respective lipophilic hormones within the aqueous media that is plasma, these proteins may have other functions independent of their transport function. For most tissues, these hormones enter the cell as the free hormone presumably by diffusion (the free hormone hypothesis), although a few tissues such as the kidney and reproductive tissues express megalin/cubilin enabling by endocytosis protein‐bound hormone to enter the cell. Measuring the free levels of these protein‐bound hormones is likely to provide a better measure of the true hormone status than measuring the total levels in situations where the levels and/or affinities of the binding proteins are altered. Methods to measure free hormone levels are problematic as the free levels can be quite low, the methods require separation of bound and free that could disturb the steady state, and the means of separating bound and free are prone to error. Calculation of free levels using existing data for association constants between the hormone and its binding protein are likewise prone to error because of assumptions of linear binding models and invariant association constants, both of which are invalid. © 2020 The Author. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: BINDING PROTEINS, CORTISOL, FREE HORMONE HYPOTHESIS, TESTOSTERONE, THYROID HORMONE, VITAMIN D
Introduction
Lipophilic hormones such as steroid hormones including vitamin D and thyroid hormone are transported in the aqueous media such as plasma by specific and nonspecific proteins. The specific proteins include vitamin D‐binding protein (DBP), which binds vitamin D and its metabolites; thyroid‐binding globulin (TBG), which binds thyroxine (T4) and triiodothyronine (T3); sex hormone‐binding globulin (SHBG), which binds testosterone and estradiol and other sex steroids; and cortisol‐binding globulin (CBG), which binds cortisol, aldosterone, and progesterone. Nonspecific‐binding proteins include albumin and lipoproteins. In the case of T4, transthyretin also contributes significantly to the binding. The fraction of hormone that is not bound to its respective carrier proteins is the free fraction. The free fraction is determined primarily by both the level and affinity of the specific binding protein, although the large amount of albumin in blood contributes to this determination. Table 1 lists the association constants that have been reported for the hormones and binding proteins discussed in this review, although these values are influenced both by clinical circumstances and mutations in the binding proteins. Of particular interest is that the free fraction varies from approximately 0.03% for T4 and 25OHD, 2% for testosterone, and 4% to 10% for cortisol. Interest in knowing the free hormone levels is based on the concept that in the case of these hormones only the free fraction is able to cross the plasma membrane into the target cells. This is the free hormone hypothesis. A corollary of this hypothesis is that measurement of the free hormone level is necessary to determine the true hormonal state. A powerful argument in support of this concept is provided by families in which the gene for the binding protein of interest is mutated rendering it null, yet the affected family members show little clinical evidence of hormone deficiency. Nevertheless, this concept has proven controversial, but at least for T4, measurement of free T4 along with thyroid‐stimulating hormone (TSH) has replaced measurement of total T4 in assessment of thyroid status. Similar arguments supporting the routine use of free hormone measurements will be presented in this review.
Table 1.
Binding Proteins for the Vitamin D Metabolites, Thyroid Hormones, Sex Steroids, and Glucocorticoids/Mineralcorticoids
| Hormone | Specific BP | Ka | % Bound | Albumin | Ka HAS | % bound | TTR | Ka TTR | % Bound | % Free | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 25OHD | DBP | 7–9 × 108 | 88 | HSA | 6 × 105 | 12 | 0.02–0.04 | Bikle et al, 1986( 42 ) | |||
| 1,25(OH)2D | DBP | 4 × 107 | 85 | HSA | 5.4 × 104 | 15 | 0.3–0.4 | Bikle et al, 1985( 44 ) | |||
| T4 | TBG | 1 × 1010 | 70 | HSA | 1.5 × 106 | 5 | TTR | 2 × 108 | 20 | 0.03 | Mimoto & Refetoff, 2020( 60 ) |
| T3 | TBG | 1 × 109 | 75 | HSA | 2 × 105 | 20 | TTR | 1 × 106 | 5 | 0.3 | Mimoto & Refetoff, 2020( 60 ) |
| Testosterone | SHBG | 2 × 109 | 44 | HSA | 4 × 104 | 50 | 2 | Heyns, 1971 | |||
| Estradiol | SHBG | 7 × 108 | 20 | HSA | 6 × 104 | 78 | 2 | Heyns, 1971( 73 ) | |||
| Cortisol | CBG | 8 × 107 | 90 | HSA | 3 × 103 | 7 | 4 | Stroupe et al, 1978( 92 ) | |||
| Aldosterone | CBG | 2 × 106 | 37 | HSA | 2 × 103 | 21 | 37 | Dunn et al, 1981( 93 ) |
This table shows the binding constants of these binding proteins for their respective hormones under normal conditions, the % of the total hormone level bound to the protein, and the % free. Numerous clinical conditions and genetic modifications alter these numbers, which are shown here for comparative purposes. BP = binding protein; CBG = cortisol‐binding globulin; DBP = vitamin D‐binding protein; HAS = human serum albumin; Ka = association constant; SHBG = sex hormone‐binding globulin; T3 = triiodothyronine; T4 = thyroxine; TBG = thyroid‐binding globulin; TTR = transthyretin.
The Free Hormone Hypothesis
The hypothesis and its modification
The free hormone hypothesis postulates that only the nonbound fraction (the free fraction) of total hormones that otherwise circulate in blood bound to their carrier proteins is able to enter cells and exert its biologic effects (Figure 1). Examples include the vitamin D metabolites, thyroid hormone, sex steroids, and cortisol. These are lipophilic hormones assumed to cross the plasma membrane by diffusion and not by an active transport mechanism. Perhaps the earliest articulation of the free hormone hypothesis was published by Recant and Riggs( 1 ); they examined thyroid function in patients with protein‐losing nephropathy. They noted that circulating thyroid hormone (measured as protein‐bound iodine) was quite low in these patients along with increased urinary losses, but with relatively little evidence for clinical hypothyroidism. They concluded that “thyroid function and the supply of hormone to the tissues in nephrosis may be normal, and that the low concentration of protein‐bound iodine in the plasma is due to the change in concentration or binding capacity of the plasma proteins in nephrosis.” Indeed, one of the strongest arguments favoring the free hormone hypothesis is that subjects with mutations in the genes for their respective binding proteins that alter the affinity or levels of the binding protein for the hormone such that total hormone levels are quite reduced have limited clinical impact, despite the low total hormone levels that might otherwise be expected in hormone deficiency. Examples will be detailed subsequently when individual binding proteins are discussed.
Fig 1.
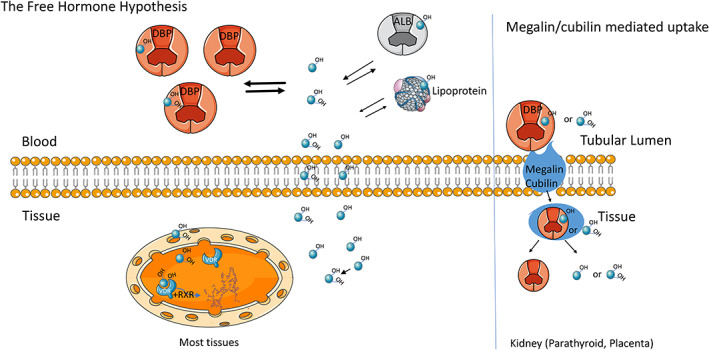
The free hormone hypothesis as illustrated for vitamin D metabolites. Vitamin D metabolites (blue circles) in the circulation are tightly bound to vitamin D‐binding protein (DBP) and to a lesser extent to albumin (ALB). For most tissues, only the unbound metabolites can cross the cell membrane. In several tissues such as the kidney and potentially the parathyroid and placenta 25OHD and 1,25(OH)2D bound to DBP may enter cells by endocytosis via megalin/cubilin and are not limited to diffusion by the free hormone. Figure reproduced from Bikle DD, Malmstroem S, Schwartz J. Current controversies: are free vitamin metabolite levels a more accurate assessment of vitamin D status than total levels? Endo Clin NA. 2017;46:901–18.
However, the free hormone concentration is not the only factor involved with the rate at which the hormone enters the cells. As articulated by Mendel,( 2 ) the movement of hormone into the cell in vivo is dependent not only on the free concentration, but also on the dissociation of hormone from its binding proteins, the rate of blood flow, the rate of uptake into the cell, and the catabolism/sequestration of hormone within the cell. These are components of the transport process that Mendel calls the free hormone transport process. Thus, the total concentration of hormone, by affecting the total amount of free hormone available to the cell, does influence the extent of transport of hormone into the cell when the rate of transport is not rate‐limiting. These considerations also reflect upon what is called the bioavailable hormone, generally that percent of hormone bound to albumin as well as free. Although the affinity constants of albumin for the hormones we are discussing are lower than that for the specific binding proteins, it is not clear that the dissociation of the hormone from albumin is not rate‐limiting for any and all target tissues, so suggesting that the so‐called bioavailable fraction is truly bioavailable is not established for any of the hormones we will discuss. Indeed at least in vitro albumin does restrict entry of these hormones into cells where examined.( 3 ) Our knowledge of most of the variables involved with transport into cells in vivo is limited, so our focus in this review will be on the free hormone itself, its measurement, and what influences this fraction of the total hormone in circulation.
The megalin/cubilin transport system
As noted above, the free hormone hypothesis postulates that only the free hormone can cross the plasma membrane. An important exception exists in some tissues for some of the protein‐bound hormones. The renal tubule differs from most other tissues in its mechanism for at least 25OHD uptake, and likely for all DBP‐bound vitamin D metabolites. The DBP–25OHD complex is filtered in the glomerulus and reabsorbed in the proximal tubule through endocytosis by the megalin/cubilin complex (Fig. 1), thereby providing 25OHD for CYP27B1 1α‐hydroxylation in the kidney tubule, as well as for the rest of the body.( 4 , 5 ) This complex is not specific for DBP, but when megalin is deleted, the major protein lost in the urine is DBP. In the mice that survive long enough, bone growth is retarded and osteopenic.( 4 ) The impact of cubilin deletion is similar, but not as severe.( 5 ) A similar mechanism may operate in the parathyroid gland and placenta, which like the renal tubule express megalin/cubilin,( 6 ) but at this point experiments to determine the impact of either megalin or cubilin deletion from the parathyroid or gland or placenta have not been reported. Similarly, megalin has been found in reproductive tissues where it enables endocytosis of sex steroids bound to SHBG. Male mice lacking megalin have undescended testes, whereas female mice have failure of vaginal opening.( 7 ) On the other hand, although there may be some uptake of DBP by immune cells, this does not appear to involve megalin/cubilin.( 8 ) The role of megalin/cubilin in the uptake of other protein bound hormones has not been described to my knowledge.
Vitamin D‐Binding Protein and Free Vitamin D Metabolites
As shown in Table 1, DBP has one of the highest association constants for 25OHD, comparable to that of TBG for T4, resulting in one of the lowest free fractions. In serum samples from normal individuals, approximately 88% of circulating 25OHD and 85% of 1,25(OH)2D are bound to DBP, whereas albumin with its substantially lower binding affinity binds only approximately 12% to 15% of these metabolites despite its 10‐fold higher concentration than DBP. Approximately 0.4% of total 1,25(OH)2D and 0.03% of total 25OHD is free in serum from normal nonpregnant individuals. The degree of saturation of DBP by the vitamin D metabolites is very low (DBP circulates at micromolar levels, whereas 25OHD concentrations are nanomolar) such that changes in vitamin D levels have little impact on the free fraction. This is not true for CBG or SHBG as will be discussed. However, vitamin D intoxication has been shown to have a modest increase in the free fraction.( 9 ) Nevertheless, changes in the level of DBP clearly do affect the free fraction as will be discussed.
Genomic regulation
The human DBP gene is located on chromosome 4q12‐q13 in close association with other members of the albuminoid family: albumin, α‐fetal protein, and afamin. It is 35 kb in length and comprised of 13 exons encoding 474 amino acids, including a 16 amino acid leader sequence, which is cleaved before release. Numerous tissues express DBP, but the liver is the major source.( 10 ) The expression of DBP is increased by estrogen, but not by androgens, (11 ) as appreciated with the rise in DBP during pregnancy( 12 , 13 ) and with oral contraceptive administration.( 14 ) Dexamethasone and certain cytokines such as IL‐6 also increase DBP production, whereas TGFβ is inhibitory.( 15 ) These cytokines and glucocorticoids are likely to play a role in the increase in DBP production following trauma( 16 ) and acute liver failure.( 17 ) Primary hyperparathyroidism, on the other hand, is associated with a reduction in DBP levels, likely contributing to the lower 25OHD levels in these patients as the free 25OHD is not reduced.( 18 ) Vitamin D itself or any of its metabolites does not regulate DBP production.( 19 )
Structure and polymorphisms
The mature human DBP is approximately 58 kD in size, although differences in glycosylation of the protein for different alleles alter the actual size. DBP is the most polymorphic gene known. Over 120 variants have been described based on electrophoretic properties( 20 ) with 1242 polymorphisms currently listed in the National Center for Biotechnology Information database.( 21 ) Of these variants, the Gc1f and Gc1s (rs7041 locus) and Gc2 (rs4588 locus) are the most common. The Gc1f and Gc1s involve two polymorphisms, one at aa 432 (416 in the mature DBP) and 436 (420 in the mature DBP). The 1f allele encodes the sequence of aa between 432–436 as DATPT, the 1s allele encodes the sequence EATPT. This subtle difference in charge makes Gc1f run faster (fast) than the Gc1s (slow) during electrophoresis. The Gc2 allele encodes DATPK, which runs slower still. Glycosylation further distinguishes the Gc1 variants from the Gc2 variant. The threonine (T) in Gc1 binds N‐acetylgalactosamine to which galactose and sialic acid bind in tandem. The lysine (K) in a comparable position in Gc2 is not glycosylated.( 22 , 23 ) This affects the conversion of DBP to DBP–MAF (macrophage activating factor), which involves a partial deglycosylation removing the galactose and sialic acid by the sequential action of sialidase and β‐galactosidase by T and B cells.( 24 ) The SNPs are in complete linkage disequilibrium, and only six haplotypes are observed with any significant frequency. Gc2 is the least abundant and Gc1f the most abundant. Gc alleles show distinct racial distribution patterns. Black and Asian populations are more likely to carry the Gc1f form and the Gc2 form is rare, whereas White populations more frequently exhibit the Gc1s and the Gc2 form. Gc1f has been stated to have the highest affinity and Gc2 the lowest affinity for vitamin D and its metabolites,( 25 ) but these differences among alleles have not been found by others, (26 ) and account for little of the variation seen among various populations that we recently reported.( 27 )
DBP is comprised of three structurally similar domains. The first domain is the binding site for the vitamin D metabolites (aa 35‐49). Fatty acid binding utilizes a single high‐affinity site for both palmitic acid and arachidonic acid, but only arachidonic acid competes with 25OHD for binding.( 28 , 29 ) The actin‐binding site is located at aa 373‐403, spanning parts of domains 2 and 3, but part of domain 1 is also involved.( 30 , 31 ) The C5a/C5a des Arg binding site is located at aa 130‐149.( 32 ) Membrane binding sites have been identified in aa 150‐172 and 379‐402.( 33 ) Less clear is the role of these sites in facilitating transport of the vitamin D metabolites into the cell, and if so whether polymorphisms in these or other sites affect that transport. In the absence of disease or pregnancy, DBP levels are relatively constant over time in adults.( 34 ) That said, various substances in the blood such as polyunsaturated fatty acids may alter the affinity of DBP for the vitamin D metabolites.( 29 )
Biologic function
The major role of DBP with respect to vitamin D is its transport function. DBP was discovered by Hirschfeld in 1959,( 35 ) and originally called group‐specific component (Gc‐globulin), but it was not until 1975 that its function as a vitamin D transport protein was appreciated.( 36 ) As noted above in normal individuals, approximately 85% of circulating vitamin D metabolites are bound to DBP, albumin binds approximately 15% of these metabolites such that approximately 0.4% of total 1,25(OH)2D3 and 0.03% of total 25(OH)D3 is free in serum from normal nonpregnant individuals. The affinity of DBP for the vitamin D2 metabolites is somewhat less than that for the vitamin D3 metabolites.( 37 ) The importance of this transport mechanism with respect to access of the vitamin D metabolite to the cell is well‐illustrated in the DBP KO mouse. In these mice, the vitamin D metabolites are presumably all free and/or bioavailable as albumin levels are normal. Tissue levels of 1,25(OH)2D were normal in the DBP KO mice, and markers of vitamin D function such as expression of intestinal TRPV6, calbindin 9k, PMCA1b, and renal TRPV5 were maintained despite having very low levels of serum 25OHD and 1,25(OH)2D and increased loss of these metabolites in the urine.( 38 ) Moreover, injection of 1,25(OH)2D into these DBP KOs showed a more rapid increase in the expression of Cyp24A1, TRPV5, and TRPV6 than in DBP intact controls.( 39 ) However, on a vitamin D‐deficient diet, they quickly developed vitamin D deficiency. Recently, a family has been reported in which the proband had a homozygous mutation in DBP with undetectable serum levels of DBP and 25OHD and very low levels of 1,25(OH)2D.( 40 ) PTH and serum calcium were normal. The proband had osteopenia and ankylosing spondylitis. Her seven siblings did not have evidence of bone disease or altered calcium metabolism, including her heterozygote brother with low levels of DBP and total levels of 25OHD, but normal levels of free 25OHD. Thus, DBP appears not to be required for access of the vitamin D metabolites to cells, consistent with the free hormone hypothesis, but serves as a circulating reservoir for the vitamin D metabolites reducing the risk of vitamin D deficiency when intake or epidermal production is limited. DBP has other important functions including actin scavenging, neutrophil recruitment, and macrophage recruitment reviewed previously, (41 ) but a discussion of which is outside the scope of this review.
Directly measured free vitamin D metabolites
The very low levels of free 25OHD and 1,25(OH)2D make their measurement a challenge. Equilibrium dialysis followed by measurement of the free hormone in the dialysate via liquid chromatography/mass spectrometry is generally considered the gold standard, but technical difficulties, labor, and cost make this an unsuitable technique to use for the vitamin D metabolites. We adopted centrifugal ultrafiltration dialysis to obtain the data enabling us to determine binding constants for DBP and albumin and the free fraction for both 25OHD and 1,25(OH)2D shown in Table 1.( 42 , 43 , 44 , 45 ) Moreover, these studies enabled us to demonstrate the changes in the free fraction associated with changes in DBP levels during liver disease and pregnancy, data indicating the important contribution that measurement of the free hormone level could make in assessing hormone status in conditions in which DBP levels varied. However, this method is not suited to high throughput, and free vitamin D measurements were not readily available until an immunometric method came onto the market. At this point, no alternative method to measure free 1,25(OH)2D has been developed.
ELISA method
A two‐step ELISA that directly measures free 25OHD levels has been developed (Future Diagnostics Solutions B.V., Wijchen, The Netherlands). In the first incubation step, an antivitamin D monoclonal antibody immobilized on a microtiter plate binds the free 25OHD in the serum sample. After washing away excess serum, the second incubation step is to add biotinylated 25OHD in a known amount to react with the unoccupied binding sites on the monoclonal antibody attached to the plate. The nonbound biotinylated 25OHD is then removed by a second washing. Thereafter, streptavidin peroxidase conjugate is added followed by the substrate 3,3′,5,5′‐tetramethylbenzidine. The bound streptavidin peroxidase can be quantified by measuring the absorbance at 450 nm generated in the reaction spectrophotometrically. The intensity is inversely proportional to the level of free 25OHD. The limit of detection is 2.8 pg/mL. This assay is dependent on the quality of the antibody used to bind the free 25OHD. The antibody in the current assay does not recognize 25(OH)D2 as well as 25(OH)D3 [77% of the 25(OH)D3 value], so it underestimates the free 25(OH)D2. However, under most situations where the predominant vitamin D metabolite is 25(OH)D3, the data compare quite well to those obtained from similar populations using the centrifugal ultrafiltration assay.( 26 , 46 )
Liquid chromatography–tandem mass spectrometry method
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) has been used to detect 25OHD in saliva, which is expected to be free of DBP and albumin and so represents free 25OHD.( 47 ) In this method, 1 mL of saliva is deproteinized with acetonitrile, purified using a Strata‐X cartridge, derivatized with 4‐phenyl‐1,2,4‐triazoline‐3,5 dione, ionized by electron spray ionization, and subjected to LC–MS/MS. This method has not received widespread use.
Calculation of free vitamin D metabolites
Because of the difficulty in measuring the free hormone, some investigators have used the association constants of DBP and albumin for the vitamin D metabolites along with measurements of total metabolite, DBP, and albumin levels to calculate the free fraction according to the following formula:
Ka is the association constant. This calculation makes a number of assumptions, including constancy of the affinity constants from individual to individual in all clinical conditions; linearity of binding to the binding proteins at all hormone concentrations; and accurate measurement of DBP, albumin, and the vitamin D metabolite of interest. All of these assumptions have proved problematic, although initial studies in normal subjects, in which DBP levels were measured with a polyclonal assay, the calculated values agreed reasonably well with the directly measured values using the centrifugal ultrafiltration dialysis method.( 42 , 45 ) However, with different populations, different assays of DBP and 25OHD, and different clinical situations the calculated levels have not correlated well with the directly measured levels, and use of this formula to calculate the free hormone level cannot be recommended.( 48 )
Free 25OHD measurements in different populations and clinical conditions
PTH and vitamin D
Directly measured free 25OHD concentrations are strongly correlated with total 25OHD concentrations and have been reported to be between 0.02% and 0.09% of total 25OHD concentrations.( 27 ) Concentrations generally range from 1.2 to 7.9 pg/mL. PTH is negatively correlated with free 25OHD, as well as total 25OHD. Serum C‐terminal telopeptide of type I collagen has been reported to have a moderate positive correlation with total and free 25OHD.( 49 ) With vitamin D supplementation, free 25OHD concentrations rise in concert with total 25OHD concentrations, 49 , 50 , 51 , 52 ) rising more steeply with vitamin D3 supplementation compared with vitamin D2. ( 53 ) With high0dose vitamin D supplementation, the changes in iPTH were significantly related to changes in free 25OHD, but not to changes in total 25OHD or changes in total 1,25(OH)2D.( 53 )
Race
Although DBP levels in a Black population were thought to be lower using a monoclonal antibody, resulting in a normal calculated free 25OHD despite the low total 25OHD, these results were subsequently shown to result from the use of a monoclonal antibody that failed to efficiently measure the dominant G1f allele in the Black population.( 54 ) Subsequent studies with a polyclonal assay found similar DBP levels between Black and White populations and consistently lower free 25OHD levels in the Black population, consistent with lower total 25OHD.( 55 , 56 ) In general, differences in DBP genotype has had little impact on free 25OHD levels, although the G2 genotype is consistently associated with slightly lower total and free 25OHD.( 27 )
Sex and pregnancy
Investigations have not found differences in free levels of 25OHD between normal men and women relative to total 25OHD levels.(27 ,51 ) Estrogen administration raises both the total and free 1,25(OH)2D levels, along with DBP levels, without significant changes in the free fraction.( 57 , 58 ) The data regarding the impact of estrogen administration on free 25OHD are less clear as they are cross sectional derived from subjects on oral contraceptives, which generally contain a progestin that, at least in one study, blocks the rise in free 1,25(OH)2D, perhaps also blocking the increase in free 25OHD.( 58 ) However, directly measured free 25OHD tends to be higher, and free 1,25(OH)2D is substantially higher in pregnant women versus comparator groups of women.( 45 , 48 ) Because these results, at least for free 1,25(OH)2D, are nonlinear with respect to the parallel increase in DBP, they suggest that the affinity of DBP for 1,25(OH)2D is decreased during pregnancy, and could account for the increased intestinal calcium transport observed during the last trimester, when demands for calcium by the developing fetus are greatest.
Liver disease
Directly measured free 25OHD and 1,25(OH)2D are higher in outpatients with cirrhosis compared with other groups, 43 , 48 ) despite lower total vitamin D metabolite concentrations. The relationship between free 25OHD and total 25OHD is both steeper and more variable in patients with liver disease than in healthy people.( 27 ) Those with the most‐severe cirrhosis and protein synthesis dysfunction have a higher percentage of free 25OHD compared with cirrhotics without protein synthesis dysfunction, but free 25OHD concentrations are similar because of the presence of both lower total 25OHD concentrations as well as lower DBP.( 59 ) Free 25OHD concentrations range between 4.5 to 8.1 pg/mL in cirrhotics with low albumin and from 6.4 to 10.6 pg/mL in those with normal albumin.
Frail elderly
Nursing home residents are older, have more medical problems, receive more medications, and are more likely to have poorer nutrition than younger people or community‐dwelling elderly. In vitamin D dose‐titration studies,( 52 ) free 25OHD levels rose in response to increases in total 25OHD, but responses appeared to be steeper than those of normal subjects despite comparable DBP levels,( 27 ) suggesting that in this group of subjects the affinity of DBP for 25OHD is reduced perhaps because of interfering medications or low‐grade inflammatory states, although this remains speculative.
Thyroid Hormone‐Binding Proteins and Free Thyroid Hormones
The status of thyroid sufficiency is generally assessed by a combination of TSH and the free levels of T4 because alterations in the thyroid hormone‐binding proteins (THBPs) are relatively common, and the free hormone levels are more accurate indicators of thyroid status than the total hormone (TH) levels. For the thyroid hormones there are three major THBPs: thyroid‐binding globulin (TBG), which has the highest binding affinity for the TH; transthyretin (TTR; formerly known as prealbumin and renamed in recognition of its role in retinol binding: TRANSport of THYroid hormone and RETINol), and albumin.( 60 ) The affinity constants, approximate amounts of T4 and T3 bound, and percent of free levels are given in Table 1. As noted previously, TBG and DBP have comparable high affinity for their respective hormones, such that the free hormone concentrations are quite low. In the case of TBG, approximately 75% of T4 and T3 are bound to the protein. Although free T4 and free T3 are recognized as the form entering the cells (free hormone hypothesis), the minor component bound to lipoproteins may have facilitated entry to cells expressing the LDL receptor.( 61 ) Unlike the vitamin D metabolites, there are known transporters for the TH in the cell membranes: monocarboxylate transporters MCT8 and 10, L‐amino acid transporters LAT1 and 2, and the organic anion transporter peptide OATP1C1.( 62 ) Although mutations in the THBPs, including null mutations in TBG, do not affect the euthyroid status of the subject despite marked changes in total TH levels consistent with the free hormone hypothesis, such mutations, at least for TTR, can cause other morbidities.
Thyroid‐binding globulin
TBG is a 54 kDa 395 amino acid monomer with a single binding site for TH. It has four glycosylation sites. Estrogen increases TBG levels primarily by increasing the sialic acid content of these carbohydrate chains increasing its half‐life in the blood. Androgens have the opposite effect.( 63 ) TBG levels are also reduced by glucocorticoids,( 64 ) hepatic failure, severe nonthyroidal illness, protein‐losing nephropathies, or gastrointestinal disorders.( 65 ) Its gene is located on the X chromosome, such that mutations typically have greater effects on males than females. The promoter has binding sites for hepatic nuclear factors 1 and 3, and the liver is the major site of production. There are 28 TBG mutations that result in complete loss of function generally from gene deletions, 18 partial deficiencies resulting primarily from missense mutations, and several gene duplications that increase TBG levels (reviewed in Mimoto and Refetoff( 60 )). Although these mutations have profound effects on total TH levels, the free TH levels are normal and the subjects are euthyroid. That said, these THBGs do appear to alter tissue distribution of TH as shown in liver perfusion studies by Mendel.( 66 )
Transthyretin
Unlike TBG, TTR is expressed in multiple tissues. Its gene is located on chromosome 18, encoding a 55 kDa 127 amino acid protein that circulates as a homo tetramer. This complex has two binding sites for TH, but the second site in under negative cooperativity such that the actual affinity for TH depends to some degree on TH concentrations. As mentioned, TTR is expressed in multiple tissues including the choroid plexus. As such, TTR is the dominant THBG in cerebrospinal fluid.( 67 ) Like TBG, a number of mutations of TTR have been reported, none of which alter free TH levels or thyroid status. However, many of these mutations lead to amyloidosis (reviewed in Mimoto and Refetoff (60 )).
Albumin
Human serum albumin (HSA) is a 66.5 kDa 585 amino acid protein made in the liver. Although HSA binds all the hormones being discussed in this review, as noted in Table 1, it is described in this section because of a common set of mutations that increase its affinity for T4, T3, or both. This results in the syndrome of familial dysalbuminemic hyperthyroxinemia (FDH).( 68 ) This leads to elevated levels of total T4 and/or T3, but normal levels in free TH. It is quite common in Hispanics (up to 1:50 of those with Puerto Rican ancestry).( 69 ) Other albumin mutations, including analbuminemia (total lack of albumin), likewise have little impact on thyroid status.( 70 )
Measurement of free total hormone
As for the measurement of the free vitamin D metabolites, the very low levels of the free TH makes this challenging. Equilibrium dialysis followed by LC–MS/MS is the gold standard, but is not practical for high throughput. Accordingly, measurements of free T4 and free T3 rely on competitive immunoassays in which the free hormone is first extracted from blood with specific antibodies, incubated with a labeled TH probe, and the unoccupied antibody binding sites determined, which are inversely proportional to the free hormone, (71 ) similar to the method described for free 25OHD. Two‐step methods are preferable because they do not require the use of T4 analogs and are less prone to nonspecific binding.( 72 ) These methods are susceptible to a number of problems with heterophile antibodies, biotin consumption by the subject, and antibody specificity. Patients with mutations in albumin that increase the binding to TH, such as FDH, can show spuriously elevated free TH levels with the automated immunoassays.( 60 )
Sex Hormone‐Binding Globulin and Free Testosterone
Sex hormone‐binding globulin (SHBG) is the most specific, highest affinity protein in blood for testosterone, dihydrotestosterone, and estrogen. It binds approximately 44% of total testosterone and 20% estradiol, whereas the lower affinity, but much more abundant, albumin binds 50% of testosterone and 78% estradiol, leaving approximately 2% free Heyns and De Moor( 72 ). However, numerous conditions affect this binding including estrogens, thyroid hormone, adiponectin, and PPARγ ligands that increase SHBG expression, and monosaccharides and proinflammatory cytokines that reduce its expression (reviewed in Hammond( 74 )). Thus, obesity (via decreased adiponectin), diabetes mellitus (via increased glucose), and inflammation can all alter SHBG levels. Similarly, liver disease and nephrotic syndrome can reduce SHBG levels via decreased production and urine losses, respectively, without obvious abnormalities in sexual phenotype. Moreover, a family has been described in which the male proband totally lacked measurable SHBG, but had normal sexual development and free testosterone levels.( 75 ) Thus, measurement of total testosterone (and estradiol) can be misleading in these circumstances. However, though free testosterone is likely to be a better assessment of androgen status,( 66 , 74 , 76 ) free testosterone measurements are at this point not totally reliable. As for the other hormones in this review, equilibrium dialysis followed by liquid chromatography/mass spectrometry is the gold standard, but it is not suitable for high throughput.( 77 ) Direct analog tracer‐based immunoassays are not considered reliable and their use not recommended.( 78 ) Calculation of the free levels based on affinity constants for SHBG and albumin depend on the invariance of these binding constants,( 79 ) an assumption that is invalid given that the two sites for testosterone on the SHBG homodimer differ and show allosteric interaction.( 80 ) Thus, the ratio of testosterone to SHBG can alter the apparent affinity of these sites.( 81 ) Moreover, such interactions in binding sites may also apply to the binding of testosterone to albumin.( 82 ) Furthermore, there are numerous polymorphisms in SHBG that influence its affinity for sex steroids (reviewed in Hammond( 74 )). Finally, a direct comparison between calculated values and those determined by equilibrium dialysis demonstrated major discrepancies.( 83 ) Therefore, the routine use of directly measured free testosterone remains desirable yet problematic and cannot be replaced by calculated values.
SHBG circulates as a 90 kDa dimer. Calcium is required for the dimerization. It is 80% saturated with testosterone in men, 20% saturated in women. The SHBG gene is located on chromosome 17. Hepatic nuclear factor 4α (HNF4α) is its main transcription factor regulator.( 84 ) Regulation of HNF4a levels appears to be the means by which proinflammatory cytokines,( 85 , 86 ) adiponectin,( 87 ) and thyroid hormone( 88 ) alter SHBG expression.
Although transport of the sex steroids is the major function of SHBG, SHBG can leave the vascular system with accumulation in extravascular tissues such as the endometrium and epididymis, but the biologic significance of this is not clear.( 89 ) As noted previously, megalin may transport SHBG into some cells,( 7 ) and megalin KO mice have abnormalities in the reproductive tract. Changes in SHBG levels have been associated with a variety of clinical conditions including osteoporosis( 90 ) and risk of diabetes mellitus,( 91 ) suggesting that SHBG may have functions beyond sex steroid transport, but these functions are not defined.
Cortisol‐Binding Globulin and Free Cortisol
CBG is the major transport protein for cortisol, binding approximately 90% of the total cortisol in blood. Albumin carries about 6% to 7% with the free levels about 4% of the total, although this ranges to 10% in some reports. CBG also transports aldosterone, but with lower affinity such that CBG accounts for 21% of bound aldosterone with albumin transporting approximately 42%, leaving 37% free.( 92 ) CBG also transports a small (17%) fraction of progesterone, which is mostly carried by albumin.( 93 ) As noted in Table 1, the affinities of CBG and albumin for these hormones are substantially lower than that of DBP and TBG for their respective hormones. As for the other hormone‐binding globulins, the role of CBG beyond hormone transport is not clear. Families null for CBG do not show major clinical syndromes indicating cortisol deficiency, although hypotension and fatigue have been noted.( 94 , 95 ) These clinical observations are consistent with the free hormone hypothesis, that CBG is not required for entry into cells. A list of mutations in CBGs affecting both levels and affinity for cortisol is found in Hammond( 74 ) and Gagliardi and colleagues,( 94 ) many of which have minimal clinical impact. However, mice with CBG null mutations appear to have increased sensitivity to an acute inflammatory challenge.( 96 ) CBG is cleaved by neutrophil elastase, conceptually providing increased free cortisol at infection sites,( 97 ) which may contribute to the altered response to inflammation in CBG null mice.
CBG is a 383 amino acid glycoprotein that circulates as a monomer of 50‐60 kDa according to its level of glycosylation. Although the liver is the main source of CBG, other tissues such as the pancreas and kidney express CBG at least in mice during development, but apparently these tissues do not contribute to the circulating levels, and the role of CBG is unclear in these tissues.( 98 ) CBG is a member of the serine protease inhibitor class located on chromosome 14 with homology to alpha 1 antitrypsin, but has no protease activity of its own.( 99 ) It has a single binding site for the steroids it transports. CBG levels are increased by estrogen( 100 ) and pregnancy,( 101 ) but are reduced by proinflammatory cytokines( 102 ) and critical illness,( 103 ) raising the concept that free cortisol measurements as in the saliva or urine might be a better way to assess cortisol status during acute illness.( 104 ) Measurement of free cortisol in blood has received little attention, nor have efforts to calculate free cortisol from published affinity constants been widely made.
Summary and Conclusions
The vitamin D metabolites, thyroid hormones, sex steroids, and glucocorticoids/mineralcorticoids are lipophilic hormones whose principal actions involve nuclear hormone receptors. Thus they need to enter cells. But because of their lipophilic nature, they are transported in the liquid milieu of the blood by proteins both specific for the hormone such as DBP, TBG, SHBG, and CBG and the nonspecific protein albumin. The free hormone hypothesis postulates that only the unbound or free hormone crosses the cell membrane. To the extent this is valid, this raises the question whether it is the free concentration that should be measured rather than the total, especially in circumstances when the levels and/or affinities of the binding proteins are altered both physiologically (eg, pregnancy), pathophysiologically (liver disease, nephrotic syndrome, acute illness), or by genetic mutations. In some cases, the binding protein may play a role in transcellular transport as in the role of megalin in the kidney to reclaim DBP bound to 25OHD in the glomerular filtrate or in SHBG bound to sex steroids in reproductive tissues. But for the most part, the free hormone hypothesis remains validated as best demonstrated by the relative lack of clinical phenotypes in families or animals lacking the specific binding protein. That said, one cannot conclude that these binding globulins have no role other than hormone transport as is well‐demonstrated by the ability of DBP to serve as an actin scavenger or as part of the macrophage activating factor, TTR to transport thyroid hormones into the cerebrospinal fluid, possible role of SHBG to transport sex steroids into the placenta, or CBG to deliver increased cortisol to sites of infection (via cleavage by neutrophil elastase). However, a major problem with strongly advocating for measurement of free hormone levels is a lack of well‐accepted means of making these measurements short of equilibrium dialysis and LC–MS of the dialysate containing the free hormone. Although FT4 is now in widespread use, other free hormone measurements remain less used because of concerns regarding their validity. This is changing, and with harmonization of the existing and future assays, I anticipate that free hormone measurements will become routinely used to assess hormone status for the vitamin D metabolites, sex hormones, and for adrenal assessment much as we use FT4 to help assess thyroid status. This will be driven by the many conditions in which total hormone measurements are misleading because of changes in the affinity and level of their binding proteins.
Disclosures
The author has nothing to disclose.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbm4.10418.
Acknowledgments
Dr Bikle is currently or recently funded by grants from the National Institutes of Health, the Veterans Administration, and Radius Pharmaceuticals.
Endnote
This table shows the binding constants of these binding proteins for their respective hormones under normal conditions, the % of the total hormone level bound to the protein, and the % free. Numerous clinical conditions and genetic modifications alter these numbers, which are shown here for comparative purposes. BP = ______; CBG = cortisol‐binding globulin;
References
- 1. Recant L, Riggs DS. Thyroid function in nephrosis. J Clin Investig. 1952;31(8):789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendel CM. The free hormone hypothesis. Distinction from the free hormone transport hypothesis. J Androl. 1992;13(2):107–16. [PubMed] [Google Scholar]
- 3. Bikle DD, Gee E. Free, and not total, 1,25‐dihydroxyvitamin D regulates 25‐hydroxyvitamin D metabolism by keratinocytes. Endocrinology. 1989;124(2):649–54. [DOI] [PubMed] [Google Scholar]
- 4. Nykjaer A, Dragun D, Walther D, et al. An endocytic pathway essential for renal uptake and activation of the steroid 25‐(OH) vitamin D3. Cell. 1999;96(4):507–15. [DOI] [PubMed] [Google Scholar]
- 5. Nykjaer A, Fyfe JC, Kozyraki R, et al. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3). Proc Natl Acad Sci U S A. 2001;98(24):13895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lundgren S, Carling T, Hjalm G, et al. Tissue distribution of human gp330/megalin, a putative Ca(2+)‐sensing protein. J Histochem Cytochem. 1997;45(3):383–92. [DOI] [PubMed] [Google Scholar]
- 7. Hammes A, Andreassen TK, Spoelgen R, et al. Role of endocytosis in cellular uptake of sex steroids. Cell. 2005;122(5):751–62. [DOI] [PubMed] [Google Scholar]
- 8. Chun RF, Lauridsen AL, Suon L, et al. Vitamin D‐binding protein directs monocyte responses to 25‐hydroxy‐ and 1,25‐dihydroxyvitamin D. J Clin Endocrinol Metab. 2010;95(7):3368–76. 10.1210/jc.2010-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pettifor JM, Bikle DD, Cavaleros M, Zachen D, Kamdar MC, Ross FP. Serum levels of free 1,25‐dihydroxyvitamin D in vitamin D toxicity. Ann Int Med. 1995;122(7):511–3. [DOI] [PubMed] [Google Scholar]
- 10. Cooke NE, McLeod JF, Wang XK, Ray K. Vitamin D binding protein: Genomic structure, functional domains, and mRNA expression in tissues. J Steroid Biochem Mol Biol. 1991;40(4–6):787–93. [DOI] [PubMed] [Google Scholar]
- 11. Hagenfeldt Y, Carlstrom K, Berlin T, Stege R. Effects of orchidectomy and different modes of high dose estrogen treatment on circulating "free" and total 1,25‐dihydroxyvitamin D in patients with prostatic cancer. J Steroid Biochem Mol Biol. 1991;39(2):155–9. [DOI] [PubMed] [Google Scholar]
- 12. Moller UK, Streym S, Heickendorff L, Mosekilde L, Rejnmark L. Effects of 25OHD concentrations on chances of pregnancy and pregnancy outcomes: A cohort study in healthy Danish women. Eur J Clin Nutr. 2012;66(7):862–8. [DOI] [PubMed] [Google Scholar]
- 13. Zhang JY, Lucey AJ, Horgan R, Kenny LC, Kiely M. Impact of pregnancy on vitamin D status: A longitudinal study. Br J Nutr. 2014;112(7):1081–7. [DOI] [PubMed] [Google Scholar]
- 14. Moller UK, Streym S, Jensen LT, et al. Increased plasma concentrations of vitamin D metabolites and vitamin D binding protein in women using hormonal contraceptives: a cross‐sectional study. Nutrients. 2013;5(9):3470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guha C, Osawa M, Werner PA, Galbraith RM, Paddock GV. Regulation of human Gc (vitamin D‐binding) protein levels: Hormonal and cytokine control of gene expression in vitro. Hepatology. 1995;21(6):1675–81. [PubMed] [Google Scholar]
- 16. Dahl B, Schiodt FV, Rudolph S, Ott P, Kiaer T, Heslet L. Trauma stimulates the synthesis of Gc‐globulin. Intensive Care Med. 2001;27(2):394–9. [DOI] [PubMed] [Google Scholar]
- 17. Schiodt FV. Gc‐globulin in liver disease. Dan Med Bull. 2008;55(3):131–46. [PubMed] [Google Scholar]
- 18. Wang X, Shapses SA, Al‐Hraishawi H. Free and bioavailable 25‐Hydroxyvitamin D levels in patients with primary hyperparathyroidism. Endocr Pract. 2017;23(1):66–71. [DOI] [PubMed] [Google Scholar]
- 19. Bjorkhem‐Bergman L, Torefalk E, Ekstrom L, Bergman P. Vitamin D binding protein is not affected by high‐dose vitamin D supplementation: A post hoc analysis of a randomised, placebo‐controlled study. BMC Res Notes. 2018;11(1):619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cleve H, Constans J. The mutants of the vitamin‐D‐binding protein: more than 120 variants of the GC/DBP system. Vox Sang. 1988;54(4):215–25. [DOI] [PubMed] [Google Scholar]
- 21. Chun RF. New perspectives on the vitamin D binding protein. Cell Biochem Funct. 2012;30(6):445–56. [DOI] [PubMed] [Google Scholar]
- 22. Malik S, Fu L, Juras DJ, et al. Common variants of the vitamin D binding protein gene and adverse health outcomes. Crit Rev Clin Lab Sci. 2013;50(1):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagasawa H, Uto Y, Sasaki H, et al. Gc protein (vitamin D‐binding protein): Gc genotyping and GcMAF precursor activity. Anticancer Res. 2005;25(6A):3689–95. [PubMed] [Google Scholar]
- 24. Uto Y, Yamamoto S, Mukai H, et al. beta‐Galactosidase treatment is a common first‐stage modification of the three major subtypes of Gc protein to GcMAF. Anticancer Res. 2012;32(6):2359–64. [PubMed] [Google Scholar]
- 25. Arnaud J, Constans J. Affinity differences for vitamin D metabolites associated with the genetic isoforms of the human serum carrier protein (DBP). Hum Genet. 1993;92(2):183–8. [DOI] [PubMed] [Google Scholar]
- 26. Bikle D, Bouillon R, Thadhani R, Schoenmakers I. Vitamin D metabolites in captivity? Should we measure free or total 25(OH)D to assess vitamin D status? J Steroid Biochem Mol Biol. 2017. Oct;173:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwartz JB, Gallagher JC, Jorde R, et al. Determination of free 25(OH)D concentrations and their relationships to Total 25(OH)D in multiple clinical populations. J Clin Endocrinol Metab. 2018;103(9):3278–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Calvo M, Ena JM. Relations between vitamin D and fatty acid binding properties of vitamin D‐binding protein. Biochem Biophys Res Commun. 1989;163(1):14–7. [DOI] [PubMed] [Google Scholar]
- 29. Bouillon R, Xiang DZ, Convents R, van Baelen H. Polyunsaturated fatty acids decrease the apparent affinity of vitamin D metabolites for human vitamin D‐binding protein. J Steroid Biochem Mol Biol. 1992;42(8):855–61. [DOI] [PubMed] [Google Scholar]
- 30. Haddad JG, Hu YZ, Kowalski MA, et al. Identification of the sterol‐ and actin‐binding domains of plasma vitamin D binding protein (Gc‐globulin). Biochemistry. 1992;31(31):7174–81. [DOI] [PubMed] [Google Scholar]
- 31. Head JF, Swamy N, Ray R. Crystal structure of the complex between Actin and human vitamin D‐binding protein at 2.5 a resolution. Biochemistry. 2002;41(29):9015–20. [DOI] [PubMed] [Google Scholar]
- 32. Zhang J, Kew RR. Identification of a region in the vitamin D‐binding protein that mediates its C5a chemotactic cofactor function. J Biolog Chem. 2004;279(51):53282–7. [DOI] [PubMed] [Google Scholar]
- 33. Zhang J, Habiel DM, Ramadass M, Kew RR. Identification of two distinct cell binding sequences in the vitamin D binding protein. Biochim Biophys Acta. 2010;1803(5):623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sonderman JS, Munro HM, Blot WJ, Signorello LB. Reproducibility of serum 25‐hydroxyvitamin d and vitamin D‐binding protein levels over time in a prospective cohort study of black and white adults. Am J Epidemiol. 2012;176(7):615–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hirschfeld J. Immune‐electrophoretic demonstration of qualitative differences in human sera and their relation to the haptoglobins. Acta Pathol Microbiol Scand. 1959;47:160–8. [DOI] [PubMed] [Google Scholar]
- 36. Daiger SP, Schanfield MS, Cavalli‐Sforza LL. Group‐specific component (Gc) proteins bind vitamin D and 25‐hydroxyvitamin D. Proc Natl Acad Sci U S A. 1975;72(6):2076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Armas LA, Hollis BW, Heaney RP. Vitamin D2 is much less effective than vitamin D3 in humans. J Clin Endocrinol Metab. 2004;89(11):5387–91. [DOI] [PubMed] [Google Scholar]
- 38. Safadi FF, Thornton P, Magiera H, et al. Osteopathy and resistance to vitamin D toxicity in mice null for vitamin D binding protein. J Clin Invest. 1999;103(2):239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zella LA, Shevde NK, Hollis BW, Cooke NE, Pike JW. Vitamin D‐binding protein influences total circulating levels of 1,25‐dihydroxyvitamin D3 but does not directly modulate the bioactive levels of the hormone in vivo. Endocrinology. 2008;149(7):3656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Henderson CM, Fink SL, Bassyouni H, et al. Vitamin D‐binding protein deficiency and homozygous deletion of the GC gene. N Engl J Med. 2019;380(12):1150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bikle DD, Schwartz J. Vitamin D binding protein, Total and free vitamin D levels in different physiological and pathophysiological conditions. Front Endocrinol (Lausanne). 2019;10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bikle DD, Gee E, Halloran B, Kowalski MA, Ryzen E, Haddad JG. Assessment of the free fraction of 25‐hydroxyvitamin D in serum and its regulation by albumin and the vitamin D‐binding protein. J Clin Endocrinol Metab. 1986;63(4):954–9. [DOI] [PubMed] [Google Scholar]
- 43. Bikle DD, Halloran BP, Gee E, Ryzen E, Haddad JG. Free 25‐hydroxyvitamin D levels are normal in subjects with liver disease and reduced total 25‐hydroxyvitamin D levels. J Clin Investig. 1986;78(3):748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bikle DD, Siiteri PK, Ryzen E, Haddad JG. Serum protein binding of 1,25‐dihydroxyvitamin D: A reevaluation by direct measurement of free metabolite levels. J Clin Endocrinol Metab. 1985;61(5):969–75. [DOI] [PubMed] [Google Scholar]
- 45. Bikle DD, Gee E, Halloran B, Haddad JG. Free 1,25‐dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. J Clin Investig. 1984;74(6):1966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwartz JB, Lai J, Lizaola B, et al. Variability in free 25(OH) vitamin D levels in clinical populations. J Steroid Biochem Mol Biol. 2014;144(Pt A):156–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Higashi T, Shibayama Y, Fuji M, Shimada K. Liquid chromatography‐tandem mass spectrometric method for the determination of salivary 25‐hydroxyvitamin D3: a noninvasive tool for the assessment of vitamin D status. Anal Bioanal Chem. 2008;391(1):229–38. [DOI] [PubMed] [Google Scholar]
- 48. Schwartz JB, Lai J, Lizaola B, et al. A comparison of measured and calculated free 25(OH) vitamin D levels in clinical populations. J Clin Endocrinol Metab. 2014;99(5):1631–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aloia J, Dhaliwal R, Mikhail M, et al. Free 25(OH)D and calcium absorption, PTH, and markers of bone turnover. J Clin Endocrinol Metab. 2015;100(11):4140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alzaman NS, Dawson‐Hughes B, Nelson J, D'Alessio D, Pittas AG. Vitamin D status of black and white Americans and changes in vitamin D metabolites after varied doses of vitamin D supplementation. Am J Clin Nutr. 2016;104(1):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sollid ST, Hutchinson MY, Berg V, et al. Effects of vitamin D binding protein phenotypes and vitamin D supplementation on serum total 25(OH)D and directly measured free 25(OH)D. Eur J Endocrinol/Eur Feder Endocr Soc. 2016;174(4):445–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schwartz JB, Kane L, Bikle D. Response of vitamin D concentration to vitamin D3 administration in older adults without sun exposure: a randomized double‐blind trial. J Am Geriatr Soc. 2016;64(1):65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shieh A, Chun RF, Ma C, et al. Effects of high‐dose vitamin D2 versus D3 on Total and free 25‐Hydroxyvitamin D and markers of calcium balance. J Clin Endocrinol Metab. 2016;101(8):3070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Powe CE, Karumanchi SA, Thadhani R. Vitamin D‐binding protein and vitamin D in blacks and whites. N Engl J Med. 2014;370(9):880–1. [DOI] [PubMed] [Google Scholar]
- 55. Nielson CM, Jones KS, Bouillon R, et al. Role of assay type in determining free 25‐hydroxyvitamin D levels in diverse populations. N Engl J Med. 2016;374(17):1695–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nielson CM, Jones KS, Chun RF, et al. Free 25‐hydroxyvitamin D: impact of vitamin D binding protein assays on racial‐genotypic associations. J Clin Endocrinol Metab. 2016;101(5):2226–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cheema C, Grant BF, Marcus R. Effects of estrogen on circulating "free" and total 1,25‐dihydroxyvitamin D and on the parathyroid‐vitamin D axis in postmenopausal women. J Clin Investig. 1989;83(2):537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bikle DD, Halloran BP, Harris ST, Portale AA. Progestin antagonism of estrogen stimulated 1,25‐dihydroxyvitamin D levels. J Clin Endocrinol Metab. 1992;75(2):519–23. [DOI] [PubMed] [Google Scholar]
- 59. Lai JC, Bikle DD, Lizaola B, Hayssen H, Terrault NA, Schwartz JB. Total 25(OH) vitamin D, free 25(OH) vitamin D and markers of bone turnover in cirrhotics with and without synthetic dysfunction. Liver Int. 2015;35(10):2294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mimoto MS, Refetoff S. Clinical recognition and evaluation of patients with inherited serum thyroid hormone‐binding protein mutations. J Endocrinol Investig. 2020;43(1):31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benvenga S, Robbins J. Enhancement of thyroxine entry into low density lipoprotein (LDL) receptor‐competent fibroblasts by LDL: An additional mode of entry of thyroxine into cells. Endocrinology. 1990;126(2):933–41. [DOI] [PubMed] [Google Scholar]
- 62. Visser WE, Friesema EC, Visser TJ. Minireview: Thyroid hormone transporters: The knowns and the unknowns. Mol Endocrinol. 2011;25(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tahboub R, Arafah BM. Sex steroids and the thyroid. Best Pract Res Clin Endocrinol Metab. 2009;23(6):769–80. [DOI] [PubMed] [Google Scholar]
- 64. Oppenheimer JH, Werner SC. Effect of prednisone on thyroxine‐binding proteins. J Clin Endocrinol Metab. 1966;26(7):715–21. [DOI] [PubMed] [Google Scholar]
- 65. Reilly CP, Wellby ML. Slow thyroxine binding globulin in the pathogenesis of increased dialysable fraction of thyroxine in nonthyroidal illnesses. J Clin Endocrinol Metab. 1983;57(1):15–8. [DOI] [PubMed] [Google Scholar]
- 66. Mendel CM. The free hormone hypothesis: A physiologically based mathematical model. Endocr Rev. 1989;10(3):232–74. [DOI] [PubMed] [Google Scholar]
- 67. Hagen GA, Elliott WJ. Transport of thyroid hormones in serum and cerebrospinal fluid. J Clin Endocrinol Metab. 1973;37(3):415–22. [DOI] [PubMed] [Google Scholar]
- 68. Hennemann G, Docter R, Krenning EP, Bos G, Otten M, Visser TJ. Raised total thyroxine and free thyroxine index but normal free thyroxine. A serum abnormality due to inherited increased affinity of iodothyronines for serum binding protein. Lancet. 1979;1(8117):639–42. [DOI] [PubMed] [Google Scholar]
- 69. DeCosimo DR, Fang SL, Braverman LE. Prevalence of familial dysalbuminemic hyperthyroxinemia in Hispanics. Ann Int Med. 1987;107(5):780–1. [DOI] [PubMed] [Google Scholar]
- 70. Pappa T, Ferrara AM, Refetoff S. Inherited defects of thyroxine‐binding proteins. Best Pract Res Clin Endocrinol Metab. 2015;29(5):735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Thienpont LM, Van Uytfanghe K, Poppe K, Velkeniers B. Determination of free thyroid hormones. Best Pract Res Clin Endocrinol Metab. 2013;27(5):689–700. [DOI] [PubMed] [Google Scholar]
- 72. Soh SB, Aw TC. Laboratory testing in thyroid conditions ‐ pitfalls and clinical utility. Ann Lab Med. 2019;39(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Heyns W, De Moor P. Kinetics of dissociation of 17beta‐hydroxysteroids from the steroid binding beta‐globulin of human plasma. J Clin Endocrinol Metab. 1971;32:147‐154 [DOI] [PubMed] [Google Scholar]
- 74. Hammond GL. Plasma steroid‐binding proteins: Primary gatekeepers of steroid hormone action. J Endocrinol. 2016;230(1):R13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vos MJ, Mijnhout GS, Rondeel JM, Baron W, Groeneveld PH. Sex hormone binding globulin deficiency due to a homozygous missense mutation. J Clin Endocrinol Metab. 2014;99(9):E1798–802. [DOI] [PubMed] [Google Scholar]
- 76. Rove KO, Crawford ED, Perachino M, et al. Maximal testosterone suppression in prostate cancer‐free vs total testosterone. Urology. 2014;83(6):1217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fiers T, Wu F, Moghetti P, Vanderschueren D, Lapauw B, Kaufman JM. Reassessing free‐testosterone calculation by liquid chromatography‐tandem mass spectrometry direct equilibrium dialysis. J Clin Endocrinol Metab. 2018;103(6):2167–74. [DOI] [PubMed] [Google Scholar]
- 78. Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: Utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab. 2007;92(2):405–13. [DOI] [PubMed] [Google Scholar]
- 79. Vermeulen A, Verdonck L, Kaufman JM. A critical evaluation of simple methods for the estimation of free testosterone in serum. J Clin Endocrinol Metab. 1999;84(10):3666–72. [DOI] [PubMed] [Google Scholar]
- 80. Goldman AL, Bhasin S, Wu FCW, Krishna M, Matsumoto AM, Jasuja R. A reappraisal of Testosterone's binding in circulation: physiological and clinical implications. Endocr Rev. 2017;38(4):302–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Heinrich‐Balard L, Zeinyeh W, Dechaud H, et al. Inverse relationship between hSHBG affinity for testosterone and hSHBG concentration revealed by surface plasmon resonance. Mol Cell Endocrinol. 2015;399:201–7. [DOI] [PubMed] [Google Scholar]
- 82. Ascenzi P, Bocedi A, Notari S, Fanali G, Fesce R, Fasano M. Allosteric modulation of drug binding to human serum albumin. Mini Rev Med Chem. 2006;6(4):483–9. [DOI] [PubMed] [Google Scholar]
- 83. Hackbarth JS, Hoyne JB, Grebe SK, Singh RJ. Accuracy of calculated free testosterone differs between equations and depends on gender and SHBG concentration. Steroids. 2011;76(1–2):48–55. [DOI] [PubMed] [Google Scholar]
- 84. Janne M, Hammond GL. Hepatocyte nuclear factor‐4 controls transcription from a TATA‐less human sex hormone‐binding globulin gene promoter. J Biol Chem. 1998;273(51):34105–14. [DOI] [PubMed] [Google Scholar]
- 85. Simo R, Barbosa‐Desongles A, Saez‐Lopez C, Lecube A, Hernandez C, Selva DM. Molecular mechanism of TNFalpha‐induced Down‐regulation of SHBG expression. Mol Endocrinol. 2012;26(3):438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Simo R, Barbosa‐Desongles A, Hernandez C, Selva DM. IL1beta down‐regulation of sex hormone‐binding globulin production by decreasing HNF‐4alpha via MEK‐1/2 and JNK MAPK pathways. Mol Endocrinol. 2012;26(11):1917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Simo R, Saez‐Lopez C, Lecube A, Hernandez C, Fort JM, Selva DM. Adiponectin upregulates SHBG production: molecular mechanisms and potential implications. Endocrinology. 2014;155(8):2820–30. [DOI] [PubMed] [Google Scholar]
- 88. Selva DM, Hammond GL. Thyroid hormones act indirectly to increase sex hormone‐binding globulin production by liver via hepatocyte nuclear factor‐4alpha. J Mol Endocrinol. 2009;43(1):19–27. [DOI] [PubMed] [Google Scholar]
- 89. Ng KM, Catalano MG, Pinos T, et al. Evidence that fibulin family members contribute to the steroid‐dependent extravascular sequestration of sex hormone‐binding globulin. J Biol Chem. 2006;281(23):15853–61. [DOI] [PubMed] [Google Scholar]
- 90. Cawthon PM, Schousboe JT, Harrison SL, et al. Sex hormones, sex hormone binding globulin, and vertebral fractures in older men. Bone. 2016;84:271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ding EL, Song Y, Manson JE, et al. Sex hormone‐binding globulin and risk of type 2 diabetes in women and men. N Engl J Med. 2009;361(12):1152–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stroupe SD, Harding GB, Forsthoefel MW, Westphal U. Kinetic and equilibrium studies on steroid interaction with human corticosteroid‐binding globulin. Biochemistry. 1978;17(1):177–82. [DOI] [PubMed] [Google Scholar]
- 93. Dunn JF, Nisula BC, Rodbard D. Transport of steroid hormones: Binding of 21 endogenous steroids to both testosterone‐binding globulin and corticosteroid‐binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53(1):58–68. [DOI] [PubMed] [Google Scholar]
- 94. Gagliardi L, Ho JT, Torpy DJ. Corticosteroid‐binding globulin: the clinical significance of altered levels and heritable mutations. Mol Cell Endocrinol. 2010;316(1):24–34. [DOI] [PubMed] [Google Scholar]
- 95. Perogamvros I, Underhill C, Henley DE, et al. Novel corticosteroid‐binding globulin variant that lacks steroid binding activity. J Clin Endocrinol Metab. 2010;95(10):E142–50. [DOI] [PubMed] [Google Scholar]
- 96. Petersen HH, Andreassen TK, Breiderhoff T, et al. Hyporesponsiveness to glucocorticoids in mice genetically deficient for the corticosteroid binding globulin. Mol Cell Biol. 2006;26(19):7236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hammond GL, Smith CL, Paterson NA, Sibbald WJ. A role for corticosteroid‐binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab. 1990;71(1):34–9. [DOI] [PubMed] [Google Scholar]
- 98. Scrocchi LA, Orava M, Smith CL, Han VK, Hammond GL. Spatial and temporal distribution of corticosteroid‐binding globulin and its messenger ribonucleic acid in embryonic and fetal mice. Endocrinology. 1993;132(2):903–9. [DOI] [PubMed] [Google Scholar]
- 99. Billingsley GD, Walter MA, Hammond GL, Cox DW. Physical mapping of four serpin genes: alpha 1‐antitrypsin, alpha 1‐antichymotrypsin, corticosteroid‐binding globulin, and protein C inhibitor, within a 280‐kb region on chromosome I4q32.1. Am J Hum Genet. 1993;52(2):343–53. [PMC free article] [PubMed] [Google Scholar]
- 100. Schwartz U, Volger H, Schneller E, Moltz L, Hammerstein J. Effects of various replacement oestrogens on hepatic transcortin synthesis in climacteric women. Acta Endocrinol (Copenh). 1983;102(1):103–6. [DOI] [PubMed] [Google Scholar]
- 101. Ho JT, Lewis JG, O'Loughlin P, et al. Reduced maternal corticosteroid‐binding globulin and cortisol levels in pre‐eclampsia and gamete recipient pregnancies. Clin Endocrinol. 2007;66(6):869–77. [DOI] [PubMed] [Google Scholar]
- 102. Bartalena L, Hammond GL, Farsetti A, Flink IL, Robbins J. Interleukin‐6 inhibits corticosteroid‐binding globulin synthesis by human hepatoblastoma‐derived (Hep G2) cells. Endocrinology. 1993;133(1):291–6. [DOI] [PubMed] [Google Scholar]
- 103. Ho JT, Al‐Musalhi H, Chapman MJ, et al. Septic shock and sepsis: a comparison of total and free plasma cortisol levels. J Clin Endocrinol Metab. 2006;91(1):105–14. [DOI] [PubMed] [Google Scholar]
- 104. Torpy DJ, Ho JT. Value of free cortisol measurement in systemic infection. Horm Metab Res. 2007;39(6):439–44. [DOI] [PubMed] [Google Scholar]