

ABSTRACT
The hormonal vitamin D metabolite, 1,25‐dihydroxyvitamin D [1,25(OH)2D], produced in kidney, acts in numerous end organs via the nuclear vitamin D receptor (VDR) to trigger molecular events that orchestrate bone mineral homeostasis. VDR is a ligand‐controlled transcription factor that obligatorily heterodimerizes with retinoid X receptor (RXR) to target vitamin D responsive elements (VDREs) in the vicinity of vitamin D‐regulated genes. Circulating 1,25(OH)2D concentrations are governed by PTH, an inducer of renal D‐hormone biosynthesis catalyzed by CYP27B1 that functions as the key player in a calcemic endocrine circuit, and by fibroblast growth factor‐23 (FGF23), a repressor of the CYP27B1 renal enzyme, creating a hypophosphatemic endocrine loop. 1,25(OH)2D/VDR–RXR acts in kidney to induce Klotho (a phosphaturic coreceptor for FGF23) to correct hyperphosphatemia, NPT2a/c to correct hypophosphatemia, and TRPV5 and CaBP28k to enhance calcium reabsorption. 1,25(OH)2D‐liganded VDR–RXR functions in osteoblasts/osteocytes by augmenting RANK‐ligand expression to paracrine signal osteoclastic bone resorption, while simultaneously inducing FGF23, SPP1, BGLP, LRP5, ANK1, ENPP1, and TNAP, and conversely repressing RUNX2 and PHEX expression, effecting localized control of mineralization to sculpt the skeleton. Herein, we document the history of 1,25(OH)2D/VDR and summarize recent advances in characterizing their physiology, biochemistry, and mechanism of action by highlighting two examples of 1,25(OH)2D/VDR molecular function. The first is VDR‐mediated primary induction of Klotho mRNA by 1,25(OH)2D in kidney via a mechanism initiated by the docking of liganded VDR–RXR on a VDRE at −35 kb in the mouse Klotho gene. In contrast, the secondary induction of FGF23 by 1,25(OH)2D in bone is proposed to involve rapid nongenomic action of 1,25(OH)2D/VDR to acutely activate PI3K, in turn signaling the induction of MZF1, a transcription factor that, in cooperation with c‐ets1‐P, binds to an enhancer element centered at −263 bp in the promoter‐proximal region of the mouse fgf23 gene. Chronically, 1,25(OH)2D‐induced osteopontin apparently potentiates MZF1. © 2020 The Authors. JBMR Plus published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research.
Keywords: PTH/VITAMIN D/FGF23, OSTEOBLASTS, OSTEOCYTES, TRANSCRIPTION FACTORS, BONE REMODELING/MOLECULAR PATHWAYS
The Vitamin D Receptor
History
The vitamin D receptor (VDR) was discovered in 1967 in the laboratory of the late Anthony W. Norman, a pioneering researcher in the vitamin D field to whom this issue is dedicated. The first author (MRH) was a graduate student in Dr. Norman's laboratory at that time; we published our observation, “The Association of a Metabolite of Vitamin D3 With Intestinal Mucosa Chromatin, In Vivo” in 1968.( 1 ) This seminal finding was significant for two reasons: (i) after intracardially injecting radioactively labeled vitamin D into vitamin D‐deficient chickens, the predominant subcellular fraction of the target small intestine containing the radioactive tag was purified nuclear chromatin, revealing a role for DNA‐driven gene transcription in the molecular response to vitamin D; and (ii) when the chromatin‐associated, labeled sterol was extracted from intestinal chromatin and analyzed chromatographically in numerous systems, it proved to be a metabolite of vitamin D more polar than 25‐hydroxyvitamin D3 [25(OH)D3] and, like 25(OH)D3, biologically active.( 1 ) These insights illuminated that vitamin D was metabolized to a hormone‐like sterol, which comprises the functional form of vitamin D located at the actual target site in the proximity of DNA. In essence, this study from the Norman laboratory constituted the first appreciation/discovery of the functionally active metabolite of vitamin D, later chemically characterized as 1α,25‐dihydroxyvtamin D3.( 2 , 3 ) Also in 1968, the more polar D metabolite first recognized in the Norman laboratory was discovered to be bound with high affinity and specificity to a protein that directed the novel D metabolite to intestinal chromatin and mediated its function in the genome. As reported in January 1969,( 4 ) a “chromosomal receptor for a vitamin D metabolite” had been brought to light, thus uncovering the missing link between this vitamin D metabolite and the genomic DNA of its biological target organ(s). This research is described in detail in the first author's (MRH) doctoral dissertation.( 5 ) For complex reasons,( 6 ) the initial work from the Norman laboratory on VDR was not widely embraced by the scientific community until Brumbaugh and colleagues( 7 , 8 , 9 , 10 , 11 , 12 ) biochemically and pharmacologically defined the vitamin D receptor in an indisputable fashion.( 13 ) VDR became recognized as a member of the nuclear receptor superfamily, specifically the subfamily of ligand‐controlled transcription factors that obligatorily heterodimerize with retinoid X receptor (RXR) to bind cognate hormone responsive elements in DNA.( 14 ) In retrospect, the original observations in the Norman laboratory were truly the scientific breakthroughs that launched VDR as one of the 48 classic nuclear receptors encoded in the human genome and portended the emergence of this family of ligand‐activated transcription factors as functional regulators of gene expression that are essential in human biology and health, while also laying the groundwork for the central role of VDR in a myriad of biological actions that are still being discovered and appreciated today.
Advances in VDR research
Subsequent to its discovery and validation, there were numerous advances in VDR research that solidified the position of VDR and its 1α,25‐dihydroxyvtamin D3 ligand in the domains of molecular and clinical endocrinology. The initial quantum leap was achieved by J. Wesley Pike and colleagues who isolated avian VDR using DNA‐cellulose affinity chromatography and generated the first serum and monoclonal antibodies to the VDR protein.( 15 , 16 ) This advance led to the molecular cloning of avian VDR by McDonnell and colleagues( 17 ) and to the cloning and expression of full‐length cDNA encoding the human vitamin D receptor.( 18 ) Thus, VDR joined the ranks of steroid and thyroid hormone/fat soluble vitamin nuclear receptors, structurally speaking, and it was the first member of this group to define the two zinc fingers as zinc atoms tetrahedrally coordinated by eight cysteine residues,( 19 ) establishing the DNA binding region of this (and all) nuclear receptors. Human VDR is depicted schematically at the top of Fig. 1A ; the double zinc finger DNA binding domain (DBD) plus its C‐terminal extension (CTE) are pictured linearly in the balance of Fig. 1A . Shaffer and Gewirth crystallized a DNA‐binding fragment of human VDR ranging from residue 16 (phenylalanine) to 125 (serine) and determined its protein structure, reporting that it contained the following three α‐helices: (i) C‐41 to K‐53, (ii) Q‐77 to I‐87, and (iii) D‐97 to R‐121.( 20 ) These three structures are positioned on the right side of zinc fingers #1, #2, and in the distal CTE, respectively. They are not demarcated in Fig. 1A , but play essential roles in the binding of VDR to the 3′ half‐element of the VDRE in vitamin D‐regulated genes as modeled in Fig. 1B . The base‐recognition VDR α‐helix, namely C‐41 to K‐53, is pictured in green in Fig. 1B , nestled in the major groove of DNA where E42, K45, R49, and R50 contact the AGGTCA 3′ half‐element of a VDRE. This α‐helix contains both the proximal box (P‐box) and the specificity box (S‐box), groups of residues that direct the binding of VDR to estrogen response element‐type half‐elements (AGGTCA) over glucocorticoid response element‐type half‐elements (AGAACA), and possess an arginine (position 49 in human VDR) that specifies heterodimeric binding to direct repeat hormone‐responsive elements, respectively.( 21 ) A final DNA sequence recognition amino acid in this VDR α‐helix is lysine‐53, which, in conjunction with the spacing of basic residue clusters in the CTE, confers selective DR3 VDRE docking capacity on VDR.( 21 ) Therefore, as can be seen in Fig. 1A , the C‐41 to K‐53 proximal α‐helix in VDR constitutes the DNA sequence‐recognition command and control nexus of the receptor protein that directs 1,25(OH)2D/VDR to vitamin D targets in the genome.
Fig 1.
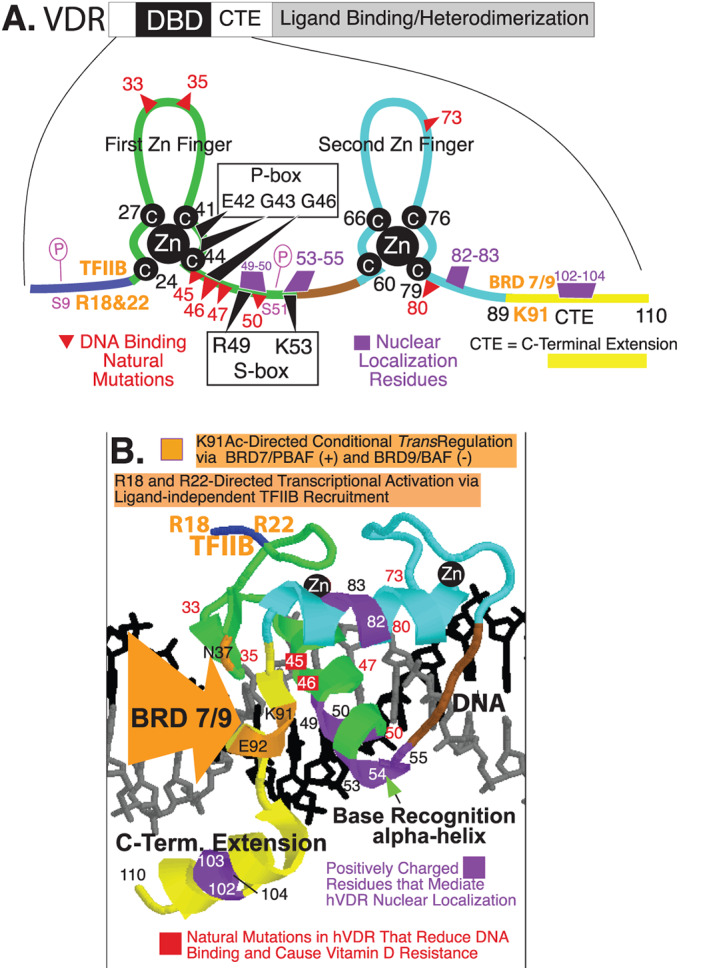
Human VDR DNA‐binding domain. (A) VDR schematic with the DBD/CTE magnified illustrating the two zinc fingers, DNA contact residues (P‐box and S‐box), DNA binding loss of function natural mutations, nuclear localization residues, transactivation residues (arginines 18 and 22; lysine 91), and phosphorylation sites (serines 9 and 51). (B) Model of VDR DBD/CTE bound to DNA. CTE = C‐terminal extension; DBD = DNA binding domain; VDR = vitamin D receptor; Zn = zinc.
As illustrated in Fig. 1B , the second α‐helix in VDR (pictured in light blue) situated on the C‐terminal side of the second zinc finger is positioned to interact with the DNA phosphate backbone, and there is extensive contact between residues in this α‐helix (specifically R73, R74, and R80) and the phosphates of DNA. The combined energy provided by the DNA base recognition and phosphate backbone binding α‐helices is required for specific VDRE binding by VDR, and this conclusion is supported by the location of VDR point mutations (highlighted in red in Fig. 1) that are found in patients with hereditary hypocalcemic vitamin D‐resistant rickets.( 22 ) To bind to DNA and regulate gene expression, transcription factors depend upon nuclear translocation after biosynthesis on the polyribosomes, and nuclear localization mandates various arrangements of basic amino acids in short stretches within the primary amino acid sequence of the protein.( 23 ) Four such segments of positively charged amino acids that mediate unliganded VDR nuclear translocation exist in the DNA‐binding domain.( 24 , 25 ) These residues align on one surface of VDR with its CTE accessible, extending along the structure of heterodimeric VDR–RXR according to calculations by Orlov and colleagues,( 26 ) creating an interaction surface for nuclear import factors initially, and subsequent to 1,25(OH)2D‐liganding and high‐affinity VDRE binding, contact with comodulators. The exception to this sequence of events is basic residues R‐49 and R‐50 in VDR, which first participate in nuclear localization, followed by VDRE base contact, and are therefore not involved in cofactor interaction. Prufer and colleagues( 27 ) have verified the essential role of arginine residues 49 and 50, as well as lysine‐53, arginine‐54, and lysine‐55 in VDR nuclear transfer, and shown that positionally equivalent basic residues in RXR are critical in nuclear localization, especially of the unliganded VDR‐RXR heterodimer.( 27 ) Thus, it appears that a quasi‐stable, hormone‐unoccupied VDR–RXR heterodimer is the species of receptor that localizes in the nucleus of vitamin D target cells, likely bound nonspecifically to DNA with relatively low affinity to slide along in open regions of chromatin (Fig. 2).
Fig 2.
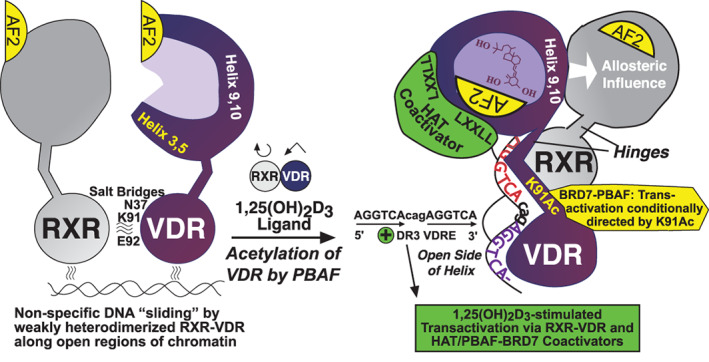
Molecular model for 1,25(OH)2D ligand‐dependent activation of VDR‐RXR, specific association of 1,25(OH)2D bound VDR‐RXR with a VDRE in DNA, followed by 1,25(OH)2D‐stimulated transactivation supported by novel comodulators. BAF = BRG1‐associated factors; CTE = C‐terminal extension; HAT = histone acetyl transferase; PBAF = polybromo‐associated BAF; RXR = retinoid X receptor; VDR = vitamin D receptor.
A revelation on VDR control of transcription rooted in the DNA‐binding domain
To summarize, pictured in Fig. 1 are the following features: (i) first zinc finger in green, (ii) second zinc finger in light blue, (iii) cysteine residues that tetrahedrally coordinate the two zinc atoms— all in white type on black circles, (iv) some of the naturally occurring mutations that reduce DNA binding and cause the phenotype of vitamin D resistance in red, (v) nuclear localization residues in purple, (vi) C‐terminal extension in yellow, and (vii) N‐terminal region in dark blue. Finally, depicted in Fig. 1 are two transactivation motifs embedded in the VDR DBD that are colored orange. The N‐terminal transcriptional activation motif consists of arginine‐18 and arginine‐22, where the basal transcription factor TFIIB binds and augments mRNA synthesis from the target gene.( 28 ) Lysine‐91 emerged early on as a pivotal residue in VDR mechanism of action studies, particularly because its point mutation abolishes the transcriptional activation function of VDR and renders the mutant a dominant‐negative receptor.( 29 ) Very recently, a novel transcriptional activation motif centered on lysine‐91 was reported in the VDR CTE.( 30 ) In their study of 1,25(OH)2D/VDR and β cells of the endocrine pancreas, the research teams of Downes and Evans( 30 ) found that 1,25(OH)2D occupation of VDR switched comodulator complexes bound to K‐91 of VDR from bromodomain‐containing protein (BRD)9/BAF (BRG1‐associated factors), a repressive complex that conferred closed chromatin when VDR was unliganded, to BRD7/PBAF (polybromo‐associated BAF) in the presence of 1,25(OH)2D, which opened chromatin to transcriptional activation of a series of target genes whose gene products preserved β‐cell integrity. In the progression of type 2 diabetes mellitus, β‐cell damage and decay resulting from inflammatory stress triggered by insulin resistance that leads to endocrine pancreas exhaustion play a significant pathological role. In a quest to understand molecular factors that contribute to β‐cell exhaustion under diabetic conditions, Wei and colleagues( 30 ) carried out a genomic CRISPR (clusters of regularly interspaced short palindromic repeats) knockout screen of human‐induced pluripotent stem cell‐derived β‐like cells employing the methodology of Yoshihara and colleagues.( 31 ) By incorporating an inducible Cas9 expression system into β‐like cells bearing a human insulin promoter‐driven green fluorescent protein (GFP) reporter, Wei and colleagues( 30 ) were able to identify genes essential for β‐like cell maintenance. Pertinent to the present review, among the many genes revealed was that encoding human VDR, thereby illuminating the vitamin D hormone receptor as an essential regulator of inflammation and β‐cell survival. Further, alternative recognition of an acetylated lysine‐91 in human VDR by BRD7 and BRD9 appears to orchestrate association of PBAF and BAF chromatin remodeling complexes, respectively, with K‐91 in the CTE. Thus, K‐91 is a VDR residue that is posttranslationally acetylated,( 30 ) and this mechanistic development adds one more feature to the incredible potential of the VDR DBD: namely ligand‐dependent transcriptional control in stem cells, opening‐up a potential treasure trove of fundamental actions of VDR that are distinguished from the well‐established transcriptional activation mechanism of action involving the C‐terminal activation function‐2 (AF‐2) domain in VDR and recruitment of histone acetyl transferase (HAT) coactivators. The fact that 1,25(OH)2D ligand promotes VDR association with PBAF to effect genomewide changes in chromatin accessibility and enhancer landscape, resulting in an anti‐inflammatory response, is a novel feature of VDR–RXR function, but it may also be relevant to understanding the uncharacterized actions of the unliganded complex, such as in hair cycling. In other words, given that hair cycling in rodents and mammals is governed by unoccupied VDR,( 32 ) it is conceivable that ligandless VDR, bound to K‐91 in the VDR CTE, recruits repressive comodulators such as BRD9/BAF and Hairless( 33 ) to govern the mammalian hair cycle. One caveat to this proposal is that Wei and colleagues( 30 ) did not actually show experimentally that unoccupied VDR–RXR was bound to genes in their genomewide screen.
VDR structure and a model for 1,25(OH)2D/VDR–RXR activation of transcription
The structure of 1,25(OH)2D‐occupied human VDR, heterodimerized with full‐length RXRα, docked on a VDRE and bound with a single coactivator, has been determined in solution via small‐angle X‐ray scattering and fluorescence resonance energy‐transfer techniques,( 34 ) as well as by cryoelectron microscopy (cryo‐EM).( 26 ) These two advances, plus additional structural investigation of the VDR–RXR heterodimer,( 35 ) permit the creation of a visual model showing how the DBD and the ligand binding/heterodimerization domains of VDR and RXR are arranged relative to one another, and how their binding to cognate ligand, DNA, and coactivators may influence one another. The left side of Fig. 2 is a hypothetical depiction of unoccupied VDR and RXR, weakly heterodimerized via salt bridges from the RXR DBD to human VDR residues N‐37, K‐91, and E‐92, having already been translocated to the nucleus as discussed above. The quasi‐stable complex shown on the left is postulated to be transiently bound and “sliding” nonspecifically along DNA in open regions of chromatin. The VDR is depicted as unoccupied on the left side of Fig. 2, with the AF‐2 α‐helix 12 in the open configuration awaiting 1,25(OH)2D ligand penetration of the hydrophobic pocket of the VDR‐ligand–binding domain. The right side of Fig. 2 illustrates in schematic fashion the spatial organization for a 1,25(OH)2D‐liganded VDR–RXR heterodimer bound to a generic VDRE DR3 element. This representation is adapted from Orlov and colleagues,( 26 ) and shows the RXR hetero‐partner unoccupied by its 9‐cis retinoic acid ligand despite the fact that Orlov and colleagues(26) included an excess of the retinoid along with 1,25(OH)2D to stabilize the protein structures for physical characterization. Our model excludes the RXR ligand based on biochemical data, indicating that 9‐cis retinoic acid suppresses rather than enhances 1,25(OH)2D‐triggered transactivation by VDR,( 36 ) and we have accumulated evidence that 1,25(OH)2D association with VDR not only amplifies dimerization with RXR, but also allosterically influences the AF‐2 helix 12 of RXR to assume the closed conformation for coactivator recruitment.( 37 , 38 ) Therefore, the key event in VDR‐mediated gene activation is the binding of the 1,25(OH)2D ligand, which generates a dramatic conformational change in the position of helix 12 at the C‐terminus of VDR, bringing it to the “closed” position to serve as part of a platform for binding the LxxLL domains of coactivators.( 39 , 40 ) The binding of coactivator, in turn, likely stabilizes the VDR–RXR heterodimer on the VDRE, and may even assist in heterodimerization by conformationally inducing the VDR LBD to face the open side of the DNA helix at the 5′ end of the VDRE (see right portion of Fig. 2). The AGGTCAcagAGGTCA DR3 VDRE is also shown in Fig. 2 as is the K‐91–containing CTE, which is positioned as contacting the cag spacer in the DR3 VDRE. The model of Orlov and colleagues(26) also portrays the CTE of the VDR DBD as facing the open side of the DNA helix opposite the side occupied by the RXR DBD, indicating that the CTE engages in coactivator recruitment.( 41 ) Finally, in addition to the HAT coactivator bound to closed helix 12 on the helix 3 and 5 platform of liganded VDR as illustrated in Fig. 2, shown is the BRD7/PBAF‐coactivator complex that associates with lysine‐91 in the VDR CTE.( 30 ) These structural alterations and protein–protein associations upon 1,25(OH)2D‐binding are made possible by the flexibility imparted on VDR via its hinge domain,( 26 ) a segment of human VDR beginning at proline‐122, which appears to be endowed with other properties beyond VDR conformational flexibility such as: (i) binding to the nuclear matrix to effect a reduction of VDR‐mediated transcription in mitotic cells,( 42 ) and (ii) phosphorylateable serine/threonine residues in which the phosphorylation status affects the transcriptional activation capacity of the VDR. A specific example of the latter is human VDR hinge residue serine‐208,( 43 ) which is phosphorylated by casein kinase II in a posttranslational modification that potentiates receptor transcriptional activity,( 44 ) probably via reduced proteolytic degradation of VDR that promotes a suprafunctional concentration of receptor.
In conclusion, the DNA‐binding region of VDR is truly a domain that commands a myriad of biological functions, not only directing 1,25(OH)2D‐liganded VDR to the many specific VDREs in the genome while engaged in DNA surveillance, but possessing amino acid sequences that impart nuclear localization plus conditional transcriptional activation, as well as likely holding the key to the actions of unliganded VDR. The DBD is clearly the “business end” of the VDR molecule that is launched by the important functions of the ligand binding/heterodimerization domain, and its properties led to the discovery of VDR in 1968/1969( 1 , 4 ) and to its purification in 1979( 45 ) and cloning in 1987.( 17 )
VDR Mediates Vitamin D Control of Bone Mineral Physiology
The calcium homeostasis endocrine loop
As illustrated in Fig. 3, the hormonal metabolite of vitamin D, 1,25(OH)2D, is produced predominantly in kidney, and acts in a variety of end organs via nuclear VDR to trigger an ensemble of molecular events that orchestrate bone mineral homeostasis. Circulating 1,25(OH)2D concentrations are governed by PTH,( 46 ) an inducer of renal D‐hormone biosynthesis catalyzed by CYP27B1 that functions as the key player in a calcemic endocrine loop, and fibroblast growth factor‐23 (FGF23), a repressor of the CYP27B1 renal enzyme that completes a novel hypophosphatemic endocrine loop.( 47 ) The calcium homeostatic loop, highlighted in Fig. 3, is triggered by low blood calcium, which signals via suboptimal‐occupation of the calcium‐sensing receptor (CaSR) to elicit the elaboration of PTH by the parathyroid glands. PTH then induces renal CYP27B1 to enhance the synthesis of 1,25(OH)2D, which acts in an autocrine manner in kidney to enhance calcium reabsorption from the glomerular filtrate via induction of TRPV5 and CaBP28K, and in endocrine fashion in the following primary vitamin D calcemic targets: intestine, bone, and parathyroid (Fig. 3). The resulting calcemia corrects calcium imbalance and closes the feedback loop through full occupation of the CaSR and silencing of PTH synthesis and secretion. In the process of correcting hypocalcemia, additional phosphate is mobilized by 1,25(OH)2D through absorption from the gut and resorption from bone, but this excess phosphate is eliminated acutely via PTH‐signaled phosphaturia and chronically through FGF23 action to inhibit renal phosphate reabsorption. Thus, bone mineral balance is maintained.
Fig 3.
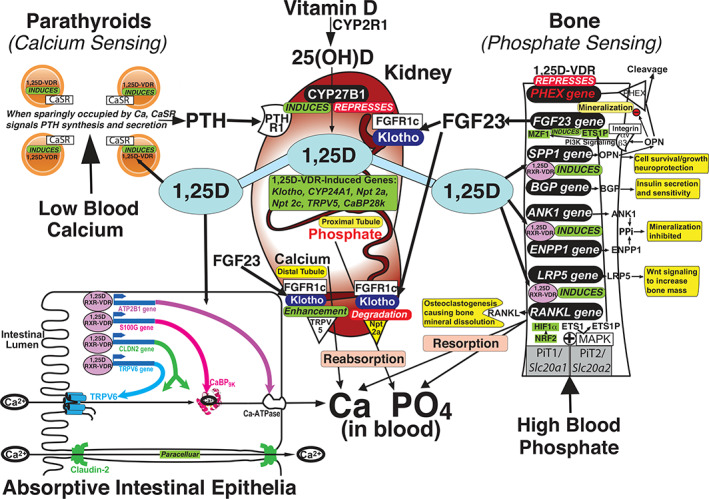
Vitamin D receptor mediates 1,25(OH)2D control of bone mineral physiology.
The phosphate homeostasis endocrine loop
FGF23 arises mainly from osteocytes,( 48 ) rendering bone a newly recognized endocrine organ that is counter‐regulatory to PTH with respect to vitamin D and calcium metabolism, although both PTH and FGF23 are hypophosphatemic in that they signal the inhibition of renal phosphate reabsorption. The essence of the phosphate homeostatic loop is phosphate sensing by bone cells that governs FGF23 synthesis and secretion. As highlighted in Fig. 3, high blood phosphate signals via a putative phosphate sensor, the PiT1/Slc20a1 and PiT2/Slc20a2 heterodimer, to initiate MAPK stimulation that catalyzes a phosphorylation cascade producing activated transcription factor intermediaries in FGF23 induction. These transcription factors remain unidentified, although we propose that NRF2 and/or HIF1α are reasonable candidates based on evidence discussed in detail in this review. Once FGF23 is elaborated by the skeleton, it functions by binding to renal FGFR1c/Klotho coreceptors to promote phosphaturia,( 49 ) calcium reclamation,( 50 ) and repress CYP27B1( 51 ) as well as induce CYP24A1( 49 , 52 ) (Fig. 3), with the latter two actions serving to curb 1,25(OH)2D levels. The resulting phosphaturia and circulating 1,25(OH)2D reduction correct the phosphate excess (while preserving serum calcium as a result of renal reabsorption) and close the feedback loop through diminished occupation of the postulated PiT1/Slc20a1‐PiT2/Slc20a2 phosphate sensor, silencing FGF23 synthesis and secretion by osteocytes.
Bone mineral endocrine physiology
Thus, through the mechanisms described above and detailed in Fig. 3, extracellular calcium and phosphate are maintained at physiologically normal levels by 1,25(OH)2D, PTH, and FGF23, facilitating appropriate bone mineralization and remodeling. 1,25(OH)2D, the vitamin D hormone, is the linchpin of an intricate endocrine network that sustains calcium and phosphate homeostasis to prevent bone disease. Notably, the reciprocity of calcium and phosphate metabolism reflects the physicochemical relationship of these two bone mineral ions, with the foundation of chronic regulation being the reciprocal control of CYP27B1 and CYP24A1 by PTH and FGF23. New mechanistic insight published by the Pike group with respect to endocrine control of 25D‐OHases, CYP27B1, (47 ) and CYP24A1,( 53 ) therefore provides a solution to the molecular puzzle of how bone mineral ions are regulated in normal physiology. In a classic converse endocrine fashion, PTH from the parathyroid glands suppresses renal CYP24A1,( 47 ) the enzyme catalyzing the first step in 1,25(OH)2D elimination, and FGF23 from bone induces renal CYP24A1.( 53 )
1,25(OH)2D/VDR–RXR actions in the parathyroid gland, small intestine, and bone
1,25(OH)2D acts primarily at the small intestinal enterocyte to induce calcium absorption, but it also functions in parathyroid glands to induce the CaSR and amplify their calcium perceiving sensitivity, in osteoblasts/osteocytes in cooperation with phosphate to induce FGF23, and in kidney proximal tubules to auto‐regulate 1,25(OH)2D by inducing CYP24A1 and repressing CYP27B1. As depicted in Fig. 3, in small intestine, 1,25(OH)2D induces a number of gene products that mediate calcium absorption, including TRPV6, CaBP9k (S100G), PMCA1b (ATP2B1), and CLDN2. 1,25(OH)2D also appears to increase expression of NPT2b in the small intestine to promote phosphate absorption, but this action on phosphate is not pictured in Fig. 3 because it may only be relevant under extreme conditions of phosphate restriction.( 54 )
As detailed in Fig. 3, 1,25(OH)2D/VDR acts directly in bone cells to impact skeletal metabolism in numerous respects, not only stimulating resorption to dramatically influence remodeling, but to modulate mineralization in far‐reaching mechanisms beyond simply supplying calcium and phosphate for mineral accretion. Accordingly, 1,25(OH)2D/VDR transcriptionally regulates a minimum of nine genes in bone, and these actions are sufficient to generate normal bone mineral physiology as currently understood. (i) 1,25(OH)2D/VDR induces RANKL( 55 ) (encoded by the TNFSF11 gene) that promotes osteoclastogenesis from osteoclast precursor cells, culminating in bone mineral resorption. (ii) 1,25(OH)2D/VDR induces LRP5,( 56 ) the gene product of which stimulates Wnt signaling to increase bone mass. (iii, iv) 1,25(OH)2D/VDR induces ANK1 and ENPP1,( 57 ) with both of their gene products enhancing pyrophosphate (PPi) concentrations that inhibit bone mineralization. (v) 1,25(OH)2D/VDR induces BGLAP,( 56 ) of which the gene product BGP/osteocalcin constitutes yet a third bone hormone (beyond FGF23 and osteopontin [OPN]) that functions systemically to increase insulin secretion by the β cells of the pancreas, insulin sensitivity of adipose and muscle, and to amplify spermatogenesis via the Leydig cells of the testes, as well as act to attenuate the parasympathetic nervous system to potentiate the “fight‐or‐flight” response. (vi) As reported in Fig. S1 C, 1,25(OH)2D/VDR induces SPP1, of which the gene product OPN functions systemically to promote cell survival and growth, as well as locally to curtail bone mineralization. This latter action of OPN is critical to the prevention of ectopic calcification, whereas the former function may elicit neuroprotection.( 58 , 59 ) Indeed, VDR agonists induce both OPN and Klotho, while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet.( 60 ) (vii) 1,25(OH)2D/VDR represses PHEX,( 61 ) which encodes the exopeptidase that possesses a loss‐of‐function mutation in X‐linked hypophosphatemic rickets and operates enzymatically to cleave and eliminate the ability of OPN to retard bone mineral accretion, as well as to putatively function as the primarily induced protein that drives the extended secondary induction of FGF23 by 1,25(OH)2D (as posited below in the subsection, Osteopontin Controls Bone Mineral Deposition in Conjunction With FGF23). (viii) 1,25(OH)2D/VDR secondarily induces FGF23( 62 ) through a proposed OPN/αvβ3integrin/PI3K/MZF1 pathway, which appears to require c‐ETS1‐P as a cooperating transcription factor (see subsection, Osteopontin Controls Bone Mineral Deposition in Conjunction With FGF23). (ix) 1,25(OH)2D/VDR represses RUNX2 ( 56 ) (not shown in Fig. 3), the master regulator of osteoblast differentiation and a controller of the expression of many bone genes.( 63 ) The bottom line is that by governing the expression of numerous genes, 1,25(OH)2D/VDR signals the sculpting and remodeling of bone mineral in situ much the way Michelangelo created the David. Finally, the above summarized pathways for endocrine control of the skeleton, and defects therein, essentially account for all molecular pathologies of bone mineral diseases that have been recognized to date. Because two major challenges faced by terrestrial animals are limited calcium availability that could compromise the endoskeleton and curtail mobility, along with an overabundance of phosphate in the diet that promotes aging and chronic disease, it is easy to comprehend the significance of the compensatory and corrective pathways executed by the vitamin D endocrine system as depicted in Fig. 3. Because VDR underpins the plethora of 1,25(OH)2D actions, the importance of this macromolecule to health and wellbeing cannot be underestimated. When liganded with 1,25(OH)2D, VDR–RXR directs a biochemical program that optimizes bone mineral usage to maintain the quality of life itself. The balance of this review of VDR will highlight the following two specific examples of its execution of 1,25(OH)2D molecular function: (i) primary induction of Klotho in kidney, and (ii) secondary induction of FGF23 in bone.
Renal Klotho Regulation by 1,25(OH)2D Occurs via a Primary Genomic Mechanism
The kidney is both the source of endocrine 1,25(OH)2D and a target for its actions
As we have emphasized in reviews over the past 20 years,( 32 , 62 , 64 , 65 , 66 , 67 , 68 , 69 ) the kidney is the nexus of the vitamin D endocrine system: both generating 1,25(OH)2D in response to hypocalcemic signals and engaging as a site for metered bone mineral elimination and/or reabsorption. Fig. 3 illustrates the central role of the kidneys in the metabolic activation of vitamin D catalyzed by CYP27B1 in response to PTH‐elicited stimulation of the renal enzyme, normally under conditions such as hypocalcemia. The effects of endocrine 1,25(OH)2D are sufficient to generate normal bone mineral physiology as we now understand it, as well as to promulgate the plethora of other biological actions now attributed to 1,25(OH)2D/VDR–RXR. As also depicted in Fig. 3, 1,25(OH)2D/VDR‐RXR drives transcriptional activation of a minimum of six genes in kidney, including Klotho (KL), NPT2a, NPT2c, TRPV5, CaBP 28K, and CYP24A1. Developed below is one focus of the current review, namely the relatively recent discovery that 1,25(OH)2D/VDR–RXR induces KL mRNA in the kidney.( 70 ) We also highlight the progress made in the last decade defining the pathobiological significance of Klotho and characterize the mechanism of its regulation by the vitamin D hormone.
Renal Klotho expression is governed by 1,25(OH)2D/VDR–RXR binding to VDREs
α‐Klotho (referred to herein as Klotho) was first reported by Kuro‐o and colleagues.( 71 ) Its disruption in mice is associated with soft tissue calcification, profound hyperphosphatemia, osteoporosis, emphysema, arteriosclerosis, skin atrophy, infertility, hypoglycemia, and a curtailed lifespan. A recessive inactivating mutation in the human KL gene elicits the phenotype of severe tumoral calcinosis.( 72 ) KL is expressed primarily in the kidneys and brain choroid plexus.( 73 ) With respect to the regulation of Klotho biosynthesis, Forster and colleagues( 70 ) reported that 1,25(OH)2D significantly induces mRNA expression of KL in a human renal (HK‐2) cell line and kl in a mouse distal convoluted tubule (mpkDCT) cell line. These findings indicate that 1,25(OH)2D is capable of both amplifying FGF23 responsiveness in the kidney by inducing the Klotho membrane coreceptor for FGF23 and of eliciting elaboration of the shed soluble Klotho hormone. To mechanistically probe regulation of KL by 1,25(OH)2D, Forster and colleagues( 70 ) performed bioinformatic analyses of both the human and mouse Klotho genes, which unveiled numerous candidate VDREs in mouse and human genes.( 70 ) When assessed for functionality by cotransfection of reporter constructs into HK‐2 cells, only one mouse VDRE (AGGTCAgagAGTTCA) located at −35,360 bp and two human VDREs (TGAACTctaCGAACC and TGAACTtctTGAACT) located on the negative strand at −47,293 bp and −32,203 bp, respectively, displayed a potency similar to the established rat osteocalcin VDRE.( 70 ) Notably, the –35 kb area of the mouse Klotho gene given in Fig. 4A is marked by VDR and RXR in vivo as deciphered by ChIP‐seq technology (J. Wesley Pike, personal communication, November 24, 2017). Furthermore, a ChIP‐seq map of the human genome( 74 ) yields an additional pair of VDREs, AGTTGAaagGGTTCC and GGAACTgcaTCCACC (negative strand), in the first intron of KL at +1536 and + 1392 bp, respectively. We thus propose that 1,25(OH)2D‐liganded VDR–RXR induces Klotho expression by binding to functional VDREs somewhat remote to the mouse and human KL structural genes, with the human VDREs apparently being separated by as much as 49 kb. Importantly, the VDREs identified in KL genes fit the pattern of possessing nearby cis‐elements that bind c/ebpβ and runx2 (Fig. 4A is an illustration of this for the mouse Klotho VDRE), a signature property of many 1,25(OH)2D/VDR‐RXR‐induced genes,( 75 ) most recently highlighted by the case of mmp13.( 76 ) Finally, as presented in Fig. 4B for comparison of VDRE environments, the promoter proximal region of the mouse spp1 gene reveals a classic VDRE that is flanked on the 5′‐side by a runx2 site and on the 3′‐side by a c/ebpβ element. In combination with the data of Tsujikawa and colleagues,( 77 ) that 1,25(OH)2D increases steady‐state Klotho mRNA levels in mouse kidney in vivo, the results reviewed herein verify that 1,25(OH)2D is the first discovered natural inducer of the Klotho longevity gene. Moreover, Tsujikawa and colleagues( 77 ) showed that renal Klotho mRNA induction after 1,25(OH)2D injection displayed identical kinetics to those of renal cyp24a1, a well‐established primary induction target for 1,25(OH)2D/VDR. In summary, Klotho is controlled by 1,25(OH)2D via a primary VDR–RXR mechanism consisting of direct binding of the hormone‐receptor complex to bona fide VDREs remote from the structural gene, but each positioned in the immediate neighborhood of cis‐associated c/ebpβ and runx2 cooperating transcription factors that apparently generate multiprotein complexes to drive induction of KL mRNA biosynthesis.
Fig 4.
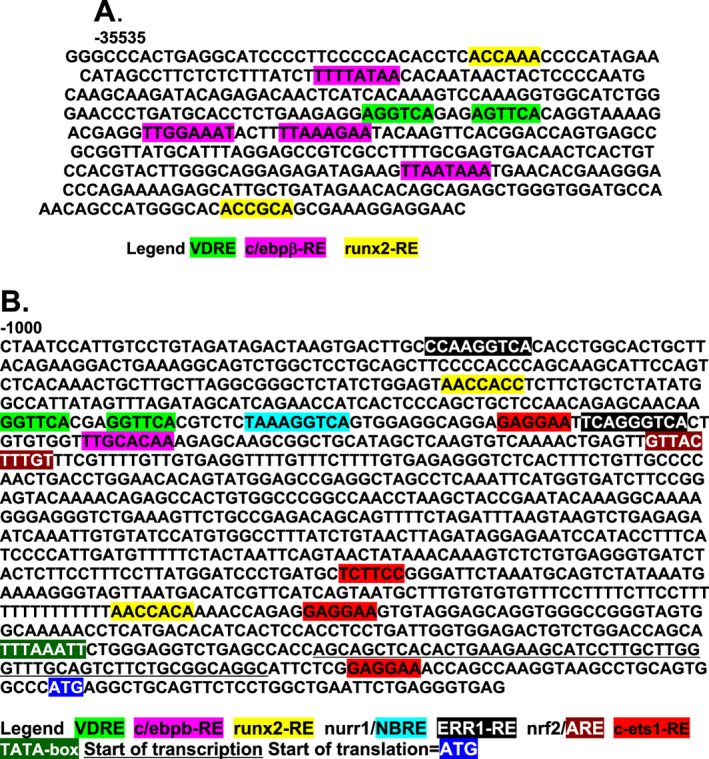
VDREs in two 1,25(OH)2D/VDR–RXR induced genes. (A) −35 kb region of the mouse klotho gene. (B) Promoter proximal region of the mouse spp1/osteopontin gene. VDREs = vitamin D responsive elements.
Klotho and its actions on ion transport in the kidney
The gene products for KL consist of multiple Klotho protein forms including a full‐length transmembrane Klotho, a proteolytically shed soluble Klotho, and a much less‐abundant secreted truncated Klotho with unknown function that may not exist in humans because of degradation of alternatively spliced Klotho mRNA.( 78 ) The transmembrane Klotho (mKL) form is cleaved by membrane‐anchored proteases including ADAM17, liberating the shed soluble Klotho (sKL) that contains both KL1 and KL2 domains functional in FGF23 ligand binding by the coreceptor complex of sKL and FGFR1c. After entering the circulation, this shed soluble Klotho form may function as a circulating hormone‐like principle.( 78 ) This circulating Klotho form has been reported to employ the glycosyl hydrolase catalytic activity to regulate the TRPV5 calcium channel( 79 ) and the renal outer medullary potassium channel 1 (ROMK1).( 80 ) However, in a recent review, Erben( 50 ) summarized the work of Chen and colleagues,( 81 ) who crystallized the ternary complex of the extracellular domain of FGFR1c, FGF23 ligand, and the Klotho ectodomain to provide compelling experimental evidence that Klotho lacks any biologically relevant glycosidase activity. Thus, there has occurred a recent paradigm shift in our understanding of Klotho to reveal that it is devoid of enzymatic activity and instead increases the abundance of TRPV5 on the renal cell membrane through FGF23 signaling (Fig. 3) via ERK1/2, SGK1, and WNK4 to depress endocytotic removal of TRPV5 from the plasma membrane.( 50 ) Thus, 1,25(OH)2D, by independently inducing TRPV5( 82 ) and Klotho,( 70 ) apparently elicits FGF23‐driven calcium retention at the kidneys. One could speculate, therefore, that 1,25(OH)2D/VDR–RXR chronically initiates calcium conservation at the kidneys over a lifetime, possibly to lower the incidence of osteoporosis. ROMK1 is likely upregulated by Klotho analogously to TRPV5 through enhancement in the apical membrane via ERK1/2, SGK1, and WNK4‐signaling, but the outcome would be increased potassium excretion.
Klotho is a FGF23 coreceptor in controlling phosphate and vitamin D metabolism
The most physiologically significant function of full‐length, membrane Klotho is to act as a renal coreceptor of FGFR1c in the feedback control of phosphate and vitamin D metabolism by bone‐derived FGF23 (Fig. 3). FGF23 signals by binding to renal FGFR1c/Klotho coreceptors and an ERK/SGK1/NHERF1 phosphorylation‐transduction pathway to cause degradation of membrane sodium phosphate cotransporter type 2a (Npt2a), with resultant downregulation of phosphate reabsorption to promote phosphaturia.( 49 ) FGF23 also signals to repress renal specific CYP27B1( 51 ) via a pathway that involves the binding of as yet unidentified transcription factors (triggered by ERK1/2) to upstream regions of the mouse cyp27b1 gene centered at approximately −5 kb and −12.5 kb.( 47 ) Finally, FGF23 further signals to induce renal CYP24A1 by binding to FGFR1c/Klotho coreceptors and promulgating an as yet unknown signal transduction pathway initiated by ERK1/2 and including unidentified transcription factors that bind to a downstream region of the mouse cyp24a1 gene centered at approximately +27 kb and possibly to the promoter proximal sequence immediately upstream of the transcription start site,( 53 ) resulting in induction of CYP24A1.( 49 , 52 ) The latter two actions serve to curb 1,25(OH)2D levels (Fig. 3). Remarkably, double knockouts of FGF23 (or its Klotho coreceptor) with either VDR( 83 ) or CYP27B1( 84 ) essentially rescue FGF23 null mice, underscoring the role of FGF23 and Klotho as counter‐regulatory hormones to 1,25(OH)2D, which appears to be the key to their health and longevity benefits. Shed, soluble Klotho may possess systemic antiaging properties independent of the phosphaturic and 1,25(OH)2D‐attenuating actions of transmembrane Klotho, but the mechanism(s) of these actions is not known.( 80 ) Conversely, although FGF23 is antiaging at the kidney by eliciting phosphate elimination and detoxifying 1,25(OH)2D, its “off‐target” actions could actually be pro‐aging in terms of coronary artery disease, as well as potential neoplastic actions in the colon,( 85 ) and it is possible that these off‐target FGF23 pathologies are “buffered” by soluble Klotho.( 86 ) Upregulation of Klotho by 1,25(OH)2D( 70 ) is thus consistent not only with potentiation of FGF23 signaling in the kidney, but also may offer protection for other cell types (eg, vascular and colon), in which the circulating soluble form of Klotho could exert beneficial actions.( 87 )
Klotho exerts numerous additional bioprotective and cellular antiaging actions
Klotho exerts antioxidative effects, some of which are reminiscent of 1,25(OH)2D actions. For example, Klotho has been reported to bind to the transient‐receptor potential canonical Ca2+ channel 1 (TRPC‐1) through its KL2 domain, and regulates TRPC‐1‐mediated Ca2+ entry to maintain endothelial integrity and prevent Ca2+‐stimulated nitric oxide synthetase formation, which contributes to the formation of potent reactive nitrogen species.( 88 ) Thus, the maintenance of Ca2+ and redox signaling at a low resting state by 1,25(OH)2D and its effectors, Nrf2 and Klotho, appears to constitute the mechanism whereby 1,25(OH)2D and Klotho prevent the ravages of oxidation. Also, Wang and colleagues( 89 ) reported that Klotho downregulates the expression of a catalytic subunit of NADPH oxidase and suppresses angiotensin II‐induced superoxide production, oxidative damage, and apoptosis through the cAMP/PKA pathway. In vivo, Klotho gene delivery similarly attenuates NADPH oxidase activity and superoxide production to prevent the progression of spontaneous hypertension and resulting renal damage.( 87 ) These findings led us to propose that 1,25(OH)2D/VDR–RXR primary induction of Klotho mRNA represents a natural pathway to maintaining healthful aging, with intracellular calcium current regulation and mitigation of oxidation being common themes.
Klotho is also known to impact Wnt signaling( 73 ) to curtail renal fibrosis and perhaps suppress tumorigenesis. Klotho‐mediated regulation of Wnt signaling was reported by Liu and colleagues,( 90 ) who showed that sKL binds to Wnt ligands to suppress downstream signal transduction, and that KL knockout enhances Wnt signaling in mice. Regarding the disease‐related consequences of Wnt signaling suppression by Klotho, activated Wnt3 signaling extends the cell cycle by arresting it at the G2/M phase, and induces fibrogenic cytokines in mouse kidney; but Klotho‐treated cells circumvent this phase and are protected against renal fibrosis.( 91 ) The loss of Klotho may therefore contribute to kidney injury by releasing the inhibition of pathogenic Wnt/β‐catenin signaling.( 92 ) Indeed, in vivo expression of Klotho decreases the activation of renal β‐catenin and diminishes renal fibrosis in chronic kidney disease.( 92 ) Conversely, reduced Klotho expression aggravates renal interstitial fibrosis,( 93 ) and overexpression of sKL abolishes the fibrogenic effects of TGF‐β1.( 92 , 94 ) In summary, Klotho overexpression or supplementation protects against fibrosis in several models of renal and cardiac fibrotic disease, with its actions appearing to be rooted in the direct inhibitory effects of circulating/soluble Klotho on TGFβ1 and Wnt signaling.( 95 )
Regarding antitumor actions of Klotho, Behera and colleagues( 96 ) found a correlation between loss of Klotho and a gain in Wnt5A expression, leading to progression of melanoma. Similarly, Abramovitz and colleagues( 97 ) have reported that both membrane and soluble Klotho serve as tumor suppressors by inhibiting tumor cell proliferation through regulation of IGF‐1 signaling. Another in vivo experiment showed that soluble Klotho possesses greater inhibitory effects on tumor cell growth than full‐length membrane Klotho.( 97 ) Therefore, through attenuation of TGFβ1‐, Wnt‐, and IGF1‐signaling pathways, Klotho also inhibits tumorigenesis. The promoter proximal region of the Klotho gene is reported to be hypermethylated in cancer, and transgenic overexpression or introduction of Klotho protein is observed to retard tumor growth in several animal models.
With respect to the antifibrotic qualities of Klotho, high concentrations of extracellular phosphate are toxic to cells, and impaired urinary phosphate excretion increases serum phosphate levels to induce a premature‐aging phenotype. Urinary phosphate levels are increased by dietary phosphate overload and might induce tubular injury and interstitial fibrosis. Extracellular phosphate exerts its cytotoxic effects by forming insoluble nanoparticles with calcium and fetuin‐A. These nanoparticles are referred to as calciprotein particles and are capable of inducing various cellular responses, including the osteogenic transformation of vascular smooth muscle cells and cell death of vascular endothelial cells and renal tubular epithelial cells. Calciprotein particles can be detected in the serum of animal models of kidney disease and in patients with chronic kidney disease (CKD) and probably contribute to the pathogenesis of CKD. This important insight provides a mechanism whereby Klotho, by preventing hyperphosphatemia, protects the vascular and renal systems, thereby prolonging lifespan. In addition, 1,25(OH)2D is thought to cooperate with Klotho in retarding vascular calcification by the induction of OPN (Fig. S1 C), a powerful antimineralization factor.( 60 ) Ironically, the two renal hormones that are deficient in patients with chronic renal failure because of renal mass loss, namely 1,25(OH)2D and Klotho, are two vital effectors of renal and vascular health, suggesting a strategy for the prevention and treatment of vascular disease, as well as CKD.
It is controversial whether Klotho affects insulin secretion and sensitivity
It has been observed that Klotho increases the plasma membrane retention of TRPV2, leading to enhanced glucose‐triggered insulin secretion from pancreatic β cells.( 98 ) Vitamin D has long been known to promote insulin secretion,( 99 ) meaning that insulin release is yet another example of the dual beneficial effects of 1,25(OH)2D and Klotho. Klotho KO mice exhibit less energy storage and expenditure compared with WT mice,( 100 ) as well as attenuated insulin production and enhanced insulin sensitivity.( 101 , 102 ) However, Anour and colleagues( 103 ) demonstrated that Klotho lacks a vitamin D‐independent physiological role in glucose homeostasis, bone turnover, and steady‐state PTH secretion, in vivo, casting doubt on Klotho alone as a bona fide regulator of any of these phenomena. In a sense, this original research by the Erben group( 103 ) was prescient and consistent with the recent opinion piece( 50 ) that “Klotho's effects on mineral homeostasis are fibroblast growth factor‐23 dependent.” Are all Klotho actions dependent upon FGFR1 signal transduction initiated by FGF23? This is the provocative question regarding Klotho in 2020! Because FGFR1 is almost universally expressed, are all of the influences of Klotho on aging actually promulgated by FGF23 signaling? In an opposing theory, Dalton and colleagues( 78 ) propose that soluble Klotho targets GM1 and GM3 sialogangliosides clustered in membrane lipid rafts which act as a Klotho “receptor” that signals control of growth factor functions. Regardless of the mechanism by which Klotho functions, the many consequences of Klotho activity, such as antioxidation, antifibrosis, antimalignancy, anticalcium transients, and antiphosphatemia, clearly collectively contribute to the antiaging potential of Klotho. Based on all these observations, Klotho can be considered a 1,25(OH)2D/VDR‐induced, organ‐protection hormone that promotes healthful aging by delaying chronic diseases, even if exclusively through beneficial signaling via FGFR1.
Accordingly, both FGF23 and Klotho have recently been implicated in maintaining male reproductive function.( 104 ) Indeed, Hansen and colleagues,( 104 ) employing mice null for either FGF23 or Klotho, found that global loss of either FGF23 or Klotho compromised testicular weight and reduced sperm count as well as motility, whereas FGF23 enhances testicular weight in WT mice. However, germ‐cell–specific knockout of Klotho elicited neither decreased sperm count nor mobility, although fewer pregnancies and Klotho heterozygous pups occurred in this group that was characterized by overexpression of testicular trpv5 and npt2b,( 104 ) indicating that testicular calcium and phosphate exchange play a role in the actions of FGF23 and Klotho in gonads, as they do in the functions of FGF23 and Klotho in kidney.
Klotho supports synaptic functioning and protects the central nervous system
In recent years, interest has arisen in the potential role of Klotho in the brain, including its possible protection of the central nervous system (CNS) and prevention of neurological diseases such as depression and the decline in cognitive function associated with aging.( 105 , 106 ) Indeed, high levels of Klotho originating in the choroid plexus are postulated to function as a gatekeeper at the interface between the brain and immune system in the choroid plexus.( 107 ) Klotho depletion in aging or disease may weaken this barrier and promote immunomediated neuropathogenesis. Experimental depletion of Klotho from the choroid plexus enhanced microglial activation in the hippocampus after peripheral injection of mice with lipopolysaccharide. In primary cultures, Klotho suppressed thioredoxin‐interacting protein‐dependent activation of the NLRP3 inflammasome in macrophages by enhancing FGF23 signaling.( 107 ) Finally, peripheral delivery of an α‐Klotho fragment acutely enhanced cognition and neural resilience in young, aging, and disease‐model mice by inducing GluN2B cleavage and increasing NMDAR‐dependent synaptic plasticity.( 108 ) Thus, as 1,25(OH)2D is hypothesized to affect the CNS, Klotho induction by 1,25(OH)2D/VDR appears to be able to accomplish similar enhancement of synaptic functions—leading to the proposal that part of the benefits to brain health afforded by 1,25(OH)2D are mediated by its primary induction of Klotho, analogous to the known improvements in renal and vascular health promulgated by 1,25(OH)2D/VDR action to boost Klotho.
Summary of the significant actions of Klotho that are potentially antipathogenic
In summary, the primary genomic induction of renal Klotho by 1,25(OH)2D/VDR–RXR leads to the following extensive list of biological consequences that benefit the entire organism because Klotho: (i) acts as a renal coreceptor for FGF23 in binding to FGFR isoforms that signal feedback control of hyperphosphatemia and excessive 1,25(OH)2D levels, both of which can lead to vascular calcification via osteogenic transformation of vascular smooth muscle cells and vascular endothelial cell death, as well as to renal tubular epithelial cell death resulting in chronic kidney disease; (ii) exerts antioxidative effects and controls intracellular calcium currents to protect tissues from the ravages of aging; (iii) blunts TGFβ1 and Wnt signaling to prevent renal and cardiac fibrotic disease, as well as tumorigenesis; (iv) acts in the CNS to enhance synaptic functions that mediate plasticity to support neural resiliency and cognition, as well as to protect against immunomediated degenerative neuropathogenesis. Although Klotho and its vitamin D hormone inducer may only play a small part in healthful aging, the sheer number of pathways impacted by this duo of renal hormones leads one to suspect that their pleiotropic actions are certainly a crucial factor in the quality and quantity of life. Because potentiation of FGF23/FGFR1 signaling is the only proven function of Klotho, the next section of this review addresses the induction of the FGF23 ligand by the 1,25(OH)2D renal hormone.
Model for Secondary Regulation of Fibroblast Growth Factor‐23 by 1,25(OH)2D/VDR
Primary and secondary regulation of gene expression by 1,25(OH)2D/VDR‐RXR
All biologic actions of 1,25(OH)2D/VDR appear to be mediated by association of liganded VDR–RXR with VDREs in target genes. In addition to the case of Klotho, discussed in detail above, other principal examples of this primary mechanism, established via ChIP‐seq results that prove the existence of this receptor protein–DNA molecular interaction, include 1,25(OH)2D/VDR‐mediated regulation of the expression of the following genes: (i) cyp24a1,( 109 ) (ii) TRPV6,( 110 ) (iii) mmp13,( 76 ) (iv) RANKL (TNFSF11),( 55 ) (v) osteocalcin (BGLAP),( 111 ) (vi) OPN (spp1),( 112 ) (vii) LRP5,( 113 ) (viii) CBS,( 114 ) and (ix) leptin (ob).( 115 ) Thus, expression of all of the afore‐cited genes is governed in a primary fashion by 1,25(OH)2D/VDR–RXR. However, there exist numerous additional genes encoding functional proteins for which no VDREs have been identified, yet their expression is modulated by vitamin D. This category of genes is likely regulated secondarily, employing mediator transcription factor proteins. Consequently, 1,25(OH)2D‐directed stimulation in this class of vitamin D‐modulated genes is sensitive to inhibition by cycloheximide (an inhibitor of protein synthesis), which is a signature property of secondary regulation of gene expression. One such gene is FGF23, the bone‐derived phosphaturic peptide hormone that is induced by 1,25(OH)2D in a cycloheximide‐sensitive fashion,( 48 ) and that is apparently devoid of proven VDREs.( 116 )
FGF2‐ and 1,25(OH)2D‐stimulated expression of the gene‐encoding FGF23
Bioinformatic analysis of the FGF23 gene reveals that the immediate proximal promoter sequence (adjacent to the TATA‐box) harbors a conserved cAMP responsive element (CRE) as well as conserved NFAT (nuclear factor of activated T cells) and c‐ets1 elements, the former two of which have been shown by point mutation to be required for induction of FGF23 by FGF2.( 117 ) Therefore, a major regulator of FGF23 is FGF2 and its receptor, FGFR1, the nuclear fragment of which integrates gene regulation of ontogeny.( 118 ) With respect to vitamin D control of FGF23 gene expression, the induction of FGF23 by 1,25(OH)2D in mouse bone in vivo was first reported by Kolek and colleagues.( 48 ) In UMR‐106 osteosarcoma cells, FGF23 mRNA upregulation was shown to be 1,25(OH)2D‐dose dependent, occurring as early as 4 hours and peaking at 24 hours after exposure to the vitamin D hormone; but the effect was abrogated in the presence of cycloheximide, indicating dependence on an intermediary transfactor protein(s).( 48 ) Subsequent experiments performed by Saini and colleagues( 119 ) verified the dramatic induction of FGF23 in UMR‐106 osteosarcoma cells and revealed that nonmalignant, normal‐outgrowth cells from rat calvariae responded to 1,25(OH)2D by displaying over a fivefold enhancement of FGF23 mRNA when treated with 10nM 1,25(OH)2D. Moreover, leptin was reported to enhance 1,25(OH)2D‐dependent FGF23 induction whereas IL‐6 elicited suppression, intimating that other transfactors impacted FGF23 expression in the presence of 1,25(OH)2D.( 119 ) One such factor, c‐ets1, for which conserved responsive elements occur in the FGF23 gene, is induced by 1,25(OH)2D, (119 ) and therefore emerges as a candidate mediator of FGF23 induction by 1,25(OH)2D. Next, Kaneko and colleagues( 120 ) dissected the mouse FGF23 promoter proximal sequence cloned by Ito and colleagues,( 121 ) discovering via truncation analysis in transfected human myelogenous leukemia K562 cells that the 1,25(OH)2D‐responsive region lies between −400 and −200 bp in vitro. Furthermore, Kaneko and colleagues(120) noted that this region contains a duo of conserved elements, namely a c‐ets1‐like and a putative nurr1 cis sequence, either of which when point mutated, eliminated transcriptional activation by 1,25(OH)2D. Next, Onal and colleagues( 122 ) employed CRISPR‐Cas9 technology to obtain evidence for a distal enhancer in the mouse fgf23 gene at −16 kb that mediates both inflammation‐ and PTH‐induced fgf23 expression. Recently, utilizing this same unbiased in vivo method, Lee and colleagues( 123 ) localized 1,25(OH)2D responsiveness in the mouse to a sequence extending from −115 to −4110 bp upstream of the FGF23 transcription start site. They also reported that this 4 kb promoter proximal region mediates fgf23 response to high phosphate challenge and contains a second LPS/inflammation‐targeted cis‐element(s). One goal in the current review is to further define the 1,25(OH)2D‐responsive region of the mouse fgf23 gene by re‐examining the proximal promoter region of the gene, and to develop a novel model of secondary regulation that reveals a heretofore unappreciated putative mediator(s) of this secondary response to 1,25(OH)2D/VDR–RXR.
Induction of cyp24a1, fgf23, OPN, and nurr1 mRNAs by 1,25(OH)2D in UMR‐106
To gain further insight into the signal transduction pathway(s) employed by 1,25(OH)2D/VDR to induce FGF23, the kinetics of FGF23 mRNA enhancement by 1,25(OH)2D in UMR‐106 osteosarcoma cells (a cell line that exhibits osteocyte‐like character) was compared with that of other vitamin D‐induced genes such as cyp24a1 (Fig. S1 A). Because it is a primary target for 1,25(OH)2D/VDR–RXR, cyp24a1 mRNA displays a rapid induction by 10nM 1,25(OH)2D, first significantly and appreciably (200‐fold) upregulated 2 hours after 1,25(OH)2D treatment and peaking 8 hours subsequent to hormone dosing, after which it decays to only a slight (14‐fold) enhancement by 48 hours. This relatively rapid mRNA time course can be considered the signature of a gene that is induced in a primary fashion by 1,25(OH)2D/VDR–RXR. As is evident in Fig. S1 B, FGF23 mRNA is much more slowly induced by exposure to 10nM 1,25(OH)2D, first appearing upregulated statistically significantly 2 hours post‐1,25(OH)2D, but very modestly (ninefold) and escalating minimally to 17‐fold at 4 hours of hormone treatment. FGF23 mRNA is not induced impressively (110‐fold) until 8 hours after 1,25(OH)2D treatment, at which point cyp24a1 mRNA enhancement has already maximized. Thus, there exists a clear temporal lag in the 1,25(OH)2D response of FGF23 compared with that of a primary gene (cyp24a1) in the same UMR‐106 target cells. As depicted in Fig. S1 B, FGF23 mRNA peaks broadly at approximately 500‐fold induction between 12 to 24 hours after hormone dosing, with the mean time of 18 hours being over twice as long as that of the appearance of the peak cyp24a1 response to 1,25(OH)2D (Fig. S1 A). FGF23 mRNA then declines much more slowly, maintaining a level of 89‐fold induction at 48 hours post‐1,25(OH)2D exposure. Therefore, the supplemental data in Fig. S1 A,B are consistent with FGF23 being induced by 1,25(OH)2D via a secondary, cycloheximide‐sensitive mechanism.
Somewhat surprising is the observation that the OPN gene, while endowed with a bona fide VDRE similar to cyp24a1, has an “intermediate” time course of induction. In Fig. S1 C, OPN (spp1) mRNA is statistically significantly induced at 2 hours (threefold), 4 hours (sixfold), and 8 hours (28‐fold) after 10nM 1,25(OH)2D treatment, with a sharp peak (91‐fold) 12 hours after hormone dosing, more reminiscent of the initial rate of FGF23 induction by 1,25(OH)2D rather than the kinetics of cyp24a1 mRNA enrichment. However, this OPN time course of 1,25(OH)2D response with a peak induction at 12 hours post‐1,25(OH)2D falls temporally between that for cyp24a1 (8 hours peak) and fgf23 (18 hours peak), instead of aligning with that for either the established primary or secondary mechanisms. That the OPN response to 1,25(OH)2D is more rapid than that of FGF23 is consistent with VDRE(s) in the spp1 gene, but the hybrid nature of OPN's temporal induction suggests that a mediator protein may be required for full stimulation of spp1 gene expression by 1,25(OH)2D. This observation implies (but does not prove) that nurr1, which we originally hypothesized,( 116 ) at least in part, mediates induction of FGF23 by 1,25(OH)2D, might comprise a significant secondary supplement to 1,25(OH)2D in the stimulation of OPN transcription, especially considering that nurr1 alone is fully capable of inducing bone‐expressed genes such as OPN( 124 ) and osteocalcin,( 125 ) and that 1,25(OH)2D synergistically enhances OPN mRNA when added with nurr1.( 124 ) As is depicted in Fig. S1 D, when UMR‐106 cells are dosed with the supraphysiologic concentration of 100nM 1,25(OH)2D, nurr1 mRNA is induced extremely rapidly, being statistically significantly upregulated (3.4‐fold) 30 minutes after sterol hormone exposure and peaking between 30 minutes and 1 hour of treatment. However, nurr1 mRNA induction decays to baseline levels by 12 hours post‐1,25(OH)2D treatment, despite the fact that the dose of 1,25(OH)2D was elevated 10‐fold to 100nM in this experiment only (Fig. S1 D). Meir and colleagues( 126 ) have similarly reported that nurr1 is induced very rapidly by 1,25(OH)2D in UMR‐106 cells with a quick (1 hour) spike in mRNA. Thus, nurr1 appears to satisfy one criterion for a 1,25(OH)2D‐dependent, primary mediator of FGF23 induction, as well as a secondary enhancer of OPN stimulation, namely that it is upregulated temporally prior to the ultimate (secondary) gene products. Finally, as documented in Fig. 4B , the proximal promoter region of the mouse spp1 gene possesses a classic VDRE that is flanked on the 5′‐side by a runx2 site and on the 3′‐side by a c/ebp‐β element, and this collection of elements is bisected by a cluster of two nurr1 sites and a c‐ets1 element. Therefore, it is not unreasonable to assume that OPN control is exercised by multiple inputs, in addition to the 1,25(OH)2D ligand, which employs transfactor mediators that signal transcriptional activation of the spp1‐enhancer module anchored by the VDRE.
However, with respect to possible secondary regulation of FGF23 by nurr1, the putative nurr1 element identified by our group,( 120 ) between −256 bp and −248 bp in the mouse fgf23 gene (Fig. 5A ), has drawbacks in that its GACAGGTCA sequence differs from the nurr1 consensus because the #1 and #3 positions are guanine and cytidine, respectively. In the original identification of the NURR1/NGFIB responsive element (NBRE), Wilson and colleagues( 127 ) reported that TATAGGTCA still binds NURR1, but not as strongly as AAAAGGTCA (the consensus). This suggests that the #1 and #3 positions in the NBRE may be “flexible,” although Murphey and colleagues( 128 ) showed that though the #1 base is “flexible,” the #3 position must be an adenine for NURR1 binding to occur. This later finding caused us to re‐examine whether the NURR1 element we originally identified( 120 ) in fact attracted a different transfactor. We also noted that this sequence in the mouse fgf23 proximal promoter region overlapped, and possibly conflicted sterically in terms of putatively bound transfactors, with a conserved upstream sequence identified as an MZF1 cis element (see Fig. 5A ). Because of these concerns for the validity of the “NURR1” element, combined with compelling precedence detailed below that MZF1 is the likely transfactor that actually mediates, along with c‐ETS1, the secondary response of FGF23 mRNA to 1,25(OH)2D/VDR, we offer an updated mechanism for how 1,25(OH)2D and other regulators actually induce fgf23 gene expression.
Fig 5.
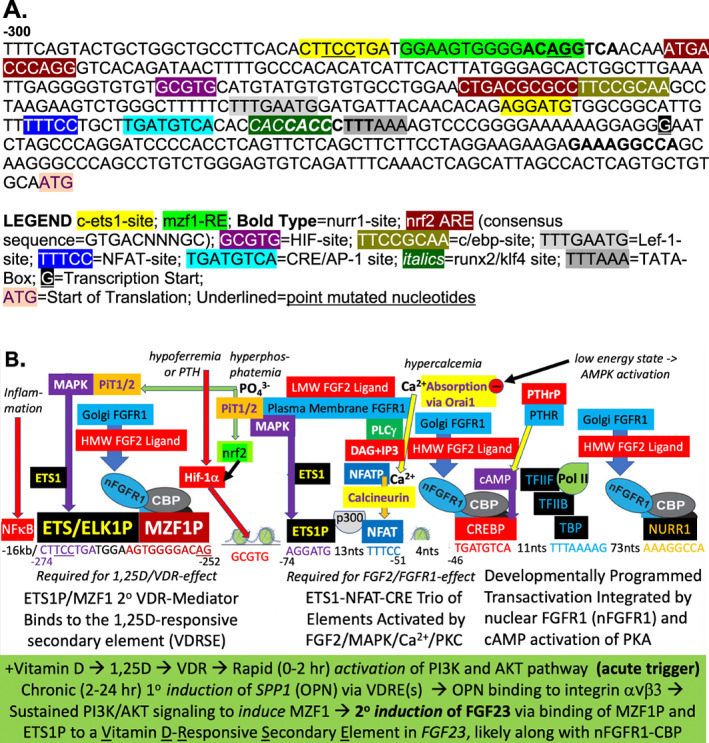
(A) Promoter proximal DNA sequence of the mouse Fgf23 gene, and (B) a model for the regulation of mouse FGF23 by 1,25(OH)2D and multiple inputs (hypoferremia, PTH, hyperphosphatemia, hypercalcemia, and low‐energy state) mediated by various transcription factors. HMW= high molecular weight; LMW = low molecular weight; NFAT = nuclear factor of activated T cells; OPN = osteopontin; VDR = vitamin D receptor; VDREs = vitamin D responsive elements.
MZF1/ETS1 proposed as transcriptional mediators of fgf23 induction by 1,25(OH)2D
The genesis of our new model is that site‐directed mutation of either the MZF1 or c‐ETS1 target sequence in the mouse fgf23 gene, between nucleotides −274 and −250 bp, as shown in Fig. 5A,B (mutated nucleotides underlined), abrogates the 1,25(OH)2D responsiveness of fgf23.( 116 ) Our interest in MZF1 and c‐ETS1 as candidate intermediaries in the induction of FGF23 by 1,25(OH)2D was fueled by the report from Yue and colleagues( 129 ) that a protein–protein complex of MZF1 and ELK1 (a member of the Ets family of transcription factors) binds to the PKCα proximal promoter region, specifically to the sequence aggaTGGGGAaggggCTTCCTgct (MZF1‐ and ELK‐responsive elements in uppercase lettering, respectively) in hepatocarcinoma. Therefore, PKCα is induced through MZF1/ELK1 mediator signaling.( 129 ) Also, PKCα is known to be required for OPN upregulation in osteoblasts exposed to excess inorganic phosphate.( 130 ) More striking is that the responsive nucleotide sequence in PKCα (defined above) dramatically resembles the 1,25(OH)2D‐responsive motif in the mouse fgf23 gene (Fig. 5A ) in that it possesses identical MZF1 (TGGGGA) and ETS1 (CTTCCT) juxtaposed core sequences, although they are 5′‐ to 3′‐switched between the two genes. This constitutes indirect evidence that the target enhancer sequence for 1,25(OH)2D identified by site‐directed mutagenesis in the mouse fgf23 gene highlighted in Fig. 5 (CTTCCTGATGGAAGTGGGGACAGG) actually responds to MZF1/ETS1 as a secondary mediator(s) of 1,25(OH)2D/VDR action in bone cells. Moreover, MZF1 is expressed in human osteoblasts and regulates the level of mRNA encoding the bone‐expressed gene, N‐cadherin.( 131 ) A MZF1‐binding motif, aagggggTGGGGAggggaggg, is present in the promoter proximal region of the human N‐cadherin gene and has been shown to cooperate with a nearby SP‐1 element, with the SP‐1/MZF1 duo of transfactors acting to stimulate N‐cadherin expression.( 131 ) Importantly, if SP‐1 is substituted by SP‐3, expression of N‐cadherin is repressed, indicating that the composition of the MZF1 partner determines the direction whereby MZF1 controls transcription. These revelations in the literature concerning the MZF1 motif provide scientific underpinning for our novel hypothesis that c‐ETS1 and MZF1 constitute dual transfactor mediators of the fgf23 mRNA response to 1,25(OH)2D/VDR. Finally, because point mutation of either of these two motifs in the mouse fgf23 promoter proximal domain ablates 1,25(OH)2D responsiveness,( 120 ) we propose herein that at least mzf1 comprises a cycloheximide‐sensitive mediator of 1,25(OH)2D's secondary action to induce fgf23 mRNA.
The present hypothesis that mzf1 is the “first‐responder” in the induction of fgf23 by 1,25(OH)2D is also supported by research results from Reiner's group.( 132 , 133 ) They have studied myeloid cell differentiation in response to 1,25(OH)2D and observed that CD11b and CD14, two monocytic differentiation markers, are dependent on MZF1 as an intermediary transfactor for their induction by 1,25(OH)2D/VDR.( 133 ) The proximal promoter regions of both genes do not contain classical VDREs, but possess consensus MZF1‐binding sites, namely AGTGGGGA, and these sites are occupied by MZF1 protein after treatment of myeloid cells with 1,25(OH)2D.( 133 ) In their earlier study, the Reiner team reported that the mechanism of action of 1,25(OH)2D in myeloid differentiation is mediated by a VDR‐PI3K signaling complex rather than via the traditional action of VDR–RXR at the genomic level.( 132 ) The evidence for this nongenomic mechanism consisted of the rapidity (5–30 minutes) of the PI3K activation response to 1,25(OH)2D, and its sensitivity to being attenuated by PI3K inhibitors, antisense oligonucleotides encoding the mRNA for the p110 catalytic subunit of PI3K, and a dominant negative mutant of PI3K.( 132 ) These data indicate that PI3K is essential for 1,25(OH)2D to signal the induction of CD11b and CD14 in differentiating myeloid cells and are consistent with our hypothesis that MZF1 constitutes an intermediary transcription factor in 1,25(OH)2D/VDR‐induced expression of genes such as fgf23. Moreover, Hmama and colleagues( 132 ) concluded, based upon rapid activation (20 minutes) of PI3K by 1,25(OH)2D and coimmunoprecipitation of VDR with PI3K, that 1,25(OH)2D‐liganded VDR is directly activating PI3K.
Nongenomic actions of 1,25(OH)2D/VDR initiate acute effects via PI3K signaling
There is considerable published evidence that 1,25(OH)2D acts nongenomically, either in combination with extranuclear VDR or putative plasma membrane 1,25(OH)2D‐binding proteins, to elicit rapid cellular effects such as activation of multiple protein kinases and phospholipases, as well as phosphatidylinositol‐3 kinase (PI3K) and p21ras.( 134 ) 1,25(OH)2D also appears to quickly generate second messengers such as Ca2+, cyclic AMP, fatty acids, and 3‐phosphoinositides, in concert with the activation of protein kinase A, src kinase, mitogen‐activated protein kinases, protein kinase C, and Ca2+‐calmodulin kinase II.( 134 ) Anthony W. Norman was a champion of nongenomic, “fast action” of 1,25(OH)2D, and made its study a major thrust in the latter stages of his career,( 66 , 135 , 136 , 137 ) after his laboratory pioneered the genomic mechanism of action as described at the beginning of this review. Specifically, Vertino and colleagues( 136 ) showed that a ligand‐binding domain‐only construct of VDR functioned in osteocytes to mediate the antiapoptotic influence of 1,25(OH)2D, and Zhang and Zanello( 137 ) reported that 1,25(OH)2D very rapidly activated PI3K/AKT antiapoptotic signaling in ROS‐17/2.8 osteoblasts. Therefore, it is of particular note that rapid nongenomic activation of PI3K may be seminal to the induction of FGF23 by 1,25(OH)2D/VDR through the MZF1 transcriptional mediator as discussed above. Moreover, by rapidly activating MAP kinases such as erk5,( 134 ) which employs c‐ETS1 as a substrate, nongenomic 1,25(OH)2D function could also contribute to fgf23 induction through phosphorylation of c‐ETS1 to c‐ETS1‐P, activating it to enter the nucleus and bind cooperatively with MZF1 to complete the duo of transintermediaries we propose herein are the key to the secondary induction of fgf23 by 1,25(OH)2D/VDR. In this fashion, rapid nongenomic effects of 1,25(OH)2D may constitute the initial molecular mechanism whereby 1,25(OH)2D/VDR, without the benefit of its RXR heteropartner, sparks the secondary induction of fgf23 via the MZF1/c‐ETS1‐P intermediaries, and this may be the first example of a gene induced by 1,25(OH)2D that “integrates” both genomic and nongenomic modes of vitamin D action.
In fact, we suggest that the mechanism put forth by Hmama and colleagues( 132 ) and reinstituted by Moeenrezakhanlou and colleagues( 133 ) may not be the complete molecular explanation for how VDR mediates induction of CD11b and CD14 and concomitant myeloid cell differentiation. Because myeloid cells also express OPN that is induced in a primary and genomic manner by 1,25(OH)2D,( 138 ) we herein put forth OPN as a candidate sustainer of the acute and likely transient [peaking at 20 minutes post‐1,25(OH)2D treatment] effect of 1,25(OH)2D to activate PI3K by chronically activating PI3K signaling to induce MZF1. By virtue of OPN's dramatic primary induction in response to 1,25(OH)2D/VDR, we propose OPN is the protein that drives MZF1 enhancement for the chronic “long‐haul.” What is the evidence that OPN is capable of upregulating MZF1?
Osteopontin controls bone mineral deposition in conjunction with FGF23
Osteopontin, which is a benchmark 1,25(OH)2D/VDR‐induced protein in bone, is a novel upstream protein that is a potent and rapid stimulator of MZF1 expression, specifically in human mesenchymal stem cells, where subsequent MZF1‐driven TGFβ signaling causes these cells to differentiate and adopt a cancer‐associated fibroblast phenotype.( 139 ) By engaging αvβ3 integrin on the cell surface, OPN activates PI3K/AKT signaling to influence numerous pathways, including those involving mTOR and β‐catenin.( 140 ) Interestingly, mTOR expression is downregulated by MZF1 as evidenced by the significant suppression of mtor promoter activity upon MZF1 overexpression in BALB/c mice.( 141 ) MZF1 overexpression also reduced MTOR expression in both fibroblasts and mouse plasmacytoma cells, with ChIP‐PCR confirming that MZF1 binds to the core cis‐element TGGGGA located at −6/−1 in the mtor promoter region.( 141 )
How does sustained MZF1 enhancement come about in cells, including osteoblasts? Although it remains possible that MZF1 is primarily/directly induced by 1,25(OH)2D via an uncharacterized VDRE, we propose, as introduced above, that MZF1 appears after 1,25(OH)2D treatment through the intermediary gene product, OPN, which is a well‐established direct 1,25(OH)2D/VDR target.( 112 ) Figure S1 C illustrates that spp1 is highly and rapidly induced by 1,25(OH)2D in UMR‐106 cells. Because OPN is a bona fide stimulator of MZF1 expression,( 139 ) we herein hypothesize that OPN is a second cycloheximide‐sensitive protein that is the key to understanding how 1,25(OH)2D induces fgf23 in a secondary fashion and maintains this induction over the long‐term. Thus, through PI3K/AKT signaling, OPN upregulates MZF1, likely via an AKT pathway to elicit the occupation of c‐myb and AP4 elements that have been shown to be functional within the MZF1 proximal promoter region.( 142 ) Notably, Chen and colleagues( 142 ) reported that enhanced MZF1 expression suppresses prostate tumor cell growth by promoting ferroportin‐driven iron egress as a result of MZF1 induction of ferroportin. The MZF1/ferroportin axis is likely tied to FGF23 because iron is a proven regulator of FGF23 through HIF1α.( 143 ) In a sense, this observation “closes the signaling circle” and further implicates MZF1 as a crucial FGF23‐modulating transcription factor. It also solidifies the position of SPP1 as a driver gene in bone and bone mineral homeostasis because the physiologic role of FGF23 is to limit bone mineralization, and FGF23 executes this task in many ways. Importantly, FGF23 retards renal phosphate reabsorption to mitigate mineralization and reduces circulating 1,25(OH)2D to lessen calcium and phosphate absorption, but it also requires the cooperation of agents such as the OPN sibling protein to act extracellularly in bone to blunt the crystallization process locally. Therefore, OPN does the local work of governing bone mineral accretion, but also apparently ensures that sufficient FGF23 is available to protect the skeleton from excessive calcification. We assert that OPN, like its partner growth factor elaborated by bone, FGF23, should be considered a novel bone hormone. After induction by 1,25(OH)2D/VDR, OPN acts as a sentinel for skeletal mineralization in several modes. It therefore makes biological sense that OPN should be the sustaining intermediary between 1,25(OH)2D/VDR and FGF23, linking a rapid nongenomic PI3K activation by 1,25(OH)2D to a continuing response of PI3K signaling driven by autocrine OPN binding to integrin αvβ3.
The conclusion is that the results published by Reiner's group( 132 , 133 ) provide a compelling analogy to the present hypothesis that 1,25(OH)2D/VDR acts secondarily to induce FGF23 in osteocytes via the following sequence of events: (i) Rapid (0–1 hour) nongenomic function of 1,25(OH)2D/VDR to activate PI3K signaling( 136 , 137 ) that produces a “starter” quantity of MZF1 to “prime the pump”; (ii) VDRE‐mediated enhancement of OPN (1–12 hours) in a primary genomic response to 1,25(OH)2D/VDR; (iii) OPN binding to αvβ3 integrin to signal protracted PI3K/AKT stimulation; (iv) PI3K/AKT signaling the sustained induction of MZF1; (v) MZF1, along with a partner transcription factor member of the c‐ETS family, binding to cognate cis elements in the proximal promoter region (−252 to −274 bp) of the murine fgf23 gene; and (vi) secondary transcriptional activation of fgf23 gene expression through the recruitment of coactivator protein(s). The fact that this analogy arose from studies in myeloid cells is reminiscent of the original observation of Ito and colleagues( 121 ) that human myelogenous leukemia K562 cells were one of the few cell types that expressed significant levels of FGF23 mRNA, most likely because of the rich background of MZF1 that occurs in myeloid leukemia cells.
FGF23 gene expression is regulated by multiple inputs beyond 1,25(OH)2D and FGF2
Figure 5A presents the proximal promoter sequence of the fgf23 gene in the mouse. Apparent are numerous transcription factor cis‐element motifs conserved across species, including the following sites: (i) TATA‐box, (ii) CRE/AP‐1, (iii) NFAT, (iv) c‐ets1, (v) Lef‐1, (vi) c/ebp, (vii) nrf2, (viii) HIF1α, (ix) nurr1, and (x) MZF1. Although any of these sites could potentially contribute to the secondary response of fgf23 expression to 1,25(OH)2D/VDR by docking a key transfactor that is influenced by 1,25(OH)2D, we have herein narrowed‐down the potential sequence that likely comprises the vitamin D responsive secondary element (VDRSE) to nucleotides −252 to −274, a sequence that contains consensus MZF1 and c‐ETS1 cis elements. To summarize and place vitamin D control of fgf23 gene expression in the general context of FGF23 developmental and physiological modulation, Fig. 5B is presented as a model that includes the 1,25(OH)2D‐dependent segment of the mouse fgf23 gene (left), with both putative transfactors, namely c‐ETS1 and MZF1, bound to their cognate elements. In Fig. 5B , the −252 bp to −274 bp sequence in fgf23 is designated as a 1,25(OH)2D‐responsive secondary element to differentiate it from the traditional 15 bp sequence of the primary VDRE. We do not yet understand exactly how c‐ETS1 participates molecularly in the enhancement of fgf23 expression by 1,25(OH)2D, other than it seems to be an obligatory partner to MZF1. Because c‐ets1 employs VDR as one of its many transcription factor cohorts,( 144 ) it is not out of the realm of possibility that c‐ets1 could attract unliganded VDR to the VDRSE as a “helper” coactivator.( 145 ) More likely, c‐ets1 cooperates with MZF1 in recruiting one or more of its documented alternative cointegrators.( 144 ) MZF1 is a transcription factor in the Kruppel family of zinc finger proteins that is expressed predominantly in myeloid progenitor cells. Indeed, MZF1 participates in controlling growth, differentiation, and apoptosis of progenitors, regulating transcription during differentiation along the myeloid lineage.( 146 ) Yet, in an important advance, Le Mee and colleagues( 131 ) discovered that MZF1 induced human N‐cadherin in osteoblasts, suggesting that MZF1 could be crucial in skeletal as well as myeloid cells. Notably, cadherins are transmembrane glycoproteins that guide calcium‐dependent cell–cell adhesion during morphogenesis and development. They also complex with catenins to govern a variety of processes including cell–cell adhesion, signal transduction, and control of gene transcription.( 147 ) Because control of FGF23 spans bone morphogenesis, development, and differentiated functions, it is intriguing that the cadherin–catenin interrelationship may be relevant to regulation of FGF23 via the MZF1 connection. Accordingly, it has been reported recently by Ko and colleagues(148) that phosphorylation‐dependent stabilization of MZF1 upregulates N‐cadherin expression during protein kinase CK2‐mediated epithelial‐mesenchymal transition in human esophageal cancer cell lines TE2 and HCE4. MZF1 is phosphorylated by CK2 at serine 27 that activates the transcription factor for DNA‐responsive element association and stabilizes it to prevent degradation via proteolysis, thereby escalating MZF1 protein concentration and in turn amplifying transcription of N‐cadherin.( 148 ) In an amazing molecular connection between induction of FGF23 by 1,25(OH)2D/VDR and CK2‐catalyzed phosphorylation, VDR is known to be phosphorylated by CK2, (43 ) and this posttranslational modification enhances the transactivation function of VDR,( 44 ) likely by stabilizing the receptor protein. Equally intriguing is the recognition of CK2, a protein kinase that participates in organ development, as a key biochemical player in fgf23 expression during the ontogeny of bone development. We postulate that CK2 may comprise an endogenous switch that modulates gene expression during cell growth and differentiation.
It is likely that endocrine control of FGF23 occurs postnatally to ensure homeostasis, whereas ontogenic modulation of FGF23 takes place during development, triggered by the FGF2/FGFR1/nFGFR1 signal cascade being activated by the FGF2 growth factor and possibly PTHrP originating in chondrocytes to stimulate bone morphogenesis (Fig. 5B, upper right ). As is the case for PTH, cAMP is the second messenger for PTHrP that functions through phospho‐CREB binding to the conserved CRE near the TATA‐box in the FGF23 gene (Fig. 5B, right center ). But as reported by Han and colleagues,( 117 ) for FGF2 to enhance FGF23 expression, other required second messengers are generated through low‐molecular‐weight (LMW) FGF2‐ligand binding to its plasma membrane FGFR1, such as diacylglycerol that activates PKC and inositol triphosphate (IP3)/Ca2+ to activate calcineurin‐catalyzed dephosphorylation of NFAT, translocating it into the nucleus to bind the NFAT element in DNA. Finally, included in the proposed model of FGF23 gene regulation is the activation of MAP kinase (MAPK) when plasma membrane FGFR1 is liganded with LMW FGF2 (Fig. 5B, center ). Phosphorylation of c‐ETS1 by MAPK is required for this transcription factor to associate with its cognate element in the fgf23 proximal promoter sequence at −74 bp, thereby allowing it to cooperate with NFAT to stimulate fgf23 transcription.( 117 )
Mineral, endocrine, and stress regulators of FGF23: Mechanisms of action
For completeness of the model proposed in Fig. 5B , several other established effectors of FGF23 homeostasis are included, namely inflammation, hypoferremia, hyperphosphatemia, PTH, hypercalcemia, and energy state. Inflammation‐triggered induction of fgf23 is initiated by LPS and cytokines such as TNF‐α and IL‐1β, with the response executed by NFκB (p65/p50) binding to the fgf23 gene in the −16 kb region( 122 ) (Fig. 5B left). Hypoferremia and likely inflammation also enhance fgf23 gene expression via HIF‐1α/HIF‐1β bound to one of several HIFREs in the fgf23 gene, and one such conserved HIFRE (GCGTG) is present in the proximal promoter sequence of the mouse fgf23 gene at −165 bp (Fig. 5A,B ). PTH also functions to enhance HIF‐1α in osteoblasts, as Frey and colleagues( 149 ) reported that HIF‐1α exerts distinct developmental functions and acts as a negative regulator of bone formation. Mice lacking HIF‐1α in osteoblasts and osteocytes formed more bone in response to intermittent PTH, which is known to be an anabolic agonist. Also, exposure of primary mouse osteoblasts to PTH resulted in the rapid induction of HIF‐1α protein levels via a post‐transcriptional mechanism. Similarly, deferoxamine, an iron chelator that inhibits the activity of the prolyl‐hydroxylase enzymes that initiate the targeting of HIF‐1α subunits for proteasomal degradation, elicits rapid induction of HIF‐1α protein levels by simulating hypoferremia, a known stimulator of fgf23 induction via HIF‐1α augmentation as described above. Therefore, as illustrated in Fig. 5B (left center), we propose that both PTH and hypoferremia act through HIF‐1α to induce fgf23 during exposure of osteoblasts to stresses of iron depletion and PTH function to diminish osteoblast mineralization, allowing for enhanced bone resorption.
Phosphate control of FGF23 is fundamental to skeletal physiology and is quite complex in nature, with high‐phosphate rapidly inducing fgf23 as well as other bone‐expressed genes including dmp1, spp1, and tnfsf11.( 123 ) Neither the molecular sensor for phosphate nor the intracellular signal transduction pathway that mediates its influence on fgf23 expression has been identified to date, although Lee and colleagues (123) recently localized the phosphate effect on FGF23 in the mouse to a sequence extending from −115 to −4110 bp upstream of the FGF23 transcription start site. The proximal promoter region of this phosphate‐responsive stretch of nucleotides is depicted in Fig. 5A , but has yet to be further dissected experimentally. Thus, to complete the model in Fig. 5B for fgf23 regulatory mechanisms propagated by phosphate, we can only speculate on the actual molecular mediators. PiT1 is induced by 1,25(OH)2D,( 150 ) signals the binding of extracellular phosphate via the ERK1/2 protein kinase cascade,( 151 ) and inhibition of phosphate uptake by PiT1 small hairpin silencing RNA in cultured vascular smooth muscle cells (VSMCs) blocks the expression of phosphate‐induced osteogenic differentiation markers such as runx2 and OPN.( 152 ) Therefore, we postulate herein that PiT1 is a reasonable candidate for a phosphate sensor/transporter role in osteoblasts and/or hypertrophic chondrocytes. Recently, Bon and colleagues( 153 ) reported that a heterodimer of high affinity sodium‐dependent phosphate transporters PiT1/Slc20a1 and PiT2/Slc20a2 determines phosphate sensing independently of phosphate influx. This raises the possibility that PiT1/2 could constitute the long‐sought phosphate sensor on bone cells. Evidence for the functional significance of PiT1 and PiT2 is provided by experiments in which ablation of PiT1 or PiT2 suppressed both phosphate‐dependent ERK1/2 (MAPK)‐catalyzed phosphorylation and subsequent induction of the mineralization inhibitors matrix Gla protein and OPN.( 153 ) Because nrf2 is one transcription factor that is upregulated in osteoblasts exposed to high‐phosphate levels,( 154 ) we put forward nrf2 as a potential candidate to mediate the effect of phosphate exposure on fgf23 gene expression (Fig. 5B ). Perhaps relevant is the identification of a conserved responsive element for nrf2 in the HIF1α gene,( 155 ) meaning that HIF1α could be the transcription factor that mediates the response of fgf23 expression to high phosphate as well as to hypoferremia and inflammation. Other candidate mediators include RUNX2, which is linked to the proposed PiT1/2 phosphate sensor by the observation that knockdown of PiT1 abrogates runx2 induction by high‐phosphate challenge.( 152 ) It is likely that other transcription factors participate in the intricate and critical regulation of FGF23 by phosphate, particularly because FGF23 is the new dominant phosphate homeostasis peptide hormone, which was previously known as “phosphatonin” (initially as a counterpoint to the naming of the rather weak calcium‐lowering hormone, calcitonin). Ironically, FGF23 is also an effective calcium‐lowering hormone by virtue of its ability to suppress the principal calcemic hormone, 1,25(OH)2D. Finally, as expected in an endocrine sense, calcium is also a modulator of FGF23, with induction of FGF23 by hypercalcemia (Fig. 5B ). David and colleagues( 156 ) showed that calcium increases FGF23 in bone, both in vivo and in cultured cells, in vitro. We demonstrated that a 1 kb mouse FGF23 promoter‐reporter construct, transfected into MC3T3‐E1 osteoblast‐like cells, responds to a high calcium challenge with a statistically significant (twofold) enhancement of transcription.( 120 ) Recently, in a preliminary experiment (data not shown) utilizing transfection of MC3T3‐E1 cells, we observed that calcium stimulation of transcription exists in the first 200 bp of the proximal promoter sequence in the mouse fgf23 gene. These data are consistent with the conclusion of Han and colleagues (117) that the NFAT transcription factor may mediate the response of fgf23 gene expression to calcium through PLCγ, IP3, and intracellular activation of calcineurin by calcium (Fig. 5B ). Finally, calcium entry via ORAI1 has recently been reported( 157 ) to be inhibited by AMPK activation in low‐energy states (Fig. 5B ), which would be accompanied by low‐phosphate levels that are not exacerbated because FGF23 expression is reduced in the absence of store‐operated calcium ion entry. Thus, in low‐energy situations, FGF23 synthesis and release are curtailed because calcineurin activation of NFAT is attenuated, thereby reducing fgf23 gene expression. Clearly, the regulation of FGF23 gene expression is managed by multiple inputs and all control systems must be in place and in perfect working order to prevent the occurrence of the many insidious chronic disorders that occur in conjunction with deranged levels of FGF23 and phosphate.
Genomic/nongenomic 1,25(OH)2D actions via PI3K in bone, brain, and immune system
In conclusion, this review posits that whereas induction of Klotho by 1,25(OH)2D in renal cells appears to be accomplished by the traditional primary mechanism of liganded VDR‐RXR binding to functional VDREs in the region of the KL gene, the mechanisms that mediate regulation of FGF23 by 1,25(OH)2D/VDR are more complicated. Herein we recognize the role of 1,25(OH)2D‐liganded VDR in rapidly activating PI3K signaling to trigger certain bioactions in a mechanism that does not involve VDR‐DNA interaction, at least initially, specifically in osteoblasts( 137 ) and osteocytes,( 136 ) where inhibition of apoptosis was the 1,25(OH)2D action studied. In this nongenomic mechanism, 1,25(OH)2D/VDR quickly (5 minutes) associates with SRC‐kinase,( 136 ) possibly activating PI3K through phosphorylation of its regulatory subunit, with phospho‐PI3K signaling downstream activation of AKT, which in turn directs antiapoptosis of these bone cells. Importantly, this overall mechanism of antiapoptosis is sensitive to inhibition by actinomycin D,( 136 ) meaning that gene transcription of DNA to mRNA is still required for full biological responsiveness. Therefore, we conclude that to complete the rapid, nongenomic action of 1,25(OH)2D in osteoblasts/osteocytes, genomic effects of 1,25(OH)2D/VDR–RXR must ultimately come into play. To resolve this conundrum, in this review we present a novel paradigm that couples nongenomic and genomic actions of 1,25(OH)2D, specifically in the form of a new molecular explanation for how 1,25(OH)2D secondarily induces FGF23 in osteoblasts/osteocytes. The common denominator is PI3K and its ability to promulgate a vast number of signaling pathways that control cell survival, proliferation, growth, metabolism, and specialized cell functions such as neurotransmission. In the case of FGF23 induction in bone cells, we postulate that OPN is the crucial 1,25(OH)2D‐induced protein that sustains PI3K activation to generate a second induced protein, the MZF1 transcription factor, which in turn directly stimulates FGF23 transcription as detailed above in this section. In a sense, by genomically inducing OPN, 1,25(OH)2D is able to exert a protracted enhancement of FGF23 production by sustaining transient PI3K activation in osteoblasts that occurs via the nongenomic route.
The same independent, yet coupled set of events appears to transpire in the CNS where 1,25(OH)2D/VDR exerts both genomic and nongenomic influences.( 158 , 159 ) Specifically, Sisley and colleagues( 160 ) demonstrated that 1,25(OH)2D acutely increases the firing frequency of paraventricular neurons through a PI3K‐dependent mechanism, while also inducing Irs2 and p85 in a genomic manner to amplify insulin signaling in hypothalamic cell culture, also via a PI3K‐dependent mechanism. In a final analogy utilizing another accepted vitamin D target, the immune system, Ferreira and colleagues( 161 ) have reported that 1,25(OH)2D/VDR advances tolerogenic human monocyte‐derived dendritic cells through stimulation of glucose metabolism via a combination of nongenomic 1,25(OH)2D/VDR activation of PI3K/Akt/mTOR signaling and genomic induction of PI3KCG, PFKFB4, and C‐MYC, with the gene products respectfully activating PI3K, glycolysis, and growth. Thus, through combined nongenomic and genomic signaling, 1,25(OH)2D/VDR dictates the creation and maintenance of the tolerogenic monocyte‐derived dendritic cell phenotype and its functions. This indicates that the immune system joins bone and brain in the group of vitamin D targets that appears to employ coupled nongenomic and genomic mechanisms of 1,25(OH)2D/VDR action.
Disclosures
All authors have nothing to disclose.
Author contributions
Mark Haussler: Conceptualization; data curation; formal analysis; funding acquisition; writing‐original draft; writing‐review and editing. Sarah Livingston: Data curation; investigation; methodology. Zhela Sabir: Data curation; investigation; methodology. Carol Haussler: Investigation; writing‐original draft; writing‐review and editing. Peter Jurutka: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; project administration; supervision; writing‐review and editing.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbm4.10432.
Supporting information
Fig. S1. Induction time‐course of mRNAs encoding bone‐expressed genes following treatment of rat osteocyte‐like UMR‐106 cells with 1,25(OH)2D. (A) cyp24a1, (B) fgf23, (C) spp1, (D) nurr1. Each value is the average of at least three experiments with triplicate biological replicates ± standard deviation.
Acknowledgments
Funding: This work was supported by the National Institutes of Health [grant numbers: DK033351 (to MRH) and CA140285 (to PWJ)].
References
- 1. Haussler MR, Myrtle JF, Norman AW. The association of a metabolite of vitamin D3 with intestinal mucosa chromatin in vivo. J Biol Chem. 1968;243:4055–64. [PubMed] [Google Scholar]
- 2. Lawson DEM, Fraser DR, Kodicek E, Morris HR, Williams DH. Identification of 1,25‐dihydroxycholecalciferol, a new kidney hormone controlling calcium metabolism. Nature. 1971;230:228–30. [DOI] [PubMed] [Google Scholar]
- 3. Holick MF, Schnoes HK, DeLuca HF, Suda T, Cousins RJ. Isolation and identification of 1,25‐dihydroxycholecalciferol, a metabolite of vitamin D active in the intestine. Biochemistry. 1971;10:2799–804. [DOI] [PubMed] [Google Scholar]
- 4. Haussler MR, Norman AW. Chromosomal receptor for a vitamin D metabolite. Proc Natl Acad Sci USA. 1969;62:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haussler MR. The specific association of a vitamin D metabolite with the genome of its target organ, in vivo. Diss Abstr B. 1969;29:4513B. [Google Scholar]
- 6. Chen TC, Weber JC, DeLuca HF. On the subcellular location of vitamin D metabolites in intestine. J Biol Chem. 1970;245(15):3776–80. [PubMed] [Google Scholar]
- 7. Brumbaugh PF, Haussler MR. 1a,25‐Dihydroxyvitamin D3 receptor: competitive binding of vitamin D analogs. Life Sci. 1973;13:1737–46. [DOI] [PubMed] [Google Scholar]
- 8. Brumbaugh PF, Haussler MR. 1a,25‐Dihydroxycholecalciferol receptors in intestine. I. Association of 1a,25‐dihydroxycholecalciferol with intestinal mucosa chromatin. J Biol Chem. 1974;249:1251–7. [PubMed] [Google Scholar]
- 9. Brumbaugh PF, Haussler MR. 1a,25‐Dihydroxycholecalciferol receptors in intestine. II. Temperature‐dependent transfer of the hormone to chromatin via a specific cytosol receptor. J Biol Chem. 1974;249:1258–62. [PubMed] [Google Scholar]
- 10. Brumbaugh PF, Haussler DH, Bressler R, Haussler MR. Radioreceptor assay for 1a,25‐dihydroxyvitamin D3 . Science. 1974;183:1089–91. [DOI] [PubMed] [Google Scholar]
- 11. Brumbaugh PF, Haussler DH, Bursac KM, Haussler MR. Filter assay for 1a,25‐dihydroxyvitamin D3: utilization of the hormone's target tissue chromatin receptor. Biochemistry. 1974;13:4097–102. [DOI] [PubMed] [Google Scholar]
- 12. Brumbaugh PF, Hughes MR, Haussler MR. Cytoplasmic and nuclear binding components for 1a,25‐dihydroxyvitamin D3 in chick parathyroid glands. Proc Natl Acad Sci USA. 1975;72:4871–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pike JW, Meyer MB, Lee SM, Onal M, Benkusky NA. The vitamin D receptor: contemporary genomic approaches reveal new basic and translational insights. J Clin Invest. 2017;127(4):1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. [DOI] [PubMed] [Google Scholar]
- 15. Pike JW, Donaldson CA, Marion SL, Haussler MR. Development of hybridomas secreting monoclonal antibodies to the chicken intestinal 1a,25‐dihydroxyvitamin D3 receptor. Proc Natl Acad Sci USA. 1982;79:7719–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pike JW, Marion SL, Donaldson CA, Haussler MR. Serum and monoclonal antibodies against the chick intestinal receptor for 1,25‐dihydroxyvitamin D3 . J Biol Chem. 1983;258(2):1289–96. [PubMed] [Google Scholar]
- 17. McDonnell DP, Mangelsdorf DJ, Pike JW, Haussler MR, O'Malley BW. Molecular cloning of complementary DNA encoding the avian receptor for vitamin D. Science. 1987;235:1214–7. [DOI] [PubMed] [Google Scholar]
- 18. Baker AR, McDonnell DP, Hughes MR, et al. Cloning and expression of full‐length cDNA encoding human vitamin D receptor. Proc Natl Acad Sci USA. 1988;85:3294–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haussler MR, Mangelsdorf DJ, Yamaoka K, et al. Molecular characterization and actions of the vitamin D hormone receptor In Ringold G, ed. Steroid Hormone Action. UCLA symposia on molecular and cellular biology, new series. 75. New York: Alan R. Liss, Inc. 1988; 247–62. [Google Scholar]
- 20. Shaffer PL, Gewirth DT. Structural basis of VDR‐DNA interactions on direct repeat response elements. EMBO J. 2002;21(9):2242–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hsieh J‐C, Whitfield GK, Jurutka PW, et al. Two basic amino acids C‐terminal of the P‐box specify functional binding of the vitamin D receptor to its rat osteocalcin DNA responsive element. Endocrinology. 2003;144:5065–80. [DOI] [PubMed] [Google Scholar]
- 22. Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25‐dihydroxyvitamin D‐resistant rickets. Endocr Rev. 1999;20(2):156–88. [DOI] [PubMed] [Google Scholar]
- 23. Cyert MS. Regulation of nuclear localization during signaling. J Biol Chem. 2001;276(24):20805–8. [DOI] [PubMed] [Google Scholar]
- 24. Hsieh J‐C, Shimizu Y, Minoshima S, et al. Novel nuclear localization signal between the two DNA‐binding zinc fingers in the human vitamin D receptor. J Cell Biochem. 1998;70(1):94–109. [PubMed] [Google Scholar]
- 25. Luo Z, Rouvinen J, Mäenpää PH. A peptide C‐terminal to the second Zn finger of human vitamin D receptor is able to specify nuclear localization. Eur J Biochem. 1994;223:381–7. [DOI] [PubMed] [Google Scholar]
- 26. Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. EMBO J. 2012;31(2):291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prufer K, Racz A, Lin GC, Barsony J. Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem. 2000;275(52):41114–23. [DOI] [PubMed] [Google Scholar]
- 28. Jurutka PW, Remus LS, Whitfield GK, et al. The polymorphic N terminus in human vitamin D receptor isoforms influences transcriptional activity by modulating interaction with transcription factor IIB. Mol Endocrinol. 2000;14(3):401–20. [DOI] [PubMed] [Google Scholar]
- 29. Hsieh JC, Jurutka PW, Selznick SH, et al. The T‐box near the zinc fingers of the human vitamin D receptor is required for heterodimeric DNA binding and transactivation. Biochem Biophys Res Commun. 1995;215(1):1–7. [DOI] [PubMed] [Google Scholar]
- 30. Wei Z, Yoshihara E, He N, et al. Vitamin D switches BAF complexes to protect beta cells. Cell. 2018;173(5):1135–49 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshihara E, Wei Z, Lin CS, et al. ERRgamma is required for the metabolic maturation of therapeutically functional glucose‐responsive beta cells. Cell Metab. 2016;23(4):622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haussler MR, Whitfield GK, Haussler CA, et al. 1,25‐Dihydroxyvitamin D and klotho: a tale of two renal hormones coming of age. Vitam Horm. 2016;100:165–230. [DOI] [PubMed] [Google Scholar]
- 33. Hsieh JC, Sisk JM, Jurutka PW, et al. Physical and functional interaction between the vitamin D receptor and hairless corepressor, two proteins required for hair cycling. J Biol Chem. 2003;278(40):38665–74. [DOI] [PubMed] [Google Scholar]
- 34. Rochel N, Ciesielski F, Godet J, et al. Common architecture of nuclear receptor heterodimers on DNA direct repeat elements with different spacings. Nat Struct Mol Biol. 2011;18(5):564–70. [DOI] [PubMed] [Google Scholar]
- 35. Zhang J, Chalmers MJ, Stayrook KR, et al. DNA binding alters coactivator interaction surfaces of the intact VDR‐RXR complex. Nat Struct Mol Biol. 2011;18(5):556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacDonald PN, Dowd DR, Nakajima S, et al. Retinoid X receptors stimulate and 9‐cis retinoic acid inhibits 1,25‐dihydroxyvitamin D3‐activated expression of the rat osteocalcin gene. Mol Cell Biol. 1993;13(9):5907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thompson PD, Jurutka PW, Haussler CA, Whitfield GK, Haussler MR. Heterodimeric DNA binding by the vitamin D receptor and retinoid X receptors is enhanced by 1,25‐dihydroxyvitamin D3 and inhibited by 9‐cis‐retinoic acid. Evidence for allosteric receptor interactions. J Biol Chem. 1998;273(14):8483–91. [DOI] [PubMed] [Google Scholar]
- 38. Thompson PD, Remus LS, Hsieh J‐C, et al. Distinct retinoid X receptor activation function‐2 residues mediate transactivation in homodimeric and vitamin D receptor heterodimeric contexts. J Mol Endocrinol. 2001;27(2):211–27. [DOI] [PubMed] [Google Scholar]
- 39. Jurutka PW, Hsieh J‐C, Remus LS, et al. Mutations in the 1,25‐dihydroxyvitamin D3 receptor identifying C‐terminal amino acids required for transcriptional activation that are functionally dissociated from hormone binding, heterodimeric DNA binding and interaction with basal transcription factor IIB, in vitro. J Biol Chem. 1997;272:14592–9. [DOI] [PubMed] [Google Scholar]
- 40. Masuyama H, Brownfield CM, St‐Arnaud R, MacDonald PN. Evidence for ligand‐dependent intramolecular folding of the AF‐2 domain in vitamin D receptor‐activated transcription and coactivator interaction. Mol Endocrinol. 1997;11(10):1507–17. [DOI] [PubMed] [Google Scholar]
- 41. Hsieh J‐C, Whitfield GK, Oza AK, et al. Characterization of unique DNA binding and transcriptional activation functions in the carboxyl‐terminal extension of the zinc finger region in the human vitamin D receptor. Biochemistry. 1999;38:16347–58. [DOI] [PubMed] [Google Scholar]
- 42. Cui X, Pertile R, Eyles DW. The vitamin D receptor (VDR) binds to the nuclear matrix via its hinge domain: a potential mechanism for the reduction in VDR mediated transcription in mitotic cells. Mol Cell Endocrinol. 2018;472:18–25. [DOI] [PubMed] [Google Scholar]
- 43. Jurutka PW, Hsieh J‐C, MacDonald PN, et al. Phosphorylation of serine 208 in the human vitamin D receptor: the predominant amino acid phosphorylated by casein kinase II, in vitro, and identification as a significant phosphorylation site in intact cells. J Biol Chem. 1993;268:6791–9. [PubMed] [Google Scholar]
- 44. Jurutka PW, Hsieh J‐C, Nakajima S, Haussler CA, Whitfield GK, Haussler MR. Human vitamin D receptor phosphorylation by casein kinase II at ser‐208 potentiates transcriptional activation. Proc Natl Acad SciU SA. 1996;93:3519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pike JW, Haussler MR. Purification of chicken intestinal receptor for 1,25‐dihydroxyvitamin D. Proc Natl Acad Sci USA. 1979;76(11):5485–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hughes MR, Brumbaugh PF, Haussler MR, Wergedal JE, Baylink DJ. Regulation of serum 1a,25‐dihydroxyvitamin D3 by calcium and phosphate in the rat. Science. 1975;190:578–80. [DOI] [PubMed] [Google Scholar]
- 47. Meyer MB, Benkusky NA, Kaufmann M, et al. A kidney‐specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J Biol Chem. 2017;292(42):17541–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kolek OI, Hines ER, Jones MD, et al. 1{alpha},25‐Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal‐gastrointestinal‐skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289(6):G1036–G42. [DOI] [PubMed] [Google Scholar]
- 49. Razzaque MS. The FGF23‐Klotho axis: endocrine regulation of phosphate homeostasis. Nat Rev Endocrinol. 2009;5(11):611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Erben RG. α‐Klotho's effects on mineral homeostasis are fibroblast growth factor‐23 dependent. Curr Opin Nephrol Hypertens. 2018;27(4):229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Perwad F, Zhang MY, Tenenhouse HS, Portale AA. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25‐hydroxyvitamin D‐1alpha‐hydroxylase expression in vitro. Am J Physiol Renal Physiol. 2007;293(5):F1577–83. [DOI] [PubMed] [Google Scholar]
- 52. Shimada T, Hasegawa H, Yamazaki Y, et al. FGF‐23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19(3):429–35. [DOI] [PubMed] [Google Scholar]
- 53. Meyer MB, Lee SM, Carlson AH, et al. A chromatin‐based mechanism controls differential regulation of the cytochrome P450 gene Cyp24a1 in renal and non‐renal tissues. J Biol Chem. 2019;294(39):14467–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Christakos S, Lieben L, Masuyama R, Carmeliet G. Vitamin D endocrine system and the intestine. Bonekey Rep. 2014;3:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Onal M, St John HC, Danielson AL, Markert JW, Riley EM, Pike JW. Unique distal enhancers linked to the mouse Tnfsf11 gene direct tissue‐specific and inflammation‐induced expression of RANKL. Endocrinology. 2016;157(2):482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barthel TK, Mathern DR, Whitfield GK, et al. 1,25‐Dihydroxyvitamin D3/VDR‐mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J Steroid Biochem Mol Biol. 2007;103(3–5):381–8. [DOI] [PubMed] [Google Scholar]
- 57. Lieben L, Carmeliet G, Masuyama R. Calcemic actions of vitamin D: effects on the intestine, kidney and bone. Best Pract Res Clin Endocrinol Metab. 2011;25(4):561–72. [DOI] [PubMed] [Google Scholar]
- 58. Schroeter M, Zickler P, Denhardt DT, Hartung HP, Jander S. Increased thalamic neurodegeneration following ischaemic cortical stroke in osteopontin‐deficient mice. Brain. 2006;129(Pt 6):1426–37. [DOI] [PubMed] [Google Scholar]
- 59. Enkhjargal B, McBride DW, Manaenko A, et al. Intranasal administration of vitamin D attenuates blood‐brain barrier disruption through endogenous upregulation of osteopontin and activation of CD44/P‐gp glycosylation signaling after subarachnoid hemorrhage in rats. J Cereb Blood Flow Metab. 2017;37(7):2555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lau WL, Leaf EM, Hu MC, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82(12):1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hines ER, Kolek OI, Jones MD, et al. 1,25‐dihydroxyvitamin D3 down‐regulation of PHEX gene expression is mediated by apparent repression of a 110 kDa transfactor that binds to a polyadenine element in the promoter. J Biol Chem. 2004;279(45):46406–14. [DOI] [PubMed] [Google Scholar]
- 62. Haussler MR, Haussler CA, Whitfield GK, et al. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the "Fountain of Youth" to mediate healthful aging. J Steroid Biochem Mol Biol. 2010;121:88–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tai PW, Wu H, Gordon JA, et al. Epigenetic landscape during osteoblastogenesis defines a differentiation‐dependent Runx2 promoter region. Gene. 2014;550(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Haussler MR, Whitfield GK, Haussler CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13(3):325–49. [DOI] [PubMed] [Google Scholar]
- 65. Haussler MR, Haussler CA, Bartik L, et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66(10 Suppl 2):S98–112. [DOI] [PubMed] [Google Scholar]
- 66. Haussler MR, Jurutka PW, Mizwicki M, Norman AW. Vitamin D receptor (VDR)‐mediated actions of 1alpha,25(OH)vitamin D: genomic and non‐genomic mechanisms. Best Pract Res Clin Endocrinol Metab. 2011;25(4):543–59. [DOI] [PubMed] [Google Scholar]
- 67. Haussler MR, Whitfield GK, Haussler CA, Hsieh J‐C, Jurutka PW. Nuclear vitamin D receptor: natural ligands, molecular structure‐function, and transcriptional control of vital genes In Feldman D, Pike JW, Adams J, eds. Vitamin D. 3rd ed San Diego: Academic Press; 2011. pp 137–70. [Google Scholar]
- 68. Haussler MR, Whitfield GK, Kaneko I, et al. The role of vitamin D in the FGF23, klotho, and phosphate bone‐kidney endocrine axis. Rev Endocr Metab Disord. 2012;13(1):57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Haussler MR, Whitfield GK, Kaneko I, et al. Molecular mechanisms of vitamin D action. Calcif Tissue Int. 2013;92(2):77–98. [DOI] [PubMed] [Google Scholar]
- 70. Forster RE, Jurutka PW, Hsieh JC, et al. Vitamin D receptor controls expression of the anti‐aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011;414(3):557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kuro‐o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. [DOI] [PubMed] [Google Scholar]
- 72. Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117(9):2684–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Y, Sun Z. Current understanding of klotho. Ageing Res Rev. 2009;8(1):43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ramagopalan SV, Heger A, Berlanga AJ, et al. A ChIP‐seq defined genome‐wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res. 2010;20(10):1352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Meyer MB, Goetsch PD, Pike JW. Genome‐wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin D hormone. J Steroid Biochem Mol Biol. 2010;121(1–2):136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Meyer MB, Benkusky NA, Pike JW. Selective distal enhancer control of the Mmp13 gene identified through clustered regularly interspaced short palindromic repeat (CRISPR) genomic deletions. J Biol Chem. 2015;290(17):11093–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17(12):2393–403. [DOI] [PubMed] [Google Scholar]
- 78. Dalton GD, Xie J, An SW, Huang CL. New insights into the mechanism of action of soluble klotho. Front Endocrinol (Lausanne). 2017;8:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The beta‐glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310(5747):490–3. [DOI] [PubMed] [Google Scholar]
- 80. Cha SK, Hu MC, Kurosu H, Kuro‐o M, Moe O, Huang CL. Regulation of renal outer medullary potassium channel and renal K(+) excretion by klotho. Mol Pharmacol. 2009;76(1):38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Chen G, Liu Y, Goetz R, et al. α‐Klotho is a non‐enzymatic molecular scaffold for FGF23 hormone signalling. Nature. 2018;553(7689):461–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schlatter E. Who wins the competition: TRPV5 or calbindin‐D28K? J Am Soc Nephrol. 2006;17(11):2954–6. [DOI] [PubMed] [Google Scholar]
- 83. Hesse M, Frohlich LF, Zeitz U, Lanske B, Erben RG. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf‐23 deficient mice. Matrix Biol. 2007;26(2):75–84. [DOI] [PubMed] [Google Scholar]
- 84. Renkema KY, Alexander RT, Bindels RJ, Hoenderop JG. Calcium and phosphate homeostasis: concerted interplay of new regulators. Ann Med. 2008;40(2):82–91. [DOI] [PubMed] [Google Scholar]
- 85. Jacobs E, Martinez ME, Buckmeier J, Lance P, May M, Jurutka P. Circulating fibroblast growth factor‐23 is associated with increased risk for metachronous colorectal adenoma. J Carcinog. 2011;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54(4):810–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kusaba T, Okigaki M, Matui A, et al. Klotho is associated with VEGF receptor‐2 and the transient receptor potential canonical‐1 Ca2+ channel to maintain endothelial integrity. Proc Natl Acad Sci USA. 2010;107(45):19308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang Y, Kuro‐o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP‐PKA pathway. Aging Cell. 2012;11(3):410–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu H, Fergusson MM, Castilho RM, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317(5839):803–6. [DOI] [PubMed] [Google Scholar]
- 91. Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Renal Physiol. 2012;303(12):F1641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta‐catenin signaling. J Am Soc Nephrol. 2013;24(5):771–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sugiura H, Yoshida T, Shiohira S, et al. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am J Physiol Renal Physiol. 2012;302(10):F1252–F64. [DOI] [PubMed] [Google Scholar]
- 94. Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor‐beta1 (TGF‐beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286(10):8655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mencke R, Olauson H, Hillebrands JL. Effects of Klotho on fibrosis and cancer: a renal focus on mechanisms and therapeutic strategies. Adv Drug Deliv Rev. 2017;121:85–100. [DOI] [PubMed] [Google Scholar]
- 96. Behera R, Kaur A, Webster MR, et al. Inhibition of age‐related therapy resistance in melanoma by rosiglitazone‐mediated induction of klotho. Clin Cancer Res. 2017;23(12):3181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Abramovitz L, Rubinek T, Ligumsky H, et al. KL1 internal repeat mediates klotho tumor suppressor activities and inhibits bFGF and IGF‐I signaling in pancreatic cancer. Clin Cancer Res. 2011;17(13):4254–66. [DOI] [PubMed] [Google Scholar]
- 98. Lin Y, Sun Z. Antiaging gene Klotho enhances glucose‐induced insulin secretion by up‐regulating plasma membrane levels of TRPV2 in MIN6 beta‐cells. Endocrinology. 2012;153(7):3029–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Norman AW, Frankel JB, Heldt AM, Grodsky GM. Vitamin D deficiency inhibits pancreatic secretion of insulin. Science. 1980;209(4458):823–5. [DOI] [PubMed] [Google Scholar]
- 100. Mori K, Yahata K, Mukoyama M, et al. Disruption of klotho gene causes an abnormal energy homeostasis in mice. Biochem Biophys Res Commun. 2000;278(3):665–70. [DOI] [PubMed] [Google Scholar]
- 101. Utsugi T, Ohno T, Ohyama Y, et al. Decreased insulin production and increased insulin sensitivity in the klotho mutant mouse, a novel animal model for human aging. Metabolism. 2000;49(9):1118–23. [DOI] [PubMed] [Google Scholar]
- 102. Wolf I, Levanon‐Cohen S, Bose S, et al. Klotho: a tumor suppressor and a modulator of the IGF‐1 and FGF pathways in human breast cancer. Oncogene. 2008;27(56):7094–105. [DOI] [PubMed] [Google Scholar]
- 103. Anour R, Andrukhova O, Ritter E, Zeitz U, Erben RG. Klotho lacks a vitamin D independent physiological role in glucose homeostasis, bone turnover, and steady‐state PTH secretion in vivo. PLoS One. 2012;7(2):e31376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hansen LB, Kaludjerovic J, Nielson JE, et al. Influence of FGF23 and Klotho on male reproduction: systemic vs direct effects. FASEB J. 2020;12436–49. [DOI] [PubMed] [Google Scholar]
- 105. Pavlatou MG, Remaley AT, Gold PW. Klotho: a humeral mediator in CSF and plasma that influences longevity and susceptibility to multiple complex disorders, including depression. Transl Psychiatry. 2016;6(8):e876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Abraham CR, Mullen PC, Tucker‐Zhou T, Chen CD, Zeldich E. Klotho is a neuroprotective and cognition‐enhancing protein. Vitam Horm. 2016;101:215–38. [DOI] [PubMed] [Google Scholar]
- 107. Zhu L, Stein LR, Kim D, et al. Klotho controls the brain‐immune system interface in the choroid plexus. Proc Natl Acad Sci USA. 2018;115(48):E11388–E96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Leon J, Moreno AJ, Garay BI, et al. Peripheral elevation of a klotho fragment enhances brain function and resilience in young, aging, and alpha‐synuclein transgenic mice. Cell Rep. 2017;20(6):1360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25‐dihydroxyvitamin D3. J Biol Chem. 2010;285(20):15599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Meyer MB, Watanuki M, Kim S, Shevde NK, Pike JW. The human transient receptor potential vanilloid type 6 distal promoter contains multiple vitamin D receptor binding sites that mediate activation by 1,25‐dihydroxyvitamin D3 in intestinal cells. Mol Endocrinol. 2006;20(6):1447–61. [DOI] [PubMed] [Google Scholar]
- 111. Ozono K, Liao J, Kerner SA, Scott RA, Pike JW. The vitamin D‐responsive element in the human osteocalcin gene: association with a nuclear proto‐oncogene enhancer. J Biol Chem. 1990;265:21881–8. [PubMed] [Google Scholar]
- 112. Noda M, Vogel RL, Craig AM, Prahl J, DeLuca HF, Denhardt DT. Identification of a DNA sequence responsible for binding of the 1,25‐dihydroxyvitamin D3 receptor and 1,25‐dihydroxyvitamin D3 enhancement of mouse secreted phosphoprotein 1 (Spp‐1 or osteopontin) gene expression. Proc Natl Acad Sci USA. 1990;87:9995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fretz JA, Zella LA, Kim S, Shevde NK, Pike JW. 1,25‐Dihydroxyvitamin D3 regulates the expression of low‐density lipoprotein receptor‐related protein 5 via deoxyribonucleic acid sequence elements located downstream of the start site of transcription. Mol Endocrinol. 2006;20(9):2215–30. [DOI] [PubMed] [Google Scholar]
- 114. Kriebitzsch C, Verlinden L, Eelen G, et al. 1,25‐dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3‐E1 cells by direct regulation of cystathionine beta‐synthase. J Bone Miner Res. 2011;26(12):2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kaneko I, Sabir MS, Dussik CM, et al. 1,25‐Dihydroxyvitamin D regulates expression of the tryptophan hydroxylase 2 and leptin genes: implication for behavioral influences of vitamin D. FASEB J. 2015;29(9):4023–35. [DOI] [PubMed] [Google Scholar]
- 116. St John HC, Bishop KA, Meyer MB, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol. 2014;28(7):1150–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Han X, Xiao Z, Quarles LD. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF‐23. J Biol Chem. 2015;290(16):10447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Stachowiak MK, Stachowiak EK. Evidence‐based theory for integrated genome regulation of ontogeny—An unprecedented role of nuclear FGFR1 signaling. J Cell Physiol. 2016;231(6):1199–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Saini RK, Kaneko I, Jurutka PW, et al. 1,25‐dihydroxyvitamin D(3) regulation of fibroblast growth factor‐23 expression in bone cells: evidence for primary and secondary mechanisms modulated by leptin and interleukin‐6. Calcif Tissue Int. 2013;92(4):339–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kaneko I, Saini RK, Griffin KP, Whitfield GK, Haussler MR, Jurutka PW. FGF23 gene regulation by 1,25‐dihydroxyvitamin D: opposing effects in adipocytes and osteocytes. J Endocrinol. 2015;226(3):155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ito M, Sakai Y, Furumoto M, et al. Vitamin D and phosphate regulate fibroblast growth factor‐23 in K‐562 cells. Am J Physiol Endocrinol Metab. 2005;288(6):E1101–9. [DOI] [PubMed] [Google Scholar]
- 122. Onal M, Carlson AH, Thostenson JD, et al. A novel distal enhancer mediates inflammation‐, PTH‐, and early onset murine kidney disease‐induced expression of the mouse Fgf23 gene. JBMR Plus. 2018;2(1):32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Lee SM, Carlson AH, Onal M, Benkusky NA, Meyer MB, Pike JW. A control region near the fibroblast growth factor 23 gene mediates response to phosphate, 1,25(OH)2D3, and LPS in vivo. Endocrinology. 2019;160(12):2877–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lammi J, Huppunen J, Aarnisalo P. Regulation of the osteopontin gene by the orphan nuclear receptor NURR1 in osteoblasts. Mol Endocrinol. 2004;18(6):1546–57. [DOI] [PubMed] [Google Scholar]
- 125. Pirih FQ, Tang A, Ozkurt IC, Nervina JM, Tetradis S. Nuclear orphan receptor Nurr1 directly transactivates the osteocalcin gene in osteoblasts. J Biol Chem. 2004;279(51):53167–74. [DOI] [PubMed] [Google Scholar]
- 126. Meir T, Durlacher K, Pan Z, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014;86(6):1106–15. [DOI] [PubMed] [Google Scholar]
- 127. Wilson TE, Fahrner TJ, Johnston M, Milbrandt J. Identification of the DNA binding site for NGFI‐B by genetic selection in yeast. Science. 1991;252(5010):1296–300. [DOI] [PubMed] [Google Scholar]
- 128. Murphy EP, Dobson AD, Keller C, Conneely OM. Differential regulation of transcription by the NURR1/NUR77 subfamily of nuclear transcription factors. Gene Expr. 1996;5(3):169–79. [PMC free article] [PubMed] [Google Scholar]
- 129. Yue CH, Huang CY, Tsai JH, et al. MZF‐1/elk‐1 complex binds to protein kinase Calpha promoter and is involved in hepatocellular carcinoma. PLoS One. 2015;10(5):e0127420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Beck GR Jr, Knecht N. Osteopontin regulation by inorganic phosphate is ERK1/2‐, protein kinase C‐, and proteasome‐dependent. J Biol Chem. 2003;278(43):41921–9. [DOI] [PubMed] [Google Scholar]
- 131. Le Mee S, Fromigue O, Marie PJ. Sp1/Sp3 and the myeloid zinc finger gene MZF1 regulate the human N‐cadherin promoter in osteoblasts. Exp Cell Res. 2005;302(1):129–42. [DOI] [PubMed] [Google Scholar]
- 132. Hmama Z, Nandan D, Sly L, Knutson KL, Herrera‐Velit P, Reiner NE. 1alpha,25‐dihydroxyvitamin D(3)‐induced myeloid cell differentiation is regulated by a vitamin D receptor‐phosphatidylinositol 3‐kinase signaling complex. J Exp Med. 1999;190(11):1583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Moeenrezakhanlou A, Shephard L, Lam L, Reiner NE. Myeloid cell differentiation in response to calcitriol for expression CD11b and CD14 is regulated by myeloid zinc finger‐1 protein downstream of phosphatidylinositol 3‐kinase. J Leukoc Biol. 2008;84(2):519–28. [DOI] [PubMed] [Google Scholar]
- 134. Hii CS, Ferrante A. The non‐genomic actions of vitamin D. Nutrients. 2016;8(3):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Norman AW, Mizwicki MT, Norman DP. Steroid‐hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov. 2004;3(1):27–41. [DOI] [PubMed] [Google Scholar]
- 136. Vertino AM, Bula CM, Chen JR, et al. Nongenotropic, anti‐apoptotic signaling of 1alpha,25(OH)2‐vitamin D3 and analogs through the ligand binding domain of the vitamin D receptor in osteoblasts and osteocytes. Mediation by Src, phosphatidylinositol 3‐, and JNK kinases. J Biol Chem. 2005;280(14):14130–7. [DOI] [PubMed] [Google Scholar]
- 137. Zhang X, Zanello LP. Vitamin D receptor‐dependent 1 alpha,25(OH)2 vitamin D3‐induced anti‐apoptotic PI3K/AKT signaling in osteoblasts. J Bone Miner Res. 2008;23(8):1238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tagliafico E, Tenedini E, Bergamaschi A, et al. Gene expression profile of Vitamin D3 treated HL60 cells shows an incomplete molecular phenotypic conversion to monocytes. Cell Death Differ. 2002;9(11):1185–95. [DOI] [PubMed] [Google Scholar]
- 139. Weber CE, Kothari AN, Wai PY, et al. Osteopontin mediates an MZF1‐TGF‐beta1‐dependent transformation of mesenchymal stem cells into cancer‐associated fibroblasts in breast cancer. Oncogene. 2015;34(37):4821–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Gimba ERP, Brum MCM, Nestal De Moraes G. Full‐length osteopontin and its splice variants as modulators of chemoresistance and radioresistance (Review). Int J Oncol. 2019;54(2):420–30. [DOI] [PubMed] [Google Scholar]
- 141. Zhang S, Shi W, Ramsay ES, et al. The transcription factor MZF1 differentially regulates murine Mtor promoter variants linked to tumor susceptibility. J Biol Chem. 2019;294(45):16756–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chen Y, Zhang Z, Yang K, Du J, Xu Y, Liu S. Myeloid zinc‐finger 1 (MZF‐1) suppresses prostate tumor growth through enforcing ferroportin‐conducted iron egress. Oncogene. 2015;34(29):3839–47. [DOI] [PubMed] [Google Scholar]
- 143. David V, Francis C, Babitt JL. Ironing out the cross talk between FGF23 and inflammation. Am J Physiol Renal Physiol. 2017;312(1):F1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dittmer J. The biology of the Ets1 proto‐oncogene. Mol Cancer. 2003;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Tolon RM, Castillo AI, Jimenez‐Lara AM, Aranda A. Association with Ets‐1 causes ligand‐ and AF2‐independent activation of nuclear receptors. Mol Cell Biol. 2000;20(23):8793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hromas R, Davis B, Rauscher FJ 3rd, et al. Hematopoietic transcriptional regulation by the myeloid zinc finger gene, MZF‐1. Curr Top Microbiol Immunol. 1996;211:159–64. [DOI] [PubMed] [Google Scholar]
- 147. Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84(3):345–57. [DOI] [PubMed] [Google Scholar]
- 148. Ko H, Kim S, Yang K, Kim K. Phosphorylation‐dependent stabilization of MZF1 upregulates N‐cadherin expression during protein kinase CK2‐mediated epithelial‐mesenchymal transition. Oncogenesis. 2018;7(3):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Frey JL, Stonko DP, Faugere MC, Riddle RC. Hypoxia‐inducible factor‐1alpha restricts the anabolic actions of parathyroid hormone. Bone Res. 2014;2:14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Tatsumi S, Segawa H, Morita K, et al. Molecular cloning and hormonal regulation of PiT‐1, a sodium‐dependent phosphate cotransporter from rat parathyroid glands. Endocrinology. 1998;139(4):1692–9. [DOI] [PubMed] [Google Scholar]
- 151. Kimata M, Michigami T, Tachikawa K, et al. Signaling of extracellular inorganic phosphate up‐regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit‐1 and Raf/MEK/ERK pathway. Bone. 2010;47(5):938–47. [DOI] [PubMed] [Google Scholar]
- 152. Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int. 2009;75(9):890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Bon N, Couasnay G, Bourgine A, et al. Phosphate (Pi)‐regulated heterodimerization of the high‐affinity sodium‐dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J Biol Chem. 2018;293(6):2102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Beck GR Jr, Moran E, Knecht N. Inorganic phosphate regulates multiple genes during osteoblast differentiation, including Nrf2. Exp Cell Res. 2003;288(2):288–300. [DOI] [PubMed] [Google Scholar]
- 155. Lacher SE, Levings DC, Freeman S, Slattery M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018;19:401–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. David V, Dai B, Martin A, Huang J, Han X, Quarles LD. Calcium regulates FGF‐23 expression in bone. Endocrinology. 2013;154(12):4469–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Glosse P, Feger M, Mutig K, et al. AMP‐activated kinase is a regulator of fibroblast growth factor 23 production. Kidney Int. 2018;94(3):491–501. [DOI] [PubMed] [Google Scholar]
- 158. Cui X, Gooch H, Petty A, McGrath JJ, Eyles D. Vitamin D and the brain: genomic and non‐genomic actions. Mol Cell Endocrinol. 2017;453:131–43. [DOI] [PubMed] [Google Scholar]
- 159. Zanatta L, Goulart PB, Goncalves R, et al. 1alpha,25‐dihydroxyvitamin D(3) mechanism of action: modulation of L‐type calcium channels leading to calcium uptake and intermediate filament phosphorylation in cerebral cortex of young rats. Biochim Biophys Acta. 2012;1823(10):1708–19. [DOI] [PubMed] [Google Scholar]
- 160. da Silva TS, Harrison K, Uzodike M, et al. Vitamin D actions in neurons require the PI3K pathway for both enhancing insulin signaling and rapid depolarizing effects. J Steroid Biochem Mol Biol. 2020;200:105690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ferreira GB, Vanherwegen AS, Eelen G, et al. Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. 2015;10(5):711–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Induction time‐course of mRNAs encoding bone‐expressed genes following treatment of rat osteocyte‐like UMR‐106 cells with 1,25(OH)2D. (A) cyp24a1, (B) fgf23, (C) spp1, (D) nurr1. Each value is the average of at least three experiments with triplicate biological replicates ± standard deviation.