

SUMMARY
Since mitochondria contribute to tumorigenesis and drug resistance in cancer, mitochondrial genetic engineering promises a new direction for cancer therapy. Here, we report the use of the perimitochondrial enzymatic noncovalent synthesis (ENS) of peptides for delivering genes selectively into the mitochondria of cancer cells for mitochondrial genetic engineering. Specifically, the micelles of peptides bind to the voltage-dependent anion channel (VDAC) on mitochondria for the proteolysis by enterokinase (ENTK), generating perimitochondrial nanofibers in cancer cells. This process, facilitating selective delivery of nucleic acid or gene vectors into mitochondria of cancer cells, enables the mitochondrial transgene expression of CRISPR/Cas9, FUNDC1, p53, and fluorescent proteins. Mechanistic investigation indicates that the interaction of the peptide assemblies with the VDAC and mitochondrial membrane potential are necessary for mitochondria targeting. This local enzymatic control of intermolecular noncovalent interactions enables selective mitochondrial genetic engineering, thus providing a strategy for targeting cancer cells.
Graphical Abstract
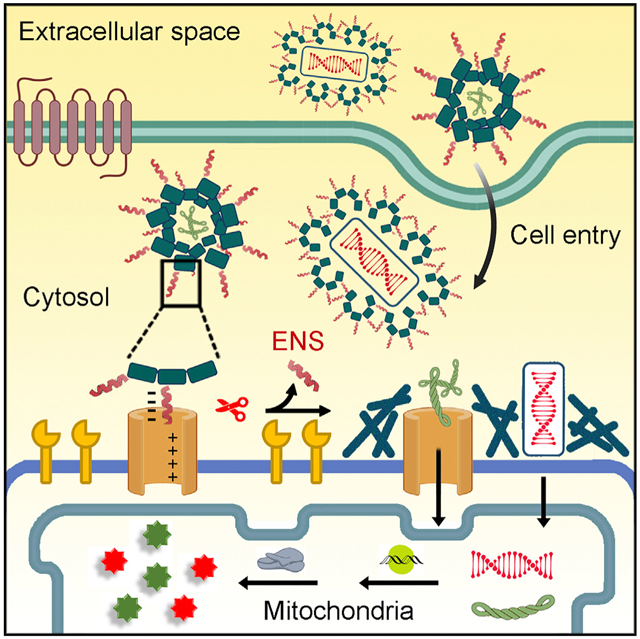
Enzymatic noncovalent synthesis (ENS), a process generating supramolecular interactions and higher-order molecular ensembles via enzymatic reactions, achieves the formation of molecular assemblies at mitochondria, which facilitates gene delivery for mitochondrial genome editing in a cancer-specific manner. Illustrating mitochondrial ENS to deliver various cargos for genetic engineering of cancer mitochondria, this work by He et al. offers a promising strategy for targeting mitochondria of cancer cells and for understanding the correlation between mitochondria and cancer.
INTRODUCTION
Cancer remains a great threat to public health in the world. Traditional cancer treatments, such as chemotherapy, are often accompanied with failures due to the recurrence of cancer, drug resistance, and the nonspecific distribution of anti-cancer agents in human body, which compromise the drug efficacy and bring in undesirable side effects on normal tissue. These challenges require approaches with the ability to differentiate between cancerous and normal cells for efficient cancer inhibition. Mitochondria, as the powerhouse of cells, participate in central roles in multiple cellular processes, especially in bioenergetic, biosynthetic, immunity, and cell death signaling. Thus, selectively targeting mitochondria of cancer cells promises an effective way to reduce drug resistance. Although many approaches have been developed to target mitochondria for cancer therapy,1-8 most of them focus on developing tight binding inhibitors for mitochondria disfunction, which are susceptible to mutations that evolve drug resistance. Thus, new strategies of targeting mitochondria become urgent and necessary.
Human mitochondria, carrying their own DNA, are one of the most important targets for genetic medicine.9,10 Additionally, the protein production machinery in mitochondria plays crucial roles in cellular metabolism,11,12 and it is promising for selectively engineering the mitochondrial genome in cancer cells to manipulate the mitochondrial metabolism for the purpose of cancer inhibition.13 Considerable progress has been made in engineering mitochondria DNA (mtDNA). For example, mitochondria replacement therapy (MRT)14 is able to repair mtDNA defects before birth.15 Re-engineered nucleases are able to edit mitochondrial genomes in cultured cells16,17 and in mouse models.18-20 These approaches, however, require further development for selectively targeting the mitochondria of cancer cells. One rational strategy is to deliver the nucleic acids or gene vectors directly to the mitochondria of cancer cells. Several labs have explored this strategy. Most reported approaches rely on positively charged peptides21 or antibodies22 but exhibit poor efficiency and selectivity.
Inspired by viral proteolytic priming (VPP),23,24 in which proteolytic cleavage changes the state (e.g., conformation, charge, or oligomerization) of viral surface proteins to facilitate viral entry of specific host cells, we decided to investigate perimitochondrial enzymatic noncovalent synthesis (ENS) of peptides for cancer-selective mitochondrial genetic engineering. ENS refers to a process that combines enzymatic reactions and noncovalent interactions for spatiotemporally constructing higher-order molecular assemblies that obtain emergent properties and functions.25-29 Although ENS has found applications in targeting cancer cells for imaging30-33 and tumor inhibition,34-37 it has yet to be explored to deliver cargo for mitochondrial genetic engineering. During our study on enzymatic gelation,38-43 we found that perimitochondrial ENS delivers small molecules or proteins into the mitochondria of cancer cells exclusively and efficiently.44 Therefore, we decided to explore perimitochondrial ENS, as a memetic of VPP, to deliver cargo for mitochondrial genetic engineering of cancer cells, with the goal of manipulating cancer metabolism. As shown in Figure 1B, perimitochondrial ENS is a multiple-step process: (1) micelles of peptide assemblies bind to receptors on the mitochondrial membrane, (2) a protease (e.g., enterokinase [ENTK]) cleaves the peptide to transform the micelles to nanofibers, and (3) such a perimitochondrial morphology/phase transition should allow gene vectors carried by the micelles to be locally positioned to mitochondria.
Figure 1. Illustration of Perimitochondrial ENS.

(A) Molecular structure of Mito-FLAG and the illustration of the ENS catalyzed by ENTK.
(B) Illustration of the proteolysis (ENTK cleaving off the FLAG-tag) of Mito-FLAG to result in supramolecular assemblies of Nap-ffk(G)y and the consequential phase/morphology transition on mitochondria to facilitate the mitochondrial genetic engineering.
Our study shows that perimitochondrial ENS enables selective genetic engineering for the mitochondria of cancer cells. Consisting of an aromatic group rich in D-tetrapeptide as the backbone and FLAG-tag,45 a substrate of ENTK,46 as the branch, the peptide (termed Mito-FLAG in this work) self-assembles into micelles, which, upon ENTK catalytically cleaving off the FLAG-tag, turn into nanofibers to result in a hydrogel. After the Mito-FLAG micelles enter cells mainly via clathrin-mediated endocytosis, the mitochondria-bound ENTK hydrolyzes the FLAG-tag, thus turning the micelles to nanofibers on mitochondria. Using mitochondria-specific codons47 further confirms the gene expression in mitochondria. This perimitochondrial ENS approach transfects gene vectors encoding CRISPR/Cas948,49 into the mitochondria of cancer cells to knock out the MT-CO1 gene, which depletes oxidative phosphorylation (OXPHOS) and re-sensitizes the cancer cells to cisplatin. Additionally, Mito-FLAG facilitates the gene expression of FUNDC1 and GFP-tagged p53 proteins in the mitochondria of cancer cells, which induces mitophagy50 and apoptosis,51 respectively. Besides DNA plasmid, combining viral gene vectors and Mito-FLAG results in the exclusive expression of non-mitochondrial proteins (e.g., GFP, RFP-LAMP1, or GFP-PTS) in cancer mitochondria. Using Rifampicin,52 an inhibitor specific to mitochondrial RNA polymerase, confirms that the delivered genes undergo transcription in mitochondria. Mechanistically, the electrostatic interaction between Mito-FLAG and voltage-dependent anion channel (VDAC) on the mitochondrial surface favors the mitochondria-specific attachment of Mito-FLAG. For the cells with a low level or knockdown of mitochondria-bound ENTK, perimitochondrial ENS is absent; therefore, the production of those non-mitochondrial proteins hardly occurs in the mitochondria. Furthermore, mitochondrial membrane potential and pH gradient are necessary for perimitochondrial ENS to target the mitochondria of the cancer cells. This work, illustrating perimitochondrial ENS as assisting genetic engineering of cancer mitochondria, offers a versatile and robust strategy for selectively targeting mitochondria of cancer cells and for the understanding the correlation between mitochondria and cancer.53
RESULTS AND DISCUSSION
ENS of the Peptide Assemblies and Targeting the Mitochondria of Cancer Cells
As reported by previous results,38,44 the FLAG-tagged branched peptide (denoted as Mito-FLAG; Figure 1A) self-assembles into micelles (18 ± 3 nm in diameter) in solution, which transform to nanofibers (27 ± 2 nm in diameter) in response to ENTK (Figure S1A). The nitrobenzoxadiazole (NBD)-labeled derivative of Mito-FLAG (denoted as NBD-Mito-FLAG; Scheme S1) not only extensively colocalizes with the mitochondria of cancer cells rather than the mitochondria of normal cells, but it also exhibits mitochondria-like morphology in cancer cells (Figure S1B). Compared to previous works21,22 in which the fluorophore-labeled mitochondria-targeting agents merely show punctate intracellular fluorescence that overlaps with mitochondria, the Mito-FLAG assemblies exhibit higher efficiency for targeting mitochondria.
Moreover, transmission electron microscopy (TEM) images confirm the micelle-to-nanofiber transition on the mitochondria of cancer cells in 24 h (Figure S1C). These results confirm that Mito-FLAG is a suitable perimitochondrial ENS candidate with cancer selectivity. The nanofibers on the mitochondria surface would increase the perimitochondrial viscosity for the retention of various entities, such as proteins,54 chemotherapy drugs,44 and gene vectors (as shown in this work). While the formation of perimitochondrial molecular assemblies is confirmed by the TEM images of the isolated whole mitochondria, it is unknown whether Mito-FLAG micelles undergo ENS inside the mitochondria. The fluorescence of NBD-Mito-FLAG in mitochondria, however, suggests the formation of molecular assemblies inside mitochondria (Figure S1) catalyzed by other mitochondrial proteases. The NBD-Mito-FLAG inside mitochondria may originate from mitochondrial protein importing machinery, but it remains to be confirmed in further study.
Delivery of Nucleic Acids into Mitochondria
As shown in Figure 1B, the nanofibers on mitochondria (Figure S1C) should increase the local viscosity, arrest the DNA cargo near the mitochondria, and promote the mitochondria-targeted gene delivery. This assumption is supported by the import of fluorescein isothiocyanate (FITC)-labeled oligonucleotides and a plasmid backbone (pMAX-DEST)55 into mitochondria. Being incubated with FITC-labeled oligonucleotides and Mito-FLAG, HeLa cells display green fluorescence mainly in mitochondria (Figure 2A) but only scattering fluorescent dots in the cytosol in the absence of Mito-FLAG (Figure S2A). We used ethidium bromide (EB)56 to generate the mtDNA-depleted (ρ°) cells. PicoGreen,57 a fluorescent dye for DNA detection, confirms that EB effectively removes mtDNA (Figures S2C-S2E). The ρ° cells incubated with the mixture of Mito-FLAG and pMAX-DEST (20 μg/mL) show multiple green fluorescent spots overlapping with MitoTracker (Figures 2B and S2E), validating that Mito-FLAG delivers DNA into mitochondria. Incubating the ρ° cells with the plasmid alone or plasmids mixed by lipofectamine hardly regains the DNA in mitochondria (Figures 2B and S2C-S2E). Imaging analysis of the mitochondria regions indicates that, on average, 89% of mitochondria contain DNA (stained by PicoGreen) after treating the ρ° cells with the pMAX plasmid and Mito-FLAG (Figures S3A and S3B). This ratio is close to that of normal HeLa cells (98%; Figures S3C and S3D). This result further supports that Mito-FLAG acts as a substrate for perimitochondrial ENS to deliver DNA into the mitochondria.
Figure 2. Perimitochondrial ENS to Deliver Oligonucleotides, Nucleic Acids, and Plasmids into Mitochondria.

(A) HeLa cells, incubated with Mito-FLAG and FITC-labeled oligonucleotides (100 nM, 24 h), display green fluorescence of FITC in mitochondria.
(B) Fluorescent images of mtDNA-depleted HeLa cells incubated with 1 mL pMAX-DEST plasmid (10 μg/mL, 24 h) mixed with or without Mito-FLAG.
(C) Gel electrophoresis analysis of pGLO plasmid in the pellet of Mito-FLAG after high-speed centrifuge.
(D) Fluorescent images of the HeLa cells incubated with pGLO and Mito-pGLO plasmid (5 μg/mL, 24 h, 0.2 wt% arabinose) in the presence of Mito-FLAG.
(E) Detection of pGLO plasmid in the mitochondria isolated from the HeLa cells treated by pGLO (5 μg/mL) and Mito-FLAG via PCR and DNA electrophoresis.
(F) Fluorescent images of the HeLa cells expressed pGLO (in mitochondria) before and after one passage. All Mito-FLAG concentrations are 200 μM.
Transfection of DNA Plasmids in Mitochondria
To validate the encapsulation of DNA plasmid in Mito-FLAG assemblies, we centrifuged the mixtures of Mito-FLAG and pGLO plasmid58 (containing GFP reporter gene). Electrophoresis shows that the pellets obtained from the mixtures of pGLO and Mito-FLAG at 400 or 200 μM clearly incorporate pGLO (Figure 2C, lanes 3 and 4). Centrifuging the solution of free pGLO hardly obtained any DNA pellets (Figures 2C, lane 5, and S4C-S4E). These results confirm that pGLO plasmid associates with the Mito-FLAG assemblies. Gel analysis using ImageJ reveals that the ratios of band intensity for lane 3/lane 2 and lane 4/lane 2 are 43% and 32%, respectively, which agrees with the encapsulation efficiency (EE%) (38.5% and 29.0%, respectively) quantified via PicoGreen methodology (Figure S4). The EE% of DNA hardly changes in fetal bovine serum (FBS; with DNase inhibitor) within 24 h (Figure S4F), suggesting that the complexes formed by pGLO plasmid and Mito-FLAG are stable in physiological condition. Moreover, the EE% of DNA increases in the solutions of high ionic strength (Figure S4G), which indicates not only that the Mito-FLAG-DNA complexes are stable at high ionic strength, but also that Mito-FLAG assemblies associate with more DNA at high ionic strength.
The HeLa cells treated by the mixture of pGLO plasmid and Mito-FLAG in the presence of arabinose (0.2 wt%) display bright green fluorescence of GFP in mitochondria, as confirmed by the extensive overlapping with the red fluorescence from MitoTracker (Figure 2D). The control (pGLO mixed with lipofectamine) displays only a few fluorescent puncta, mainly in the cytosol (Figure S5A). To validate that the GFP is expressed in mitochondria rather than being trafficked into mitochondria after the protein expression by cytosolic ribosomes, we created a mutated pGLO plasmid (Mito-pGLO) that uses mitochondrial-specific codons (TGA for tryptophan and AGG for the stop; Figure S5B) in the GFP gene. Thus, the GFP encoded by Mito-pGLO should be expressed only in mitochondria. Like pGLO, the HeLa cells incubated with the mixture of Mito-pGLO and Mito-FLAG exhibit green fluorescence that extensively overlaps with the red signal from MitoTracker (Figure 2D). The high expression of GFP in mitochondria also results in the export of some GFP from mitochondria59 into the cytosol (Figure 2D), indicating the efficient transfection achieved by Mito-FLAG-based perimitochondrial ENS. As a control, the intracellular delivery of Mito-pGLO using lipofectamine to the HeLa cells generates few fluorescent puncta in mitochondria (Figure S5A). These results confirm that the perimitochondrial ENS delivers the pGLO or Mito-pGLO gene into the mitochondria of HeLa cells. Polymerase chain reaction (PCR) of the isolated DNA (Figure 2E) from the mitochondria of the HeLa cells further proves that Mito-FLAG efficiently delivers DNA plasmid to mitochondria for gene expression. The GFP fluorescent intensity in the mitochondria of transfected HeLa cells decreases (Figure 2F) after one passage of the cells. Although it indicates that the transfection is largely transient, this result further confirms the transfection of the pGLO gene to the mitochondria of the HeLa cells by Mito-FLAG perimitochondrial ENS, as illustrated in Figure 1.
CRISPR/Cas9 for Knocking Out MT-CO1 in the mtDNA
Since the mitochondrially encoded proteins, such as mitochondrial cytochrome c oxidase subunit I (MT-CO1), are critical for OXPHOS, knocking out the proteins would disrupt mitochondrial energetics, reduce ATP supply, and dysfunction efflux pumps, which are essential for drug resistance in cancer. This assumption is supported by the Mito-FLAG-facilitated delivery of LentiCRISPRV2 plasmid encoding CRISPR/Cas9 and the guide RNA (gRNA) of MT-CO1 (termed CRISPR-MT-CO1; Figure S6A) into mitochondria. To confirm the delivery and gene expression in mitochondria, we incubated HeLa cells with the mixture of Mito-FLAG and CRISPR-MT-CO1-mCherry plasmid, which encodes mCherry as the indicator of gene expression, in addition to CRISPR/Cas9 and the gRNA of MT-CO1. As shown in Figure 3A, intensive fluorescence from mCherry presents in the mitochondria of HeLa cells incubated with Mito-FLAG and CRISPR-MT-CO1-mCherry plasmid, validating the mitochondria-targeted gene delivery via Mito-FLAG perimitochondrial ENS and the gene expression in mitochondria. To examine the efficiency of mitochondria genetic engineering through CRISPR/Cas9, we evaluated the protein level of MT-CO1 in cells. Western blot and immunofluorescence staining analysis reveal a significantly reduced MT-CO1 level in the HeLa cells incubated with the mixture of Mito-FLAG and CRISPR-MT-CO1 plasmid (third day), compared to those treated by solvent control (PBS), free plasmid, or Mito-FLAG alone (Figures 3B and S6B). Moreover, PCR experiments (starting with equal dosages of whole-cell DNA) using the primers flanking the targeted cleavage site generate drastically decreased product from the HeLa cells that undergo the incubation with Mito-FLAG and CRISPR-MT-CO1 plasmid (third day; Figure 3C). However, the PCR products from the cells treated by PBS, plain plasmid, or Mito-FLAG are mostly identical (Figure 3C). Replacing the gRNA in CRISPR-MT-CO1 plasmid with random gRNA hardly results in the knockout (KO) of MT-CO1 (Figure S6C). These results suggest that the Mito-FLAG perimitochondrial ENS delivers the CRISPR-MT-CO1 plasmid into the mitochondria of HeLa cells, where the Cas9 protein and gRNA are made. The CRISPR/Cas9 complex in mitochondria cleaves the target gene, leading to the KO of MT-CO1.
Figure 3. Perimitochondrial ENS Delivers CRISPR/Cas9 Components into the Mitochondria of Cancer Cells for Mitochondrial Genome Editing.

(A) Fluorescent images of HeLa cells incubated with plasmid encoding CRISPR-Cas9-mCherry and siRNA for MT-CO1. The red fluorescence from mCherry in mitochondria suggests the gene delivery and expression in mitochondria.
(B) Western blot analysis of MT-CO1 for HeLa cells treated by solvent control (PBS), free CRISPR-MT-CO1 plasmid, Mito-FLAG, and the mixture of Mito-FLAG and CRISPR-MT-CO1 plasmid. The reduced level of MT-CO1 protein in the combination treatment suggest the KO of the MT-CO1 gene.
(C) PCR analysis of the MT-CO1 gene for HeLa cells treated by solvent control (PBS), free CRISPR-MT-CO1 plasmid, Mito-FLAG, and the mixture of Mito-FLAG and CRISPR-MT-CO1 plasmid. The band of MT-CO1 gene in the combination group is hardly observed.
(D) ATP (red) visualization in HeLa cells incubated with solvent control (PBS), free CRISPR-MT-CO1 plasmid, Mito-FLAG, and the mixture of Mito-FLAG and CRISPR-MT-CO1 plasmid. The cells in the combination group exhibit little fluorescence from ATP-Red in mitochondria, indicating the disfunction of OXPHOS (MT-CO1 is the catalytic site of cytochrome c oxidase).
(E–G) The KO of MT-CO1 (E) leads to reduced efficiency in MDR, (F) sensitizes the cancer cell to cisplatin, and (G) reduces mitochondrial membrane potential. Data are presented as mean ± standard deviation.
Because MT-CO1 plays a critical role in OXPHOS, the KO of MT-CO1 in HeLa cells results in deficient synthesis of ATP (visualized by an ATP probe; Figure 3D) in the mitochondria. The ATP insufficiency decreases the efficiency of the ATP-dependent drug efflux pumps and multidrug resistance (MDR) in cancer cells (Figures 3E and S6D). Since the MDR is a major factor in the failure of many forms of chemotherapy, the MT-CO1-KO cancer cells, being MDR ineffective, become more sensitive to chemotherapeutic agents, such as cisplatin, than the wild type (WT; Figure 3F). Moreover, because the maintenance of the mitochondrial membrane potential largely relies on cytochrome c oxidase,60 the deletion of the subunit 1 in this protein also generates less polarized mitochondria in cells (Figures 3G and S6E).
Mitochondrial Transgene Expression of FUNDC1 to Induce Mitophagy
Because the overexpression of FUN14 domain containing 1 (FUNDC1) in mitochondria results in mitophagy,50 we incubated HeLa cells with a mixture of Mito-FLAG and the plasmids encoding Myc-tagged FUNDC1 protein (FUNDC1-Myc)50 in mitochondria for inducing mitophagy. Western blot experiments reveal that the HeLa cells incubated with the mixture of Mito-FLAG (200 μM) and FUNDC1 plasmid (5 μg/mL, 3 days) exhibit higher FUNDC1 and LC3B (a maker protein for autophagosome) levels in the mitochondrial and cytosolic fractions, respectively (Figure 4A), compared to the controls, indicating the gene expression of FUNDC1 plasmid in mitochondria and the initiation of mitophagy in cells. Fluorescent microscopy reveals elongated mitochondria in HeLa cells treated by Mito-FLAG only but globular mitochondria in the cells incubated with the mixture of Mito-FLAG and FUNDC1-Myc plasmids (Figure 4B), suggesting the engulfment of mitochondria by autophagosomes.61 Immunochemical staining of LC3B in FUNDC1-plasmid-transfected HeLa cells (via Mito-FLAG perimitochondrial ENS) clearly shows the redistribution of nucleus-pool LC3 to the cytoplasm62 and the co-localization with mitochondria (Figure 4C, indicated by arrows), confirming mitophagy. Given that FUNDC1 associates with LC3 on the mitochondrial outer membrane (MOM) for mitophagy, these results also suggest that the FUNDC1-Myc protein, although produced in the mitochondrial matrix, can move out to the MOM via protein export,59 where it interacts with LC3. Little co-localization between LC3 and mitochondria presents in the control HeLa cells (Figure 4C).
Figure 4. Perimitochondrial ENS for Mitochondrial Transgene Expression to Induce Mitophagy or Apoptosis.

(A) Western blot analysis of FUNDC1 and LC3B levels in the mitochondria fraction and cytosolic fraction, respectively, of HeLa cells incubated with conditions of interest. VDAC1 serves as mitochondria protein loading control. FUNDC1 plasmid, 5 μg/mL; Mito-FLAG, 200 μM. Time: 2 days.
(B) Mito-FLAG perimitochondrial ENS delivers the FUNDC1 gene into mitochondria to induce mitochondrial morphology changes within 2 days.
(C) Detection of mitophagy via immunofluorescent staining of autophagy marker LC3 (second day).
(D) Fluorescent images of HeLa cells incubated with free GFP-TP53 plasmid and the plasmid mixed with Mito-FLAG (200 μM, 24 h).
(E) Cell viability of HeLa, Saos-2, and HS-5 cells incubated with free GFP-TP53 plasmid (5 μg/mL), Mito-FLAG (200 μM), and the plasmid (5 μg/mL) mixed with Mito-FLAG (200 μM, third day). Data are presented as mean ± standard deviation.
(F and G) Apoptosis analysis of HeLa (F) and HS-5 (G) cells incubated with GFP-TP53 plasmid (5 μg/mL) mixed by Mito-FLAG (200 μM, third day) via PI staining.
Mitochondrial Transgene Expression of p53 to Induce Apoptosis
Tumor suppressor p53 is a protein that directly participates in the intrinsic apoptosis pathway by inducing mitochondrial outer membrane permeabilization. Herein, we transfect the plasmid encoding GFP-tagged p53 (GFP-TP53)63 into mitochondria for imaging mitochondrial p53 and direct apoptosis initiation. HeLa cells incubated with the mixture of GFP-TP53 plasmid and Mito-FLAG (24 h) exhibit bright green fluorescence in mitochondria (Figure 4D), suggesting the import of GFP-TP53 plasmid and the generation of p53 protein in mitochondria. In contrast, HeLa cells treated by free plasmid show sporadic puncta in the cytosol with no colocalization with the mitochondria (Figure 4D). Generally, HeLa cells escape from p53-induced apoptosis via E6 protein-mediated p53 degradation.64 After the mitochondrial transfection of GFP-TP53 plasmid, the viability of HeLa cells reduces significantly (Figure 4E), which agrees with the apoptosis analysis via propidium iodide (PI; Figure 4F), suggesting the p53-induced apoptosis. Incubating free GFP-TP53 plasmid or Mito-FLAG with HeLa cells hardly causes cell death (Figures 4E, 4F, and S7A). Western blot analysis shows more cytosolic cytochrome c (cyt c) in the transfected HeLa cells (by Mito-FLAG, third day) than in the control cells (Figure S7B). These results agree with the p53-induced mitochondrial permeabilization and cyt c release in the intrinsic apoptosis pathway.65 The perimitochondrial ENS of Mito-FLAG also delivers GFP-TP53 plasmid into the mitochondria of Saos-2 cell (Figure S7C), another TP53 null cancer cell line, which induces significant cell death (Figures 4E and S7D), although free GFP-TP53 plasmid and Mito-FLAG are innoxious (Figures 4E and S7E). Since the association of p53 with the proteins from the bcl-2 family at the MOM is essential for triggering mitochondrial permeabilization, again, the cell death suggests that the mitochondrially encoded p53 may move out, via protein export,59 to the MOM. On the contrary, neither free GFP-TP53 plasmid nor the mixture of Mito-FLAG and GFP-TP53 plasmid impairs the viability of HS-5 cells (Figures 4E and 4G), which agrees with the observed specificity of Mito-FLAG perimitochondrial ENS for cancer cells (Figures 5 and 6).
Figure 5. Perimitochondrial ENS Combines with Viral Gene Vectors for Mitochondrial Transgene Expression.

(A) TEM images of free baculovirus.
(B) TEM images of the mixture of baculovirus and Mito-FLAG micelles (arrow) before and after adding ENTK.
(C) Fluorescent images of HeLa cells incubated with baculoviral RFP-LAMP1 for 24 h.
(D) Fluorescence from RFP-LAMP1 in HeLa cells treated by the mixture of baculovirus and Mito-FLAG localizes in mitochondria. Mitochondria are stained by MitoTracker.
(E) Western blot of RFP-LAMP1 protein in mitochondria isolated from the HeLa cells treated by baculoviral RFP-LAMP1 with or without Mito-FLAG for 24 h.
(F) Fluorescent images of virus-transfected (RFP-LAMP1) HeLa cells before and after cell passage.
(G) Localizations of RFP-LAMP1 in the mitochondria of the HeLa cells were treated by processes 1 and 2. Red, RFP; green, MitoTracker.
(H) Rifampycin inhibits the protein expression of RFP-LAMP1 in mitochondria. Red, RFP; green = MitoTracker.
(I–K) Saos-2, HepG2, and HS-5 cells are incubated with baculoviral RFP-LAMP1 mixed with Mito-FLAG. All Mito-FLAG concentrations are 200 μM.
Figure 6. Mechanistic Insights.

(A) The cellular uptake of NBD-Mito-FLAG (200 μM for 2 h, a fluorescent analog of Mito-FLAG) without and with CPZ (5 μM, clathrin-dependent endocytosis inhibitor).
(B) Confocal fluorescence images of HeLa cells incubated with NBD-Mito-FLAG (200 μM) and chloroquine (10 μM).
(C) Western blot analysis of ENTK in mitochondria isolated from HeLa and HS-5 cells confirms higher ENTK expression in HeLa than in HS-5 cells.
(D) TEM images of mitochondria isolated from HeLa, HEK293, and HS-5 cells that incubated with 200 μM Mito-FLAG for 24 h. Results indicate that ENS-induced phase transition on mitochondria is critical for mitochondria-specific gene transfection.
(E and F) Fluorescent images of NBD-Mito-FLAG and RFP-LAMP1 in ENTK-knockdown HeLa cells. The reduced mitochondrial fluorescence and colocalization (indicated by arrows) suggest that ENTK is essential for the mitochondrial accumulation of FLAG-tagged peptides.
(G and H) Fluorescent images of the HeLa cells incubated with (G) NBD-Mito-FLAG (200 μM, 2 h) or (H) the mixture of virus (RFP-LAMP1) and Mito-FLAG (200 μM, 24 h) in the presence of erastin of different concentrations. The non-mitochondria fluorescence (lysosome) in (H) is indicated by a white arrow.
(I) Fluorescent images of HeLa cells incubated with NBD-Mito-FLAG (200 μM, 2 h) after the pretreatment by Ndz (10 μM, 1 h).
Perimitochondrial ENS Combining with Viral Vectors for Mitochondrial Transgene Expression
We used Mito-FLAG to pack baculovirus vectors because liposomes enhance transfection of viral vectors.66 While the TEM of the solution of baculovirus shows rod-like viruses (Figure 5A), the TEM of the mixture reveals that the micelles made of Mito-FLAG adhere to the viruses, generating clusters of Mito-FLAG-coated virus (Figure 5B). Upon the addition of the enzyme ENTK, the Mito-FLAG-virus clusters break into uncoated viruses entangled by nanofibers of 6 ± 1 nm (Figure 5B). Being incubated with the baculovirus encoding RFP-LAMP1 or GFP-PTS, the HeLa cells display fluorescence only in the intended organelles (i.e., lysosome or peroxisome, respectively) (Figures 5C and S8A). However, the transfection with Mito-FLAG perimitochondrial ENS results in the fluorescence of RFP-LAMP1 or GFP-PTS largely overlapping with the fluorescence from MitoTracker (Figures 5D and S8A), though some fluorescence appears in the cytosol, likely due to the baculovirus without encapsulation by Mito-FLAG or protein export from mitochondria.59 Moreover, Mito-FLAG results in more cells to express RFP-LAMP1 or GFP-PTS compared to the control (free baculoviral vectors; Figures S8B and S8C), indicating efficient transfection achieved with Mito-FLAG perimitochondrial ENS. Western blot analysis of the proteins in the isolated mitochondria (Figure 5E) confirms that the mitochondrial expression of RFP-LAMP1 occurs only with the baculoviral vectors plus Mito-FLAG perimitochondrial ENS.
Both the fluorescence of GFP-PTS and RFP-LAMP1 in mitochondria of the HeLa cells decreases after subculture (Figures 5F and S8D), with more significant reduction for GFP-PTS—indicating the dilution of the baculoviral DNAs in mitochondria during cell proliferation—and a higher proteolytic resistance for RFP-LAMP1 than GFP-PTS against mitochondrial proteases. We also treated the cells by two different orders: (1) concurrent incubation and (2) sequential incubation. As shown in Figure 5G, while the concurrent incubation achieves the gene expression in mitochondria, the sequential incubation results in the localization of GFP-PTS (Figure S8E) and RFP-LAMP1 merely in their intended organelles. These results eliminate the possibility that the non-mitochondrial proteins observed in mitochondria originate from the Mito-FLAG-induced protein redistribution to mitochondria after normal gene expression (in the cytosol or endoplasmic reticulum [ER]).54
Moreover, rifampicin (80 μg/mL), a mitochondrial RNA polymerase inhibitor,52 significantly weakens the mitochondrial fluorescence of HeLa cells treated by Mito-FLAG mixed with baculovirus (RFP-LAMP1) or pGLO plasmid (Figures 5H, S9A, and S9B) but hardly affects the cell viability and protein expression in cells undergoing conventional transfection (Figures S9C and S9D), confirming that the genes delivered by perimitochondrial ENS undergo transcription in mitochondria.
Besides baculoviral vectors, the combination of Mito-FLAG with adeno-associated virus (AAV) or lentivirus (LTV) encoding GFP or RFP, respectively, also results in mitochondrial specific gene expression (Figure S10). Without the addition of Mito-FLAG, the expressed GFP diffuses in entire cells. These results suggest that the Mito-FLAG perimitochondrial ENS is suitable for a variety of gene vectors for mitochondria-targeting transfection.
Mitochondrial Transgene Expression in a Cell-Specific Manner
We applied the Mito-FLAG for mitochondrial transgene expression in Saos-2, HepG2, HS-5, and HEK293 cells. Like the observation in HeLa cells, cells incubated with the baculoviral vectors alone produce fluorescence at the intended organelles of the encoding proteins (i.e., RFP-LAMP1 and GFP-PTS at lysosome and peroxisome, respectively) (Figure S11). Notably, mixing Mito-FLAG with the baculoviral vectors results in the gene expression exclusively in the mitochondria of two cancer cell lines, Saos-2 and HepG2 cells (Figures 5I and 5J). For the two noncancerous cell lines, HS-5 (Figures 5K and S12) and HEK293 (Figure S12), mixing Mito-FLAG with the gene vectors (virus or plasmid) hardly yields mitochondrial-exclusive gene expression. These results indicate that the Mito-FLAG perimitochondrial ENS enables mitochondrial transgene expression in a cancer-cell-specific manner.
Endocytosis and Endosomal Escape
While M-β-CD67 hardly affects the uptake of NBD-Mito-FLAG (Figure S13), chlorpromazine (CPZ)68 significantly decreases the intracellular fluorescence of NBD-Mito-FLAG, indicating that the uptake of Mito-FLAG mainly relies on clathrin-dependent endocytosis (Figure 6A). The pretreatment of chloroquine, an inhibitor of endosomal acidification, results in significantly reduced mitochondrial localization of NBD-Mito-FLAG in HeLa cells (Figure 6B), indicating that the pH-buffering effect69 of the carboxylic groups in FLAG-tag facilitates the endosomal escape.
Specificity to Cancer Cells
We examined the amount of ENTK on the mitochondria isolated from HeLa, HS-5, and HEK293 cells (Figures 6C, S14A, and S14B). ENTK presents in the mitochondria isolated from these three cell lines, but HeLa mitochondria have much a higher level of ENTK than those of HS-5 and HEK293 cells. This result agrees with the cancer-cell specificity of the mitochondria-localized nanoparticle-to-nanofiber transition (Figure 6D) and gene transfection. We generated ENTK-knockdown HeLa cells via small interfering RNA (siRNA)-mediated gene silencing. The knockdown of mitochondrial ENTK in HeLa cells (Figures S14C and S14D) substantially reduces the accumulation of NBD-Mito-FLAG in the mitochondria of the HeLa cells (Figure 6E), compared to the cells treated by the siRNA for non-target control. Besides, the ENTK-knockdown HeLa cells yield much more non-mitochondrial fluorescence after Mito-FLAG-assisted baculoviral transfection compared to those in the control (Figure 6F), suggesting a diminished mitochondrial specificity.
Interacting with Mitochondrial Surface
Besides enzymatic conversion of the micelles of Mito-FLAG to nanofibers, the electrostatic interaction between Mito-FLAG and the VDAC on the mitochondrial surface favors the mitochondria-specific attachment, similar to the tubulin-VDAC binding through the negatively charged C-terminal tails of tubulin.70 Two experiments, indeed, indicate that blocking the VDAC antagonizes the Mito-FLAG from approaching mitochondria: Erastin, a VDAC-targeting blocker, decreases the accumulation of NBD-Mito-FLAG and reduces the localization of RFP-LAMP1 (transfected by Mito-FLAG) in mitochondria (Figures 6G and 6H). Cells incubated with nocodazole (Ndz), a microtubule destabilizer to increase free tubulin, exhibit weakened mitochondrial fluorescence from NBD-Mito-FLAG (Figure 6I). Moreover, the pretreatment of FCCP,71 a drug that dissipates mitochondrial membrane potential and proton gradient, diminishes the mitochondria-specific localization and gene transfection by Mito-FLAG perimitochondrial ENS (Figure S15), indicating that mitochondrial membrane potential and proton gradient contribute to the mitochondrial targeting of Mito-FLAG perimitochondrial ENS.
Acting differently than the cationic-cell-penetrating peptides,72 Mito-FLAG, as an enzyme-responsive, negatively charged peptide, forms micelles and undergoes ENTK-induced morphology/phase transition on mitochondria. Such a perimitochondrial ENS process promotes DNA import into mitochondria. Notably, a local enzymatic reaction converting micelles of Mito-FLAG to nanofibers for mitochondrial targeting sidesteps the need for mitochondrial protein import sequence (MTS). Additionally, the cancer-cell-specific mitochondria DNA import via the Mito-FLAG perimitochondrial ENS implies a new non-genetic difference in the mitochondria between cancer and normal cells. This strategy promises the development of new genetic therapeutics for cancer. Thus, the stability of the Mito-FLAG micelles in more complex environments, such as in vivo, is worth further study in future works. Illustrating perimitochondrial ENS for facilitating the genetic engineering of mitochondria in cancer, this work offers a versatile and robust strategy for selectively targeting the mitochondria of cancer cells and for manipulating the metabolism of cancer cells.
EXPERIMENTAL PROCEDURES
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bing Xu (bxu@brandeis.edu).
Materials Availability
This study did not generate new unique materials.
Data and Code Availability
All the key data supporting the findings of this study are presented within the article and the Supplemental Information.
Peptide Synthesis
All amino acid derivatives involved in the synthesis were purchased from GL Biochem (Shanghai) Ltd. The synthesis of all peptide fragments was based on solid-phase peptide synthesis (SPPS). The branched peptides were made via the combination of SPPS and liquid phase synthesis.44 All crude compounds were purified by high-performance liquid chromatography (HPLC) with the yield of 70%–80%.
Cell Culture
HeLa, HepG2, Saos-2, and HS-5 cells were purchased from American Type Culture Collection (ATCC). HEK293 cells were provided by Prof. Chris Miller from Brandeis University. All the cell lines used in this work were authenticated by CellCheck 9-human (9 Marker STR Profile and Inter-species Contamination Test, IDEXX), confirming a 100% match of the cell identity. HeLa, HepG2, and HEK293 cells were cultured in minimum essential medium (MEM) supplemented with 10% (vol/vol) FBS, 0.5% (vol/vol) penicillin (10,000 unit), and 0.5% (vol/vol) streptomycin (10,000 unit). Saos-2 cells were cultured in McCoy’s 5A with l-glutamine supplemented with 15% (vol/vol) FBS, 10% (vol/vol) FBS, 0.5% (vol/vol) penicillin (10,000 unit), and 0.5% (vol/vol) streptomycin (10,000 unit). HS-5 cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% (vol/vol) FBS, 0.5% (vol/vol) penicillin (10,000 unit), and 0.5% (vol/vol) streptomycin (10,000 unit). All cells were maintained at 37°C in a 5% CO2 incubator.
Cell Viability Assay
All cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay via standard protocol.73
Plasmid Import
For Mito-FLAG-assisted delivery of plasmids (pGLO58; [Bio-Rad], Mito-pGLO [ordered from Invitrogen; the final construct was verified by sequencing], pMAX-DEST [Addgene, CAT#: 37631], FUNDC1-Myc ([OriGene, CAT#: RC208211], and GFP-TP53 [Addgene, CAT# 12091]), the plasmids were dissolved in culture medium (with 10% FBS) followed by the addition of Mito-FLAG. The mixtures were incubated in room temperature for 20 min before adding to cells (already adhered). Lipofectamine 3000 (Invitrogen) was used for the general transfection of plasmids following the manufacturer’s instruction. Arabinose (0.2 wt%) was added in the delivery of pGLO and Mito-pGLO. Cells were incubated at 37°C in a 5% CO2 incubator before further analysis. Unless specially mentioned, the final concentration of Mito-FLAG is 200 μM. The dose of plasmids is 5 μg per 80,000 cells. The recommended concentration of plasmid (in medium) is 5 μg/mL. The incubation time is 48 h.
Virus Import
For Mito-FLAG-assisted delivery of viral vectors (baculovirus74,75; [Thermo Fisher Scientific, CAT#C10604 and CAT#C10597], AAV [Addgene, CAT#: 37825-AAV5], and LTV [abm, CAT# LVP691]), the viruses were mixed in culture medium (with 10% FBS) followed by the addition of Mito-FLAG. The mixtures were incubated at room temperature for 20 min before adding to cells (already adhered). Following the manufacturer’s protocol, cells were directly incubated with the virus in culture medium for the general transfection. Cells were incubated at 37°C in a 5% CO2 incubator before further analysis. Unless specially mentioned, the final concentration of Mito-FLAG is 200 μM.
Immunofluorescence
Cells were plated on confocal dishes (CellVis) and fixed in 4 wt% paraformaldehyde for 15 min and permeabilized with 1% BSA and 0.1% Tween 20. Fixed cells were incubated in primary antibody at 4°C overnight, washed three times for 5 min each, incubated in secondary antibody for 1 h, and washed three times for 5 min each.
Confocal Microscopy
In total, 80,000 cells were seeded in 32-mm confocal dishes (CellVis) and incubated in a cell incubator at standard condition for 24 h for adherence. After being treated by the condition of interest, cells were analyzed by ZEISS LSM 880 Confocal Microscope, and images were taken.
Intracellular Fluorescence Quantification
Cells were seeded in Petri dishes and incubated in standard condition for 12 h before experiment. After being treated by the condition of interest, all cells were washed by PBS (three times for 5 min). After the washing step, cells were detached from the dishes using trypsin. The trypsin was removed by centrifuge and washing with PBS. The cell suspensions (in PBS) were diluted to 105 cell/mL. Then, 100 μL diluted cell suspension was added in a 96-well plate (at least 5 wells per sample). The intracellular fluorescence intensity was immediately determined using a DTX 880 Multimode Detector (Beckman Coulter Inc.). The experiments were repeated, and the intracellular fluorescence intensity was averaged (n > 3).
TEM
Samples of interest were dropped on copper grids and dried. Uranyl acetate was used as negative stain. All TEM images were taken by FEI Morgagni Transmission Electron Microscope.
Mitochondria Isolation
Mitochondria were isolated from cells treated by conditions of interest using a mitochondria isolation kit (Thermo Fisher Scientific, CAT#89874) following the manufacturer’s instruction.
Extraction of Mitochondrial DNA
The mitochondrial DNA was extracted by a mitochondrial DNA isolation kit (Abcam, CAT#ab65321) following the manufacturer’s protocol.
Western Blots of Mitochondrial Proteins
Mitochondria were isolated from cells treated by conditions of interest using a mitochondria isolation kit (Thermo Fisher Scientific). Total mitochondrial protein extracts were prepared in lysis buffer (Cell Signaling Technology, with 1X protease inhibitor cocktail [Abcam]) followed by five freeze-thaw circles and 30 min of sonication. Protein concentration was determined by the Coomassie Blue method. Protein extracts (20 μg per lane) were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Western blotting was performed according to standard protocols. Gel analysis was conducted using ImageJ. All antibodies were purchased from Abcam.
ENTK Gene Knockdown
For each transfection, the siRNA duplex was diluted into 100 μL antibiotic-free and FBS-free siRNA transfection medium (Santa Cruz Biotechnology, sc-36868). The recommended final siRNA concentration is 0.1 μM. For each transfection, 6 μL of siRNA transfection reagent (Santa Cruz Biotechnology, sc-29528) was diluted into 100 μL siRNA transfection medium. The siRNA duplex solution was mixed directly into the dilute transfection reagent using a pipette. The mixture was incubated at room temperature for 30 min. Then, 0.8 mL siRNA transfection medium was added into the 0.2 mL mixture of siRNA and transfection reagent (total volume 1 mL). Cells (80,000 per transfection, already adhered) were washed once with 2 mL of siRNA transfection medium and incubated with the 1-mL mixture of siRNA and transfection reagent for 8 h at 37°C in a CO2 incubator. Then, 1 mL of normal growth medium containing 2 times FBS and antibiotics concentration was added to the cells without removing the transfection mixture. The cells incubated for an additional 24 h, followed by aspirating the medium and replacing with fresh 1 × normal growth medium. Cells were incubated for another 48 h before further assay.
mtDNA Depletion and the Imaging of Mitochondrial DNA
Cells were incubated with 1 μM ethidium bromide for 24 h or longer for mtDNA depletion. PicoGreen was used to check the depletion of mtDNA.57 After that, cells were washed by PBS three times for 5 min. After the washing step, the mtDNA-depleted cells were incubated with culture medium in the presence of free pMAX-DEST (Addgene), the mixture of Mito-FLAG and pMAX-DEST, or the mixture of lipofectamine and pMAX-DEST for another 24 h. The mtDNA-depleted cells incubated with only culture medium were used as the mtDNA-negative control. Cells were then washed by PBS three times for 5 min and stained by PicoGreen (in culture medium) for 2 h before further analysis.
PCR Experiment
The MT-CO1 gene was amplified by Phusion DNA polymerase (New England Biolabs, CAT#M0530) via corresponding primers (forward: TAAGCACCCTAATCAACTGGC, reverse: GCCTCCACTATAGCAGATGCG) according to the manufacturer’s protocol. The products were examined by DNA electrophoresis.
DNA Encapsulation Efficiency
To prove that plasmids are associated with Mito-FLAG, free pGLO plasmid (2 μg/mL) and the mixture of Mito-FLAG (200 μM) and pGLO plasmid (2 μg/mL) was centrifuged by an Eppendorf 5430 R centrifuge (14,000 rpm, 1 h). The supernatant was transferred to new centrifuge tubes, and the pellets were resuspended in 20 μL tris-borate-EDTA buffer (TBE) buffer. The presence of pGLO plasmid in pellets was determined by DNA electrophoresis. All samples (20 μL per well) for DNA electrophoresis were loaded in 1 wt% agarose. The gel was run at 120 V for 1.5 h. EB was used for DNA staining in gel. The amount of pGLO plasmid in the supernatant was quantified by PicoGreen assay.76 EE% was calculated as EE% = ([DNA added – free unentrapped DNA]/DNA added) × 100%.
Statistical Analysis and Assembly
Data presented are means ± SD (except in the semi-quantification of fluorescent images). All tests were analyzed from n > 9 cells from n > 3 independent experiments per condition. Fluorescent images semi-quantification was conducted using ImageJ. All images were assembled using Photoshop.
Supplementary Material
HIGHLIGHTS.
Mitochondrial molecular assembly enables efficient gene delivery into mitochondria
Mitochondrial genome editing manipulates the cellular functions of cancer cells
Mitochondrial molecular assembly redirects viral gene vectors to mitochondria
Local enzymatic reaction targets cancer selectively
ACKNOWLEDGMENTS
We thank Dr. Adam E. Osborne and Prof. Zachary D. Nagel at Harvard University for helping the PCR experiment of pGLO plasmid. We also thank Prof. Chris Miller and Dr. Ming-Feng Tsai for sharing HEK293 cells. This work was supported by the NIH (CA142746, CA252364) and NSF (DMR-2011846).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrp.2020.100270.
DECLARATION OF INTERESTS
Brandeis University has filed a patent application on using Mito-Flag for drug delivery.
REFERENCES
- 1.Fulda S, Galluzzi L, and Kroemer G (2010). Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov 9, 447–464. [DOI] [PubMed] [Google Scholar]
- 2.Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, Scroggins B, Neckers L, and Altieri DC (2009). Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J. Clin. Invest 119, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hendrick JP, and Hartl F-U (1993). Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem 62, 349–384. [DOI] [PubMed] [Google Scholar]
- 4.Horton KL, Stewart KM, Fonseca SB, Guo Q, and Kelley SO (2008). Mitochondria-penetrating peptides. Chem. Biol 15, 375–382. [DOI] [PubMed] [Google Scholar]
- 5.Jean SR, Ahmed M, Lei EK, Wisnovsky SP, and Kelley SO (2016). Peptide-Mediated Delivery of Chemical Probes and Therapeutics to Mitochondria. Acc. Chem. Res 49, 1893–1902. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Feng Z, Wang Y, Zhou R, Yang Z, and Xu B (2016). Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc 138, 16046–16055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi Y, Lim SK, Liang Q, Iyer SV, Wang HY, Wang Z, Xie X, Sun D, Chen YJ, Tabar V, et al. (2019). Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He H, Lin X, Guo J, Wang J, and Xu B (2020). Perimitochondrial Enzymatic Self-Assembly for Selective Targeting the Mitochondria of Cancer Cells. ACS Nano 14, 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace DC (2018). Mitochondrial genetic medicine. Nat. Genet 50, 1642–1649. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen DN, Green JJ, Chan JM, Longer R, and Anderson DG (2009). Polymeric materials for gene delivery and DNA vaccination. Adv. Mater 21, 847–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng N, Wei D, Dai B, Zheng L, Zhao M, Xin N, Chi Z, Zhao Y, Ma T, Jahane R, and Sun L (2017). Mitochondrial genome encoded proteins expression disorder, the possible mechanism of the heart disease in metabolic syndrome. Cell. Physiol. Biochem 43, 959–968. [DOI] [PubMed] [Google Scholar]
- 12.Reinecke F, Smeitink JA, and van der Westhuizen FH (2009). OXPHOS gene expression and control in mitochondrial disorders. Biochim. Biophys. Acta 1792, 1113–1121. [DOI] [PubMed] [Google Scholar]
- 13.Weinberg SE, and Chandel NS (2015). Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol 11, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. (2010). Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature 465, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brooks HR (2018). Mitochondria: Finding the Power to Change. Cell 175, 891–893. [DOI] [PubMed] [Google Scholar]
- 16.Minczuk M, Papworth MA, Kolasinska P, Murphy MP, and Klug A (2006). Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. USA 103, 19689–19694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mok BY, de Moraes MH, Zeng J, Bosch DE, Kotrys AV, Raguram A, Hsu F, Radey MC, Peterson SB, Mootha VK, et al. (2020). A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacman SR, Kauppila JHK, Pereira CV, Nissanka N, Miranda M, Pinto M, Williams SL, Larsson N-G, Stewart JB, and Moraes CT (2018). MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med 24, 1696–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kauppila JHK, Baines HL, Bratic A, Simard M-L, Freyer C, Mourier A, Stamp C, Filograna R, Larsson N-G, Greaves LC, and Stewart JB (2016). A Phenotype-Driven Approach to Generate Mouse Models with Pathogenic mtDNA Mutations Causing Mitochondrial Disease. Cell Rep. 16, 2980–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gammage PA, Viscomi C, Simard M-L, Costa ASH, Gaude E, Powell CA, Van Haute L, McCann BJ, Rebelo-Guiomar P, Cerutti R, et al. (2018). Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med 24, 1691–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan SM, and Bennett JP Jr. (2004). Development of mitochondrial gene replacement therapy. J. Bioenerg. Biomembr 36, 387–393. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa T, Somiya K, Munechika R, Harashima H, and Yamada Y (2018). Mitochondrial transgene expression via an artificial mitochondrial DNA vector in cells from a patient with a mitochondrial disease. J. Control. Release 274, 109–117. [DOI] [PubMed] [Google Scholar]
- 23.Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, and White JM (2009). The primed ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J. Virol. 83, 2883–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann M, Hofmann-Winkler H, and Pöhlmann S (2018). Priming time: How cellular proteases arm coronavirus spike proteins Activation of Viruses by Host Proteases (Springer International Publishing; ), pp. 71–98. [Google Scholar]
- 25.Yang Z, Gu H, Fu D, Gao P, Lam JK, and Xu B (2004). Enzymatic formation of supramolecular hydrogels. Avd. Mater 16, 1440–1444. [Google Scholar]
- 26.Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, and Pashkuleva I (2015). Controlling cancer cell fate using localized biocatalytic self-assembly of an aromatic carbohydrate amphiphile. J. Am. Chem. Soc 137, 576–579. [DOI] [PubMed] [Google Scholar]
- 27.Gao J, Wang H, Wang L, Wang J, Kong D, and Yang Z (2009). Enzyme promotes the hydrogelation from a hydrophobic small molecule. J. Am. Chem. Soc 131, 11286–11287. [DOI] [PubMed] [Google Scholar]
- 28.Shi J, and Schneider JP (2019). De novo Design of Selective Membrane-Active Peptides by Enzymatic Control of Their Conformational Bias on the Cell Surface. Angew. Chem. Int. Ed. Engl 58, 13706–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He H, Tan W, Guo J, Yi M, Shy AN, and Xu B (2020). Enzymatic Noncovalent Synthesis. Chem. Rev 120, 9994–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao Y, Shi J, Yuan D, and Xu B (2012). Imaging enzyme-triggered self-assembly of small molecules inside live cells. Nat. Commun 3, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, and Xu B (2016). Enzyme-instructed self-assembly for spatiotemporal profiling of the activities of alkaline phosphatases on live cells. Chem 1, 246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng Z, Wang H, Wang F, Oh Y, Berciu C, Cui Q, Egelman EH, and Xu B (2020). Artificial Intracellular Filaments. Cell Rep. Phys. Sci 1, 100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Zhang B, Kuang Y, Gao Y, Shi J, Zhang XX, and Xu B (2013). A redox responsive, fluorescent supramolecular metallohydrogel consists of nanofibers with single-molecule width. J. Am. Chem. Soc 135, 5008–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen P, Ma Y, Zheng Z, Wu C, Wang Y, and Liang G (2019). Facile syntheses of conjugated polymers for photothermal tumour therapy. Nat. Commun 10, 1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang C, Zheng D, Shi F, Xu T, Yang C, Liu J, Wang L, and Yang Z (2017). Enzyme-assisted peptide folding, assembly and anti-cancer properties. Nanoscale 9, 11987–11993. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Feng Z, Yang C, Liu J, Medina JE, Aghvami SA, Dinulescu DM, Liu J, Fraden S, and Xu B (2019). Unraveling the cellular mechanism of assembling cholesterols for selective cancer cell death. Mol. Cancer Res 17, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng Z, Han X, Wang H, Tang T, and Xu B (2019). Enzyme-instructed peptide assemblies selectively inhibit bone tumors. Chem 5, 2442–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He H, Wang H, Zhou N, Yang D, and Xu B (2017). Branched peptides for enzymatic supramolecular hydrogelation. Chem. Commun. (Camb.) 54, 86–89. [DOI] [PubMed] [Google Scholar]
- 39.Rodon Fores J, Criado-Gonzalez M, Chaumont A, Carvalho A, Blanck C, Schmutz M, Serra CA, Boulmedais F, Schaaf P, and Jierry L (2019). Supported Catalytically Active Supramolecular Hydrogels for Continuous Flow Chemistry. Angew. Chem. Int. Ed. Engl 58, 18817–18822. [DOI] [PubMed] [Google Scholar]
- 40.Shi J, Fichman G, and Schneider JP (2018). Enzymatic Control of the Conformational Landscape of Self-Assembling Peptides. Angew. Chem. Int. Ed. Engl 57, 11188–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wada A, Tamaru S, Ikeda M, and Hamachi I (2009). MCM-enzyme-supramolecular hydrogel hybrid as a fluorescence sensing material for polyanions of biological significance. J. Am. Chem. Soc 131, 5321–5330. [DOI] [PubMed] [Google Scholar]
- 42.Toledano S, Williams RJ, Jayawarna V, and Ulijn RV (2006). Enzyme-triggered self-assembly of peptide hydrogels via reversed hydrolysis. J. Am. Chem. Soc 128, 1070–1071. [DOI] [PubMed] [Google Scholar]
- 43.He H, Liu S, Wu D, and Xu B (2020). Enzymatically Formed Peptide Assemblies Sequestrate Proteins and Relocate Inhibitors to Selectively Kill Cancer Cells. Angew. Chem. Int. Ed 59, 16445–16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He H, Wang J, Wang H, Zhou N, Yang D, Green DR, and Xu B (2018). Enzymatic cleavage of branched peptides for targeting mitochondria. J. Am. Chem. Soc 140, 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hopp TP, Prickett KS, Price VL, Libby RT, March CJ, Cerretti DP, Urdal DL, and Conlon PJ (1988). A short polypeptide marker sequence useful for recombinant protein identification and purification. Nat. Biotechnol 6, 1204. [Google Scholar]
- 46.Pavlov IP, and Thompson WH (1902). The work of the digestive glands (Charles Griffin). [Google Scholar]
- 47.Jukes TH, and Osawa S (1993). Evolutionary changes in the genetic code. Comp. Biochem. Physiol. B 106, 489–494. [DOI] [PubMed] [Google Scholar]
- 48.Glass Z, Lee M, Li Y, and Xu Q (2018). Engineering the delivery system for CRISPR-based genome editing. Trends Biotechnol. 36, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glass Z, Li Y, and Xu Q (2017). Nanoparticles for CRISPR-Cas9 delivery. Nat. Biomed. Eng 1, 854–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol 14, 177–185. [DOI] [PubMed] [Google Scholar]
- 51.Green DR, and Kroemer G (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. [DOI] [PubMed] [Google Scholar]
- 52.Gadaleta MN, Greco M, and Saccone C (1970). The effect of rifampicin on mitochondrial RNA polymerase from rat liver. FEBS Lett. 10, 54–56. [DOI] [PubMed] [Google Scholar]
- 53.Wallace DC (2012). Mitochondria and cancer. Nat. Rev. Cancer 12, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He H, Guo J, Lin X, and Xu B (2020). Enzyme-Instructed Assemblies Enable Mitochondria Localization of Histone H2B in Cancer Cells (Angew Chem Int). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klezovitch O, Risk M, Coleman I, Lucas IM, Null M, True LD, Nelson PS, and Vasioukhin V (2008). A causal role for ERG in neoplastic transformation of prostate epithelium. Proc. Natl. Acad. Sci. USA 105, 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X, Zhang N, and Niu R (2007). Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol. Lett 170, 83–93. [DOI] [PubMed] [Google Scholar]
- 57.Ashley N, Harris D, and Poulton J (2005). Detection of mitochondrial DNA depletion in living human cells using PicoGreen staining. Exp. Cell Res 303, 432–446. [DOI] [PubMed] [Google Scholar]
- 58.Bassiri EA (2011). pGLO mutagenesis: a laboratory procedure in molecular biology for biology students. Biochem. Mol. Biol. Educ 39, 432–439. [DOI] [PubMed] [Google Scholar]
- 59.Poyton RO, Duhl DM, and Clarkson GH (1992). Protein export from the mitochondrial matrix. Trends Cell Biol. 2, 369–375. [DOI] [PubMed] [Google Scholar]
- 60.Pacelli C, Latorre D, Cocco T, Capuano F, Kukat C, Seibel P, and Villani G (2011). Tight control of mitochondrial membrane potential by cytochrome c oxidase. Mitochondrion 11, 334–341. [DOI] [PubMed] [Google Scholar]
- 61.Gomes LC, and Scorrano L (2013). Mitochondrial morphology in mitophagy and macroautophagy. Biochim. Biophys. Acta 1833, 205–212. [DOI] [PubMed] [Google Scholar]
- 62.Huang R, and Liu W (2015). Identifying an essential role of nuclear LC3 for autophagy. Autophagy 11, 852–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boyd SD, Tsai KY, and Jacks T (2000). An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat. Cell Biol 2, 563–568. [DOI] [PubMed] [Google Scholar]
- 64.Węsierska-Gądek J, Schloffer D, Kotala V, and Horky M (2002). Escape of p53 protein from E6-mediated degradation in HeLa cells after cisplatin therapy. Int. J. Cancer 101, 128–136. [DOI] [PubMed] [Google Scholar]
- 65.Haupt S, Berger M, Goldberg Z, and Haupt Y (2003). Apoptosis - the p53 network. J. Cell Sci 116, 4077–4085. [DOI] [PubMed] [Google Scholar]
- 66.Fraley R, Subramani S, Berg P, and Papahadjopoulos D (1980). Introduction of liposome-encapsulated SV40 DNA into cells. J. Biol. Chem 255, 10431–10435. [PubMed] [Google Scholar]
- 67.Le PU, Guay G, Altschuler Y, and Nabi IR (2002). Caveolin-1 is a negative regulator of caveolae-mediated endocytosis to the endoplasmic reticulum. J. Biol. Chem 277, 3371–3379. [DOI] [PubMed] [Google Scholar]
- 68.Wang L-H, Rothberg KG, and Anderson RG (1993). Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol 123, 1107–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varkouhi AK, Scholte M, Storm G, and Haisma HJ (2011). Endosomal escape pathways for delivery of biologicals. J. Control. Release 151, 220–228. [DOI] [PubMed] [Google Scholar]
- 70.Rostovtseva TK, Sheldon KL, Hassanzadeh E, Monge C, Saks V, Bezrukov SM, and Sackett DL (2008). Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 105, 18746–18751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson LV, Walsh ML, Bockus BJ, and Chen LB (1981). Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol 88, 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ross MF, Filipovska A, Smith RA, Gait MJ, and Murphy MP (2004). Cell-penetrating peptides do not cross mitochondrial membranes even when conjugated to a lipophilic cation: evidence against direct passage through phospholipid bilayers. Biochem. J 383, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gerlier D, and Thomasset N (1986). Use of MTT colorimetric assay to measure cell activation. J. Immunol. Methods 94, 57–63. [DOI] [PubMed] [Google Scholar]
- 74.Goo MS, Sancho L, Slepak N, Boassa D, Deerinck TJ, Ellisman MH, Bloodgood BL, and Patrick GN (2017). Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol 216, 2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nilsen T, Slagsvold T, Skjerpen CS, Brech A, Stenmark H, and Olsnes S (2004). Peroxisomal targeting as a tool for assaying potein-protein interactions in the living cell: cytokine-independent survival kinase (CISK) binds PDK-1 in vivo in a phosphorylation-dependent manner. J. Biol. Chem 279, 4794–4801. [DOI] [PubMed] [Google Scholar]
- 76.Ahn SJ, Costa J, and Emanuel JR (1996). PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res. 24, 2623–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the key data supporting the findings of this study are presented within the article and the Supplemental Information.