

Abstract
Cellular metal homeostasis is a critical process for all organisms, requiring tight regulation. In the major pathogen Helicobacter pylori, the acquisition of nickel is an essential virulence determinant as this metal is a cofactor for the acid-resistance enzyme, urease. Nickel uptake relies on the NixA permease and the NiuBDE ABC transporter. Till now, bacterial metal transporters were reported to be controlled at their transcriptional level. Here we uncovered post-translational regulation of the essential Niu transporter in H. pylori. Indeed, we demonstrate that SlyD, a protein combining peptidyl-prolyl isomerase (PPIase), chaperone, and metal-binding properties, is required for the activity of the Niu transporter. Using two-hybrid assays, we found that SlyD directly interacts with the NiuD permease subunit and identified a motif critical for this contact. Mutants of the different SlyD functional domains were constructed and used to perform in vitro PPIase activity assays and four different in vivo tests measuring nickel intracellular accumulation or transport in H. pylori. In vitro, SlyD PPIase activity is down-regulated by nickel, independently of its C-terminal region reported to bind metals. In vivo, a role of SlyD PPIase function was only revealed upon exposure to high nickel concentrations. Most importantly, the IF chaperone domain of SlyD was shown to be mandatory for Niu activation under all in vivo conditions. These data suggest that SlyD is required for the active functional conformation of the Niu permease and regulates its activity through a novel mechanism implying direct protein interaction, thereby acting as a gatekeeper of nickel uptake. Finally, in agreement with a central role of SlyD, this protein is essential for the colonization of the mouse model by H. pylori.
Author summary
Metal ions are essential for the viability of all living organisms. Indeed, more than one-third of all proteins need metal cofactors for their function. Intracellular metal concentrations require tight control as non-physiological amounts are very toxic. In particular, nickel plays a unique role in Helicobacter pylori, a bacterial pathogen that colonizes the stomach of about half of the human population worldwide and is associated with the development of gastric cancer. Nickel is essential for H. pylori as it is the cofactor of urease, an enzyme indispensable for resistance to the gastric acidity of the stomach and thus for in vivo colonization. To import nickel despite its scarcity in the human body, H. pylori requires efficient uptake mechanisms. Till now, control of nickel uptake was only reported to rely on transcriptional regulators. In the present study, we uncovered a novel mechanism of regulation of nickel acquisition. SlyD, a multifunctional enzyme was found to control, by direct protein interaction, the activity of an essential nickel uptake system in H. pylori. We revealed that the SlyD chaperone activity is mandatory for the active conformation and thus functionality of the nickel permease.
Introduction
Metal ions are essential for the viability of all living organisms. Metals are known to be involved in over 40% of enzymatic reactions, and metal-binding proteins carry out at least one step in almost all biological pathways, notably in essential processes such as metabolism, respiration and photosynthesis [1]. In cells, the amount and distribution of each metal must be finely tuned, to prevent toxic effects of some metal ions and to ensure that metalloproteins bind their cognate metal ion, thereby preventing mis-metalation [2].
The allocation of transition metal ions has also been associated with bacterial virulence [3]. To protect themselves, the infected hosts combat bacterial colonization by limiting the bioavailability of metal ions, a process known as nutritional immunity [4]. Conversely, the hosts are well known to exploit the potential toxicity of metal ions to intoxicate invading pathogens [5]. To counter the host protective strategies and maintain proper cytoplasmic metal ion abundance, pathogenic bacteria have evolved a network of metalloregulatory processes that control import and efflux, as well as storage and intracellular trafficking. Many efforts have gone into understanding iron homeostasis and trafficking. Less information is known for nickel although it is a cofactor of at least nine enzymes important for metabolism or virulence [6,7]. We report here an original mechanism by which Helicobacter pylori controls nickel import.
The bacterium H. pylori is a pathogen that infects the stomach of about half of the human population and is associated with the development of gastritis, peptic ulcer disease and adenocarcinoma causing the death of approximately 800,000 people each year in the world [8]. H. pylori is a model bacterium for the study of metal metabolism because its survival depends on two nickel enzymes, urease and [NiFe]-hydrogenase, both essential for colonization of the stomach and important for the virulence of the bacterium [9]. Urease catalyzes the hydrolysis of urea to ammonium, which serves as a buffer that allows H. pylori to survive the acidity of the stomach. Urease represents about 6% of the soluble proteins of H. pylori and contains 24 nickel ions per active complex [10]. The [NiFe]-hydrogenase is also essential for colonization by allowing the bacterium to utilize molecular hydrogen as an energy substrate [11]. Therefore, H. pylori needs a constant and significant flow of nickel to ensure its survival, but in the host stomach, the nickel concentration is very low (about 0.5 nM). Accordingly, we previously observed that H. pylori and other gastric Helicobacter species have, over the course of evolution, acquired several genes that encode factors involved in the transport and the storage of nickel, highlighting the importance of nickel import in the adaptation of bacteria to the hostile environment of the stomach [12,13].
In H. pylori, the import of nickel through the outer membrane is carried out by the FrpB4 TonB-dependent transporter (TBDT) [14]. The metal is then transported into the cytoplasm by one of the two sole nickel uptake systems of H. pylori, NixA and Niu [13,15]. The NixA permease is a member of the NiCoT family that uses the physicochemical gradient of the cytoplasmic membrane as an energy source. The NiuBDE (in short Niu) ABC transporter energized by NTP has recently been identified in our team [13,16]. For now, the only regulation of nickel uptake that was reported in H. pylori relies on transcriptional repression by NikR, a pleiotropic nickel responsive regulator [17,18]. Both nickel transporters are required for efficient colonization of the mouse model, with Niu even being essential for the process [13]. To date, only one pathway of nickel export has been reported in H. pylori, the proton-driven metal efflux pump, CznABC [19]. H. pylori also expresses several unique proteins able to bind nickel in the cytoplasm. First, HspA the sole homologue of the chaperone protein GroES in H. pylori, has a C-terminal histidine and cysteine-rich extension, absent from non-Helicobacter bacteria that behaves like a nickel sequestration domain [20]. Second, H. pylori produces two small histidine-rich proteins, Hpn and Hpn-2 [21–23]. HspA and Hpn/Hpn-2 contribute to nickel storage but also to the control of hydrogenase and urease activity, respectively [12,20]. One other nickel-binding protein in the H. pylori cytoplasm is SlyD [24], but the physiological function of this protein was not defined and was investigated here.
SlyD is a member of the FK506-binding protein (FKBP) family with peptidyl-prolyl cis-trans isomerase activity (PPIase) [25,26]. These enzymes catalyze the cis/trans-isomerization of peptidyl-prolyl (XAA-Pro) bonds which can occur spontaneously but is often a rate-limiting step for protein folding, thus modulating protein activity, interaction with protein partners or other protein signaling [27,28]. SlyD belongs to a subfamily of PPIases that is found in bacteria and archaea, characterized by the insertion of a chaperone domain into the FKBP domain "insert-in-flap (IF)" which enables it to also function as an efficient molecular chaperone, thus preventing protein aggregation [29,30]. Although distinct, the IF and the FKBP domains are mechanistically linked [31,32]. Furthermore, bacterial SlyD homologues have a C-terminal extension varying from 2 to 50 residues in length [24,33] which binds divalent cations. In E. coli, the extension contains 13 histidine and 6 cysteine residues, along with multiple carboxylate amino acids, and can bind a variety of divalent cations including up to 7 nickel ions per SlyD molecule [25,34]. Notably, nickel binding to the C-terminal region was shown to down regulate the E. coli SlyD PPIase activity [25,35]. The N-terminal domain was also found to harbor an additional nickel binding site [33].
The SlyD protein was first studied in E. coli where it was characterized as a host factor during bacteriophage ΦX174 infection, stabilizing the viral lysis protein E [36]. However, the extent of the cellular function and the physiological substrates of SlyD are still not clear. It has been shown that the chaperone IF domain can bind the TAT (Twin-Arginine Translocation) secretion signal sequences, facilitating the translocation of folded proteins from the cytoplasm to the periplasm [37]. E. coli SlyD also plays a role in the maturation of the [NiFe]-hydrogenase and slyD mutant strains display two-to-ten times lower [NiFe]-hydrogenase activity compared to that of the wild-type bacteria [38,39]. An in vitro interaction between the SlyD IF domain and the HypB hydrogenase accessory protein appears to be required for nickel insertion and maturation of the hydrogenase complex, a process that depends on the C-terminal nickel-binding region [40–42].
Much less is known about the role of SlyD protein in H. pylori. The structure of its C-terminus truncated form in solution has been determined [24]. The H. pylori SlyD C-terminal extension contains multiple metal-binding residues (5 histidines and 5 cysteines), allowing the purified protein to bind divalent cations, such as particularly nickel [24]. Previous interactomic studies suggested that SlyD is part of a complex comprising the UreA urease subunit and the hydrogenase maturation accessory protein HypB [43,44]. It was reported later that H. pylori SlyD binds, through its IF domain, to HypB and to the TAT signal peptide of the periplasmic HydA [NiFe]-hydrogenase subunit [24,45]. Additionally, it has been shown that the concomitant deletion of slyD and hypA resulted in diminished urease activity [46]. However, the actual role of SlyD in urease activity in this context is not fully understood.
In the present study, we explored for the first time the role of the SlyD protein in nickel metabolism and virulence of H. pylori and demonstrated that SlyD is required for colonization of a mouse model by H. pylori. By combining assays to measure metal resistance, nickel transport and accumulation, we provide evidence that SlyD controls nickel uptake in H. pylori. In addition, we show that SlyD directly interacts with the NiuD permease subunit of the NiuBDE ABC transporter. Altogether, the results indicate that SlyD performs an essential role in H. pylori, and support a model of a novel mode of regulation of nickel acquisition.
Results
Mutagenesis of the Helicobacter pylori cytoplasmic SlyD protein
To study the role of SlyD in H. pylori, we first constructed a mutant with a complete deletion of the slyD open reading frame in strain B128 [47,48] and a complemented strain in which the wild type slyD gene was reintroduced at the native locus in the ΔslyD mutant. To dissect the contributions of the different activities of the SlyD protein, a series of strains expressing mutant versions of SlyD from the native chromosomal locus under the control of the WT promoter was also constructed (Fig 1). Structural modeling based on the NMR structures of the SlyD proteins from E. coli [49] and from H. pylori strain 26695 [24] along with analysis of conserved residues in the H. pylori SlyD protein (S1A Fig), allowed us to predict the locations of the functional domains of SlyD (Figs 1A and S1B). The mutants are represented in Fig 1B. The first mutant designated SlyD-PPI, carries substitutions of three residues that are predicted to take part in the peptidyl-prolyl isomerase activity, I47S, Y73A and F137Y. The second mutant, designated SlyD-ΔIF carries a deletion of 56 residues encompassing the IF chaperone domain. In the last mutant strain, designated SlyD-ΔCter, the C-terminal His and Cys region-rich starting from residue 155 was removed. The strains expressing the SlyD mutants were viable and presented negligible growth and viability defects (S2A Fig).
Fig 1. Illustration of the H. pylori SlyD wild type and mutant proteins.

A. Schematic representation of the functional domains of the SlyD protein of H. pylori strain B128, with indication of the corresponding encompassing residues. The regions required for the peptidyl-prolyl isomerase activity (PPIase) are colored in blue, the "inserted in Flap" IF chaperone domain is colored in pink and the C-terminal metal-binding region is colored in green. B. Illustration of the different SlyD mutants, SlyD-PPI, SlyD-ΔIF and SlyD-ΔCter. The three residues (I47S, Y73A and F137Y) changed in the SlyD-PPI mutant are marked by a black arrow.
To evaluate the expression of each SlyD construct in H. pylori, we produced specific anti-SlyD antibodies and performed Western analysis under reducing conditions (with DTT), revealing production of all SlyD variants, although at lower levels for the SlyD-ΔIF mutants (Fig 2A). The subcellular localization of SlyD was also analyzed by using a validated H. pylori fractionation procedure (S2B Fig). We found that SlyD protein is exclusively present in the soluble fraction strongly suggesting that it is a cytoplasmic protein.
Fig 2. Analysis of the production of SlyD wild type and mutant proteins in H. pylori.

A. Western blot of equal amounts of total extracts, under reducing conditions, from H. pylori B128 WT strain and B128-derived mutants carrying the following mutations ΔslyD, ΔslyD c-slyD (complemented mutant), slyD-PPI, slyD-ΔIF, and slyD-ΔCter strain, that were probed with specific anti-SlyD polyclonal antibodies prepared during this study. An arrow shows the position of the SlyD protein. B. Western blot of purified recombinant SlyD proteins (WT, SlyD-PPI, SlyD-ΔIF and SlyD-ΔCter) probed with specific anti-SlyD polyclonal antibodies. An arrow shows the position of the monomeric SlyD proteins, red stars and blue circles highlight SlyD dimers and trimers, respectively.
Peptidyl-prolyl isomerase (PPIase) activity and nickel regulation of the H. pylori SlyD protein
To investigate the biochemical activities of the H. pylori SlyD variants, the corresponding recombinant proteins were expressed and purified from E. coli. Under non-reducing conditions, we observed that the SlyD proteins form multimers (probably dimers and trimers) that are not observed upon analysis of the SlyD-ΔCter mutant (Fig 2B) or under reducing conditions (Fig 2A). These forms likely result from the spontaneous formation of disulfide bounds between the cysteine residues present in the C-terminal domain of SlyD (Fig 1B).
Circular dichroism spectroscopy analysis of the different SlyD proteins showed that the mutations did not result in major secondary structure changes (S3A Fig). Next, the PPIase activities of the purified wild type and mutant SlyD proteins were assayed by using an in vitro assay in which cis to trans prolyl isomerization of a tetrapeptide substrate is followed by monitoring the electronic absorption at 314 nm [50]. Purified E. coli SlyD protein served as a positive control (Fig 3). Our measurements revealed that the PPIase activity of wild type H. pylori SlyD protein is similar (84%) to that of the E. coli protein. The H. pylori SlyD-ΔIF and SlyD-ΔCter mutants retained even higher activity than that of the WT protein (145% and 150%, respectively) (Fig 3). In contrast the SlyD-PPI mutant had, as anticipated, completely lost detectable PPIase activity.
Fig 3. In vitro PPIase activity and nickel regulation of H. pylori wild type and mutant SlyD proteins.
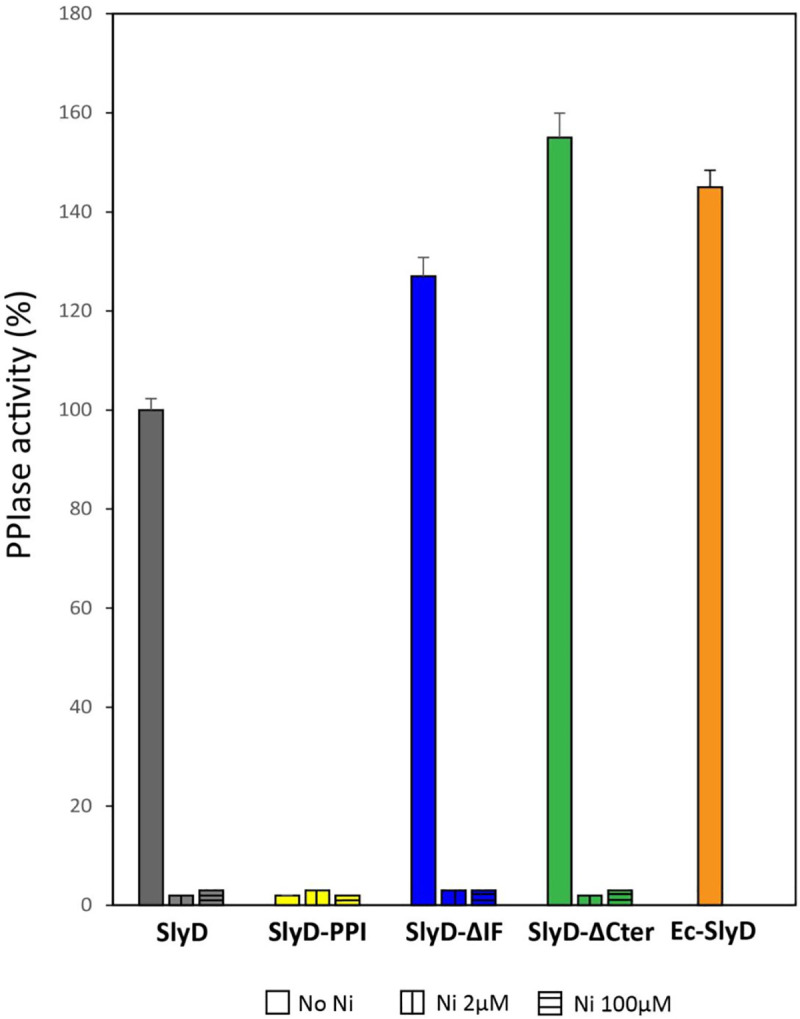
PPIase activities of WT and mutant SlyD proteins, measured without or with 2 μM or 100 μM NiSO4. The PPIase activity of purified E. coli SlyD (EcSlyD) is also presented as a control. The data (S3B Fig) were fit to second-order rate equations and the PPIase activities are expressed as a percentage of the activity of the wild type H. pylori SlyD protein. Grey bars correspond to the wild type protein, yellow to the SlyD-PPI mutant, blue to the SlyD-ΔIF mutant and green to the SlyD-ΔCter mutant. The orange bar corresponds to the E. coli SlyD protein. The values are the averages from three replicates and error bars represent the standard deviation.
In vitro PPIase activity of E. coli SlyD was reported to be negatively affected upon nickel binding [25]. Purified H. pylori SlyD protein was previously reported to bind nickel ions with a Kd value of 2.74 ± 0.26 μM [24]. Therefore, the PPIase activities of purified SlyD proteins (2 μM) were assayed in the presence of 2 μM or 100 μM NiSO4. It was found that both nickel concentrations totally inhibit the PPIase activity of all the SlyD variants (Figs 3 and S3B–S3C).
These data established the PPIase activity of the H. pylori SlyD protein and validate the importance of the three predicted active site residues. In addition, they demonstrate that neither the chaperone domain nor the C-terminal region is required for PPIase activity in vitro and that stoichiometric nickel inhibits this activity even in the absence of the C-terminal metal-binding region.
SlyD inactivation increases H. pylori tolerance to high nickel concentrations
The nickel-dependent regulation of the SlyD PPIase activity, along with its established nickel-binding properties [24], prompted us to examine whether this protein plays a role in nickel metabolism, transport and/or trafficking in H. pylori. We first tested whether SlyD is required for the activation of the nickel-dependent enzymes of H. pylori, urease and hydrogenase. We found that urease and hydrogenase activities are not affected in the ΔslyD mutant as compared to those of the wild type strain (S1 Table). In the closely related bacterium Campylobacter jejuni, wild type hydrogenase activity was also measured in the ΔslyD mutant [51]. These results demonstrated that hydrogenase activity, in these two epsilon proteobacteria, does not require SlyD, in contrast to the situation in E. coli [41].
Then, the tolerance of the wild type strain and slyD mutants to toxic nickel exposure was evaluated by measuring their growth at neutral pH in liquid BB medium after 24 h incubation with 1.5 mM NiCl2 (Fig 4). Enhanced tolerance might result from several mechanisms, among which decreased metal uptake, as we showed to be the case for an H. pylori mutant lacking nickel transporters [13].
Fig 4. Tolerance of H. pylori wild type and mutant strains to nickel exposure.
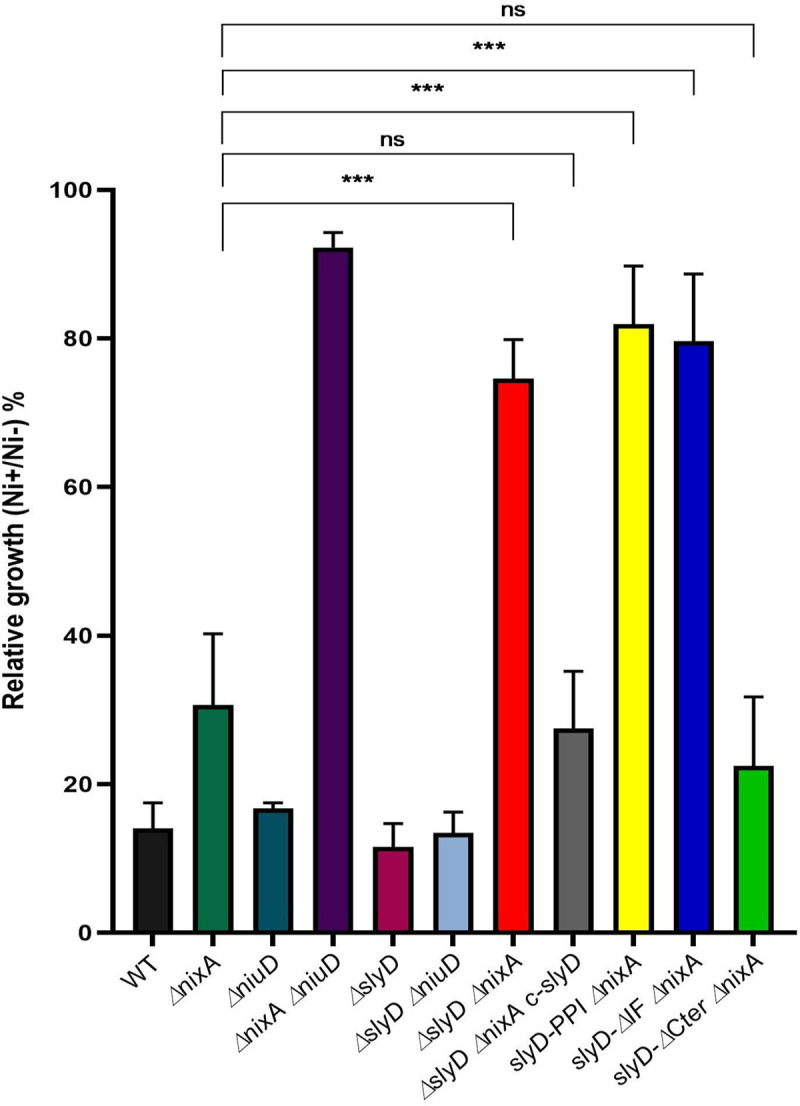
Growth of H. pylori B128 wild type strain, isogenic slyD mutants and a complemented strain (c-slyD) was measured after 24h in the presence of 1.5 mM NiCl2 or without added metal. The results are presented as the percentage of growth in the presence of nickel relative to growth in its absence. In this figure as well as in Figs 5, 6 and 7, the same color codes were used for the bars corresponding to each strain or mutant which name is indicated below each bar. Black bars correspond to the wild type strain, dark green to the ΔnixA mutant, dark blue to the ΔniuD mutant, violet to the ΔnixA ΔniuD mutant, dark red to the ΔslyD mutant, light blue to the ΔslyD ΔniuD mutant, bright red to ΔslyD ΔnixA mutant, light grey to the ΔslyD ΔnixA c-slyD mutant, yellow to the slyD-PPI ΔnixA mutant, bright blue to the slyD-ΔIF ΔnixA mutant and light green to the slyD-ΔCter ΔnixA mutant. The data correspond to the mean value of three independent experiments. Error bars represent the standard deviation. Statistics are presented only for the comparison with the ΔnixA mutant: *** corresponds to p<0.001 and "ns" for non-significant. S9 Fig presents the complete statistical analysis of these data.
Here, we first found that the ΔslyD mutant presented no difference in nickel tolerance. We next examined the effect of the ΔslyD mutation in combination with deletions of the genes encoding the nickel uptake systems, NixA or NiuD (Fig 4). The single and double ΔnixA and ΔniuD mutants behaved as we previously reported; the mutant deficient in both nickel transporters being strongly tolerant to nickel [13]. The nickel tolerance of the ΔslyD ΔniuD double mutant was similar to that of the single ΔniuD mutant. In contrast, it was found that the ΔslyD ΔnixA mutant was as highly tolerant to nickel as the ΔnixA ΔniuD mutant, suggesting that nickel uptake is strongly impaired in the ΔslyD ΔnixA strain. This phenotype could be complemented by the re-introduction of a wild type slyD copy (c-slyD), indeed the ΔslyD ΔnixA c-slyD strain recovered nickel tolerance levels comparable to that of the parental ΔnixA mutant (Fig 4). Then, the effect of mutations in the slyD gene was tested in combination with ΔnixA (Fig 4). Deletion of the C-terminal domain of SlyD (strain slyD-ΔCter ΔnixA) did not impact the tolerance of bacteria to high nickel concentrations. However, the mutants carrying either the SlyD-PPI mutation (slyD-PPI ΔnixA) or the deletion of its chaperone domain (slyD-ΔIF ΔnixA) were insensitive to 1.5 mM nickel exposure just like the ΔslyD ΔnixA mutant, attesting of the lack of activity of slyD variants in this assay.
These results indicate that H. pylori SlyD plays an essential role in nickel transport and/or metabolism. In addition, under these test conditions, the C-terminal domain of SlyD is not essential for this activity but both the PPIase isomerase activity and chaperone domain of SlyD are required for its function related to nickel uptake or metabolism.
SlyD inactivation results in reduced intracellular nickel availability
One possible explanation for these results is that SlyD modulates nickel accumulation into H. pylori. To test this hypothesis, we used an assay to indirectly evaluate the intracellular nickel content of different mutants by measuring the expression of fecA3, a gene known to be repressed by the transcriptional regulator NikR, in response to intracellular nickel concentrations [18]. First, we introduced in our mutant strains, a stable plasmid carrying a reporter gene fusion PfecA3::lacZ that we previously validated as a reporter of intracellular nickel bioavailability [13]. Fig 5 presents the ratio of ß-galactosidase activities of strains grown for 24 h in media supplemented with 100 μM NiCl2 versus without additional nickel. Lower ratios indicate stronger PfecA3::lacZ repression and thus higher intracellular nickel availability. The wild type strain, as well as the ΔslyD and ΔnixA individual mutants present ratios of about 15–20%, attesting of proper nickel-dependent repression and therefore efficient nickel uptake whereas the ΔniuD ΔnixA mutant presents a ratio of 70% as expected from its inability to import nickel [13]. The ΔslyD ΔnixA double mutant presents a ratio of 50% that is significantly different from that of the single nixA mutant, and could be complemented by the reintroduction of a wild type slyD gene. The phenotype of the three targeted slyD mutants in combination with ΔnixA was next analyzed. The slyD-PPI ΔnixA and slyD-ΔIF ΔnixA mutants presented a weak reduction in the repression by nickel while the deletion of the SlyD C-terminal domain did not prevent its activity. In conclusion, these results revealed an essential contribution of SlyD to the accumulation of intracellular nickel and demonstrated that the C-terminal domain is not required for this activity.
Fig 5. Evaluation of intracellular nickel availability of H. pylori wild type strain and isogenic mutants with a PfecA3::lacZ reporter fusion.
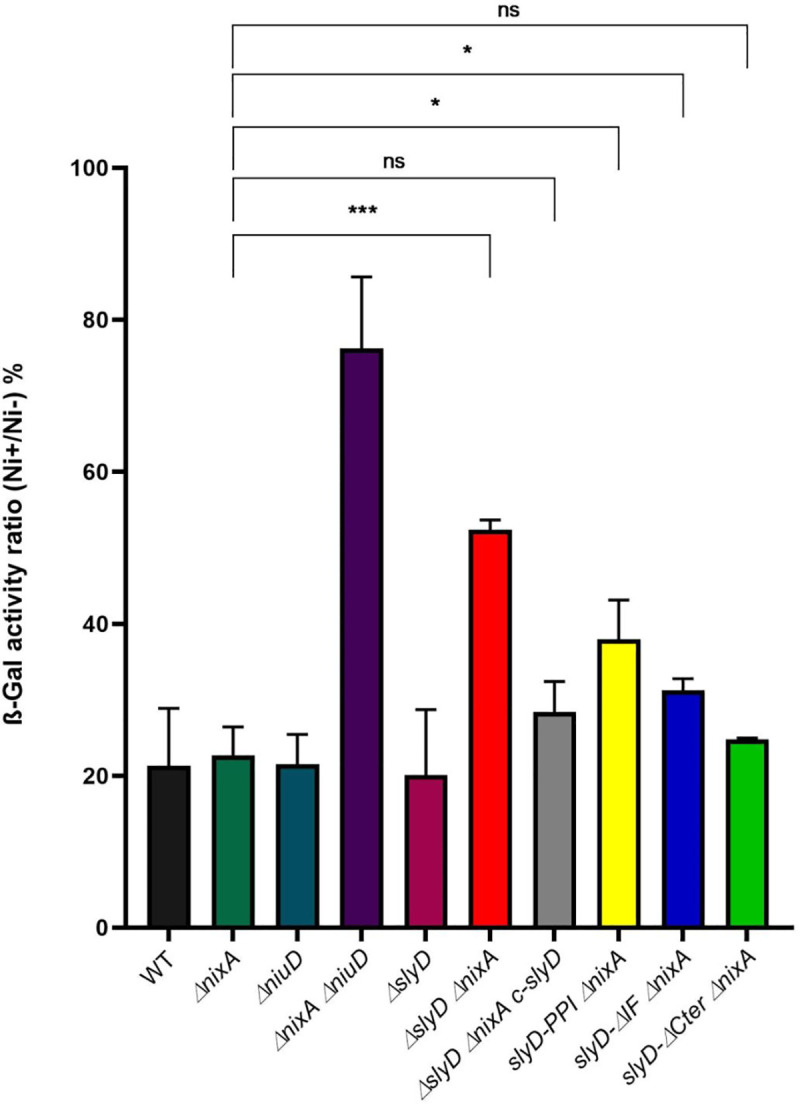
ß-galactosidase activity of a PfecA3::lacZ reporter fusion expressed from a plasmid in different H. pylori B128-derived strains, after 24H exposure to 100 μM NiCl2. The expression of the fusion decreases in a NikR-dependent manner with increasing intracellular nickel concentration. In medium without added nickel, the ß-galactosidase activities of the different strains were found to be comparable (about 6,000 miller units). ß-galactosidase activities are presented as the ratio of activity measured in strains grown in the presence of 100 μM NiCl2 or in the absence of nickel supplementation, expressed as a percentage. Color codes of the bars are as in Fig 4. The data correspond to the mean value of three independent experiments (S5 Table). Error bars represent the standard deviation. Statistics are presented only for the comparison with the ΔnixA mutant. * corresponds to p<0.05, *** to p<0.001 and "ns" for non-significant. S9 Fig presents the complete statistical analysis of these data.
Reduced intracellular nickel content of a ΔslyD ΔnixA mutant
To evaluate the role of SlyD in nickel accumulation more precisely, we measured the total intracellular nickel content of our collection of mutants grown 24h in the presence of 100 μM NiCl2, by Inductively-coupled plasma optical emission spectrometry (ICP-OES) as previously reported [13] (Fig 6). Strains grown without supplemented nickel had metal levels below the detection limit. In agreement with our previous publication [13], the nickel content of the ΔnixA and ΔnixA ΔniuD mutants was reduced 2.4-fold and 15-fold, respectively as compared to the wild type strain. Consistent with the data presented above, the ΔslyD mutant accumulated nickel as efficiently as the wild type strain. However, when the ΔslyD mutation was combined with ΔnixA, we measured a significant 1.5-fold and 3.5-fold reduction in nickel content as compared with the ΔnixA single mutant and the wild type strain, respectively. This reduction was clearly restored in the complemented ΔslyD ΔnixA c-slyD strain. When compared to the full-length deletion of slyD, only the slyD-IF mutant was significantly deficient in nickel accumulation. These data confirm that SlyD is indeed involved in nickel accumulation in H. pylori and underline a major role of its chaperon function associated with the IF domain.
Fig 6. Measurement of the intracellular nickel content of H. pylori wild type strain and isogenic mutants by ICP-OES.
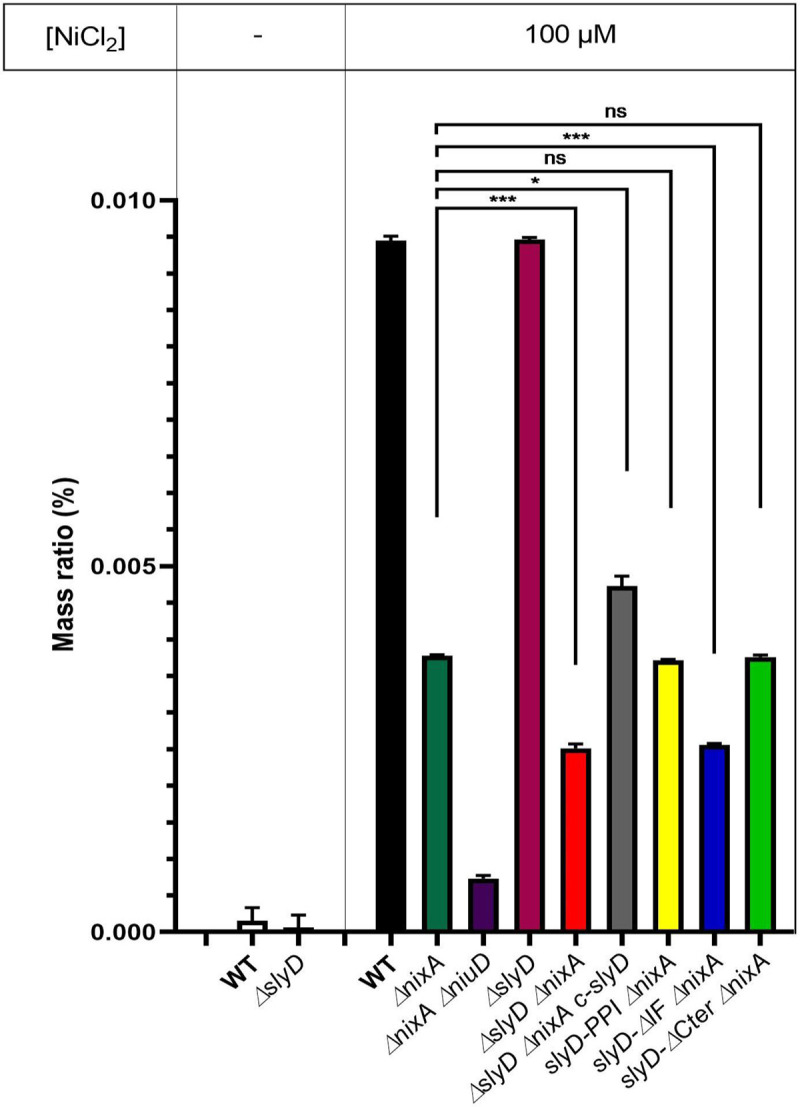
Nickel amounts were measured by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) and are expressed as the percentage of the ratio of nickel mass versus total sample mass. Strains were grown either without added nickel or with 100 μM NiCl2. Color codes of the bars are as in Fig 4. The measurement of each strain under each condition was performed in triplicates in two experiments. Statistics are presented only for the comparison with the ΔnixA mutant. * corresponds to p<0.05, *** to p<0.001 and "ns" for non-significant. S9 Fig presents the complete statistical analysis of these data.
SlyD is required for the activity of the nickel ABC transporter Niu
Increased tolerance to nickel toxicity associated with decreased nickel availability and accumulation could result from impaired metal import, a reduction in intracellular nickel storage capacity or enhanced export. Concerning nickel efflux, the only published system is Czn [19]. However, in our strain and under our conditions, we did not detect any increase in nickel sensitivity in mutant strains lacking this system alone (ΔcznABC) or in combination with the ΔslyD deletion (ΔcznABC ΔslyD). This excludes a role of the Czn efflux system in our nickel tolerance phenotypes.
To define the role of SlyD in nickel import, the uptake rates of radioactive 63NiCl2 were measured in the wild type and mutant strains at pH 5 during 30 min (Fig 7). The nickel uptake rate is moderately reduced in the ΔnixA mutant and strongly decreased in the ΔnixA ΔniuD mutant [13]. No significant change in the nickel uptake rate was seen in the ΔslyD mutant compared to that of the wild type bacteria. However, the ΔslyD ΔnixA mutant presented a 50% reduction in 63Ni uptake rate, which was significantly different from the rate of the ΔnixA mutant and could be complemented with wild-type slyD. The three strains carrying targeted slyD mutations in the ΔnixA background were also tested. The slyD-ΔIF ΔnixA strain exhibited a reduced uptake rate that phenocopied that of the ΔslyD ΔnixA mutant. While the slyD-PPI ΔnixA behaved like the ΔnixA mutant, we observed that nickel uptake of strain slyD-ΔCter ΔnixA was more robust than the ΔnixA mutant and closer to the level of the WT strain.
Fig 7. SlyD is required for the uptake of radioactive nickel by the Niu nickel transporter.
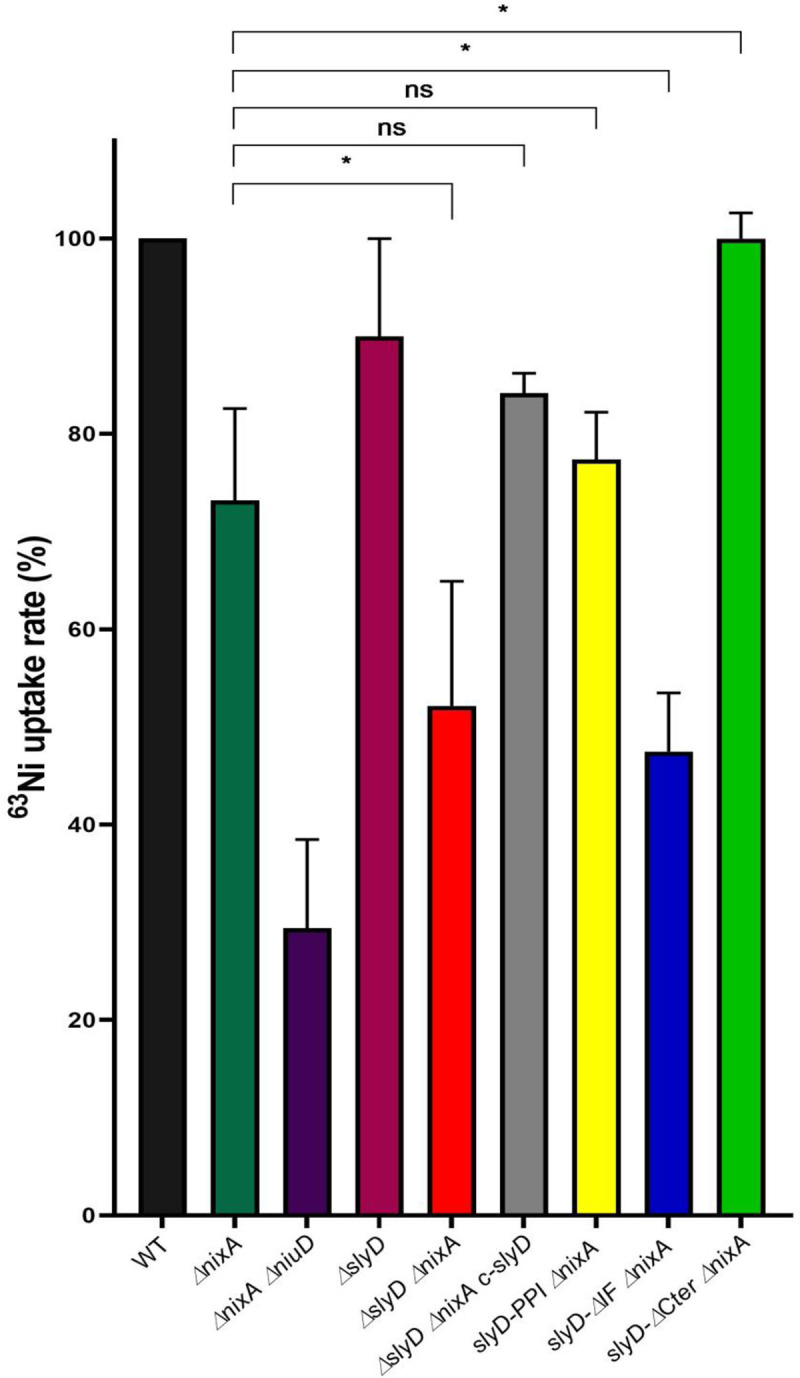
Measurements of radioactive nickel uptake rates in H. pylori B128 wild type strain and isogenic mutant strains in the presence of 10 μM of 63NiCl2. Uptake rates are normalized to the rate of wild type H. pylori strain. Color codes of the bars are as in Fig 4. Error bars represent the standard deviation. Statistics are presented only for the comparison with the ΔnixA mutant. * corresponds to p<0.05 and "ns" for non-significant. S9 Fig presents the complete statistical analysis of these data.
These results demonstrate that, under these test conditions, SlyD is required for optimal nickel uptake by the Niu transporter and only its chaperone domain plays a crucial role in this process.
By which mechanism does SlyD impact the activity of the Niu transport system?
The finding that a PPIase like SlyD regulates a metal transport activity has, to our knowledge, never been previously reported. Therefore, several tests were performed in order to investigate how SlyD regulates the Niu transport system. First, we examined whether SlyD controls the expression of the niu genes. Expression of the niuD and niuB1 genes (respectively, the first gene of the niuDE operon and the first gene of the niuB1-niuB2 operon encoding the two NiuB paralogous proteins [13]), as monitored by RT-qPCR, were similar in the wild type and ΔslyD mutant (S4 Fig). This indicates that SlyD does not regulate the Niu transporter at the transcriptional level.
We then examined whether SlyD impacts the protein levels of components of the Niu system or their subcellular localization (S5 Fig). To this aim, the NiuB1 protein was purified and used to produce specific polyclonal antibodies. Contrary to NiuB1, attempts to produce anti-NiuD antibodies were unsuccessful. Therefore, a C-terminal fusion between the niuD gene and a V5-tag was constructed and introduced at its original locus into the wild type strain and the ΔslyD mutant. The amounts and localization of NiuD and NiuB were analyzed by Western blot after fractionation of crude extracts of bacteria grown without or with the addition of 100 μM NiCl2, a non-toxic nickel amount (S5 Fig). As previously reported, production of the NiuB and NiuD proteins was repressed by nickel [52,53]. Notably, SlyD impacted neither the amounts nor the localization of the two proteins. NiuB was found in both the soluble extract and inner membrane fractions in a NiuD-independent manner. As expected, NiuD was exclusively detected in the inner membrane.
These results show that SlyD impacts neither the protein levels nor the subcellular localization of NiuD and NiuB. This suggests that SlyD might regulate the activity of the Niu transporter by direct protein interaction.
SlyD interacts with NiuD but not with NiuB or NiuE
We hypothesized that the control of the Niu transporter by SlyD might involve direct interaction between SlyD and one or more components of the Niu system. This possibility was examined by using the Bacterial Adenylate Cyclase Two-Hybrid system (BACTH) and testing pairwise interactions between protein fusions in E. coli [54]. The slyD gene was fused at its 5’ or 3’-extremity with a fragment encoding the T25 domain of the Bordetella pertussis adenylate cyclase into the pKT25 or pNKT25 vector, respectively. The niuB1, niuE, and niuD genes were fused at their 3’-extremities with the T18 fragment adenylate cyclase into the pUT18 vector. Fusions with the 5'-extremity of NiuD could not be obtained as they were toxic in E. coli.
No interaction was detected between SlyD and either the periplasmic nickel-binding protein NiuB1 or the cytoplasmic NTP-binding protein NiuE. In contrast, combinations of pNKT(slyD) with pUT18(niuD) (in which the 3'-extremity of niuD is fused to the T18 sequence) scored positive in the ß-galactosidase assay revealing an interaction between SlyD and the transmembrane permease subunit NiuD (Fig 8A). As a control, we verified that NiuD was correctly targeted to the inner membrane in E. coli (S6 Fig). To delineate the region of interaction of NiuD with SlyD, we constructed plasmids expressing T18 fusions of NiuD proteins with progressive C-terminal truncations (Figs 8A and 8B and S7). The first NiuD deletions (Δ1, Δ2, Δ3) abolished its interaction with SlyD and larger deletions (Δ4, Δ5, Δ6, Δ7, Δ8, Δ9) restored the interaction with SlyD. These results can be interpreted in light of a predictive model of the NiuD protein folding and trans-membrane segments (S7 Fig). Indeed, this model suggests that the Δ1, Δ2, Δ3 fusions are predicted to expose the T18 fused domain into the periplasm, a localization that prevents its interaction with the cytoplasmic SlyD-T25 fusions. In contrast, the larger deletions (Δ4, Δ5, Δ6, Δ7, Δ8, Δ9) are compatible with a cytoplasmic exposure of the T18 domain. These interpretations also reinforce our confidence in the specificity of the NiuD-SlyD interaction. When we further deleted the NiuD protein (Δ10, Δ11, Δ12), the interaction was lost. The comparison between Δ9 (positive for interaction) and Δ10 (negative for interaction) allowed us to identify, at the NiuD C-terminus, a predicted cytoplasmic loop with three residues (Arg208-Trp209-Arg210) that are essential for NiuD interaction with SlyD.
Fig 8. SlyD interacts with NiuD, the membrane permease of the Niu nickel uptake system.

Bacterial two-hybrid BACTH was used to analyze, in E. coli strain BTH101, the interaction between SlyD and the NiuD permease. The values and standard deviation for each strain and controls are available in S6 Table, and each measurement was performed three times. A. ß-galactosidase activities of pairwise combinations of WT SlyD with different truncated and mutant versions of the NiuD protein. B. Illustration of the truncated NiuD protein versions and sequence of the NiuD region surrounding the RWR motif in the region that is required for the interaction with SlyD. C. ß-galactosidase activities of pairwise combinations of wild type and mutant SlyD proteins (PPI, ΔIF and ΔCter) with wild type and mutant NiuD proteins.
To further determine which NiuD residues are important for its interaction with SlyD, we introduced mutations into plasmid pUT18(NiuDΔ7) that targeted the region defined above (Figs 8A and 8B and S7). First, when we introduced a triple exchange of Gly for residues Arg208-Trp209-Arg210, the ß-galactosidase activity dramatically dropped, confirming the importance of this region for NiuD interaction with SlyD. In the cases of single mutants Pro203 to Val, and Trp209 to Phe or Gly, the ß-galactosidase activity decreased but was still significant, indicating that these residues are not crucial for the NiuD interaction with SlyD. In contrast, the interaction was abolished when we introduced into NiuD the Arg208 to Gln and Arg210 to Gly mutations, indicating that these two arginine residues are essential for the NiuD interaction with SlyD.
Finally, we checked the impact of SlyD mutations on the interaction with full-length NiuD and Δ7, which generated comparable results, and used Δ1 and the triple RWR mutant as negative controls (Fig 8C). For the NiuD and Δ1 constructs, the interaction is maintained with the SlyD-PPI mutant and decreased with SlyD-IF. For the SlyD-Cter mutant, although the ß-galactosidase activity was diminished, the interaction was still detectable above baseline, and the reduced signal may be due to the reduced expression of this fusion given that the stability of this SlyD mutant protein is reduced (S6 Fig). Unexpectedly, the SlyD-IF mutant displayed significant interaction with the NiuDΔ1 and triple mutant negative controls suggesting that the IF chaperone region of SlyD contributes to the specificity to its protein interaction.
These data revealed a physical interaction between the SlyD protein and the NiuD permease subunit and defined regions of the transporter that are critical for this direct contact.
SlyD is essential for the mouse stomach colonization
The role of SlyD during gastric colonization was evaluated using the mouse model of infection by H. pylori. Different slyD mutations were introduced into the H. pylori mouse-adapted strain SS1 [55]; these included ΔslyD, the complemented strain ΔslyD-c-slyD and the slyD-PPI, slyD-ΔIF and slyD-ΔCter mutations. The expression of the SlyD protein in the WT and mutant strains was validated by western blot using anti-SlyD antibodies (S8 Fig). We orogastrically inoculated 109 bacteria of WT SS1 and every mutant strain in seven NMRI mice each. One month later, colonization was assessed by quantitative cultures of stomach homogenates. As presented in Fig 9, the ΔslyD mutant was completely deficient in its capacity to colonize the mouse stomach. The complemented strain SS1 ΔslyD+c-slyD recovered partial capacity of colonization. This was also the case for the SS1 slyD-PPI and SS1 slyD-ΔCter mutants. In contrast slyD-ΔIF mutant was completely unable to colonize the mouse stomach just like the ΔslyD mutant.
Fig 9. SlyD is essential for mouse colonization by H. pylori SS1 strain.

Each diamond corresponds to the colonization load of one mouse one month after infection with the H. pylori strain indicated below. Each strain was tested in a group of seven mice. The color codes for the different strains are as in Fig 4. Horizontal bars represent the geometric means of the colonization load for the wild type bacteria, each mutant and the chromosomally complemented ΔslyD mutant (designated c-slyD). A dashed line shows the detection limit. The ΔslyD and isogenic mutants exhibited a statistically significant colonization defect when compared to the WT or ΔslyD c-slyD strains, respectively (**p<0.01, ***p<0.001 and "ns" non-significant).
These results show that slyD is indispensable for colonization of the mouse stomach. The PPI and C-terminus domains are not essential for in vivo colonization. In contrast, the chaperone domain plays a major role in gastric colonization.
Discussion
Control of essential metal homeostasis is a complex and critical process. Bacteria have developed two major strategies to maintain this equilibrium. First, bacteria produce in the cytoplasm and/or the periplasm several "sequestering" proteins that specifically bind and store metals, preventing their association with unwanted targets [2]. The second strategy is control of metal import and/or efflux. In the vast majority of cases, expression of the metal transport systems is under the control of specific transcriptional regulators which have activities proportional to intracellular metal concentrations [2]. However, this type of regulation involves several steps as compared to post-translational regulation that was, to our knowledge, never reported for metal transporters in bacteria.
Here, such a novel mode of regulation was demonstrated; we found that the SlyD metallochaperone is required for the activity and thus active conformation of Niu, an essential nickel ABC transporter of H. pylori. In addition, SlyD is crucial for colonization of a mouse model by this pathogen. Our findings represent the first example of post-translational regulation of a metal transporter that relies on a peptidyl-prolyl isomerase (PPIase)-chaperone SlyD protein. To our knowledge, there is only one previous report of cis-trans Proline isomerization being required for activation of an ABC transporter [56]. In that instance, it was found that the periplasmic binding protein component of the system requires isomerization but the corresponding PPIase was not identified. Enzymes that combine chaperone and PPIase activities have mainly been characterized in E. coli for which nine such proteins were reported [25]. Their general function is to accelerate protein folding, but actual substrates are mostly unknown. One exception is the cytoplasmic Trigger factor (TF), a ribosome-associated molecular chaperone that, by acting as a foldase on numerous nascent polypeptides, protects them from misfolding and aggregation. TF mutants reported so far did not discriminate between the chaperone and PPIase activities and the PPIase activity is thought not to be central in bulk TF function [57]. In H. pylori, only three chaperones with PPIase activity are found, the periplasmic SurA homologue (HP0175) [58] and two cytoplasmic proteins: a TF homologue [59] and SlyD [24].
Here, we explored the in vivo role of H. pylori SlyD, a multifaceted protein that combines chaperone, PPIase and metal-binding properties. Using a collection of H. pylori slyD mutants, we characterized the function of this protein both in vitro and in vivo. We found that, in vitro, the SlyD PPIase activity was comparable to that of E. coli SlyD, and validated three essential active site residues. In our test, deletion of the IF chaperone domain did not affect PPIase activity. This result contrasts with previous studies that found that the two domains act synergistically, with the IF region being required for PPIase catalytic efficiency [24,60]. This discrepancy can be attributed to differences between the assays used. In these latter reports, protein refolding was measured, which reflects both chaperone and PPIase activities, while our test, using an isolated proline-containing tetrapeptide, specifically assays PPIase activity.
Using four different in vivo tests, we consistently demonstrated that the SlyD protein is important for the activity of the NiuBDE nickel ABC transporter. Using two-hybrid experiments, we found a direct interaction between the NiuD permease subunit and SlyD. Multicomponent ABC transporters generally require molecular chaperones for correct insertion within membranes in order to prevent aggregation or accumulation of toxic "dead-end" intermediates [61]. However, we observed no significant change of NiuD protein levels in the H. pylori inner membrane of the ΔslyD mutant, suggesting that SlyD does not affect synthesis and membrane insertion of NiuD but that it likely acts beyond this step. We found that the chaperone activity of SlyD, located in the IF domain, is required for NiuD activity, probably by helping the permease to acquire an active conformation. Indeed, in our tests, the SlyD-ΔIF mutant phenocopied the ΔslyD mutant with respect to nickel uptake; this is not due to a defect in PPIase activity, since deletion of the IF domain (SlyD-ΔIF) does not affect the PPIase activity in vitro. In two-hybrid tests, the interaction between the SlyD-ΔIF mutant and NiuD was reduced and the SlyD-ΔIF mutant presented, with negative controls, presumably non-specific interactions. We conclude that the SlyD IF domain is required for a productive NiuD-SlyD interaction and activation and, more generally, that the IF domain might be important for SlyD target recognition and discrimination. The function of the SlyD PPIase activity in the activation of Niu is more intriguing. Indeed, among the four tests that we performed, the slyD-PPI mutant only robustly phenocopied the ΔslyD mutation in the assay of tolerance to the high 1.5 mM nickel concentration. In the other tests, where nickel concentration was much lower (100 μM and 10 μM), the SlyD-PPI mutant did not display a major loss of Niu activation. In addition, as suggested by the results of the two-hybrid assays, the nickel tolerance phenotype of the SlyD-PPI mutant is not a consequence of a loss of physical interaction between SlyD-PPI and NiuD. Thus, we conclude that the PPIase function of SlyD only becomes critical in vivo under conditions of nickel overload, possibly through regulation of the NiuD conformation under these conditions (for instance through isomerization of the bonds of two proline residues close to the interacting region that we defined by two-hybrid) but it might also result from a role in the function of other nickel-resistance factors.
We also investigated the role of the C-terminal nickel-binding region of SlyD in Niu activation. This was particularly interesting since nickel binding at the SlyD C-terminal region was reported to inhibit PPIase activity of the E. coli protein [25] and Cheng et al. [24] observed conformational changes around the FKBP/PPIase domain after nickel binding to H. pylori SlyD. In vitro, we found that removal of the SlyD C-terminal region resulted in increased in vitro PPIase activity. Furthermore, addition of an equivalent of nickel completely inhibited the PPIase activity of the wild type SlyD protein and this inhibition was preserved in the SlyD mutant lacking a C-terminal region. This result indicates that down-regulation by nickel is not only relying on the C-terminal region but also on other binding sites in the SlyD N-terminal domains. In future experiments, it will be interesting to define which residue(s) of the N-terminal SlyD domains is(are) required for this C-terminal independent inhibition.
Analysis of the phenotype of the SlyD C-terminal deletion mutant in H. pylori reveals that this region of the protein is not required for the impact of SlyD on nickel metabolism. Thus, the diminished ß-galactosidase activity measured when SlyD-ΔCter was tested with NiuD in two-hybrid assays was unexpected. However, we could attribute this result to a lower level of expression of the SlyD-ΔCter mutant under these test conditions. Interestingly, the SlyD-ΔCter mutant presented slightly higher radioactive nickel uptake rates. We speculate that the C-terminal SlyD region could negatively regulate its chaperone activity, an aspect that we intend to analyze in future studies.
The two-hybrid assays allowed us to define regions of NiuD that are important for its interaction with SlyD (Figs 8 and S7). We defined a three amino-acid long motif of NiuD (RWR), predicted to be located within a cytoplasmic loop between transmembrane helices 6 and 7, that is essential for its interaction with SlyD. Interestingly, this loop (and this RWR motif) is surrounded by two proline residues (Pro203, Pro228) that might be substrates for the PPIase SlyD activity. We speculate that isomerization of the corresponding AA-proline bonds might induce conformational changes either in the cytoplasmic loop containing the RWR motif, or in the flanking transmembrane helices, which could positively impact NiuD activity.
Finally, we demonstrated that SlyD is essential for colonization of the mouse stomach by H. pylori. PPIases belonging to other families have been associated with the virulence of several bacteria [62,63]. In the mouse model, we found that the chaperone function of SlyD is essential and observed minor effects of the PPI and ΔCter mutations. We previously published that the Niu transporter is essential for colonization [13]. Thus, it is likely that the essentiality of SlyD in vivo is, at least partially, due to defective nickel uptake by the Niu system. However, it is possible that additional in vivo essential functions are disturbed in the absence of SlyD.
We propose a model for the regulation of the NiuD nickel permease by SlyD that implies direct protein interaction (Fig 10). The SlyD chaperone activity is essential for the correct folding of NiuD or its association with its partners and thus for the functioning of the Niu transporter. SlyD could, in addition, play the role of a sensor of intracellular nickel concentration and act as a "gatekeeper" of nickel uptake allowing a shut down or activation of the Niu transporter when nickel bio-availability increases or decreases, respectively.
Fig 10. Model for the regulation of nickel uptake by the SlyD protein.

In H. pylori, nickel ions (small blue dots) are transported across the outer membrane by FrpB4 (blue), a TonB-dependent transporter. Once in the periplasm, uptake of nickel through the inner membrane can be performed by the NixA permease (violet) or the ABC-transporter, NiuBDE (orange, grey and yellow). NiuB1-2 (orange) are the periplasmic nickel shuttles that deliver the metal to the NiuD permease, which activity is energized by the NiuE NTPase. The function of the Niu transporter requires activation by SlyD (red), a regulation that relies on direct interaction between SlyD and the NiuD permease. This is illustrated on the left panel, presenting conditions of low nickel availability. On the right panel, when nickel is available, binding of nickel to SlyD regulates its PPIase activity. This allows SlyD to sense the intracellular nickel concentration and to act as a gate keeper to control nickel entry.
In conclusion, the role of SlyD in H. pylori is reminiscent of the multiple signaling roles of eukaryotic PPIases in cellular processes including immune response, neuronal differentiation and cell cycle control. We anticipate that bacterial PPIases have similar important properties that have, till now, been largely underestimated.
Material and methods
Ethics statement
Experiments in mice were carried out in strict accordance with the recommendations in the Specific Guide for the Care and the Use of Laboratory Animals of the Institut Pasteur, according to the European Directive (2010/63/UE) and the corresponding French law on animal experimentation (Arrêtés de 1988). The protocol has been approved by the Committee of Central Animal Facility Board of the Institut Pasteur (#08–551). To follow the new European directives, the project was approved by the CETEA, Comité d’éthique en Expérimentation Animale of the Institut Pasteur (#2013–0051) and by the Ministère de l’Enseignement Supérieur et de la recherche (#751501).
Bacterial strains and growth conditions
The H. pylori strains used in this study are derivatives of B128 [47,48] and SS1 [55] (S2 Table). They were grown at 37°C under microaerophilic conditions (6% O2, 10% CO2, 84% N2) on Blood Agar Base 2 (Oxoid) plates supplemented with 10% defibrinated horse blood or Brucella broth agar (BD Difco) plates (designated BB) supplemented with 10% fetal calf serum (FCS, Eurobio). For liquid cultures, we used Brucella broth (BD Difco), designated BB, supplemented with 10% fetal calf serum (Eurobio) or with 0.2% β-cyclodextrin (Sigma), designated BBß. All plates and liquid cultures were supplemented with the following antibiotics-anti-fungal cocktail: amphotericin B 2.5 μg.mL-1, polymyxin B 0.31 μg.mL-1, trimethoprim 6.25 μg.mL-1 and vancomycin 12.5 μg.mL-1. Selection of H. pylori mutants and transformants was performed using kanamycin 20 μg.mL-1, chloramphenicol 6 μg.mL-1, streptomycin 10 μg.mL-1 or apramycin 10 μg.mL-1. Escherichia coli XL1-Blue bacteria grown on solid or liquid Luria-Bertani (LB) medium was used for subcloning and as a host for the preparation of the plasmids employed to transform H. pylori. E. coli strain BTH101 was used for Bacterial Two Hybrid and BL21(DE3) ΔslyD::apra for protein overexpression and purification (S2 Table). LB medium was supplemented with chloramphenicol 30 μg.mL-1, ampicillin 100 μg.mL-1 or kanamycin 40 μg.mL-1 when required.
Molecular technics
Molecular biology experiments were performed according to standard procedures and the supplier (Fermentas) recommendations. NucleoBond Xtra Midi Kit (Macherey-Nagel) and QIAamp DNA Mini Kit (Qiagen) were used for plasmid preparations and H. pylori genomic DNA extractions, respectively. PCR was performed either with DreamTaq DNA polymerase (ThermoFisher), Q5 DNA polymerase (Biolabs) or with PrimeSTAR Max DNA polymerase (Takara) when the product required high fidelity polymerase. The pGEMT vector (Promega, S3 Table) was used to construct, in E. coli, the suicide plasmids that served for mutagenesis in H. pylori.
Construction of H. pylori slyD mutants and of the niuD-V5 fusion strain
An unmarked slyD deletion mutant of H. pylori strain B128 (S2 Table) was constructed by allelic exchange as previously described [13]. We used a H. pylori suicide plasmid derived from pGEMT, in which about 500 bp of the 5’-end and the 3’-end regions immediately flanking the open reading frame of slyD gene were cloned on each side of a difH-cat-rpsL-difH cassette amplified with primers difH-rpsL-cat1 and difH-rpsL-cat2 (primers are listed in S4 Table). This plasmid (S3 Table) was used to naturally transform H. pylori strain B128 that we made Streptomycin resistant. The insertion of the cassette by homologous recombination was selected on blood agar plates containing chloramphenicol 6 μg.mL-1. Removal of the cassette was achieved by plating the CmR clones on blood agar plates containing streptomycin 10 μg.mL-1. The Gibson assembly method was used to obtain a PCR product designed to introduce a slyD deletion in H. pylori strain SS1 (S2 Table). This PCR product carried a Cm resistance cassette flanked by 500 bp upstream and downstream slyD PCR fragments. Deletion of the slyD gene was verified in both genetic backgrounds by PCR and sequencing of the gene region. Plasmids used for these constructs are listed in S3 Table. The slyD deletions of strains B128 and SS1 were complemented by reintroducing a wild type slyD copy at its original locus on the chromosome under the control of its own promotor. For that, a PCR product was generated with the Gibson assembly procedure. This PCR product comprised the slyD gene followed by the apramycin resistance cassette and flanked by the 500 bp upstream and downstream flanking region of the slyD gene. The final PCR-amplified product was used to directly transform in B128 ΔslyD or SS1 ΔslyD::Km strain resulting in a strain in which the wild type slyD gene and the apra cassette integrated by homologous recombination between flanking regions of the slyD locus. The same procedure was used to introduce slyD versions carrying mutations (PPI, ΔIF, ΔCter) in strain B128 and SS1. Sequencing was performed in both genetic backgrounds to verify the deletions and reintroduction of wild type or mutated versions of slyD at the correct locus. The expression of the mutated SlyD versions in B128 and SS1 was validated by western blot.
The NiuD-V5 fusion was obtained by gene synthesis (Eurofins). A PCR fragment carrying this fusion, a kanamycin resistance cassette and 500 bp downstream the niuD gene were fused in this order using the Gibson assembly procedure. The final PCR product was directly naturally transformed into H. pylori and allelic exchange was selected on kanamycin. Correct insertion of the fusion and cassette were verified by PCR and sequencing.
Purification of recombinant H. pylori SlyD proteins and circular dichroism spectroscopy analysis
SlyD wild-type (WT) and mutants (PPI, ΔIF and ΔCter) proteins were expressed according to the method previously described [38] in a ΔslyD::apra BL21(DE3) strain of E. coli using pET28(a)+ vector (Novagen). SlyD proteins were purified by using a nickel-nitrilotriacetic acid (Ni-NTA, Qiagen) column and the presence of SlyD in the protein fractions collected at each step was verified by SDS-PAGE analysis. Cleavage with thrombin was initiated by the addition of 0.3 U thrombin protease (Novagen) per mg target protein. The purified proteins were dialyzed overnight in thrombin cleavage buffer (20 mM Tris-HCl, pH 8.4, 150 mM NaCl, 2.5 mM CaCl2) for 12 hours at 4°C. This step was followed by anion exchange on a MonoQ column (GE Healthcare) in Tris-HCl 20mM + 200mM NaCl, 1mM TCEP buffer. Selected fractions were pooled and used for verification by Mass Spectrometry analysis (Department of Chemistry, University of Toronto) and further activity assays.
CD spectra were recorded on an Olis rapid scanning monochromator at room temperature. Approximately 15 or 25 μM protein was used in 5 mM Phosphate buffer, pH 7.6. CD spectra were collected by scanning the wavelength range of 200–260 nm using a step size of 1 nm and an integration time of 2 s. Five scans were averaged for each sample, and each CD spectrum was normalized to mean residue ellipticity [θ]mre (deg cm2 dmol–1) using the equation [θ]mre = [(MM/N– 1) × θ]/(c × l × 10)], where MM is the molecular mass of the protein in Da, N is the number of residues, θ is the measured ellipticity (degrees), c is the total protein concentration in g/mL, and l is the cell path length. The averaged spectra were smoothed by using a three-period moving average. The concentrations of the SlyD samples were confirmed after the scans.
Peptidyl-prolyl isomerase activity assays
An uncoupled protease-free assay was used to measure the PPIase activity of SlyD and the variants [40,50]. The substrate, succinyl-Ala-Phe-Pro-Phe-4-pNa (Bachem Bioscience), was dissolved in trifluoroethanol (dried over sieves) and 0.47 M LiCl (dried in 220°C oven overnight). The reactions contained 35 mM HEPES, pH 7.6, and 2 μM of each purified SlyD protein (wild type or mutants) and were incubated at 10°C before the addition of 71 μM substrate with/out 2 or 100μM of NiSO4. Isomerization was monitored at 314 nm at 10°C on a Cintra 404 spectrophotometer and fit to a single exponential decay. The time course of the reversible first-order prolyl isomerization was measured during 180 seconds, and the apparent second-order rate constant was calculated.
Metal sensitivity assay and evaluation of intracellular nickel content
The effect of metal exposure on H. pylori growth was tested by inoculating bacteria at OD600 0.1, in 10 mL liquid medium (BB with FCS) without or with 1.5 mM NiCl2. Bacterial growth was monitored 24 hours later by measuring their OD600. The data correspond to at least three independent experiments.
For the evaluation of the intracellular nickel content, we used a PfecA3 promoter fusion as a reporter, as previously validated [13]. The PfecA3 promoter is under the control of the nickel-responsive transcriptional regulator of H. pylori, NikR and its activity is thus proportional to the intracellular nickel concentration. A pILL2157(PfecA3::lacZ) fusion plasmid (S3 Table) was transformed into strain B128 and its isogenic mutants. To measure the activity of the reporter gene, H. pylori bacteria were grown on blood agar plates for 24 hours, then inoculated at OD600 0.05 in BB FCS liquid medium and grown overnight. This preculture was used to inoculate the bacteria at OD600 0.1 in liquid BB FCS without or with the addition of 100 μM NiCl2. After 24h, the β-galactosidase activity of these cultures was measured to monitor the response to nickel of the reporter fusion, and thus NikR activity (the values are available in S5 Table).
Nickel content measurements by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES)
Overnight liquid cultures of H. pylori strain were diluted and grown until OD600 0.1 at 37°C in 15 ml Brucella-Broth with FCS, then 100 μM NiCl2 were added and the cultures were left to grow until OD600 6 after 24h. Then, the cultures were washed once with PBS-1X prior to be resuspended in cold PBS-1X with EDTA 1mM and adjusted at OD600 10. Six mL of this culture preparation were centrifuged at 4,000 g at 4°C for 25 min through 400 μL of a 1:2 mixture of the silicone oils AR20/AR200 (Wacker) in order to separate the cells from the medium. Pellet were dried by speed-vac for 2h at 60°C. Ten mg of the pellet were mineralized overnight with a solution mix of 500μL nitric acid 69% (EMSURE) and 500 μL sulfuric acid 96% (Alfa Aesar). After mineralization, MiliQ water was added in each sample to a final volume of 20 mL. Nickel content was measured by ICP-OES with an Agilent 720 Series with axially-viewed plasma and with a Ni calibration curve of 10–1,000 ppb at “Institut Lavoisier de Versailles”. The content of Ni(II) was determined using a curve established with certified ICP grade nickel-standards. The measurement of each strain under each condition was performed in triplicates in two experiments. The results are presented as the percentage of the ratio of nickel mass versus total sample mass.
Transport of radioactive nickel
The procedure was adapted from our previously published protocol [13]. The preculture of B128 wild type and isogenic mutants was used to inoculate, at OD600 0.1, 10 mL of fresh BB medium supplemented with 10% FCS and incubated under microaerophilic conditions with shaking at 37°C. When the cultures reached OD600 0.5, cells were harvested, washed and resuspended in the same volume of BBβ and shaken during 20 min under microaerophilic conditions at 37°C. Radioactive 63NiCl2 (3.953 mCi/mL), was isotopically diluted 10-fold with cold NiCl2 and added at a final concentration of 10 μM. 63NiCl2 was supplied by the Eckert & Ziegler Isotope Products (Valencia, CA USA). Kinetics were performed for 30 minutes. Aliquots of 1 mL were withdrawn, immediately vacuum filtered through 0.45 μm pore-size cm filters (diameter = 2.5; Millipore) and washed with 10 mL of 50 mM HEPES buffer (pH 7.0), 1 mM cold NiCl2. Two series of experiments were performed and each time point was measured in duplicates. Uptake rates were calculated as CPM of accumulated 63Ni as a function of time.
Urease and [NiFe] hydrogenase activity measurements in H. pylori strains
Urease activity of whole H. pylori cells was assayed by measuring the ammonia production using the Ammonia-Assay kit (Sigma) as described [13]. The NH3 concentration in the supernatant was measured with the ammonia-assay kit according to the manufacturer’s (Sigma) instructions. Hydrogen uptake activity was determined spectrophotometrically at 604 nm by following the color change of methyl viologen (MV) from a colorless oxidized form to a dark-violet reduced form as described in [20]. The data correspond to at least three independent experiments with two technical replicates each time.
Bacterial Two-Hybrid assays
The Bacterial Two-Hybrid (BACTH) test is based on the reconstitution of adenylate cyclase activity in a cya- E. coli strain as a result of the interaction between two proteins: a bait and a prey fused to two separate catalytic domains (T18 and T25) of the Bordetella pertussis adenylate cyclase. Empty pNKT25 an pUT18 vectors served as controls of background adenylate cyclase activity [54]. To detect interactions between the SlyD and Niu proteins, slyD and niu genes were amplified by PCR using primers listed in S4 Table and chromosomal DNA from B128 H. pylori strain as a template.
Several plasmids were constructed (S3 Table) expressing either an N-terminal or a C-terminal fusion of these proteins with the T25 catalytic domain (derived from vectors pKNT25 and pKT25, respectively) or either a N-terminal or a C-terminal fusion with the T18 catalytic domain (derived from vectors pUT18 and pUT18C, respectively). All inserts were digested by XhoI and EcoRI, and were then cloned into plasmids pUT18, pNKT25, pUT18C and pKT25 (PCR primers listed in S4 Table). The two plasmids expressing fusions to be tested were co-transformed in E. coli strain BTH101 and transformants were selected in Luria-Bertani agar plates containing kanamycin and ampicillin at 30°C. To avoid toxic effect of transformation of the plasmid pUT18(niuD+) into the recipient strain BTH101, 1% glucose was added to the media. Five mL of LB medium supplemented with antibiotics and IPTG 10−3 M were inoculated with the transformants clones and incubated overnight at 30°C. Quantification of the interactions in strains carrying each plasmid combination was obtained by measurement of the β-galactosidase activity expressed in Miller units that was performed in at least 5 replicates as in [54] (S6 Table).
RNA extraction and cDNA synthesis
A total of 30 ml of three independent cultures grown at pH7 for 24 h with/out 100 μM NiCl2 was centrifuged for 15 min at 4000 g, treated with RNA protect solution (Qiagen) and stored at -80°C. Cells were lysed and RNA was extracted with the Nucleospin miRNA kit (Macherey-Nagel). RNA was incubated for 30 minutes at 37°C with 2 U/μL of Turbo DNase-free enzyme (Ambion). Synthesis of cDNA reactions were carried out following the manufacturer’s protocol using SuperScript IV First-Strand Synthesis System (ThermoFisher), starting with 1 μg total RNA. cDNA was final diluted to 10 ng/μl in Nuclease-free water. qRT-PCR Mix was performed with Power SYBR Green PCR Master Mix (Applied Biosystems), 900 nM of each primer (S4 Table), and 30 ng of total cDNA. PCR products were amplified and detected with an Applied Biosystem (Thermofisher) machine. The cycling conditions were as follows: one cycle at 95°C for 10min, 45 cycles at 95°C for 15 s and 60°C for 2 min, and 80 cycles at 55°C for 30 s with a 0.5°C increase every 30 s. The transcript levels were normalized to the level of the housekeeping ppK (encoding polyphosphate kinase, HP1010) as previously validated [18]. The data correspond at least two independent experiments with two technical replicates each time.
Fractionation and western blots
Recombinant H. pylori SlyD protein was purified from E. coli and used to raise polyclonal antibodies that were validated by Western blot under reducing conditions (with DTT) (Fig 2A). The cellular fractionation protocol was adapted from [64]. H. pylori cells were grown to an OD600 of 0.8–1, then harvested by centrifugation and washed twice in PBS prior to be resuspended at OD600 10 and disrupted by sonication in a lysis buffer containing 10 mM Tris-HCl pH7.5 (buffer A) and Complete Protease Inhibitor Cocktail (Roche). Cell debris was removed by centrifugation at 20,000g at 4°C for 15 minutes and supernatants were collected as total extracts. The supernatants were transferred to ultracentrifugation tubes (Polyallomer, Beckman Coulter) of 1.5 mL and then centrifuged 45 min at 100,000g at 4°C. The supernatant contains soluble fraction and the pellet total membranes. The pellet was washed once with buffer A and then resuspended in 10 mM tris-HCl pH 7.5 + 1% N-lauroyl-sarcosin (Sigma-Aldrich) and Complete Protease Inhibitor Complete (buffer B). After another ultracentrifugation step, the supernatant contains the inner membrane and pellet outer membrane.
Western blots were performed with 20 μg of proteins loaded and separated on a 4–20% Mini-Protean TGX Stain-Free precast protein gel (BioRad) and subsequently electrotransferred on a polyvinylidene difluoride (PVDF) membrane (Biorad) by TransBlot Turbo system (Biorad). The H. pylori SlyD and NiuB proteins were detected with rabbit polyclonal antibodies anti-SlyD and anti-NiuB at the respective dilutions of 1:5,000, 1:3,000. Goat anti-rabbit IgG-HRP (Promega) were used as secondary antibodies at 1:5,000 dilution and the detection were achieved with the ECL reagent (Thermo Fisher). V5 tag was detected with an antibody anti-V5 coupled with HRP (SantaCruz) at 1:10,000 dilution. The separation of the soluble and the membrane fractions was validated with a control Western Blot with antibodies against the cytoplasmic AmiE protein [65].
Mouse model of colonization
NMRI-specific pathogen-free mice (Charles River Laboratories) were orogastrically inoculated with 109 CFU of SS1 H. pylori WT and mutants prepared in 100 μL of peptone broth. One month after inoculation, mice were sacrificed and stomachs were harvested and crushed in peptone broth. Viable H. pylori colonizing the stomach were enumerated by the culture of serial dilutions of homogenized tissue on blood agar plates containing in addition bacitracin (200 μg.mL-1) and nalidixic acid (10 μg.mL-1).
Statistical analysis
The Student's t-test was used to determine the significance of the means of the data. The Mann-Whitney test was used for mouse colonization assay to compare colonization loads.
Supporting information
A. The alignment of the aminoacid sequence of a selection of SlyD proteins from 10 different H. pylori strains highlights a strong sequence conservation. Protein sequences were extracted from Kegg database and multiple alignment was made with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). B. Prediction of the tridimensional structures of H. pylori mutant SlyD proteins. The NMR structure of E. coli SlyDΔCter protein (2KFW PDB [49]) served to represent its structure. When relevant, the C-terminal metal-binding region of unknown structure has been added for clarity (in green). The NMR structure of H. pylori SlyDΔCter protein [2KFW PDB [24]] served to represent its structure. Structure models of SlyD mutants was obtained with the Phyre2 software and modelized using PyMol. The PPIase domain is represented in blue, chaperone IF domain in pink and C-terminal region in green.
(PPTX)
A. Growth curve and colony forming units (CFU) counts of WT and slyD mutant strains grown in BB medium at 37°C and followed during 1,500 min. Growth was followed by measuring OD600 and viability by CFU counting. The graphics represent a mean of three independent experiments and show that slyD deletion or mutations does not significantly impact H. pylori growth. B. Analysis of the subcellular localization of the SlyD protein in H. pylori by fractionation. Western blot was probed with anti-SlyD polyclonal antibodies on total extracts (T), soluble extract (S) and inner membrane (M) fractions prepared from H. pylori B128 wild type strain, a ΔslyD mutant and a ΔslyD-c slyD complemented strain. SlyD is exclusively detected in the soluble fraction, testifying of its cytosolic localization.
(PPTX)
A. Circular Dichroism spectra of SlyD wild type and mutant proteins were analyzed in 5 mM phosphate buffer (pH = 7.6). Five scans were averaged for each sample. B and C. A protease-free assay was used to measure PPIase activity of purified WT and mutant SlyD proteins without nickel or after addition of NiSO4 at 2 μM (panel B) or 100 μM (panel C). PPIase activity of purified E. coli SlyD was measured as a control. The time course of the reversible cis to trans first-order prolyl isomerization of a tetrapeptide substrate was followed by the decrease of absorbance recorded at 314 nm and represented as ln(A/A0). The average of three independent experiments is represented.
(PPTX)
qRT-PCR normalized fold changes of the expression of niuD and niuB1 genes in a B128 wild type strain and ΔslyD mutant. ppK was used as the housekeeping gene for normalization of the values. These results are the means with the standard deviations of two independent experiments.
(PPTX)
Western blot analysis of total extracts (T), soluble extract (S) and inner membrane (M) fractions prepared from H. pylori B128 wild type strain and ΔslyD mutant, each expressing NiuD-V5 fusion, grown without or with 100 μM of NiCl2. Western blots were revealed with anti-NiuB (panel A) and anti-V5 antibodies (panel B). The amount of both NiuD and NiuB1 proteins was diminished in the presence of nickel as expected. NiuB protein (35 kDa) was found in both the soluble (that comprises the periplasmic fraction) and inner membrane fractions in similar amounts in the WT, ΔslyD and ΔniuD strains. NiuD-V5 protein (35 kDa) is exclusively localized in the inner membrane in similar amounts of WT strain and ΔslyD mutant.
(PPTX)
Western blot analysis of total extracts (T), soluble extract (SE) and inner membrane (IM) fractions prepared from E. coli BTH101 co-transformed with pU18::niuDΔ5 and pNKT25::slyD or pNKT25::slyD-PPI. Western blot was revealed with anti-Cya antibodies. NiuDΔ5 fused to the T18 Cya fragment is located in the E. coli inner membrane. B. Western blot analysis of total extracts prepared from E. coli BTH101 carrying SlyD wild type and mutant proteins fused to the T25 fragment and revealed with anti-SlyD antibodies. The production of SlyD-ΔCter-T25 fusion is sharply diminished in comparison with WT SlyD-T25 fusion protein.
(PPTX)
A. Schematic representation of the predicated topology of a NiuD monomer as determined by the TMHMM program (Upper panel). N denotes the N-terminus, C the C-terminus of the protein; cylinders represent the 10 transmembrane helices (TMH) of NiuD and L1-L9 the periplasmic (L1, 3, 5, 7 and 9) and cytoplasmic (L2, 4, 6 and 8) loops (L) between TMHs. First and last residues flanking TMHs are indicated (numbering). L6’s sequence is indicated with the two proline residues in red and the RWR motif highlighted in blue; the RWR residues are depicted by blue circles on the scheme. Lower panel shows the structure prediction of a NiuD monomer (Phyre2 program, cartoon representation). TMH6 and 7 are green and L6 is orange, with the RWR motif (residues represented with blue sticks), and the two proline residues (spheres) flanking the loop in red. B. Schematic representation of NiuD’s and NiuD’s mutants (Δ1 to Δ12) topology. Each mutant’s name is indicated on the left and numbers indicate TMHs. The yellow circle depicts the T18 two-hybrid tag. In some cases, this tag might adopt alternative orientations/topologies that are indicated with grey cartoons. The periplasm is located above each figure, while the cytoplasm is located below. When T18 lies in or orientates towards the periplasmic side, it cannot participate to the interaction with SlyD-T25 that is located in the cytoplasm. Results of two-hybrid assays (SlyD-T25 and NiuD-T18 interactions) are indicated in each case with a color code.
(PPTX)
H. pylori SS1 WT strain and SS1-derived mutants carrying the following mutations ΔslyD, ΔslyD c-slyD (complemented strain), slyD-PPI, slyD-ΔIF, and slyD-ΔCter strain, that were probed with specific anti-SlyD polyclonal antibodies prepared during this study.
(PPTX)
The Student's t-test was used to determine significant differences between every pair of mean values from Figs 4, 5, 6 and 7. A color code indicates the p values for each comparison. A. Statistical analysis of the values of tolerance to toxic nickel exposure of H. pylori wild type and mutants from Fig 4. B. Statistical analysis of the values of the nickel content reporter of H. pylori wild type and mutants from Fig 5. C. Statistical analysis of the values of the ICP-OES nickel content measurements of H. pylori wild type and mutants from Fig 6. D. Statistical analysis of the values of radioactive nickel uptake rates of H. pylori wild type and mutants from Fig 7.
(PPTX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
This publication is dedicated to the memory of Professor Deborah B Zamble, an outstanding scientist and wonderful friend.
We thank Charles Calmettes (INRS-Institut Armand-Frappier Canada) for his precious help in the analysis of the predicted SlyD structure and the gift of the NiuB-expressing plasmid. We thank Flavien Bourdreux from the Institut Lavoisier, UMR CNRS 8180 (Université de Versailles-Saint-Quentin en Yvelines) for the ICP-OES measurements. We are grateful to Christine Cavazza (Université Grenoble Alpes, CEA, CNRS, IRIG, CBM, Grenoble) for hydrogenase activity measurements and her comments on the manuscript. We are also thankful to Sumith Kumar for his interest in this work, his advice and careful reading of the manuscript.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
The study was supported by Projet transversal of the Institut Pasteur, grant number: PTR#73 -17 to HDR; Janssen donation to HDR; Agence Nationale de la Recherche (ANR16-CE18-0026-03) to HDR; Fondation de la recherche médicale for the grant DBF20161136767 to HDR and Canadian Institutes of Health Research (MOP-142421) to DBZ. The funders had no role in study, design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Chandrangsu P, Rensing C, Helmann JD. Metal homeostasis and resistance in bacteria. Nat Rev Microbiol. 2017;15(6):338–50. 10.1038/nrmicro.2017.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldron KJ, Rutherford JC, Ford D, Robinson NJ. Metalloproteins and metal sensing. Nature. 2009;460(7257):823–30. 10.1038/nature08300 [DOI] [PubMed] [Google Scholar]
- 3.Palmer LD, Skaar EP. Transition metals and virulence in bacteria. Annu Rev Genet. 2016;50(1):67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchetti M, De Bei O, Bettati S, Campanini B, Kovachka S, Gianquinto E, et al. Iron metabolism at the interface between host and pathogen: From nutritional immunity to antibacterial development. Int J Mol Sci. 2020;21(6). 10.3390/ijms21062145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Begg SL. The role of metal ions in the virulence and viability of bacterial pathogens. Biochem Soc Trans. 2019;47(1):77–87. 10.1042/BST20180275 [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Zamble DB. Nickel homeostasis and nickel regulation: An overview. Chem Rev. 2009;109(10):4617–43. 10.1021/cr900010n [DOI] [PubMed] [Google Scholar]
- 7.Alfano M, Cavazza C. Structure, function, and biosynthesis of nickel-dependent enzymes. Protein Sci. 2020;(January):1071–89. 10.1002/pro.3836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–39. 10.1128/CMR.00011-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Reuse H, Vinella D, Cavazza C. Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of Helicobacter pylori. Vol. 3, Frontiers in Cellular and Infection Microbiology. Front Cell Infect Microbiol; 2013. [cited 2020 Oct 23]. 10.3389/fcimb.2013.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stingl K, De Reuse H. Staying alive overdosed: How does Helicobacter pylori control urease activity. Int J Med Microbiol. 2005;295(5):307–15. 10.1016/j.ijmm.2005.06.006 [DOI] [PubMed] [Google Scholar]
- 11.Olson JW, Maier RJ. Molecular hydrogen as an energy source for Helicobacter pylori. Science. (80-). 2002;298(5599):1788–90. 10.1126/science.1077123 [DOI] [PubMed] [Google Scholar]
- 12.Vinella D, Fischer F, Vorontsov E, Gallaud J, Malosse C, Michel V, et al. Evolution of Helicobacter: Acquisition by gastric species of two histidine-rich proteins essential for colonization. PLoS Pathog. 2015;11(12):1–32. 10.1371/journal.ppat.1005312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer F, Robbe-Saule M, Turlin E, Mancuso F, Michel V, Richaud P, et al. Characterization in Helicobacter pylori of a nickel transporter essential for colonization that was acquired during evolution by gastric Helicobacter species. PLoS Pathog. 2016;12(12):1–31. 10.1371/journal.ppat.1006018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schauer K, Gouget B, Carrière M, Labigne A, De Reuse H. Novel nickel transport mechanism across the bacterial outer membrane energized by the TonB/ExbB/ExbD machinery. Mol Microbiol. 2007;63(4):1054–68. 10.1111/j.1365-2958.2006.05578.x [DOI] [PubMed] [Google Scholar]
- 15.Fulkerson JF, Mobley HLT. Membrane topology of the NixA nickel transporter of Helicobacter priori: Two nickel transport-specific motifs within transmembrane helices II and III. J Bacteriol. 2000;182(6):1722–30. 10.1128/jb.182.6.1722-1730.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaik MM, Cendron L, Salamina M, Ruzzene M, Zanotti G. Helicobacter pylori periplasmic receptor CeuE (HP1561) modulates its nickel affinity via organic metallophores. Mol Microbiol. 2014;91(4):724–35. 10.1111/mmi.12487 [DOI] [PubMed] [Google Scholar]
- 17.Ernst FD, Stoof J, Horrevoets WM, Kuipers EJ, Kusters JG, Van Vliet AHM. NikR mediates nickel-responsive transcriptional repression of the Helicobacter pylori outer membrane proteins FecA3 (HP1400) and FrpB4 (HP1512). Infect Immun. 2006;74(12):6821–8. 10.1128/IAI.01196-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller C, Bahlawane C, Aubert S, Delay CM, Schauer K, Michaud-Soret I, et al. Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res. 2011;39(17):7564–75. 10.1093/nar/gkr460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stähler FN, Odenbreit S, Haas R, Wilrich J, Van Vliet AHM, Kusters JG, et al. The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect Immun. 2006;74(7):3845–52. 10.1128/IAI.02025-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schauer K, Muller C, Carrière M, Labigne A, Cavazza C, De Reuse H. The Helicobacter pylori GroES cochaperonin HspA functions as a specialized nickel chaperone and sequestration protein through its unique C-terminal extension. J Bacteriol. 2010;192(5):1231–7. 10.1128/JB.01216-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seshadri S, Benoit SL, Maier RJ. Roles of His-rich hpn and hpn-like proteins in Helicobacter pylori nickel physiology. J Bacteriol. 2007. June;189(11):4120–6. 10.1128/JB.01245-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng Y-B, Yang N, Sun H. Metal-Binding Properties of an Hpn-Like Histidine-Rich Protein. Chem. 2011. May 16;17(21):5852–60. 10.1002/chem.201100279 [DOI] [PubMed] [Google Scholar]
- 23.Cun S, Sun H. A zinc-binding site by negative selection induces metallodrug susceptibility in an essential chaperonin. Proc Natl Acad Sci. 2010. March 16 107(11):4943–8. 10.1073/pnas.0913970107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng T, Li H, Xia W, Sun H. Multifaceted SlyD from Helicobacter pylori: Implication in [NiFe] hydrogenase maturation. J Biol Inorg Chem. 2012;17(3):331–43. 10.1007/s00775-011-0855-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hottenrott S, Schumann T, Plückthun A, Fischer G, Rahfeld JU. The Escherichia coli SlyD is a metal ion-regulated peptidyl-prolyl cis/trans-isomerase. J Biol Chem. 1997;272(25):15697–701. 10.1074/jbc.272.25.15697 [DOI] [PubMed] [Google Scholar]
- 26.Scholz C, Eckert B, Hagn F, Schaarschmidt P, Balbach J, Schmid FX. SlyD proteins from different species exhibit high prolyl isomerase and chaperone activities. Biochemistry. 2006;45(1):20–33. 10.1021/bi051922n [DOI] [PubMed] [Google Scholar]
- 27.Theuerkorn M, Fischer G, Schiene-Fischer C. Prolyl cis/trans isomerase signalling pathways in cancer. Curr Opin Pharmacol. 2011;11(4):281–7. 10.1016/j.coph.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 28.Fanghänel J, Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci. 2004;9(October 2004):3453–78. 10.2741/1494 [DOI] [PubMed] [Google Scholar]
- 29.Wulfing C, Lombardero J, Pluckthun A. An Escherichia coli protein consisting of a domain homologous to FK506- binding proteins (FKBP) and a new metal binding motif. J Biol Chem. 1994;269(4):2895–901. [PubMed] [Google Scholar]
- 30.Scholz C, Schaarschmidt P, Engel AM, Andres H, Schmitt U, Faatz E, et al. Functional solubilization of aggregation-prone HIV envelope proteins by covalent fusion with chaperone modules. J Mol Biol. 2005;345(5):1229–41. 10.1016/j.jmb.2004.10.091 [DOI] [PubMed] [Google Scholar]
- 31.Haupt C, Weininger U, Kovermann M, Balbach J. Local and coupled thermodynamic stability of the two-domain and bifunctional enzyme SlyD from Escherichia coli. Biochemistry. 2011;50(34):7321–9. 10.1021/bi2000627 [DOI] [PubMed] [Google Scholar]
- 32.Kahra D, Kovermann M, Löw C, Hirschfeld V, Haupt C, Balbach J, et al. Conformational plasticity and dynamics in the generic protein folding catalyst SlyD unraveled by single-molecule FRET. J Mol Biol. 2011;411(4):781–90. 10.1016/j.jmb.2011.05.002 [DOI] [PubMed] [Google Scholar]
- 33.Kovermann M, Schmid FX, Balbach J. Molecular function of the prolyl cis/trans isomerase and metallochaperone SlyD. Biol Chem. 2013;394(8):965–75. 10.1515/hsz-2013-0137 [DOI] [PubMed] [Google Scholar]
- 34.Kaluarachchi H, Sutherland DEK, Young A, Pickering IJ, Stillman MJ, Zamble DB. The Ni(II)-Binding Properties of the Metallochaperone SlyD. J Am Chem Soc. 2009. December 30;131(51):18489–500. 10.1021/ja9081765 [DOI] [PubMed] [Google Scholar]
- 35.Martino L, Kelly G, Conte MR. Letter to the Editor: Resonance assignment of SlyD from E. coli. Biomol NMR Assign. 2009;3(2):235–7. 10.1007/s12104-009-9183-3 [DOI] [PubMed] [Google Scholar]
- 36.Roof WD, Horne SM, Young KD, Young R. slyD, a host gene required for φX174 lysis, is related to the FK506- binding protein family of peptidyl-prolyl cis-trans-isomerases. J Biol Chem. 1994;269(4):2902–10. [PubMed] [Google Scholar]
- 37.Graubner W, Schierhorn A, Brüser T. DnaK plays a pivotal role in Tat targeting of CueO and functions beside SlyD as a general Tat signal binding chaperone. J Biol Chem. 2007;282(10):7116–24. 10.1074/jbc.M608235200 [DOI] [PubMed] [Google Scholar]
- 38.Zhang JW, Butland G, Greenblatt JF, Emili A, Zamble DB. A role for SlyD in the Escherichia coli hydrogenase biosynthetic pathway. J Biol Chem. 2005;280(6):4360–6. 10.1074/jbc.M411799200 [DOI] [PubMed] [Google Scholar]
- 39.Pinske C, Sargent F, Sawers RG. SlyD-dependent nickel delivery limits maturation of [NiFe]-hydrogenases in late-stationary phase Escherichia coli cells. Metallomics. 2015;7(4):683–90. 10.1039/c5mt00019j [DOI] [PubMed] [Google Scholar]
- 40.Kaluarachchi H, Siebel JF, Kaluarachchi-Duffy S, Krecisz S, Sutherland DEK, Stillman MJ, et al. Metal selectivity of the Escherichia coli nickel metallochaperone, SlyD. Biochemistry. 2011;50(49):10666–77. 10.1021/bi2014882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaluarachchi H, Altenstein M, Sugumar SR, Balbach J, Zamble DB, Haupt C. Nickel binding and [NiFe]-hydrogenase maturation by the metallochaperone SlyD with a single metal-binding site in Escherichia coli. J Mol Biol. 2012;417(1–2):28–35. 10.1016/j.jmb.2012.01.037 [DOI] [PubMed] [Google Scholar]
- 42.Leach MR, Jie WZ, Zamble DB. The role of complex formation between the Escherichia coli hydrogenase accessory factors HypB and SlyD. J Biol Chem. 2007;282(22):16177–86. 10.1074/jbc.M610834200 [DOI] [PubMed] [Google Scholar]
- 43.Stingl K, Schauer K, Ecobichon C, Labigne A, Lenormand P, Rousselle JC, et al. In vivo interactome of Helicobacter pylori urease revealed by tandem affinity purification. Mol Cell Proteomics. 2008;7(12):2429–41. 10.1074/mcp.M800160-MCP200 [DOI] [PubMed] [Google Scholar]
- 44.Cheng T, Li H, Yang X, Xia W, Sun H. Interaction of SlyD with HypB of Helicobacter pylori facilitates nickel trafficking. Metallomics. 2013:804–7. 10.1039/c3mt00014a [DOI] [PubMed] [Google Scholar]
- 45.Hu L, Cheng T, He B, Li L, Wang Y, Lai YT, et al. Identification of metal-associated proteins in cells by using continuous-flow gel electrophoresis and inductively coupled plasma mass spectrometry. Angew Chemie—Int Ed. 2013;52(18):4916–20. 10.1002/anie.201300794 [DOI] [PubMed] [Google Scholar]
- 46.Benanti EL, Chivers PT. An intact urease assembly pathway is required to compete with NikR for nickel ions in Helicobacter pylori. J Bacteriol. 2009;191(7):2405–8. 10.1128/JB.01657-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McClain MS, Shaffer CL, Israel DA, Peek RM, Cover TL. Genome sequence analysis of Helicobacter pylori strains associated with gastric ulceration and gastric cancer. BMC Genomics. 2009;10(1):1–14. 10.1186/1471-2164-10-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farnbacher M, Jahns T, Willrodt D, Daniel R, Haas R, Goesmann A, et al. Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics. 2010;11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martino L, He Y, Hands-Taylor KLD, Valentine ER, Kelly G, Giancola C, et al. The interaction of the Escherichia coli protein SlyD with nickel ions illuminates the mechanism of regulation of its peptidyl-prolyl isomerase activity. FEBS J. 2009;276(16):4529–44. 10.1111/j.1742-4658.2009.07159.x [DOI] [PubMed] [Google Scholar]
- 50.Janowski B, Wöllner S, Schutkowski M, Fischer G. A protease-free assay for peptidyl prolyl cis/trans isomerases using standard peptide substrates. Anal Biochem. 1997;252(2):299–307. 10.1006/abio.1997.2330 [DOI] [PubMed] [Google Scholar]
- 51.Howlett RM, Hughes BM, Hitchcock A, Kelly DJ. Hydrogenase activity in the foodborne pathogen Campylobacter jejuni depends upon a novel ABC-type nickel transporter (NikZYXWV) and is SlyD-independent. Microbiol. 2012;158(6):1645–55. [DOI] [PubMed] [Google Scholar]
- 52.Danielli A, Scarlato V. Regulatory circuits in Helicobacter pylori: Network motifs and regulators involved in metal-dependent responses. FEMS Microbiol Rev. 2010;34(5):738–52. 10.1111/j.1574-6976.2010.00233.x [DOI] [PubMed] [Google Scholar]
- 53.Jones MD, Ademi I, Yin X, Gong Y, Zamble DB. Nickel-responsive regulation of two novel Helicobacter pylori NikR-targeted genes. Metallomics. 2015;7(4):662–73. 10.1039/c4mt00210e [DOI] [PubMed] [Google Scholar]
- 54.Karimova G, Ullmann A, Ladant D. A bacterial two-hybrid system that exploits a cAMP signaling cascade in Escherichia coli. Methods Enzymol. 2000;328(1988):59–73. 10.1016/s0076-6879(00)28390-0 [DOI] [PubMed] [Google Scholar]
- 55.Lee A, O’Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, Dixon MF. A standardized mouse model of Helicobacter pylori infection: Introducing the Sydney strain. Gastroenterology. 1997;112(4):1386–97. 10.1016/s0016-5085(97)70155-0 [DOI] [PubMed] [Google Scholar]
- 56.Cortes-Hernandez P, Domínguez-Ramírez L. Role of cis-trans proline isomerization in the function of pathogenic enterobacterial Periplasmic Binding Proteins. PLoS One. 2017;12(11):1–16. 10.1371/journal.pone.0188935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maillard J, Genevaux P, Holliger C. Redundancy and specificity of multiple trigger factor chaperones in Desulfitobacteria. Microbiology. 2011;157(8):2410–21. 10.1099/mic.0.050880-0 [DOI] [PubMed] [Google Scholar]
- 58.Naveen V, Chu CH, Chen BW, Tsai YC, Hsiao CD, Sun YJ. Helicobacter pylori cell binding factor 2: Insights into domain motion. J Struct Biol. 2016;194(1):90–101. 10.1016/j.jsb.2016.02.002 [DOI] [PubMed] [Google Scholar]
- 59.Fan D, Zhou Q, Liu C, Zhang J. Functional characterization of the Helicobacter pylori chaperone protein HP0795. Microbiol Res. 2016;193:11–9. 10.1016/j.micres.2016.08.012 [DOI] [PubMed] [Google Scholar]
- 60.Löw C, Neumann P, Tidow H, Weininger U, Haupt C, Friedrich-Epler B, et al. Crystal structure determination and functional characterization of the metallochaperone SlyD from Thermus thermophilus. J Mol Biol. 2010;398(3):375–90. 10.1016/j.jmb.2010.03.014 [DOI] [PubMed] [Google Scholar]
- 61.Daley DO. The assembly of membrane proteins into complexes. Curr Opin Struct Biol. 2008;18(4):420–4. 10.1016/j.sbi.2008.04.006 [DOI] [PubMed] [Google Scholar]
- 62.Obi IR, Nordfelth R, Francis MS. Varying dependency of periplasmic peptidylprolyl cis-trans isomerases in promoting Yersinia pseudotuberculosis stress tolerance and pathogenicity. Biochem J. 2011;439(2):321–32. 10.1042/BJ20110767 [DOI] [PubMed] [Google Scholar]
- 63.Bzdyl NM, Scott NE, Norville IH, Scott AE, Atkins T, Pang S, et al. Peptidyl-prolyl isomerase ppiB is essential for proteome homeostasis and virulence in Burkholderia pseudomallei. Infect Immun. 2019. October 1;87(10):e00528–19. 10.1128/IAI.00528-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kutter S, Buhrdorf R, Haas J, Schneider-Brachert W, Haas R, Fischer W. Protein subassemblies of the Helicobacter pylori cag type IV secretion system revealed by localization and interaction studies. J Bacteriol. 2008;190(6):2161–71. 10.1128/JB.01341-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Skouloubris S, Labigne A, De Reuse H. The AmiE aliphatic amidase and AmiF formamidase of Helicobacter pylori: Natural evolution of two enzyme paralogues. Mol Microbiol. 2001;40(3):596–609. 10.1046/j.1365-2958.2001.02400.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. The alignment of the aminoacid sequence of a selection of SlyD proteins from 10 different H. pylori strains highlights a strong sequence conservation. Protein sequences were extracted from Kegg database and multiple alignment was made with Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). B. Prediction of the tridimensional structures of H. pylori mutant SlyD proteins. The NMR structure of E. coli SlyDΔCter protein (2KFW PDB [49]) served to represent its structure. When relevant, the C-terminal metal-binding region of unknown structure has been added for clarity (in green). The NMR structure of H. pylori SlyDΔCter protein [2KFW PDB [24]] served to represent its structure. Structure models of SlyD mutants was obtained with the Phyre2 software and modelized using PyMol. The PPIase domain is represented in blue, chaperone IF domain in pink and C-terminal region in green.
(PPTX)
A. Growth curve and colony forming units (CFU) counts of WT and slyD mutant strains grown in BB medium at 37°C and followed during 1,500 min. Growth was followed by measuring OD600 and viability by CFU counting. The graphics represent a mean of three independent experiments and show that slyD deletion or mutations does not significantly impact H. pylori growth. B. Analysis of the subcellular localization of the SlyD protein in H. pylori by fractionation. Western blot was probed with anti-SlyD polyclonal antibodies on total extracts (T), soluble extract (S) and inner membrane (M) fractions prepared from H. pylori B128 wild type strain, a ΔslyD mutant and a ΔslyD-c slyD complemented strain. SlyD is exclusively detected in the soluble fraction, testifying of its cytosolic localization.
(PPTX)
A. Circular Dichroism spectra of SlyD wild type and mutant proteins were analyzed in 5 mM phosphate buffer (pH = 7.6). Five scans were averaged for each sample. B and C. A protease-free assay was used to measure PPIase activity of purified WT and mutant SlyD proteins without nickel or after addition of NiSO4 at 2 μM (panel B) or 100 μM (panel C). PPIase activity of purified E. coli SlyD was measured as a control. The time course of the reversible cis to trans first-order prolyl isomerization of a tetrapeptide substrate was followed by the decrease of absorbance recorded at 314 nm and represented as ln(A/A0). The average of three independent experiments is represented.
(PPTX)
qRT-PCR normalized fold changes of the expression of niuD and niuB1 genes in a B128 wild type strain and ΔslyD mutant. ppK was used as the housekeeping gene for normalization of the values. These results are the means with the standard deviations of two independent experiments.
(PPTX)
Western blot analysis of total extracts (T), soluble extract (S) and inner membrane (M) fractions prepared from H. pylori B128 wild type strain and ΔslyD mutant, each expressing NiuD-V5 fusion, grown without or with 100 μM of NiCl2. Western blots were revealed with anti-NiuB (panel A) and anti-V5 antibodies (panel B). The amount of both NiuD and NiuB1 proteins was diminished in the presence of nickel as expected. NiuB protein (35 kDa) was found in both the soluble (that comprises the periplasmic fraction) and inner membrane fractions in similar amounts in the WT, ΔslyD and ΔniuD strains. NiuD-V5 protein (35 kDa) is exclusively localized in the inner membrane in similar amounts of WT strain and ΔslyD mutant.
(PPTX)
Western blot analysis of total extracts (T), soluble extract (SE) and inner membrane (IM) fractions prepared from E. coli BTH101 co-transformed with pU18::niuDΔ5 and pNKT25::slyD or pNKT25::slyD-PPI. Western blot was revealed with anti-Cya antibodies. NiuDΔ5 fused to the T18 Cya fragment is located in the E. coli inner membrane. B. Western blot analysis of total extracts prepared from E. coli BTH101 carrying SlyD wild type and mutant proteins fused to the T25 fragment and revealed with anti-SlyD antibodies. The production of SlyD-ΔCter-T25 fusion is sharply diminished in comparison with WT SlyD-T25 fusion protein.
(PPTX)
A. Schematic representation of the predicated topology of a NiuD monomer as determined by the TMHMM program (Upper panel). N denotes the N-terminus, C the C-terminus of the protein; cylinders represent the 10 transmembrane helices (TMH) of NiuD and L1-L9 the periplasmic (L1, 3, 5, 7 and 9) and cytoplasmic (L2, 4, 6 and 8) loops (L) between TMHs. First and last residues flanking TMHs are indicated (numbering). L6’s sequence is indicated with the two proline residues in red and the RWR motif highlighted in blue; the RWR residues are depicted by blue circles on the scheme. Lower panel shows the structure prediction of a NiuD monomer (Phyre2 program, cartoon representation). TMH6 and 7 are green and L6 is orange, with the RWR motif (residues represented with blue sticks), and the two proline residues (spheres) flanking the loop in red. B. Schematic representation of NiuD’s and NiuD’s mutants (Δ1 to Δ12) topology. Each mutant’s name is indicated on the left and numbers indicate TMHs. The yellow circle depicts the T18 two-hybrid tag. In some cases, this tag might adopt alternative orientations/topologies that are indicated with grey cartoons. The periplasm is located above each figure, while the cytoplasm is located below. When T18 lies in or orientates towards the periplasmic side, it cannot participate to the interaction with SlyD-T25 that is located in the cytoplasm. Results of two-hybrid assays (SlyD-T25 and NiuD-T18 interactions) are indicated in each case with a color code.
(PPTX)
H. pylori SS1 WT strain and SS1-derived mutants carrying the following mutations ΔslyD, ΔslyD c-slyD (complemented strain), slyD-PPI, slyD-ΔIF, and slyD-ΔCter strain, that were probed with specific anti-SlyD polyclonal antibodies prepared during this study.
(PPTX)
The Student's t-test was used to determine significant differences between every pair of mean values from Figs 4, 5, 6 and 7. A color code indicates the p values for each comparison. A. Statistical analysis of the values of tolerance to toxic nickel exposure of H. pylori wild type and mutants from Fig 4. B. Statistical analysis of the values of the nickel content reporter of H. pylori wild type and mutants from Fig 5. C. Statistical analysis of the values of the ICP-OES nickel content measurements of H. pylori wild type and mutants from Fig 6. D. Statistical analysis of the values of radioactive nickel uptake rates of H. pylori wild type and mutants from Fig 7.
(PPTX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.