

Keratin defects associated with blistering skin diseases alter force transmission to the nucleus.
Abstract
The keratin network of intermediate filaments provides keratinocytes with essential mechanical strength and resilience, but the contribution to mechanosensing remains poorly understood. Here, we investigated the role of the keratin cytoskeleton in the response to altered matrix rigidity. We found that keratinocytes adapted to increasing matrix stiffness by forming a rigid, interconnected network of keratin bundles, in conjunction with F-actin stress fiber formation and increased cell stiffness. Disruption of keratin stability by overexpression of the dominant keratin 14 mutation R416P inhibited the normal mechanical response to substrate rigidity, reducing F-actin stress fibers and cell stiffness. The R416P mutation also impaired mechanotransduction to the nuclear lamina, which mediated stiffness-dependent chromatin remodeling. By contrast, depletion of the cytolinker plectin had the opposite effect and promoted increased mechanoresponsiveness and up-regulation of lamin A/C. Together, these results demonstrate that the keratin cytoskeleton plays a key role in matrix rigidity sensing and downstream signal transduction.
INTRODUCTION
The epidermis of the skin provides a tough and resilient physical barrier from the external environment, and its mechanical properties are largely determined by the resident keratinocytes, which express high levels of keratin intermediate filaments. Like all intermediate filaments, keratins assemble by end-to-end linkage of tetramers to create protofilaments, which then form 8- to 12-nm intermediate filaments consisting of eight protofilaments (1). Keratin intermediate filaments can further bundle together and undergo posttranslational phosphorylation and disulfide bonding to generate a complex cytoskeletal network (1). Keratins are the most abundant structural protein in keratinocytes and the primary determinant of the cells’ mechanical properties (2, 3). The keratin cytoskeleton is also physically connected with cell-matrix adhesions, cell-cell adhesions, and the actin cytoskeleton (4, 5), thereby mechanically integrating it with the external environment and maintaining structural integrity of the epidermis.
The mechanical importance of the keratin cytoskeleton is underscored by the development of blistering skin diseases, such as epidermolysis bullosa simplex (EBS), which are typically caused by dominant mutations in the basal keratin K5 or K14 (6, 7). The Dowling Meara subtype of EBS is the most severe and results in clusters of intraepidermal blisters all over the body upon mild mechanical stress (8). Mutations usually occur in the conserved helical motifs of the rod domain and lead to filament instability and formation of protein aggregates (9). Recessive mutations in the cytolinker plectin, which links keratin filaments to hemidesmosomes, other intermediate filaments, and F-actin microfilaments, also give rise to a specific subtype of EBS associated with muscular dystrophy (EBS-MD) and causes intraepidermal blistering (10, 11).
In addition to their critical structural functions within the epidermis, keratinocytes are also highly mechanoresponsive cells and able to sense a diverse range of biophysical cues such as extracellular matrix topography (12, 13), stiffness (14, 15), and dynamic strain (16, 17). Keratinocyte mechanosensing regulates many basic cellular functions, including proliferation (14), differentiation (17, 18), and migration (15), and contributes to the maintenance of normal tissue homeostasis and wound repair (19). To date, the actin cytoskeleton has been identified as a key mediator of keratinocyte mechanosensing and signal transduction to downstream pathways, such as serum response factor (18), epidermal growth factor receptor (14), and yes-associated protein (YAP) (20, 21). In addition, the nucleus has emerged as a major mechanosensing element within the cell and can modulate gene expression programs through remodeling of the nuclear lamina and chromatin architecture (17, 22, 23).
While much work has focused on the function of the actin cytoskeleton in cellular mechanotransduction, the contribution of intermediate filaments remains poorly understood. One recent study has shown that hemidesmosome–intermediate filament interactions in keratinocytes dampen focal adhesion assembly, cytoskeletal tension, and YAP signaling (24). In addition, we previously demonstrated that loss of plectin disrupts normal keratin organization and leads to increased deformability of the nucleus (25). Although these findings suggest that the keratin cytoskeleton buffers the cell from mechanical signaling, the direct role of keratins in mechanotransduction and the response to altered mechanical environments are still unclear. In the present study, we therefore investigated how the keratin cytoskeleton adapts to changes in matrix stiffness and mediates signal transduction to the nucleus. We show that increased matrix stiffness promotes the formation of a rigid, interconnected meshwork of keratin bundles through cross-talk with the actin cytoskeleton. Using cellular models of two different types of EBS, we further demonstrate that impaired K14 stability and plectin deficiency have distinct effects on stiffness-dependent remodeling of the nuclear lamina. Thus, these findings provide previously unidentified insights into the complex role of the keratin cytoskeleton in mechanosensing and signal transduction.
RESULTS
The keratin cytoskeleton adapts to altered matrix rigidity
To determine the role of the keratin cytoskeleton in cellular mechanosensing, we investigated the response of human keratinocyte lines to altered matrix rigidity using polyacrylamide (PA) hydrogels as a model system. HaCaT keratinocytes were cultured on collagen-coated PA gels with elastic moduli of 8, 70, and 214 kPa (fig. S1), mimicking the stiffness of the underlying dermis in normal skin, moderately stiffened scar tissue, and highly stiffened scar tissue, respectively (14). Keratin organization in cells on the different PA gels was examined using Airyscan super-resolution microscopy and quantitative image analysis of K14, which, with K5, forms the primary pair of keratin filaments in basal keratinocytes. We observed clear remodeling of the keratin bundles in conjunction with increased cell spreading on the stiffer substrates (Fig. 1, A and B). K14 organization within the central cytoplasmic region was analyzed by quantifying mean bundle thickness and spacing, while overall alignment was measured by the dispersion of bundle orientation angles (low dispersion = highly aligned). In addition, intersections of K14 bundles were estimated by measuring the density of junctions in skeletonized images (Fig. 1C).
Fig. 1. Matrix rigidity regulates structural remodeling of the keratin cytoskeleton.
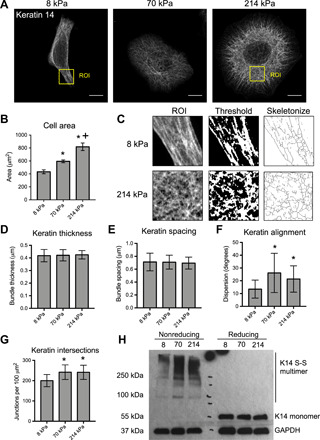
(A) Representative immunofluorescence images of K14 in HaCaT keratinocytes cultured for 24 hours on PA gels with elastic moduli of 8, 70, and 214 kPa. Images were acquired using Zeiss Airyscan confocal microscopy. Scale bars, 10 μm. (B) Quantification of average cell area on PA hydrogels. Data represent means ± SEM of N = 3 experiments (40 to 50 cells per experiment). *P < 0.05 compared to 8 kPa. +P < 0.05 compared to 70 kPa. (C) Schematic of the ImageJ analysis protocol for K14 bundles, including selection of a region of interest [ROI; labeled in (A)], thresholding, and skeletonization. (D) Quantification of average K14 bundle thickness and (E) spacing was performed on ROIs within the cytoplasm using the BoneJ plug-in. (F) Keratin alignment was quantified by the mean dispersion (SD) of K14 bundle angles within the whole cell using the Directionality plug-in. (G) Keratin bundle intersection was estimated by quantifying the mean density of junctions within skeletonized images of K14 using the Skeleton plug-in. All keratin data represent means ± SD of N = 30 cells from three experiments. *P < 0.05 compared to 8 kPa. (H) Western blot analysis of keratin cross-linking for cell lysates prepared with (reducing) or without (nonreducing) β-mercaptoethanol and probed for K14 and GAPDH.
Overall, we observed no effects of substrate stiffness on bundle thickness or spacing, but stiffness did affect bundle alignment and intersections (Fig. 1, D to G). On the soft 8 kPa gels, keratinocytes displayed a polarized morphology and a more aligned and less intersecting organization of K14 bundles. By contrast, cells on the 70 and 214 kPa gels were more spread, and the keratin bundles were more randomly oriented and interconnected. Similar responses to matrix stiffness were found for primary human keratinocytes, confirming the validity of the HaCaT line as a model of keratinocyte mechanosensing (fig. S2A).
To further validate the image-based methods and assess the biochemical changes in keratin cross-linking, we performed Western blot analysis of K14 under reducing and nonreducing conditions. Increased levels of disulfide-bonded K14 multimers could be observed on the 70 and 214 kPa gels compared to the softer 8 kPa gels under nonreducing conditions, while there were similar amounts of total K14 across all substrates under reducing conditions (Fig. 1H). Treatment with the serine/threonine phosphatase inhibitor, okadaic acid, confirmed that formation of these K14 multimers depended on phosphorylation (fig. S2, B and C). Recent studies have demonstrated the importance of disulfide-bonded multimers in the formation of the perinuclear keratin cage (26). Although we observed no major differences in organization of K14 around the nucleus, stiffness-dependent changes in disulfide bonding may still influence perinuclear cage mechanics. Together, these findings indicate that the keratin cytoskeleton structurally and biochemically adapts to changes in matrix rigidity, forming a more interconnected meshwork on stiffer substrates.
Matrix rigidity regulates keratin and cell mechanics
As the keratin cytoskeleton is a major determinant of keratinocyte biomechanics (2, 3), we next investigated how cell mechanics changed in conjunction with keratin remodeling on PA gels of different stiffness. We combined live-cell epifluorescence imaging with atomic force microscopy (AFM) to measure the deformability of the keratin cytoskeleton when indented with the AFM tip (Fig. 2A). Particle image velocimetry of keratinocytes expressing K14 tagged with green fluorescent protein (K14-GFP) during indentation revealed less deformation of the keratin cytoskeleton in cells cultured on the stiff PA gels (70 and 214 kPa) compared to the softer gels (8 kPa; Fig. 2, B and C). Furthermore, analysis of the Young’s modulus from whole-cell AFM measurements demonstrated that overall keratinocyte stiffness progressively increases on increasingly stiffer substrates (Fig. 2D). Similar trends were also observed when the Young’s moduli measurements were separated into the cytoplasmic and nuclear regions (fig. S3, A to C). These results indicate that remodeling of the keratin cytoskeleton on stiff matrices corresponds with reduced deformability of the network and overall stiffening of the cell and suggest that the keratin cytoskeleton may play a role in the cellular adaptation to altered mechanical environments.
Fig. 2. Matrix rigidity regulates keratin deformability and cell mechanics.
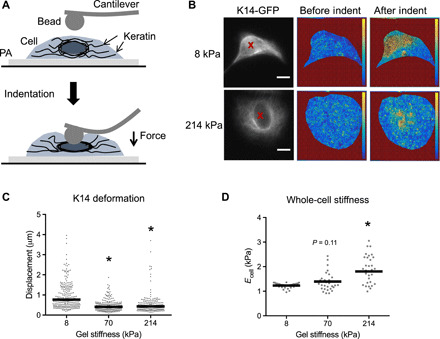
(A) Schematic of AFM indentation of single cells on PA gels with a round cantilever tip and imaging of K14-GFP. (B) Representative fluorescence images of cells expressing K14-GFP on 8 and 214 kPa gels, with red X marking the indentation point. Corresponding heat maps of displacement vector magnitudes before and after indentation were calculated using PIV. (C) Quantification of mean local keratin displacement during indentation within a 12.5-μm radius of the indentation point. Data points represent individual indentations, and bars indicate the overall mean displacement for N = 55 to 60 cells from three experiments. *P < 0.05 compared to 8 kPa. (D) Quantification of the Young’s modulus for whole cells using force-displacement data from AFM indentation. Data points represent individual cells, and bars indicate the mean modulus for N = 35 to 39 cells. *P < 0.05 compared to 8 kPa. P = 0.1 for Young’s modulus on 70 versus 8 kPa.
Although there were no differences in keratin remodeling between keratinocytes on PA gels of 70 and 214 kPa, cells still displayed increased spreading and Young’s moduli over this range of substrate stiffness, suggesting that other cytoskeletal elements may be involved. Immunofluorescence analysis of the actin cytoskeleton and paxillin-containing focal adhesions revealed that keratinocytes formed more prominent F-actin stress fibers and focal adhesions around the periphery of the cells on the stiffest substrates (Fig. 3A). These changes were quantified by significant increases in the number of both F-actin filaments and focal adhesions (Fig. 3, B and C). We also observed that the most peripheral K14 bundles on the 214 kPa gels were oriented radially toward the focal adhesions (Fig. 3A). This association of keratin bundles with focal adhesions was consistent with previous studies (27) and suggested that cross-talk between the two networks could be involved in keratinocyte mechanosensing. Disruption of actomyosin contractility in keratinocytes on stiff PA gels by treating with blebbistatin led to a significant decrease in the formation of an intersecting keratin network but had no effect on cell area or keratin alignment (Fig. 3, D to G). Together, these findings demonstrate that tension within the actin cytoskeleton modulates keratin remodeling.
Fig. 3. Cross-talk with the F-actin cytoskeleton mediates keratin remodeling.
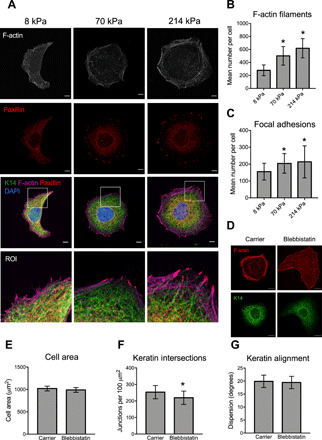
(A) Representative immunofluorescence images of F-actin, paxillin, and K14 in keratinocytes cultured on PA gels. Lower panel shows the zoomed ROI from the periphery of the cell. Scale bars, 5 μm. (B) Quantification of the mean number of F-actin filaments and (C) focal adhesions per cell using IN Cell Developer. Data represent means ± SD of N = 25 to 27 cells from three experiments. *P < 0.05 compared to 8 kPa. (D) Representative immunofluorescence images of K14 and F-actin in keratinocytes cultured on stiff PA gels and treated with the carrier control (0.1% DMSO) or 50 μM blebbistatin for 24 hours. (E) Quantification of cell area, (F) keratin intersections, and (G) keratin alignment in cells treated with carrier or blebbistatin. Data represent means ± SD of N = 30 cells from three experiments. *P < 0.05 compared to carrier.
To understand the direct relationship between cell shape and keratin remodeling, we also analyzed K14 organization on rigid micropatterned substrates consisting of circular islands of type I collagen with diameters of 30 or 50 μm. Cells cultured on large micropatterns displayed K14 bundles with increased numbers of intersections, suggesting that cell shape can influence keratin remodeling independently of substrate stiffness (fig. S4, A and B). Nevertheless, the lack of correlation between cell spreading and keratin remodeling between cells on 70 and 214 kPa gels (fig. S4C) and the effects of blebbistatin on K14 organization but not cell shape (Fig. 3, D to G) further suggest that cell shape and keratin organization are not strictly coupled and can be affected by additional factors.
Disruption of keratin stability impairs cell mechanics and stiffness-dependent F-actin remodeling
To determine the functional role of the keratin cytoskeleton in mediating cell mechanics and mechanosensing, we next generated HaCaT keratinocyte lines expressing either wild-type (WT) K14 or K14 with the dominant negative R416P point mutation, which causes the severe Dowling-Meara form of EBS (28). The R416P mutant K14 can be assembled into intermediate filaments, but due to destabilization of the central rod domain, the filaments will collapse into aggregates under mild stress. We confirmed that the GFP-tagged keratins colocalized with the total immunostained K14 and could therefore be incorporated into filaments along with the endogenous K14 (Fig. 4A). Similar levels of K14 expression in both cell lines were verified by Western blot analysis, with GFP-tagged keratin comprising approximately 30% of the endogenous keratin, and there appeared to be no compensation in the stress-associated type I keratin, K16 (Fig. 4B). Cells expressing K14 WT displayed a normal keratin meshwork (Fig. 4A). In the K14 R416P cells, however, expression of the mutant keratin led to the formation of a more disorganized network, quantified by reduced keratin intersections, and in many cells, K14 formed dense aggregates at the periphery (Fig. 4, A and C). Live imaging of the K14 R416P cells further revealed that these aggregates form following the spontaneous collapse of the keratin network (movie S1). Last, AFM analysis of cell mechanics indicated that the Young’s moduli of K14 R416P keratinocytes were significantly reduced compared to those of K14 WT cells (Fig. 4D). We therefore conclude that destabilization of K14 through the R416P mutation causes structural remodeling of the keratin cytoskeleton and affects overall cell mechanics.
Fig. 4. K14 R416P mutation disrupts cytoskeletal organization and cell mechanics.
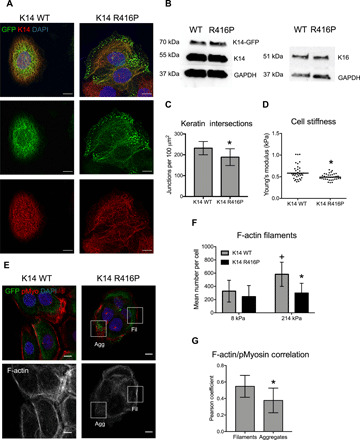
(A) Representative fluorescence images of total immunostained K14 and GFP-tagged K14 in HaCaT cells expressing either WT K14-GFP or R416P mutant K14-GFP. Scale bars, 10 μm. (B) Western blot analysis of expression levels of endogenous (55 kDa), GFP-tagged (70 kDa) K14, and K16 in cells expressing K14 WT or K14 R416P. GAPDH was used as a loading control. (C) Keratin bundle intersections were estimated in K14 WT or K14 R416P cells on stiff PA gels by quantifying the mean density of junctions within skeletonized images of immunostained K14. Data represent means ± SD of N = 12 to 20 cells from three experiments. *P < 0.05 compared to K14 WT. (D) AFM analysis of the Young’s modulus in K14 WT and K14 R416P cells on glass. Data points represent individual cells, and bars indicate the mean modulus for N = 35 cells from three experiments. *P < 0.05 compared to K14 WT. (E) Representative fluorescence images of F-actin, phospho-myosin, and K14-GFP in K14 WT and K14 R416P cells on 214 kPa PA gels. ROI notes mislocalization of phospho-myosin around K14 aggregates (Agg) or associated with F-actin around intact K14 filaments (Fil). Scale bars, 10 μm. (F) Quantification of the mean number of F-actin filaments per cell on different PA gels. Data represent means ± SD of N = 23 to 56 cells from three experiments. *P < 0.05 compared to K14 WT at same stiffness; +P < 0.05 compared to 8 kPa for the same cell type. (G) Quantification of Pearson correlation coefficient for colocalization of F-actin and pMyosin in ROIs associated with K14 R416P aggregates or intact filaments. Data represent means ± SD of N = 30 cells. *P < 0.05 compare to Fil.
As inhibition of actomyosin contractility influenced keratin remodeling, we also investigated whether keratin instability reciprocally affected cell spreading and the actin cytoskeleton. Although there were no differences in stiffness-dependent cell spreading (fig. S4D), K14 R416P–expressing keratinocytes displayed fewer F-actin stress fibers compared to K14 WT cells when cultured on the stiff 214 kPa PA gels (Fig. 4, E and F). Moreover, F-actin stress fibers in WT cells on stiff gels colocalized with phospho-myosin staining, whereas at sites of K14 aggregates in the R416P cells, phospho-myosin appeared more disorganized with reduced colocalization with F-actin fibers (Fig. 4, E and G). These findings indicate that keratin stability feeds back to the F-actin cytoskeleton and suggest a bidirectional cross-talk between the two networks in the response to matrix rigidity.
The keratin cytoskeleton regulates mechanotransduction to the nuclear lamina
Downstream of cell-matrix adhesions and cytoskeletal remodeling, the nucleus is a central mechanosensing element within the cell and responsible for transducing diverse mechanical signals into cellular responses (17, 22, 23). We next examined how disruption of keratin stability affected nuclear morphology and expression of lamin A/C, a key mediator of nuclear mechanics and signal transduction (22, 23). Immunofluorescence staining of lamin A/C in K14 WT and R416P cells cultured on PA gels revealed that the cross-sectional area of the nucleus did not depend on substrate stiffness or keratin stability (Fig. 5A and fig. S5A). However, lamin A/C expression on stiffer gels was significantly reduced in K14 R416P cells compared to WT cells (Fig. 5B), and this response was confirmed by Western blot analysis (Fig. 5C). The effects of substrate stiffness on lamin A/C expression were similarly observed in primary human keratinocytes (fig. S5B), and there were no differences in lamin B1 expression between WT and R416P cells (fig. S5, C and D), suggesting that the effects are specific to A-type lamins.
Fig. 5. Impaired K14 stability inhibits nuclear mechanotransduction.
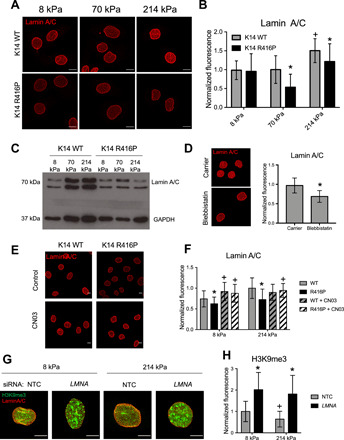
(A) Representative images of lamin A/C in HaCaT keratinocytes expressing K14 WT or K14 R416P cultured on PA gels. Scale bars, 10 μm. (B) Quantification of total lamin A/C fluorescence per cell. Data represent means ± SD for N = 30 to 50 cells from three experiments. *P < 0.05 compared to K14 WT at same stiffness; +P < 0.05 compared to 8 kPa for the same cell type. (C) Western blot analysis of lamin A/C and GAPDH expression in K14 WT and K14 R416P cells cultured on different PA gels. (D) Representative images and quantification of lamin A/C in the parental HaCaT line on 214 kPa gels following treatment with either carrier (0.1% DMSO) or 50 μM blebbistatin for 24 hours. Scale bars, 10 μm. Data represent means ± SD for N = 30 to 40 cells from three experiments. (E) Representative images of lamin A/C in K14 WT and K14 R416P cells on 214-kPa gels treated with Rho-activating peptide CN03 (5 μg/ml) for 24 hours. Scale bars, 10 μm. (F) Quantification of total lamin A/C fluorescence in cells on 8 and 214 kPa gels with or without CN03 treatment. Data represent means ± SD for N = 64 to 103 cells from three experiments. +P < 0.05 compared to untreated for the same gel and cell line; *P < 0.05 compared to WT for the same gel and medium. (G) Representative images of lamin A/C and H3K9me3 fluorescence in HaCaT cells were transfected with nontargeting control (NTC) or LMNA siRNA and cultured on 8 or 214 kPa gels for 24 hours. Scale bars, 5 μm. (H) Quantification of total H3K9me3 fluorescence per cell. Data represent means ± SD for N = 30 cells from three experiments. +P < 0.05 compared to 8 kPa for the same siRNA treatment; *P < 0.05 compared to NTC for the same gel stiffness.
As the F-actin cytoskeleton is known to regulate lamin A/C expression in other cell types (23) and is dysregulated in K14 R416P cells, we also tested its role in keratinocyte nuclear mechanotransduction. Inhibition of actomyosin contractility with blebbistatin in the parental HaCaT line phenocopied the K14 R416P mutation and reduced expression of lamin A/C on stiff substrates (Fig. 5D). In addition, stimulation of Rho-A activity and stress fiber formation with the CN03 peptide up-regulated lamin A/C on soft gels and rescued the effects of the K14 R416P mutation on stiff gels (Fig. 5, E and F, and fig. S5E). Together, these results indicate that the keratin and F-actin cytoskeletal networks mediate nuclear mechanotransduction, and that disruption of K14 stability impairs stiffness-dependent adaptation of the nuclear lamina through down-regulation of actomyosin contractility.
To understand the downstream impact of altered lamin A/C expression, we also examined the expression of the heterochromatin marker histone H3 lysine 9 trimethylation (H3K9me3), which has recently been implicated in the nuclear softening response to mechanical strain (29). Keratinocytes cultured on stiff PA gels displayed lower H3K9me3 levels compared to cells on soft gels, and small interfering RNA (siRNA) knockdown of lamin A/C blocked this response and resulted in a significant up-regulation of H3K9me3 (Fig. 5, G and H, and fig. S5F). We therefore conclude that stiffness-dependent changes in lamin A/C expression regulate downstream heterochromatin remodeling.
Plectin deficiency differentially affects stiffness-dependent lamin A/C expression
To explore how other forms of EBS and keratin dysregulation influence nuclear mechanotransduction, we examined the response of plectin-deficient (Plec KO) mouse keratinocytes to altered substrate stiffness. Mice lacking the Plec gene mimic human EBS-MD, and keratinocytes derived from these animals display a more open and widely spaced network of keratin bundles (30, 31). Consistent with previous findings, we also observed increased spacing of keratin bundles in Plec KO cells compared to WT cells on stiff PA gels (Fig. 6, A and B). In contrast to the K14 R416P mutation, however, Plec KO cells spread more on stiff gels and displayed larger nuclei, compared to WT keratinocytes (Fig. 6, C and D). These results indicate that loss of plectin enhances keratinocyte sensitivity to matrix stiffness, and this response is consistent with our previous studies using micropatterned substrates (25).
Fig. 6. Plectin deficiency differentially regulates nuclear mechanotransduction.
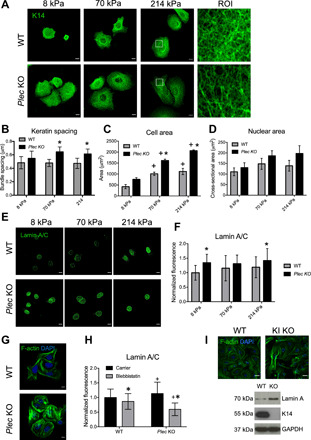
(A) Representative immunofluorescence images of K14 in WT or Plec KO mouse keratinocytes cultured on PA gels. ROI panels represent the boxed areas in 214 kPa images. Scale bars, 10 μm. (B) Quantification of keratin bundle spacing. Data represent means ± SD for N = 12 to 14 cells from three experiments. *P < 0.05 compared to WT for the same stiffness. (C) Quantification of cell area and (D) nuclear cross-sectional area in WT and Plec KO cells cultured on different PA gels. Data represent means ± SEM for N = 3 experiments. *P < 0.05 compared to WT for the same stiffness; +P < 0.05 compared to 8 kPa for the same cell type. (E) Representative immunofluorescence images of lamin A/C in WT or Plec KO cells cultured on PA gels. Scale bars, 10 μm. (F) Quantification of total lamin A/C fluorescence per cell in WT and Plec KO cells on different PA gels. Data represent means ± SD for N = 30 cells from three experiments. *P < 0.05 compared to WT for the same stiffness. (G) Representative immunofluorescence images of F-actin in WT or Plec KO cells cultured on collagen-coated coverslips. Scale bars, 10 μm. (H) Quantification of total lamin A/C fluorescence in WT and Plec KO cells on collagen-coated coverslips treated with carrier (0.1% DMSO) or 50 μM blebbistatin. Data represent means ± SD for N = 63 to 88 cells from three experiments. +P < 0.05 compared to WT for the same treatment; *P < 0.05 compared to carrier for the same cell type. (I) Immunofluorescence images of F-actin in WT and type I keratin (KI) KO and Western blot analysis of lamin A/C, K14, and GAPDH in WT and KI KO cells. Scale bars, 10 μm.
We next assessed the downstream effects of plectin deficiency on lamin A/C expression. We observed a significant increase in lamin A/C levels in Plec KO keratinocytes compared to WT keratinocytes (Fig. 6, E and F). In addition, Plec KO keratinocytes displayed thickened F-actin stress fibers consistent with previous findings (24, 31) (Fig. 6G). Up-regulation of lamin A/C in Plec KO cells was in direct contrast to the effects of the K14 R416P mutation and indicates that these two perturbations differentially affect cellular mechanosensing. To test whether plectin deficiency also mediated nuclear mechanotransduction via F-actin tension, we attempted to reverse lamin A/C up-regulation in Plec KO keratinocytes by inhibition of actomyosin contractility. Here, Plec KO cells were more sensitive to blebbistatin treatment than WT keratinocytes and displayed a greater reduction in lamin A/C expression (Fig. 6H). This result therefore demonstrates that tension within the actin cytoskeleton plays a central role in force transduction to the nucleus and mediates the differential effects of the K14 R416P mutation and plectin deficiency on the nuclear lamina.
As plectin is an essential linker between integrins and keratin filaments, we also explored the influences of substrate stiffness and EBS mutations on hemidesmosome assembly. Immunostaining for integrin α6 revealed no apparent effects of substrate stiffness on the levels or organization of hemidesmosomes on type I collagen–coated PA gels (fig. S6, A and B). Under these experimental conditions, plectin localized along the K14 bundles (fig. S6, A and C). While this pattern was not affected by introduction of the K14 R416P mutation, plectin was absent from the K14 R416P aggregates (fig. S6D). Likewise, there were no observable differences in integrin α6 expression between WT and Plec KO keratinocytes (fig. S6E). As all of our studies were carried out on sparse keratinocytes cultured on collagen-coated substrates, these responses likely reflect a wound healing environment, and in future studies, it will be interesting to explore similar mechanotransduction processes in confluent epithelial sheets with substrates coated with basement membrane extracellular matrix.
Last, we compared the disparate responses of K14 R416P mutation and plectin deficiency to the effects of a total loss of keratin using mouse keratinocytes lacking the entire type I keratin locus (32). Similar to the Plec KO keratinocytes, total keratin depletion led to an up-regulation of F-actin stress fibers and lamin A/C levels in cells cultured on rigid glass or plastic substrates (Fig. 6I). These results further confirm that distinct perturbations of the keratin cytoskeleton can differentially affect nuclear mechanotransduction.
DISCUSSION
The overall findings of this study provide previously unidentified insights into how the keratin cytoskeleton structurally and mechanically adapts to changes in the cell’s mechanical environment and the downstream effects on signal transduction to the nucleus. We show that increasing matrix stiffness promotes the formation of a more highly cross-linked and intersecting keratin network, which is less deformable and corresponds to increased stiffness of the cell as a whole. This response also depends on cross-talk with the F-actin cytoskeleton, suggesting that actomyosin contractility helps to direct keratin remodeling. Notably, the keratin cytoskeleton mediates downstream mechanotransduction to the nucleus. While K14 R416P overexpression disrupted keratin stability and down-regulated lamin A/C expression, loss of either the cytolinker plectin or the entire type I keratin locus led to an up-regulation of lamin A/C. The distinct effects of these different perturbations to the keratin cytoskeleton highlight the complexity of cellular mechanotransduction in keratinocytes and suggest that specific subtypes of EBS differentially affect keratinocyte mechanosensing.
On the basis of our current work and recent findings from other groups (24, 33, 34), we propose that tension within the actin cytoskeleton is a key determinant of the differential effects of EBS mutations on the nucleus. In Plec KO keratinocytes, increased lamin A/C was associated with increased formation of F-actin stress fibers. These results are consistent with previous reports in which disruption of hemidesmosomes by loss of either the β4 integrin or plectin led to increased focal adhesion assembly, stress fiber formation, and traction force generation (24). Likewise, live-cell imaging of keratin dynamics has demonstrated associations between focal adhesions and keratin assembly (27) and between the flows of keratin and actin in migrating cells (35). By contrast, the K14 R416P mutation reduced stress fiber formation and resulted in mislocalized phospho-myosin, indicating that this mutation has a negative impact on actomyosin contractility. Analogous destabilizing mutations in K14 have recently been shown to have a similar effect on focal adhesions, F-actin, and traction force generation (33), and in the context of EBS, K14 instability can also have a distinct and a more severe impact on desmosomal adhesions and cell mechanics compared to either K5 mutations or total loss of keratin (34). One potential explanation for these responses could involve sequestration of F-actin regulators, such as receptor for activated c-kinase 1 (RACK1) (36, 37) or the guanine nucleotide exchange factor Solo (38), within keratin aggregates, thereby inhibiting the ability to generate F-actin tension. Alternatively, aggregate formation may stimulate a more general misfolded protein or stress response that inhibits contractility (39, 40). Dissecting these unique mechanisms of mechanotransduction in dominant EBS mutations will therefore be an important area of investigation in future studies.
The actin cytoskeleton directly connects to the nucleus through the linker of the cytoskeleton and nucleoskeleton complex (LINC) and applies forces to the nuclear lamina (41). Lamin A/C, in particular, can sense and adapt to F-actin tension to maintain the structure and integrity of the nucleus in other cell types (23, 42). We show here that lamin A/C expression in keratinocytes similarly increases with increasing matrix stiffness. While inhibition of actomyosin activity reduced lamin A/C levels, Rho A activation rescued expression on soft substrates or in K14 R416P mutants. This line of evidence supports a central role for F-actin tension in mediating the response to the different EBS mutations. While this study focused exclusively on the interplay between actin and keratin, it is also important to recognize potential interactions with the microtubule network, which control keratinocyte polarity and asymmetric cell division (43) and could contribute to the biomechanical regulation of nuclear positioning via LINC components, such as Nesprin-4 (44).
As the nuclear lamina regulates a wide range of nuclear functions including chromatin remodeling (17), DNA damage repair (42, 45), and activity of specific transcription factors (46), it has the potential to have a significant impact on the response of keratinocytes to altered matrix stiffness. In particular, our findings implicate lamin A/C in the regulation of stiffness-dependent heterochromatin remodeling. The distinct effects of the K14 R416P mutation and plectin deficiency on lamin A/C levels could therefore lead to differential changes in gene expression, which, in turn, could affect disease pathogenesis. A major area for future investigations will be mapping out the specific downstream changes in gene expression associated with the different mutations and their sensitivity to various mechanical stimuli beyond matrix stiffness. Understanding of the unique signaling pathways could ultimately open up new opportunities for targeted and EBS subtype–specific therapies.
MATERIALS AND METHODS
PA gel fabrication
All chemicals and reagents were from Sigma-Aldrich unless otherwise noted. PA gels were prepared on the basis of previously published protocols. Briefly, 13-mm-diameter round or 22 mm × 60 mm rectangular glass coverslips were first cleaned by three 15-min sonication cycles in ethanol and left to dry overnight. A silane coating was then applied by immersing the coverslips in 5% (v/v) 3-aminopropyltrimethylsiloxane in water solution for 30 min under agitation. After six washes with water, coverslips were incubated with a 0.5% (v/v) glutaraldehyde solution in phosphate-buffered saline (PBS) for 15 min. Coverslips were washed three times with deionized water and left to dry overnight. To prepare PA gels with varied rigidities, different stock solutions were prepared with a ratio variation between PA and bis-acrylamide (fig. S1A). These stock solutions were degased for 5 min at room temperature and further stored at 4°C until use. To create a thin layer of PA on the coverslip surface, 1 ml of each stock solution was mixed with 4 μl of Tetramethylethylenediamine and 6 μl of 10% (v/v) ammonium persulfate (APS) in water. Drops of 10 μl of the final mixture were placed on a Sigmacote-coated microscope slide and then covered by the silane-coated coverslips. Gels were allowed to cross-link for 5 min at room temperature. After this period, each slide was immersed in a 50 mM Hepes solution in water. Once hydrated, the gels were gently peeled off the microscope slide and stored at 4°C in 50 mM Hepes solution until use.
To promote cell attachment, PA gels were functionalized with type I collagen from rat tail (BD Biosciences). PA gels were first activated by incubation with the cross-linker Sulfo-SANPAH (0.2 mg/ml) (BioVision) and exposure to 365-nm ultraviolet (UV) light for 5 min. Gels were washed twice with deionized water, and the reaction was repeated a second time. Gels were then washed twice with PBS, incubated with collagen (50 μg/ml) for 3 hours at room temperature, and washed twice more with PBS. Before cell seeding, gels were sterilized by exposure to 254-nm UV light for 3 min in a UVP 95-0339-02 cross-linking drawer (John Morris Group) followed by two sterile PBS washes, a brief rinse with 70% (v/v) ethanol, and two final washes with PBS. PA substrates were incubated with cell culture medium for 30 min immediately before cell seeding.
Microindentation of PA gels
To characterize PA gel stiffness, microindentation was performed using ElectroPuls E1000 (Instron) fitted with a 10-N load cell. A preload of 0.3 mN was first applied, and samples were displaced to 20% strain at a rate of 1%/s, and the resultant force was acquired. A 10-fold slower strain rate of 0.1%/s was also applied to all samples, and the obtained traces were identical, confirming their elastic properties. The linear region of the force-displacement curve was further used to calculate the tangent modulus, as previously described (47).
Micropatterned substrate fabrication
Micropatterned substrates were fabricated by microcontact printing and surface-initiated polymerization of (oligo ethylene glycol methacrylate) (POEGMA) brushes as previously described (18). Briefly, master silicon molds were created by photolithography and used to cast poly-dimethylsiloxane stamps. The micropatterned stamps were inked with the thiol initiator, ω-mercaptoundecyl bromoisobutyrate, and brought into conformal contact with gold-coated coverslips for 15 s to deposit the initiator as a self-assembled monolayer. Atom transfer radical polymerization of the monomeric solution oligo(ethylene glycol) methyl ether methacrylate (Mn 300) was carried out in a water/ethanol (4:1) solution of OEGMA (1.6 M), Cu(II)Br (3.3 mM), 2,2-bipyridine (82 mM), and Cu(I)Cl (33 mM). The reaction was performed at room temperature for 15 min and rinsed twice with water. Following sterilization with 70% ethanol, patterned substrates were coated with rat type I collagen (20 μg/ml) (BD Biosciences) in PBS for 1 hour at 37°C. Substrates were rinsed three times with 1 mM HCl and twice with PBS. All chemicals were from Sigma-Aldrich unless otherwise indicated.
Cell culture
All cell culture supplies were from Thermo Fisher Scientific unless otherwise stated. The HaCaT keratinocyte line was maintained in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 1% (v/v) penicillin/streptomycin (P/S) and 10% (v/v) fetal bovine serum (FBS) (Biosera). WT and complete Plec KO (lacking all isoforms) mouse keratinocytes were provided by G. Wiche’s laboratory (Vienna, Austria) (25), cultivated in EpiLife medium, and supplemented with 1× Human Keratinocyte Growth Supplement and 1% (v/v) P/S. Keratinocytes from WT mice and mice lacking the entire type I keratin cluster (KI KO) were provided by T. Magin’s laboratory (Leipzig, Germany) (34) and maintained in DMEM/F12 1:3 supplemented with 10% chelex-treated FBS, 0.18 mM adenine (Sigma-Aldrich), hydrocortisone (0.5 mg/ml), 100 pM cholera toxin (Sigma-Aldrich), epidermal growth factor (10 ng/ml; PeproTech), insulin (0.5 μg/ml; Sigma-Aldrich), and 1% P/S. For passaging, all cell lines were detached through a 5- to 10-min incubation with 0.05% (v/v) trypsin/EDTA and further resuspended in either complete DMEM (HaCaTs) or EpiLife + 10% FBS (v/v) (mouse keratinocytes). Cells were then centrifuged at 1200 rpm for 5 min. The pellet was homogenized using 10 ml of fresh medium, and cells were replated. Mouse keratinocytes were specifically replated on type I collagen–coated cell culture dishes. All cell lines were maintained in 5% CO2 at 37°C.
To generate keratinocytes with stable expression of WT and R416P mutant K14, HaCaT cells were transduced with retroviral plasmids encoding the WT and mutant genes. pLPC_NMyc vector, a gift from T. de Lange (Addgene plasmid #12540), was reengineered to replace NMyc with AcGFP. Human K14 complementary DNA (cDNA) was amplified and cloned in Eco RI and Bam HI restriction sites. Site-directed mutagenesis was carried out to introduce mutation in K14 cDNA using Phusion DNA polymerase Taq DNA ligase both from NEB. Amphotropic retrovirus supernatant was prepared as described previously (48). Cells were incubated in DMEM with polybrene (5 μg/ml) for 15 min and then replaced by DMEM supplemented with fresh polybrene and virus (1:1 diluted virus supernatant) mix. Cells were centrifuged for 1 hour, at 1000 rpm and 32°C to aid transduction, followed by a medium change and 37°C incubation until confluency was achieved. Cell selection was further performed for 10 days using G418 sulfate (500 μg/ml) (active concentration). After 10 days of selection, GFP-positive cells were further purified by flow sorting (BD FACSAria IIIu) and replated in DMEM supplemented with 10% FBS (v/v) and 1% P/S (v/v). Further culture of transduced cells was carried out in the presence of G418 sulfate (500 μg/ml). Before seeding onto PA gels, cells were incubated without G418 sulfate for 24 hours, and all experiments on PA gels did not include G418 sulfate.
Primary human keratinocytes from neonatal foreskin were cultured on a feeder layer of J2-3T3 fibroblasts as previously described (49). Fibroblasts were maintained in DMEM GlutaMAX, supplemented with 10% bovine serum and 1% (v/v) P/S, and an inactivated monolayer was prepared by treatment with mitomycin C (0.4 mg/ml; Sigma-Aldrich) 24 hours before plating the keratinocytes.
Primary keratinocytes were cultured in FAD medium consisting of one part Ham’s F12, three parts DMEM, 100 nM adenine, 10% FBS, 1% P/S, hydrocortisone (0.5 μg/ml), insulin (5 μg/ml), 0.1 nM cholera toxin, and epidermal growth factor (10 ng/ml). Primary keratinocytes were passaged at 60 to 70% confluency by first removing the fibroblasts with Versene, followed by detachment of the keratinocytes with 0.05% (v/v) trypsin/EDTA solution.
For immunostaining, cells were seeded at a density of 15,000 cells/cm2 on PA-coated 13-mm-diameter coverslips. For Western blot analysis, cells were seeded onto rectangular (20 mm x 60 mm) PA-coated coverslips at a density of 56,800 cells/cm2. To improve cell seeding efficiency and uniformity, rectangular substrates were placed in 10-cm dishes coated with polydimethlysiloxane (PDMS; Sylgard 184, Farnell), which provided a hydrophobic coating and concentrated the cell suspension on the PA gels. All analyses were performed 24 hours after seeding onto PA gels. For inhibitor treatments, cells were allowed to adhere for 1 hour before treatment with carrier control medium [0.1% dimethyl sulfoxide (DMSO)], 50 μM blebbistatin (Sigma-Aldrich), or CN03 (5 μg/ml; Rho Activator II, Cytoskeleton).
siRNA transfection
HaCaT keratinocytes were seeded into T25 culture flasks at a density of 8 × 105 cells per flask and cultured for 24 hours before transfection. Cells were then transfected with 20 nM of the nontargeting control RNA (Silencer Select Negative Control, 4390843, Thermo Fisher Scientific) or LMNA targeting siRNA (Silencer Select, s8221, Thermo Fisher Scientific) using the jetPRIME reagent (Polyplus Transfection) according to the manufacturer’s instructions. To improve knockdown, cells were transfected a second time 24 hours later and then seeded onto PA gels 72 hours after the first transfection.
Western blot analysis
For total cell lysates, each large coverslip (22 mm × 60 mm) was washed with PBS and placed in a new petri dish on ice. Cells were incubated with radioimmunoprecipitation assay (RIPA) buffer (7 μl/cm2) with 10% (v/v) phosphatase inhibitor (Roche) and 0.5% (v/v) protease inhibitor (Sigma-Aldrich) for 5 min, then gently scraped, and transferred to microcentrifuge tubes. Samples were sonicated at high intensity for 30-s intervals for 5 min, and the supernatant was collected and used on further analysis. Protein concentration was measured using the BCA (Bicinchoninic Acid) Assay (Pierce) according to the manufacturer’s instructions. For loading samples, total protein was adjusted using 4× sample buffer, β-mercaptoethanol (Sigma-Aldrich), and water. When using nonreduced samples, β-mercaptoethanol was not added to the final loading mix.
All lysates were denatured at 95° for 5 min before loading into an 8% (w/v) PA running gel. Electrophoresis was performed using the Bio-Rad Mini-Protean Tetra Cell System (Bio-Rad) at 100 V for 90 min. Protein was transferred to a nitrocellulose membrane (GE Healthcare) at 300 mA for 1 hour at 4°C. Each membrane was incubated for 1 min with Ponceau S (Sigma-Aldrich) to confirm uniform and complete transfer. Membranes were further incubated for 1 hour with a 5% nonfat dry milk (Marvel) solution in tris-buffered saline with 0.5% Tween (TBS-T) under agitation. Membranes were then incubated with selected primary antibodies overnight at 4°C. The following day, membranes were washed three times with TBS-T and further incubated for 1 hour at room temperature with the secondary antibody [horseradish peroxidase–conjugated anti-rabbit or anti-mouse antibodies (1:5000, Dako)]. Membranes were washed three times with TBS-T and developed with an enhanced chemiluminescence (ECL) solution (Millipore) for 1 min followed by exposure to x-ray film (GE Healthcare). Primary antibodies included anti-K14 [clone LL002, Abcam (1:5000)], anti-K16 [ab76416, Abcam Rb (1:5000)], glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [ab9485, Abcam Rb (1:2000)], lamin A [ab26300, Abcam (1:1000)], and lamin A/C [clone 636, Santa Cruz Biotechnology (1:1000)].
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde in PBS (w/v) for 10 min at room temperature. After two PBS washes, cells were permeabilized for 10 min with 0.2% Triton X-100 in PBS (v/v), washed twice more with PBS, and blocked for 30 min in a solution of 10% FBS (v/v) and 0.25% fish skin gelatin (v/v) in PBS. Samples were incubated with the selected primary antibodies in blocking solution at 4°C overnight. Primary antibodies for immunostaining included anti-K14 [clone LL002, Abcam (1:500)], lamin A/C [clone 636, Santa Cruz Biotechnology (1:500)], lamin A [ab26300, Abcam (1:500)], paxillin [clone 177, BD Biosciences (1:500)], H3K9me3 [ab8898, Abcam (1:500)], plectin [clone 10F6, Santa Cruz Biotechnology (1:200)], integrin α6 [clone 4F10, eBioscience (1:200)], and phospho-myosin light chain [T18/S19, Cell Signaling Technology (1:200)]. Cells were washed three times in PBS and incubated with secondary antibodies and/or dyes for 1 hour at room temperature. Secondary antibodies included anti-mouse [Alexa Fluor 488 and Alexa Fluor 568, Molecular Probes (1:1000)] and anti-rabbit [Alexa Fluor 568, Molecular Probes (1:1000)]. Phalloidin [Alexa Fluor 488 and Alexa Fluor 633, Molecular Probes (1:200)] and 4′,6-diamidino-2-phenylindole [DAPI; Biotium (1:1000)] were used to label the F-actin cytoskeleton and nuclei, respectively. Samples were washed a final time with PBS and deionized water. For mounting, 10-μl drops of Mowiol were placed on small square coverslips, and stained samples were mounted on top of the drop and sealed with nail polish. The reverse side of the stained coverslip was then mounted on top of a standard glass microscope slide with another 10-μl drop of Mowiol.
Image acquisition and analysis
Immunofluorescence imaging was performed using either the DM4000 epifluorescence microscope (Leica) with a 40× objective or the LSM 880 confocal microscope with Airyscan (Zeiss) and a 63× objective (Plan-Apochromatic, 1.4 oil), driven by the Zen Black 2.3 software. Three-dimensional Z-stack images were acquired with the Airsycan 32 GaAsP array detector with the laser lines HeNe 633 nm, IDPSS 561 to 10 nm, diode 405 to 30 nm, and argon 488 nm, beam splitters 488/561/633 nm and 405 nm, and bandpass filter 570 to 620 nm plus low-pass filter 670 nm for the far red channel and bandpass filters 420 to 480 nm plus 495 to 620 nm for the red channel, 420 to 480 nm plus 495 to 550 nm for the green channel, and 420 to 480 nm plus 495 to 550 nm for the blue channel. Images were combined into maximum intensity projections for image analysis.
Overall cell and nuclear areas were measured from epifluorescence images of the K14 and DAPI signals, respectively, and quantified using Cell Profiler based on intensity and area segmentation. Analysis of the keratin cytoskeleton was performed using ImageJ. For bundle thickness, spacing, and branching measurements, 80-μm2 regions of interest within the cytoplasm were selected (avoiding the nucleus), enhanced using the Contrast Limited Adaptive Histogram Equalization (CLAHE) filter, and converted to a binary image using a fixed contrast and brightness threshold. Thickness and spacing were analyzed using the BoneJ plug-in, and branching was estimated by skeletonizing the image and measuring the number of junctions using the Skeleton plug-in. At least three regions of interest were measured per cell. Overall alignment of keratin bundles was analyzed using the Alignment plug-in to measure the dispersion of bundle angles in nonthresholded images on whole cells. Colocalization of F-actin and pMyosin was measured within regions of interest associated with K14 R416P aggregates or intact filaments using the Coloc2 tool in ImageJ and measuring the Pearson correlation coefficient between the F-actin and pMyosin channels. Similar analysis was performed for plectin and K14 correlation within cytoplasmic regions of interest in the parental HaCaT line.
Actin filament, focal adhesion, and lamin A/C analysis was performed using the IN Cell Developer Toolbox (GE). Here, single cells were automatically identified by segmentation of the DAPI and phalloidin signals and separated using a clump breaking step. Individual F-actin filaments in each cell were then detected using intensity and size segmentation for the phalloidin images. Similarly, focal adhesions at the periphery of the cells were identified on the basis of intensity size segmentation for the paxillin images. Total lamin A/C fluorescence was measured within the nuclear region of each cell.
Cell mechanics analysis
AFM was used to measure the biophysical properties of cells. The AFM System (NanoWizard4, JPK) was mounted on an epifluorescence microscope (AxioObserver Z.1, Zeiss). Images of live keratinocytes were scanned under liquid conditions (DMEM medium with 25 mM Hepes supplemented with 10% FBS and 1% P/S) at 37°C with a V-shaped silicon nitride cantilever (MSNL-10 Bruker) in contact mode. The cantilever had a spring constant of 0.03 N/m, length of 225 μm, and width of 20 μm. The spring constant of the cantilever was calibrated using the thermal fluctuations method based on sensitivity calculation on the glass. Force maps of the cells were taken in quantitative imaging mode at a resolution of 32 × 32 pixels, using a ramp length of 6000 nm, ramp speeds of 300 μm/s, and a force setpoint of 4 nN. The scan area was 80 μm x 80 μm to include an entire cell. The Young’s modulus (E) was determined from the force-distance curves. The force-distance curves were analyzed using the “Composite Cell-Substrate” (CoCS) model for adherent cells over soft substrates (50) and a custom pipeline written in MATLAB. The 1024 pixels acquired for each quantitative image map were split into categories “background” or “cell” by thresholding the histogram of the pixel height values using Otsu’s method. The pixels categorized as “cell” were further split into categories “cytoplasm” (measured pixel height within 20 to 55% of total cell height) or “nucleus” (measured pixel height within 65 to 100% of total cell height). For each probed cell, values reported for Ecell, Ecytoplasm, and Enucleus correspond to the median values of Young’s modulus of all pixels in an image map that had been categorized as cell, cytoplasm, or nucleus, respectively.
AFM and live fluorescence imaging was used to measure the keratin displacement of HaCaT cells expressing K14-GFP in parallel with indentation measurements. A V-shaped silicon nitride tipless cantilever (MLCT-O10, Bruker) with silica beads (6 to 8 μm, Spherotech) attached had a spring constant of 0.6 N/m, length of 75 μm, and width of 18 μm. Indentation of the cells was taken in force spectroscopy mode using the setpoint of 50 nN with extend speed of 2 μm/s and extend delay of 3 s. Each cell had two indentations on different points. In parallel, fluorescence images of K14-GFP cells were acquired using the epifluorescence microscope with a 20× objective. The image sequences were processed using particle image velocimetry (PIV) with the PIVlab app for MATLAB. Image pairs were analyzed 2 s apart. Images were preprocessed with a CLAHE filter with a window size of 20 pixels and denoised with a Wiener denoise filter with a kernel size of 3. Noncell areas were masked, and PIV with a fast Fourier transform window deformation was applied over multiple passes with window sizes decreasing by a power of 2. The smallest window size was 16 pixels. Velocity limits were manually applied to exclude noise, and the vector field was filtered using an SD threshold of 7 and a median threshold of 5. The displacement maps corresponding to the indentation events were identified by finding the displacement fields with the largest mean displacement distributed at an even period through the image sequence. On these selected displacement maps, the local displacement at the indentation site was measured as the mean of the displacements in a 12.5-μm radius around the indentation point.
Statistical analysis
All data were analyzed with either a one-way or two-way analysis of variance (ANOVA) and Tukey’s test for post-hoc analysis using Prism (GraphPad). For whole-cell morphology measurements including cell area, nuclear area, and percentage of cells with aggregates, data were analyzed as means ± SEM of three independent experiments. For cell mechanics and subcellular analysis of the cytoskeleton, adhesions, and nuclear lamina, data were analyzed as means ± SD of single cells pooled from three independent experiments. Significance was determined for P < 0.05.
Acknowledgments
We would like to thank J. Soetaert for assistance with the Airyscan superresolution imaging, L. Gammon for assistance with the IN Cell Developer Toolbox, and R. Delaine-Smith for assistance with the microindentation measurements. Funding: This study was funded by the Biotechnology and Biological Sciences Research Council (BB/P006108/1), the Queen Mary Institute of Bioengineering PhD studentship for ACL, and the Rosetrees Trust (M312-F1) to A.W. Author contributions: A.C.L. carried out most of the cell culture experiments, analyzed data, and cowrote the manuscript. K.S., M.C.K., and N.G. carried out AFM experiments on cell mechanics and analyzed the data. N.G. cowrote the manuscript. O.J.P. performed micropattern experiments and analyzed chromatin remodeling. R.R. prepared PA gels and carried out Western blot analysis. D.A. and A.W. generated the K14 WT and R416P constructs and assisted with cell line generation. J.T.C. carried out inhibitor and Rho activation experiments, analyzed data, and cowrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd6187/DC1
REFERENCES AND NOTES
- 1.Jacob J. T., Coulombe P. A., Kwan R., Omary M. B., Types I and II keratin intermediate filaments. Cold Spring Harb. Perspect. Biol. 10, a018275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramms L., Fabris G., Windoffer R., Schwarz N., Springer R., Zhou C., Lazar J., Stiefel S., Hersch N., Schnakenberg U., Magin T. M., Leube R. E., Merkel R., Hoffmann B., Keratins as the main component for the mechanical integrity of keratinocytes. Proc. Natl. Acad. Sci. U.S.A. 110, 18513–18518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seltmann K., Fritsch A. W., Käs J. A., Magin T. M., Keratins significantly contribute to cell stiffness and impact invasive behavior. Proc. Natl. Acad. Sci. U.S.A. 110, 18507–18512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walko G., Castañón M. J., Wiche G., Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 360, 529–544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broussard J. A., Getsios S., Green K. J., Desmosome regulation and signaling in disease. Cell Tissue Res. 360, 501–512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulombe P. A., Kerns M. L., Fuchs E., Epidermolysis bullosa simplex: A paradigm for disorders of tissue fragility. J. Clin. Invest. 119, 1784–1793 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulombe P. A., Hutton M. E., Letai A., Hebert A., Paller A. S., Fuchs E., Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: Genetic and functional analyses. Cell 66, 1301–1311 (1991). [DOI] [PubMed] [Google Scholar]
- 8.McGrath J. A., Ishida-Yamamoto A., Tidman M. J., Heagerty A. H., Schofield O. M., Eady R. A., Epidermolysis bullosa simplex (Dowling-Meara). A clinicopathological review. Br. J. Dermatol. 126, 421–430 (1992). [DOI] [PubMed] [Google Scholar]
- 9.Ishida-Yamamoto A., McGrath J. A., Chapman S. J., Leigh I. M., Lane E. B., Eady R. A., Epidermolysis bullosa simplex (Dowling-Meara type) is a genetic disease characterized by an abnormal keratin-filament network involving keratins K5 and K14. J. Invest. Dermatol. 97, 959–968 (1991). [DOI] [PubMed] [Google Scholar]
- 10.McLean W. H., Pulkkinen L., Smith F. J., Rugg E. L., Lane E. B., Bullrich F., Burgeson R. E., Amano S., Hudson D. L., Owaribe K., McGrath J. A., McMillan J. R., Eady R. A., Leigh I. M., Christiano A. M., Uitto J., Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev. 10, 1724–1735 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Smith F. J., Eady R. A., Leigh I. M., McMillan J. R., Rugg E. L., Kelsell D. P., Bryant S. P., Spurr N. K., Geddes J. F., Kirtschig G., Milana G., de Bono A. G., Owaribe K., Wiche G., Pulkkinen L., Uitto J., McLean W. H., Lane E. B., Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat. Genet. 13, 450–457 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Trappmann B., Gautrot J. E., Connelly J. T., Strange D. G., Li Y., Oyen M. L., Cohen Stuart M. A., Boehm H., Li B., Vogel V., Spatz J. P., Watt F. M., Huck W. T., Extracellular-matrix tethering regulates stem-cell fate. Nat. Mater. 11, 642–649 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Gautrot J. E., Malmström J., Sundh M., Margadant C., Sonnenberg A., Sutherland D. S., The nanoscale geometrical maturation of focal adhesions controls stem cell differentiation and mechanotransduction. Nano Lett. 14, 3945–3952 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Kenny F. N., Drymoussi Z., Delaine-Smith R., Kao A. P., Laly A. C., Knight M. M., Philpott M. P., Connelly J. T., Tissue stiffening promotes keratinocyte proliferation through activation of epidermal growth factor signaling. J. Cell Sci. 131, jcs215780 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Wickert L. E., Pomerenke S., Mitchell I., Masters K. S., Kreeger P. K., Hierarchy of cellular decisions in collective behavior: Implications for wound healing. Sci. Rep. 6, 20139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takei T., Han O., Ikeda M., Male P., Mills I., Sumpio B. E., Cyclic strain stimulates isoform-specific PKC activation and translocation in cultured human keratinocytes. J. Cell. Biochem. 67, 327–337 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Le H. Q., Ghatak S., Yeung C.-Y. C., Tellkamp F., Günschmann C., Dieterich C., Yeroslaviz A., Habermann B., Pombo A., Niessen C. M., Wickström S. A., Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Connelly J. T., Gautrot J. E., Trappmann B., Tan D. W., Donati G., Huck W. T., Watt F. M., Actin and serum response factor transduce physical cues from the microenvironment to regulate epidermal stem cell fate decisions. Nat. Cell Biol. 12, 711–718 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Miroshnikova Y. A., Le H. Q., Schneider D., Thalheim T., Rübsam M., Bremicker N., Polleux J., Kamprad N., Tarantola M., Wang I., Balland M., Niessen C. M., Galle J., Wickström S. A., Adhesion forces and cortical tension couple cell proliferation and differentiation to drive epidermal stratification. Nat. Cell Biol. 20, 69–80 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Walko G., Woodhouse S., Pisco A. O., Rognoni E., Liakath-Ali K., Lichtenberger B. M., Mishra A., Telerman S. B., Viswanathan P., Logtenberg M., Renz L. M., Donati G., Quist S. R., Watt F. M., A genome-wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat. Commun. 8, 14744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Totaro A., Castellan M., Battilana G., Zanconato F., Azzolin L., Giulitti S., Cordenonsi M., Piccolo S., YAP/TAZ link cell mechanics to Notch signalling to control epidermal stem cell fate. Nat. Commun. 8, 15206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lammerding J., Schulze P. C., Takahashi T., Kozlov S., Sullivan T., Kamm R. D., Stewart C. L., Lee R. T., Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 113, 370–378 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swift J., Ivanovska I. L., Buxboim A., Harada T., Dingal P. C. D. P., Pinter J., Pajerowski J. D., Spinler K. R., Shin J.-W., Tewari M., Rehfeldt F., Speicher D. W., Discher D. E., Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W., Zuidema A., Te Molder L., Nahidiazar L., Hoekman L., Schmidt T., Coppola S., Sonnenberg A., Hemidesmosomes modulate force generation via focal adhesions. J. Cell Biol. 219, e201904137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida F. V., Walko G., McMillan J. R., McGrath J. A., Wiche G., Barber A. H., Connelly J. T., The cytolinker plectin regulates nuclear mechanotransduction in keratinocytes. J. Cell Sci. 128, 4475–4486 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Lee C.-H., Kim M.-S., Chung B. M., Leahy D. J., Coulombe P. A., Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat. Struct. Mol. Biol. 19, 707–715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kölsch A., Windoffer R., Würflinger T., Aach T., Leube R. E., The keratin-filament cycle of assembly and disassembly. J. Cell Sci. 123, 2266–2272 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Wood P., Baty D. U., Lane E. B., McLean W. H. I., Long-range polymerase chain reaction for specific full-length amplification of the human keratin 14 gene and novel keratin 14 mutations in epidermolysis bullosa simplex patients. J. Invest. Dermatol. 120, 495–497 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Nava M. M., Miroshnikova Y. A., Biggs L. C., Whitefield D. B., Metge F., Boucas J., Vihinen H., Jokitalo E., Li X., García Arcos J. M., Hoffmann B., Merkel R., Niessen C. M., Dahl K. N., Wickström S. A., Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ackerl R., Walko G., Fuchs P., Fischer I., Schmuth M., Wiche G., Conditional targeting of plectin in prenatal and adult mouse stratified epithelia causes keratinocyte fragility and lesional epidermal barrier defects. J. Cell Sci. 120, 2435–2443 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Osmanagic-Myers S., Gregor M., Walko G., Burgstaller G., Reipert S., Wiche G., Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 174, 557–568 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bär J., Kumar V., Roth W., Schwarz N., Richter M., Leube R. E., Magin T. M., Skin fragility and impaired desmosomal adhesion in mice lacking all keratins. J. Invest. Dermatol. 134, 1012–1022 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Fujiwara S., Deguchi S., Magin T. M., Disease-associated keratin mutations reduce traction forces and compromise adhesion and collective migration. J. Cell Sci. 133, jcs243956 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Homberg M., Ramms L., Schwarz N., Dreissen G., Leube R. E., Merkel R., Hoffmann B., Magin T. M., Distinct impact of two keratin mutations causing epidermolysis bullosa simplex on keratinocyte adhesion and stiffness. J. Invest. Dermatol. 135, 2437–2445 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Pora A., Yoon S., Dreissen G., Hoffmann B., Merkel R., Windoffer R., Leube R. E., Regulation of keratin network dynamics by the mechanical properties of the environment in migrating cells. Sci. Rep. 10, 4574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kröger C., Loschke F., Schwarz N., Windoffer R., Leube R. E., Magin T. M., Keratins control intercellular adhesion involving PKC-α-mediated desmoplakin phosphorylation. J. Cell Biol. 201, 681–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leube R. E., Moch M., Windoffer R., Intermediate filaments and the regulation of focal adhesion. Curr. Opin. Cell Biol. 32, 13–20 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Fujiwara S., Ohashi K., Mashiko T., Kondo H., Mizuno K., Interplay between Solo and keratin filaments is crucial for mechanical force-induced stress fiber reinforcement. Mol. Biol. Cell 27, 954–966 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Alessandro M., Russell D., Morley S. M., Davies A. M., Lane E. B., Keratin mutations of epidermolysis bullosa simplex alter the kinetics of stress response to osmotic shock. J. Cell Sci. 115, 4341–4351 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Chamcheu J. C., Navsaria H., Pihl-Lundin I., Liovic M., Vahlquist A., Törmä H., Chemical chaperones protect epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation: Involvement of heat shock proteins and MAP kinases. J. Invest. Dermatol. 131, 1684–1691 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Crisp M., Liu Q., Roux K., Rattner J. B., Shanahan C., Burke B., Stahl P. D., Hodzic D., Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 172, 41–53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho S., Vashisth M., Abbas A., Majkut S., Vogel K., Xia Y., Ivanovska I. L., Irianto J., Tewari M., Zhu K., Tichy E. D., Mourkioti F., Tang H.-Y., Greenberg R. A., Prosser B. L., Discher D. E., Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev. Cell 49, 920–935.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lechler T., Fuchs E., Asymmetric cell divisions promote stratification and differentiation of mammalian skin. Nature 437, 275–280 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roux K. J., Crisp M. L., Liu Q., Kim D., Kozlov S., Stewart C. L., Burke B., Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. U.S.A. 106, 2194–2199 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Earle A. J., Kirby T. J., Fedorchak G. R., Isermann P., Patel J., Iruvanti S., Moore S. A., Bonne G., Wallrath L. L., Lammerding J., Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 19, 464–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ho C. Y., Jaalouk D. E., Vartiainen M. K., Lammerding J., Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497, 507–511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaine-Smith R. M., Burney S., Balkwill F. R., Knight M. M., Experimental validation of a flat punch indentation methodology calibrated against unconfined compression tests for determination of soft tissue biomechanics. J. Mech. Behav. Biomed. Mater. 60, 401–415 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Aldehlawi H., Niemiec K. A., Avisetti D. R., Lalli A., Teh M.-T., Waseem A., The monoclonal antibody EPR1614Y against the stem cell biomarker keratin K15 lacks specificity and reacts with other keratins. Sci. Rep. 9, 1943 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rheinwald J. G., Green H., Epidermal growth factor and the multiplication of cultured human epidermal keratinocytes. Nature 265, 421–424 (1977). [DOI] [PubMed] [Google Scholar]
- 50.Rheinlaender J., Dimitracopoulos A., Wallmeyer B., Kronenberg N. M., Chalut K. J., Gather M. C., Betz T., Charras G., Franze K., Cortical cell stiffness is independent of substrate mechanics. Nat. Mater. 19, 1019–1025 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd6187/DC1