

An oligoamidine with two bacteria-specific mechanisms of action showed broad-spectrum, resistance-resistant antimicrobial effect.
Abstract
Antibiotic resistance is now a major threat to human health, and one approach to combating this threat is to develop resistance-resistant antibiotics. Synthetic antimicrobial polymers are generally resistance resistant, having good activity with low resistance rates but usually with low therapeutic indices. Here, we report our solution to this problem by introducing dual-selective mechanisms of action to a short amidine-rich polymer, which can simultaneously disrupt bacterial membranes and bind to bacterial DNA. The oligoamidine shows unobservable resistance generation but high therapeutic indices against many bacterial types, such as ESKAPE strains and clinical isolates resistant to multiple drugs, including colistin. The oligomer exhibited excellent effectiveness in various model systems, killing extracellular or intracellular bacteria in the presence of mammalian cells, removing all bacteria from Caenorhabditis elegans, and rescuing mice with severe infections. This “dual mechanisms of action” approach may be a general strategy for future development of antimicrobial polymers.
INTRODUCTION
Antibiotic resistance is an emerging threat to public health and to the global economy (1, 2). For this reason, there is interest in the development of resistance-resistant antibiotics (3–5), compounds having good efficacy in killing pathogens with acquired resistance, as well as having low rates of resistance generation. In general, antibiotics targeting a single enzyme have a high likelihood of generating resistance because a small change in the drug target can result in a large decrease in the drug’s potency (6), and successful monotherapeutic antibiotics are often those that have more than one enzyme target or are those with a more “physical” mechanism of action, such as disrupting bacterial cell membranes (7–9). Hence, membrane-targeting antimicrobial polymers are of interest since there are numerous naturally occurring examples—polypeptides and proteins—and their discovery has stimulated the development of synthetic mimics. These polymers frequently contain both cationic and hydrophobic regions (Fig. 1A) (10–17), with previous studies mainly focused on their structural diversification to achieve a better therapeutic index (TI). These studies have been fruitful but have been limited by the structural similarity of mammalian and bacterial cell membranes (13). A multitargeting antimicrobial exhibiting a second mechanism of action might reasonably be considered even more effective than a monotherapeutic agent targeting a single molecule or a membrane. Such an agent might additionally provide a path for future development of antimicrobial polymers. The question is how might such multitargeting be achieved?
Fig. 1. Design, synthesis, and characterization of the antimicrobial oligoamidine.
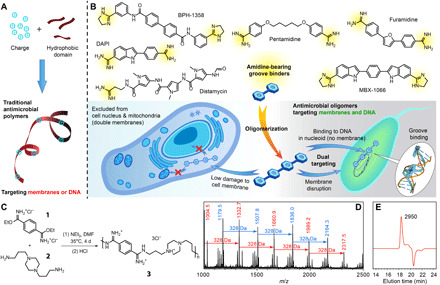
(A) Structural features of traditional antimicrobial polymers. (B) Structural illustration of groove binders, including pentamidine, the U.S. Food and Drug Administration–approved antiprotozoal agent and an antibiotic sensitizer; furamidine, an antitrypanosomal agent in clinical trials; distamycin, a polyamide-based antibiotic; 4′,6-diamidino-2-phenylindole (DAPI) and Hoechst 33342, commonly used DNA-staining dyes; as well as the two recently reported DNA-targeting antimicrobials, BPH-1358 and MBX-1066. The proposed mechanisms of the designed oligoamidine antimicrobial agent from the polymeric approach are also shown. (C) Synthetic scheme of the oligoamidine studied in this work. (D) MALDI-TOF (matrix-assisted laser desorption/ionization–time-of-flight) spectrum of oligomer 3. (E) Gel permeation chromatogram of oligomer 3 with marked molecular weight in dalton, calculated from a poly(ethylene glycol)–based calibration curve. The negative peak is due to the solvent. m/z, mass/charge ratio.
In this context, we were stimulated by the recent discovery that the wound dressing agent polyhexamethylene biguanide (PHMB), previously thought to function by membrane targeting, was actually killing bacteria through bacterial DNA condensation (18–20). Such macromolecular DNA binders are few (21–25) but are of interest because they suggest the possibility of using DNA binding as a second mechanism for antimicrobial polymers. Targeting DNA is, however, not without risk because DNA binders might also attack host DNA. PHMB only showed preferential binding to bacterial DNA over eukaryotic DNA in some instances (18–20), and no other reported DNA targeting macromolecules have demonstrated selectivity between bacterial and mammalian DNA (21–25). Nevertheless, it should be noted that bacterial DNA is located in nucleoids and is more vulnerable than its eukaryotic counterpart, which is protected by the nuclear envelope, a sieve that excludes large molecules (26). In addition, many polymers taken up by cells have been observed to be trapped in endosomes, further limiting their interactions with eukaryotic DNA. These observations lay the foundation for achieving selectivity for bacterial DNA binding, which, in combination with selective bacterial membrane disruption, is expected to lead to even higher potency, lower rates of resistance and, potentially, improved toxicity profiles. Here, we report a rationally designed polymer, an oligoamidine, that selectively targets the integrity of bacterial cell membranes and binds only to bacterial, but not mammalian, DNA. As the first example of an antimicrobial homopolymer with a dual-selective mechanism of action, the oligomer showed resistance-resistant and broad-spectrum efficacy against numerous bacteria, excellent therapeutic indices due to its selective disruption of bacterial membranes and inability to enter the eukaryotic nucleus (Fig. 1B), very low levels of resistance generation, and excellent in vivo activity.
RESULTS
Rational design of the oligomer
Among the DNA binding molecules with different binding modes, i.e., intercalation, alkylation, strand cleaving, and groove binding (27, 28), groove binders appeared to be the most suitable for the development of dual-selective antimicrobial polymers because they usually bear hydrophobic and cationic groups (29), resembling the structures of most antimicrobial polymer repeat units. This binding mode can also be easily made multivalent for stronger affinity (30–32) since an oligomerized groove binder can have high binding affinity to DNA due to the continuous DNA groove that the ligand can wind along. Given these considerations, we elected to construct oligomeric groove binders based on amidine structures. The amidine structure is both a cationicity provider and a well-recognized groove binding element (28, 29, 33), and some small-molecule examples are shown in Fig. 1B. In addition, the simplicity in constructing amidine linkages is also a key for the realization of this oligomeric design (Fig. 1C).
We chose to avoid the introduction of highly hydrophobic domains by putting cations and short alkyl chains directly on the oligomer backbone, forming a charge-on-backbone structure, expected to reduce cytotoxicity by avoiding surfactant-like features of segregated charge and hydrophobic domains (12, 34, 35). This side chain–free structure and the small benzamidine motif were also considered necessary for groove binding, to prevent undesirable steric hindrance that would reduce DNA binding affinity. The oligoamidine synthesis route featured the relatively slow coupling reaction between imidate esters and amines, as well as the less-soluble oligomeric products in N,N′-dimethylformamide (DMF), which put an upper limit on the products’ molecular weight (ca. 2 to 2.5 kDa; Fig. 1, D and E, and fig. S1), potentially lowering its cytotoxicity (12). However, the oligoamidine was still large enough to be excluded from the eukaryotic nucleus (26). Following this strategy, a series of oligoamidines was synthesized from the polycondensation of p-ethylphthalimidate (1) and various diamines (2 and others; table S1). Diamine linkers with different hydrophobicity, flexibility, and charge were chosen because these factors are known to affect DNA binding (29, 36, 37) and cell membrane binding (11, 12). As these bindings are mostly based on hydrogen bonding, electrostatic interactions, and hydrophobic forces, a screening of oligomers from these diamines could be a convenient route to a hit. In an initial screening, oligoamidine 3 (formed from 1 and 2) showed the best performance against Enterococcus faecalis, Staphylococcus aureus, Escherichia coli, and Bacillus subtilis and was chosen for further investigation.
Oligomer 3 has broad-spectrum antimicrobial activity and low hemolytic activity
We investigated the activity of 3 against B. subtilis, E. faecalis, Mycobacterium smegmatis, Mycobacterium tuberculosis, and all of the ESKAPE pathogens (Table 1, in bold), including multiple multidrug-resistant (MDR) clinical isolates. Table 1 lists the minimum inhibitory concentration (MIC) values of 3 against these bacteria, and the MIC values of standard antibiotics are also summarized for comparison. It was clear that some clinical isolates exhibited very high resistance against colistin, one of the antibiotics of last resort, but they remained highly susceptible to 3. 3 showed high serum resistance, killing MDR clinical isolates in medium with 50% fetal bovine serum (FBS) or in whole sheep blood (fig. S2A). In addition, considering that a metabolically repressed, antibiotic-tolerant state is of importance for antibiotic persistence and tolerance (38–40), we also evaluated the killing kinetics of 3 against MDR Acinetobacter baumannii in Dulbecco’s modified Eagle’s medium (DMEM; with 10% FBS), a nutrient-free medium, to examine 3’s effectiveness against such dormant bacteria. Results indicated that 3 was strongly bactericidal in DMEM, with a 15-min minimal bactericidal concentration (MBC) of 8 μg/ml, 90-min MBC of 2 μg/ml, and 24-hour MBC of ≤0.25 μg/ml against A. b -1 (fig. S2B). For comparison, ciprofloxacin’s 1.5- and 24-hour MBC under the same evaluation condition was >32 μg/ml. Similar killing kinetics were observed with M. smegmatis, which is highly resistant to many antibiotics due to its thick cell wall and slow growth (fig. S2C). Therefore, 3 not only has rapid killing kinetics but also eradicates bacteria in metabolically repressed states, representing a substantial improvement over the majority of conventional antibiotics. Notably, the compound derived from the constitutional repeating unit of oligomer 3 [DB213, a reported HIV-1 viral RNA groove binder (41); table S1] showed no antibacterial activity at 128 μg/ml, indicating the necessity of more than just groove binding to a single site, in bacterial killing.
Table 1. MIC and therapeutic indices of 3 and various antibiotics.
Different Gram-positive, Gram-negative bacteria, and mycobacteria strains, including laboratory strains and clinical isolates with multidrug resistance, were covered in the evaluation. B. s, Bacillus subtilis; S. a, Staphylococcus aureus; E. c, Escherichia coli; E. fa, Enterococcus faecalis; E. fi, Enterococcus faecium; K. p, Klebsiella pneumoniae; A. b, Acinetobacter baumannii; P. a, Pseudomonas aeruginosa; M. s, Mycobacterium smegmatis; M. tb, Mycobacterium tuberculosis; and E. cl, Enterobacter cloacae. USA300 and USA400 are methicillin-resistant S. aureus strains. ND, not determined.
| Bacteria* |
MIC of 3 (μg/ml) |
MIC† of antibiotics (μg/ml) |
Therapeutic index‡ of 3 (HC50/MIC) |
||||||
| Ampicillin | Gentamycin | Erythromycin | Ciprofloxacin | Trimethoprim | Colistin | ||||
| Gram positive | S. a | 0.5 | <1 | <1 | <1 | <1 | >128 | >128 | >10,000 |
| S. a USA300 | 2 | ND | >2500 | ||||||
| S. a USA400 | 2 | >2500 | |||||||
| S. a -1 | 4 | >128 | 32 | >128 | 16 | >128 | >128 | >1250 | |
| S. a -2 | 4 | >128 | 32 | 128 | 16 | >128 | >128 | >1250 | |
| B. s | 4 | ND | >1250 | ||||||
| E. fa | 4 | <1 | 8 | <1 | <1 | >128 | >128 | >1250 | |
| E. fi -1 | 2 | >128 | ND | >128 | 32 | >128 | >128 | >2500 | |
| Gram negative | K. p | 4 | >128 | <1 | 64 | <1 | 32 | <1 | >1250 |
| K. p -1 | 2 | >128 | >128 | 32 | <1 | >128 | <1 | >2500 | |
| K. p -2 | 2 | >128 | <1 | 64 | <1 | <1 | <1 | >2500 | |
| K. p -3 | 2 | >32 | >32 | >32 | <1 | >32 | 32 | >2500 | |
| K. p -4 | 4 | >32 | >32 | >32 | 32 | >32 | >32 | >1250 | |
| K. p -5 | 4 | >32 | >32 | >32 | 16 | >32 | 32 | >1250 | |
| K. p -6 | 2 | >32 | >32 | >32 | 32 | >32 | 32 | >2500 | |
| A. b | 2 | >128 | 32 | 16 | <1 | 16 | <1 | >2500 | |
| A. b -1 | 2 | >128 | >128 | >128 | 32 | 128 | <1 | >2500 | |
| A. b -2 | 2 | >128 | <1 | 16 | <1 | 16 | <1 | >2500 | |
| P. a | 4 | 128 | <1 | 128 | <1 | >128 | <1 | >1250 | |
| P. a -1 | 4 | >128 | 4 | >128 | <1 | >128 | <1 | >1250 | |
| P. a -2 | 4 | >128 | 4 | 32 | <1 | >128 | <1 | >1250 | |
| E. cl -1 | 2 | >128 | <1 | 128 | <1 | 8 | <1 | >2500 | |
| E. cl -2 | 4 | >128 | <1 | 128 | <1 | 64 | >128 | >1250 | |
| E. cl -3 | 4 | >32 | >32 | >32 | 2 | >32 | 16 | >1250 | |
| E. cl -4 | 2 | >32 | >32 | >32 | >32 | >32 | 8 | >2500 | |
| E. cl -5 | 4 | >32 | 4 | >32 | >32 | >32 | 16 | >1250 | |
| E. c (K12) | 2 | 16 | <1 | 32 | <1 | >128 | >128 | >2500 | |
| E. c -1 | 2 | >128 | <1 | 64 | 4 | <1 | <1 | >2500 | |
| E. c -2 | 4 | >128 | 64 | 64 | 16 | >128 | <1 | >1250 | |
| Mycobacterium | M. s | 2.5 | 62 | ND | 15 | 0.5 | ND | ND | >2000 |
| M. tb H37Rv | 25 | 160 | 4 | 128 | 2.5 | >128 | 16 | >200 | |
| M. tb Erdman | 25 | ND | >200 | ||||||
*Strains having their names ending with -x are clinical isolates with multidrug resistance, and ESKAPE strains are shown in bold.
†Minimal inhibitory concentration.
‡The HC50 measured for 3 was >5000 μg/ml.
The half maximal hemolytic concentration (HC50) value of a compound denotes the concentration required to lyse 50% of red blood cells (RBCs) in 1 hour and is commonly used to represent the eukaryotic cytotoxicity of antimicrobial polymers. Oligomer 3 showed low hemolytic activity with an HC50 > 5000 μg/ml (table S1). For comparison, the only U.S. Food and Drug Administration–approved antimicrobial polymer PHMB has an HC50 of 102 μg/ml, which is >50-fold worse than 3. Also, at its MIC (2 to 4 μg/ml), 3-treated mammalian cells [human embryonic kidney (HEK) 293 and NIH/3T3] showed ≥90% viability at 24 hours. The IC50 (half maximal inhibitory concentration) measured for 3 (HEK293, 24 hours) was 25 μg/ml. Using the commonly adopted TI calculation (TI = HC50/MIC or HC50/MBC), 3 had a TI of >2500 for most bacteria and >20000 for A. b -1 in DMEM. We also studied the hemolytic activity of 3 in an extended time period (24× longer than used in commonly adopted protocols) and observed negligible RBC hemolysis (fig. S2D). These results indicated that the in vitro performance of 3 was quite promising.
Oligomer 3 disrupts bacterial cell membrane selectively
The oligomer’s membrane activity was then visualized by using scanning electron microscopy (SEM), with fragments and wrinkled surfaces being observed for 3-treated bacteria (Fig. 2A and fig. S3A). We then performed a cytoplasmic membrane depolarization assay using S. aureus or A. baumannii and the DiSC3(5) (3,3′-dipropylthiadicarbocyanine iodide) probe, a dye that exhibits intense fluorescence when the membrane potential is collapsed. We observed a large increase in fluorescence upon treatment with 3 indicating that membrane disruption contributes to the bactericidal effect of 3 (fig. S3B). Bactericidal antibiotics are also known to stimulate the production of toxic, reactive oxygen species (ROS) (42, 43), and ROS generation in 3-treated E. coli was indeed observed using a dichlorofluorescein diacetate (DCFH-DA) probe and flow cytometry (fig. S3C). We also found that addition of bacterial lipopolysaccharide (LPS) greatly inhibited the antimicrobial activity of 3 (fig. S3D), suggesting that 3 might initially bind to LPS in Gram-negative bacteria, leading to membrane perturbation, the mechanism of action reported for another bisamidine, pentamidine (44).
Fig. 2. Evidences for oligoamidine 3’s two distinct antimicrobial mechanisms of action, membrane disruption, and DNA binding.
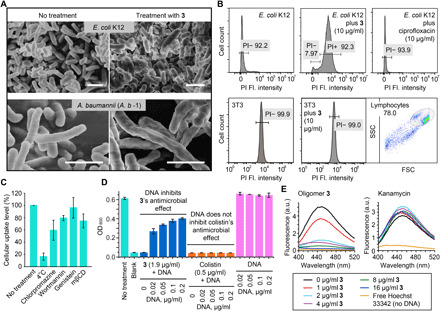
(A) 3-treated E. coli (K12) (top) and MDR A. b -1 (bottom) cells showing clear evidence of membrane damage, exhibiting fragments and wrinkled surfaces, similar to previous reports on surfactant-based antimicrobial polymers. Scale bars, 4 μm. (B) Results of membrane permeability assay using E. coli, 3T3 cells and propidium iodide (PI), showing bacterial cell membrane disruption by the addition of 3. PI is a fluorescent dye that can only penetrate compromised cell membranes, so its uptake into cells indicated membrane disruption. Compound treatment time was 4 hours for E. coli. The membrane of the NIH/3T3 cells were not affected by 3 even over an extended time period (24 hours). Fl., fluorescence. (C) Exploration of 3’s cellular internalization mechanism. Chlorpromazine, wortmannin, mβCD/genistein, and 4°C condition, respectively, inhibit clathrin-mediated endocytosis, macropinocytosis, caveolae-mediated endocytosis, and energy-dependent endocytosis in general. (D) Inhibition of 3’s activity by DNA. Externally added DNA substantially inhibited oligomer 3’s antimicrobial effect against MDR A. b -1, whereas colistin’s efficacy was minimally affected by added DNA. (E) Fluorophotometric studies showing that 2 μg/ml of 3 was able to displace >80% of the minor groove binding dye, Hoechst 33342 (4 μg/ml), bound to double-stranded DNA (dsDNA), while kanamycin was not able to incur any change in Hoechst’s fluorescence. a.u., arbitrary units.
In contrast, 3 showed only weak membrane binding to mammalian cells. When treated with oligomer 3, there were large increases (~60 mV) in surface ζ potential with A. baumannii and S. aureus but not with RAW 264.7 cells or NIH/3T3 cells (fig. S3E). Multivalence was essential for such large changes in potential (fig. S3F). Because large changes in surface zeta potential have been correlated with membrane destabilization and increased membrane permeability (45), these observations are consistent with membrane disruption by 3 in bacterial cells but no such effect in mammalian cells. We also used flow cytometry and confocal with propidium iodide (PI) to further verify this membrane selectivity. As shown in Fig. 2B and fig. S4 (A and B), there was a major increase in PI uptake for 3-treated E. coli and M. smegmatis but no effect for bacteria treated with ciprofloxacin, which is not membrane active. In contrast, there were negligible permeability changes observed for 3-treated NIH/3T3 cells, even after 24 hours. Yet, 3 itself could enter cells. Mechanistic study showed that 3’s entrance to the cells was inhibited by low temperature and various endocytic inhibitors, revealing that the uptake of 3 was mainly through an energy-dependent, complex endocytosis pathway, which explains its intracellular appearance while showing low membrane disruption (Fig. 2C). 3 selectively disrupts bacterial cell membranes, due perhaps to the absence or low percentage of negatively charged lipids and other species on mammalian cell surfaces, such as LPS and teichoic acids (46, 47), cardiolipins, phosphatidylserine, and phosphatidylinositol (47). In addition, many negatively charged lipids are localized to the inner leaflet of the mammalian cell membrane, while the outer leaflet is more neutral (47, 48). Such direct and selective membrane targeting is an attractive feature for an antibacterial agent targeting persistent infections caused by slow-growing or nongrowing bacteria, which are not susceptible to treatment with antibiotics that target biosynthetic pathways in growing cells (9).
Evidence for DNA binding antimicrobial mechanism
The molecular design of 3 was based on the hypothesis that it would target both bacterial membranes and DNA. The results shown above support selective membrane disruption by 3, but what about bacterial versus mammalian DNA targeting? As shown in fig. S4C, dynamic light scattering (DLS) showed that 3 promoted the aggregation of genomic DNA from A. baumannii, forming oligomer-DNA polyplexes. The same phenomenon was observed with both plasmid DNA and linear DNA, showing sequence-independent double-stranded DNA (dsDNA) complexation (fig. S4D). The antimicrobial activity of 3 could be suppressed by the addition of genomic DNA (from A. b -1), whereas the membrane-targeting small-molecule antibiotic, colistin, showed no such effect upon DNA addition (Fig. 2D). That is, the strong binding of 3 to exogenous DNA resulted in polyplex formation and deactivation of the oligomer, but no such effect is observed with non–DNA-targeting colistin.
Computational docking results using a fragment of 3 and dsDNA indicate minor groove binding (fig. S4E), as seen with many amidine-based DNA binders (49). In addition, gel retardation assay results using 3 and a plasmid DNA labeled with Gel-Red [a DNA intercalator (50)] showed that 3 could retard DNA migration but not displace Gel-Red (fig. S4F), implying a nonintercalating DNA binding mode. A fluorophotometric titration assay indicated that 3, but not the non–DNA-targeting kanamycin, led to a dose-dependent displacement of Hoechst 33342 (Fig. 2E), suggesting that 3 binds to DNA more tightly than does Hoechst 33342, through a Hoechst-like (minor groove) binding mechanism.
Oligomer 3 selectively binds genomic DNA in bacterial nucleoids
To test the hypothesis that 3 binds to DNA in bacterial nucleoids, we labeled 3 with either fluorescein isothiocyanate (FITC) or rhodamine B isothiocyanate (RhB-ITC) and incubated the labeled 3 with bacterial or mammalian cells. The confocal microscope images indicate that staining with FITC-3 (green) matched well with the results from nucleoid staining (blue) in E. coli, B. subtilis, and M. smegmatis (Fig. 3A and fig. S5). In the latter two cases, the staining of FITC-3 also colocalized with the staining of FM4-64, a membrane-staining dye, and this phenomenon served as a direct evidence for the dual mechanism of action of 3. In mammalian cells, RhB-3 did not enter the nucleus but stayed in endosomes. This localization is likely a result of the challenges to endosomal escape and the sieve-like property of the nuclear envelope (Fig. 3A and fig. S6A) (26). The exclusion of 3 from the mammalian cell nucleus is an important factor in lowering cytotoxicity to mammalian cells and complements the selective disruption by 3 of bacterial membranes. Also of interest in this context is that RhB-3 did not enter the mitochondrion in NIH/3T3 cells and stain mitochondrial DNA (fig. S6B), possibly because mitochondria, similar to the nucleus, have a double-membrane bilayer structure.
Fig. 3. Evidences for oligoamidine 3’s selectivity for bacteria over eukaryotic cells on both of its mechanisms of action.
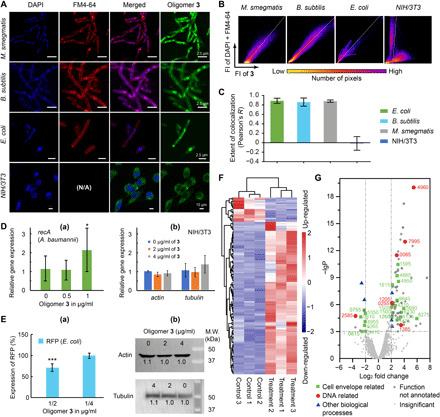
(A) Overlays of confocal microscopic images of bacteria and mammalian cells stained by DAPI, FM4-64, and 3-FITC. N/A, not applicable. (B) Representative two-dimensional fluorescence intensity histograms of stained bacteria and mammalian cells. (C) Pearson’s correlation coefficient, R, for membrane and DNA staining versus staining with 3-FITC. The statistics covered >20 randomly picked cells in each category. (D) Selective activation of housekeeping gene in bacteria cells by 3. (a) 3 activated recA transcription in A. baumannii at its sub-MIC concentration. (b) The transcription of two housekeeping genes in NIH/3T3 cells, actin and tubulin, was not affected at an even higher concentration of 3. (E) Selective inhibition of protein expression in bacteria cells by 3. (a) Oligomer 3 reduced the expression of protein (RFP) in its sub-MIC concentration. (b) Protein expression of Tubulin and Actin in NIH/3T3 cells was not significantly affected. (F) Heatmap of differential expression analysis showing gene regulation changes in A. baumannii treated with 3. Control and treatment groups each had three replicates. (G) Volcano plot of the transcriptome results using 3-treated A. baumannii. -lgP, -log10 (P of the corresponding protein). P is the octanol-water partition coefficient.
More quantitatively, we analyzed our confocal images using Coloc 2 (Fiji-ImageJ) to generate two-dimensional fluorescence intensity histograms and calculated the Pearson correlation coefficients, R. A combined channel of 4′,6-diamidino-2-phenylindole (DAPI) and FM4-64 was used versus the FITC-3 channel to obtain histograms, and representative results for E. coli, B. subtilis, M. smegmatis, and NIH/3T3 cells are shown in Fig. 3B. A single population group for all the bacteria histograms was observed, indicating colocalization of the two channels. In contrast, pixels on the NIH/3T3 histograms clearly separated into two groups, indicating that the two channels were not colocalized. In addition, Pearson’s R is used to describe the correlation of the intensity distribution between channels, and its range is from 1.0 to −1.0, with 1 representing a total positive correlation, 0 for no correlation, and −1 for a total negative correlation. The calculated R values (Fig. 3C) were ~0.9 for all three bacteria but ~0 for the NIH/3T3 cells. These results show in a quantitative manner that 3 selectively targets bacterial DNA and bacterial membranes.
Oligomer 3 affects RNA and protein expression differently in bacterial and mammalian cells
We next determined the effects of 3 on transcription and protein expression in bacterial or mammalian cells. As shown in Fig. 3D, a quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay showed that 3 induced increased recA gene expression in A. baumannii at a sub-MIC concentration (1 μg/ml). This observation is consistent with the DNA binding of 3 and the fact that recA is a DNA repair gene that codes for RecA, a protein essential for the repair and maintenance of DNA, as well as for the induction of the SOS response (51). The activation of recA indicated that 3 damaged bacterial DNA and caused bacteria to execute their repair mechanism. The generation of cellular ROS (fig. S3C) might also cause oxidative DNA damage and contribute to the up-regulation of recA as a SOS response. In contrast, in NIH/3T3 cells, gene expression of key skeletal proteins such as Actin and Tubulin remained stable upon treatment with 3 at even higher concentrations (Fig. 3D), because its chromosomal DNA was not targeted by 3. For protein expression changes, plasmid-encoded red fluorescent protein (RFP) and Tubulin/Actin were chosen as markers in E. coli and NIH/3T3, respectively. As shown in Fig. 3E, treatment of low-dose 3 for 24 hours (0.5 μg/ml) resulted in a 30% reduction of RFP compared to the control group. In contrast, expression levels of Tubulin and Actin were essentially unchanged upon treatment with 3 at an even higher concentration for 24 hours. On the basis of these results, we conclude that 3 only affects transcription and translation in bacterial cells, due to its selective binding to bacterial DNA.
Oligomer 3 significantly changes gene expression patterns in bacteria
To gain further insight into the mechanism of action of 3, we performed a transcriptome study using 3-treated A. baumannii. The fragments per kilobase per million mapped reads for each gene were compared between 3-treated and untreated bacteria (data file S1). Up- or down-regulated expression was defined with a cutoff value of |log2 (fold change)| ≥ 2 and P value of <0.001. The heatmap result of the transcriptome study indicated differential expression patterns in the control and treatment groups (Fig. 3F), supporting the hypothesis that 3 affects gene expression leading to misregulation of essential genes and subsequent cell death. We also performed a comparative analysis of various genes and classified them into cell envelope–related, DNA-related, and other biological process–related genes (data file S1). As shown in the volcano plot (Fig. 3G), 63% of the up- or down-regulated functional genes were associated with cell envelope/membrane structure and function, whereas 20% were related to DNA binding and damage repair. Consistent with the qRT-PCR result showing recA up-regulation, the same gene (IX87_RS21205) was up-regulated by 4.3-fold. One of the most up-regulated genes (IX87_RS04960) is annotated as an adenosine triphosphatase (ATPase) in the family of ATPases associated with diverse cellular activities (AAA) (data file S1). ATPases in the AAA family are known to be involved in DNA replication, chromatin remodeling, ribosomal RNA (rRNA) processing, membrane fusion, and many other essential cellular processes (52, 53). A protein structure prediction algorithm (54) indicates that IX87_RS04960 has a high degree of similarity (>98% confidence of prediction) to the ATPase domain of RecA, which is also a member of the AAA family (55). It is thus very likely that the highly up-regulated IX87_RS04960 gene was a homolog of the annotated recA gene (IX87_RS21205) and also a response to the DNA damage induced by 3. The other highly up-regulated gene, IX87_RS08275, is predicted to be a membrane protein (56), consistent with the membrane disruption mechanism of 3.
Resistance generation rate of 3
Traditional antimicrobial polymers typically have a low resistance generation rate because they target and physically disrupt bacterial membranes. However, binding of small molecules to DNA as a target may lead to high resistance rates, as shown in fig. S7; E. coli could quickly acquire resistance to DNA binding small molecules such as Hoechst, DAPI, and mitomycin, possibly because of efflux pump or other deactivation mechanisms. With both membrane disruption and DNA binding mechanisms, 3 caused no resistance generation (Fig. 4, A and B) when treating M. smegmatis or E. coli, whereas rifampin and kanamycin led to notable resistance development. Even for one of the last-resort antibiotics, the membrane-targeting colistin, there was a fourfold increase in MIC after 274 generations, in M. smegmatis (Fig. 4A). Note also that colistin was much less effective against multiple bacteria strains compared to 3, even before resistance generation. These results again indicated the resistance-resistant nature of 3.
Fig. 4. The resistance-resistant nature of 3 and its performance at the presence of eukaryotic cells.
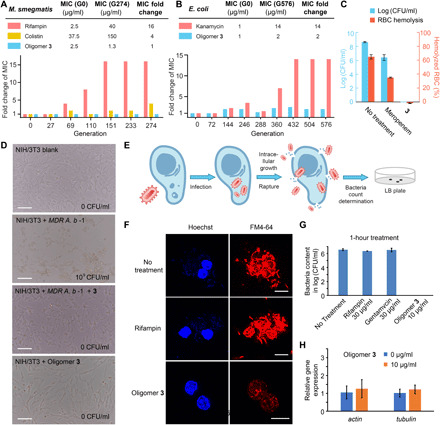
(A) Resistance generation rate comparison between rifampin, colistin, and 3 in M. smegmatis. (B) Resistance generation rate comparison between kanamycin and 3 in E. coli. (C) Rescue of MDR bacteria-infected RBCs by 3. Oligomer 3 significantly outperformed meropenem by killing all the MDR A. b -1 in the presence of RBCs without causing hemolysis. (D) Rescue of MDR bacteria-infected eukaryotic (NIH/3T3) cells by 3. Scale bars, 100 μm. (E) Cartoon illustration of the intracellular bacteria model study. (F) Confocal microscopic images of M. smegmatis–infected RAW 264.7 cells with or without treatment of rifampin (30 μg/ml) or 3 (10 μg/ml). Only cells treated by 3 survived, with bacteria eliminated. Scale bars, 10 μm. (G) Bacteria burden determination of M. smegmatis–infected RAW 264.7 cells after 1 hour using different treatments. (H) The transcription of actin and tubulin in RAW 264.7 cells (10 μg/ml, 1 hour) was not affected at the condition of the intracellular antimicrobial study.
Bacteria killing by 3 in the presence of mammalian cells
We next investigated the killing of bacteria by 3 in the presence of mammalian cells. We first used RBCs and the highly virulent A. b -1 as the model system, because large amounts of A. baumannii could lead to hemolysis of RBCs, giving a detectable color change (57). Thus, RBCs and A. b -1 were cocultured, and meropenem, 3, or a blank was added to the coculture. We observed that 3 (8 μg/ml) killed all bacteria without any sign of RBC hemolysis at 24 hours, whereas meropenem only reduced the amount of live bacteria by 30%, and there was 35% hemolysis observed (Fig. 4C). Similarly, with A. b -1 cocultured with NIH/3T3 cells, large amounts of A. baumannii can induce apoptosis of the mammalian cells (57). The addition of 3 (10 μg/ml) into the coculture resulted in the killing of all bacteria, whereas the mammalian cells were unaffected (Fig. 4D). As a control, there was no cytotoxic effect of 3 on NIH/3T3 cells at 10 μg/ml. These results clearly indicate that this 3 has excellent antimicrobial efficiency against an MDR bacterium and excellent biocompatibility at its working concentration.
Intracellular bacteria killing by oligomer 3
Mammalian cell membrane permeability is a major problem for several classes of small-molecule antibiotics (58). To be resistance resistant, good membrane permeability is a must for combating persistent infections (9) and intracellular bacterial pathogens such as M. tuberculosis, reducing treatment time and survival chance of bacterial persisters, thus decreasing the chance of resistance generation. Given the observation that 3 could be endocytosed into eukaryotic cells, we hypothesized that 3 might have good intracellular efficacy. We thus established a model system using RAW 264.7 cells, a transformed murine macrophage line, infected with M. smegmatis, a commonly used model for M. tuberculosis, as illustrated in Fig. 4E. We compared the effects of rifampin and 3 on bacterial cell burden and host cell morphology. Using confocal microscopy, we found that 3 effectively killed the bacteria and had no effect on host cell morphology, whereas rifampin was ineffective (Fig. 4F and fig. S8). Colony-forming unit (CFU) quantification of the samples at different time points showed a fast-killing kinetic profile for 3, but rifampin and gentamycin were ineffective, due to poor cell permeability (Fig. 4G). RT-PCR study showed that the transcription of actin and tubulin in RAW 264.7 cells was not affected under the condition of the above intracellular antimicrobial study (Fig. 4H).
Efficacy evaluation using a Caenorhabditis elegans infection model
Encouraged by the in vitro results with 3, we moved to a simple in vivo model, C. elegans. We infected C. elegans with S. aureus, A. baumannii, or Pseudomonas aeruginosa, including one MDR clinical isolate, and the infected worms were treated with phosphate-buffered saline (PBS), antibiotic, or 3. C. elegans were later lysed, and the viability of bacteria was measured (Fig. 5A). Notably, 3 (10 to 20 μg/ml) resulted in the complete eradication of bacteria, including the MDR clinical isolate, in all experiments. In contrast, small-molecule antibiotics showed mediocre to good performance for the laboratory strains, but their effectiveness against MDR bacteria was poor. No sign of toxicity was observed for healthy C. elegans treated with 3 at the same concentration, and the LD50 (median lethal dose) measured for 3 was >512 μg/ml. Despite the simplicity of the model, these results show the efficacy of 3 in eradicating bacterial infections, especially when MDR bacteria are involved.
Fig. 5. In vivo efficacy and biocompatibility evaluation of oligoamidine 3.
(A) Complete eradication of bacteria by oligomer 3 in infected C. elegans models, outperforming several commercial antibiotics. C. elegans were infected with different bacteria as labeled above. (B) Increased survival rate for mice treated with 3 in the mice cutaneous abscess infection model study. (C) Survival analysis result showing that 3 reduced mouse mortality rate from ~55 to <10% in the mice excision wound model study. (D) Significant reduction of the number of bacteria on the infected areas by oligomer 3 in the mice excision wound model study. (E) Comparison of organ bacterial burden in the infected mouse. The results suggest that oligomer 3 should have substantially suppressed the dissemination of bacteria into various organs in infected mice. (F) Tissue immunohistology section images. The subcutaneous treatment of 3 did not damage mice organs at its effective concentration (40 mg/kg).
Effectiveness evaluation using mouse models
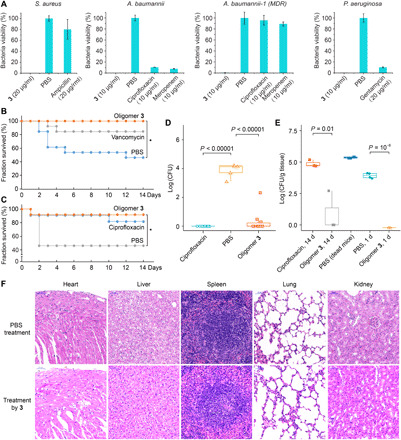
Dermis wound infections, such as burns, surgical site infections, and diabetic foot ulcers, are enormous issues in health care (59). Given that P. aeruginosa and S. aureus are the two most common causes of chronic wound infections (60, 61), we constructed two mouse infection models with S. aureus and P. aeruginosa, to evaluate the in vivo efficacy of 3. First, a mouse cutaneous abscess infection model was established by creating artificial cutaneous abscesses with subcutaneous injection of S. aureus (5 × 107 CFU) in mice, and treatments with 3, vancomycin, or PBS were performed. After 14 days, we observed a full rescue in the group treated with 3, whereas mice in the PBS group had final survival rate of 46% (Fig. 5B). Oligomer 3 also outperformed vancomycin despite the fact that this S. aureus strain is vancomycin sensitive, presumably because of vancomycin’s ineffectiveness in killing intracellular pathogens. Toxicity evaluation using healthy mice showed no apparent weight loss by 3 at the treatment dosage (fig. S9A), and this low toxicity profile was supported by the obtained immunohistology section images of the mice organs (fig. S9B). We then moved to a mouse excision wound model, in which large surface wounds were created and the wound areas were infected with a highly virulent P. aeruginosa strain (reference strain 14, 108 CFU). The wound area was treated with 3, ciprofloxacin, or PBS over 5 days. The results shown in Fig. 5C clearly indicate that 3 significantly improved the survival rate of mice, reducing mortality from ~55 to <10%. Bacteria on the wounds were almost fully eliminated by 3 and ciprofloxacin in 2 days (Fig. 5D and fig. S9C). The treatments also promoted healing of the wounds, with 3 outperforming ciprofloxacin in achieving 50% healing (fig. S9, D and E).
Oligomer 3 suppressed dissemination of bacteria to systemic infection
The observed high mortality rate for PBS-treated groups in the above model studies suggested that the surface infections could develop into fatal systemic infections. Dead mice in the excision wound model study showed high bacteria loading (~108 CFU/g of tissue) in multiple organs [Fig. 5E, PBS (dead mice)]. We also noticed that although mice treated with ciprofloxacin or 3 showed no statistical difference in survival, the residual organ bacterial loads for 3-treated mice was significantly lower than those for ciprofloxacin-treated mice (Fig. 5E; ciprofloxacin, 14 days, versus oligomer 3, 14 days). To further evaluate the effectiveness of 3 in the early suppression of bacteria dissemination, a separate set of experiments was established using the same conditions as in the mouse excision wound model, in which mice were treated with 3 or PBS once. Mice were euthanized 1 day after injection, and the results (Fig. 5E, PBS, 1 day) clearly showed that infections of major organs started within 24 hours of P. aeruginosa inoculation. Oligomer 3 rapidly reduced the bacteria content in multiple organs (Fig. 5E, oligomer 3, 1 day), which might be a decisive factor for mortality reduction. These results suggest that early treatment may be essential to prevent systemic infection. It is also of note that 3 did not show any evidence of toxicity toward the organs, as can be seen in the tissue immunohistology section images (Fig. 5F).
DISCUSSION
Successful monotherapeutic antibiotics are often those having multiple targets, as with the fluoroquinolones, or they target bacterial membranes, as with daptomycin and colistin, but resistance still develops hence previously unexplored targets or mechanisms of action are needed. One approach, stimulated by the discovery of naturally occurring antimicrobial peptides and proteins, has been the development of resistance-resistant antimicrobial polymers, compounds with high efficacy against MDR bacteria while simultaneously having a low resistance generating rate, but their cytotoxicity and hemolytic activity remain an issue. These antimicrobial polymers function in one of two ways. In most cases, they bind to and disrupt the function of bacterial but not human cell membranes, while in few other cases, they bind to and disrupt the function of bacterial cytoplasmic targets. The latter mechanism of action is much less explored, but considering the trail of development of small molecular antibiotics, bringing in more targets and mechanisms of action is likely an approach for solving the problems brought by the membrane-disruption mechanism of the antimicrobial polymers.
In this work, through rational design and screening, we used a polymeric approach to connect amidine and hydrophobic moieties and obtained a “charge-on-backbone” oligoamidine that targets bacterial membranes and DNA simultaneously and selectively. The small, “doubly selective” oligomer is made in a single-step reaction and exhibits not only high and broad-spectrum therapeutic indices against various bacteria, including MDR clinical isolates, but also much lower resistance generation than current antibiotics, as well as fast-killing kinetics. This oligomeric antimicrobial killed bacteria efficiently and selectively in the presence of RBCs or mammalian cells and also killed intracellular bacteria without affecting host cells. The lead compound 3 also showed very good in vivo efficacy in a C. elegans model and in mouse cutaneous abscess and excision wound infection models, comparable to or better than that of current antibiotics. Given the ease of synthesis of 3 and the possibility of structural diversification by changing the linker, charged moiety, binding moiety, and polymer chain length, there is considerable room for future performance optimization. On the basis of the efficacy of 3, we consider this “dual mechanisms of action” approach a potentially general strategy to achieve high performance as well as low toxicity while retaining the resistance-resistant feature of antimicrobial polymers—both natural and synthetic.
MATERIALS AND METHODS
Chemicals, bacteria, and cell lines
All reagents were purchased from Sigma-Aldrich, Acros Organics, TCI America, Macklin Inc., Adamas Reagent, and Beyotime Biotechnology and used without further purification unless otherwise noted. Water was obtained from a Milli-Q purification system. Other solvents were dried using 4 Å molecular sieves.
The following bacteria were purchased from the American Type Culture Collection (ATCC, Manassas, VA): S. aureus Newman strain (ATCC 25904), S. aureus USA300 (ATCC BAA-1717), S. aureus USA400 (ATCC BAA-1707), B. subtilis from subsp. subtilis (Ehrenberg) Cohn (ATCC 6051), E. faecalis (Andrewes and Horder) Schleifer and Kilpper-Balz (ATCC 19433), K. pneumoniae subsp. pneumoniae Schroeter Trevisan (ATCC 27736), A. baumannii Bouvet and Grimont (ATCC 19606), P. aeruginosa PA01 (ATCC 47085), E. coli K12 (ATCC 29425), M. smegmatis MC2 155 (ATCC 700084), M. tuberculosis H37Rv (ATCC 27294), and M. tuberculosis Erdman (ATCC 35801). E. coli BL21 was from TsingKe Biological Technology, Beijing. P. aeruginosa reference strain 14 was obtained from BEI Resources. Bacterial clinical isolates were from Shenzhen People’s Hospital and Peking University People’s Hospital. All bacteria strains were confirmed by 16S ribosomal DNA sequencing. Homo sapiens embryonic kidney cell line HEK293 (ATCC CRL-3216), murine macrophage cell line RAW 264.7 (ATCC TIB71), and murine embryonic fibroblast cell line NIH/3T3 (ATCC CRL-1658) were purchased from ATCC. Bacteria names without an affix or note are laboratory strains.
Synthesis of the oligoamidines
The synthetic route to 3 is shown in Fig. 1C. Under N2, the two monomers, diethyl terephthalimidate dihydrochloride (1; 43.9 mg, 0.15 mmol, 1 eq.) and 1,4-bis(3-aminopropyl)piperazine (2; 30.0 mg, 0.15 mmol, 1 eq.) were mixed in 1.5 ml of anhydrous DMF in a 7-ml glass vial with a silicone-top screw cap. Triethylamine (83.7 μl, 0.6 mmol, 4 eq.) was added, and the reaction was kept at 35°C with stirring for 96 hours. Aqueous HCl (3.0 M, 2 ml) was then added, and the solution was dialyzed [molecular weight cutoff (MWCO) = 1 kDa] against water for 14 hours, with water changed every 2 hours. The purified solution was lyophilized to give a white solid (36 mg). This material was characterized on a Bruker Avance III HD 400-MHz spectrometer equipped with an autosampler and on a gel permeation chromatography system equipped with a Waters 1515 isocratic pump and a Waters 2414 refractive index detector. Separations were performed at 50°C using aqueous NaNO3 (0.2 M) and NaH2PO4 (0.01 M) as the mobile phase.
For the labeling of oligoamidine 3 with FITC or RhB-ITC, oligomer 3 (10 mg) was added to phosphate buffer [(pH 8.5) 2 ml and 0.05 M]. Five micrograms (excess) of FITC or RhB-ITC was dissolved in dimethyl sulfoxide (100 μl), and the solution was added to the solution of 3. The mixture was stirred for 2 hours, and concentrated HCl was carefully added to acidify the mixture to pH = 1, resulting in a clear solution. The solution was dialyzed using Spectra/Por 7 regenerated cellulose dialysis tubing (pretreated; MWCO = 1 kDa) against water for 10 hours to remove HCl and excess dyes. Labeled oligomer 3 in solid form was then obtained by freeze-drying in the dark.
Other oligomers summarized in table S1 were synthesized following the same procedure using the same equivalents for starting materials. 13C nuclear magnetic resonance (NMR) spectrum of oligomer 3 and the 1H NMR spectra of all the other oligomers are provided in data file S2.
Time-kill assay
An overnight starter culture of bacteria in DMEM plus 10% FBS was diluted 1000-fold in the same media and grown at 37°C to reach an OD600 (optical density at 600 nm) of approximately 0.3. This log-phase culture was then diluted into fresh DMEM with 10% FBS to generate the working solution containing bacteria (106 CFU/ml). The working solution was treated with serially diluted compounds of interest, and the time at which each compound was added was defined as 0 hours. At each designated time point, aliquots of the suspension (10 μl) were 10-fold serially diluted and plated onto LB agar plates. After overnight cultivation at 37°C, colonies were counted, and CFU per milliliter was calculated. Experiments were performed in triplicate or more.
Morphology study by SEM imaging
The morphology of A. b -1 (clinical isolate), E. coli (K12), and M. smegmatis before and after treatment with 3 was observed by using SEM. A suspension of bacteria in exponential growth phase was incubated with 3 (12 μg/ml for A. b -1, 8 μg/ml for E. coli, and 20 μg/ml for M. smegmatis) for 1.5 or 3 hours. A bacterial suspension without any treatment was used as the negative control. Suspensions of the experiment and control groups were centrifuged (1500g, 12 min), and the supernatants were removed. Bacteria were fixed after an overnight incubation at 4°C with PBS containing 2.5% glutaraldehyde. Samples were washed three times with PBS and then dehydrated using a series of ethanol solutions (30, 50, 70, 90, and 100% in Milli-Q purified water). The samples were mounted on a copper tape, air-dried, and sputter-coated with gold for observation using a Hitachi S-4800 field emission scanning electron microscope.
ζ potential measurement
Bacteria were cultured in cation-adjusted Mueller-Hinton broth (CAMHB) at 37°C overnight. Samples of each culture were diluted 40-fold in fresh CAMHB and incubated at 37°C for 1.5 to 3 hours. Bacteria were harvested by centrifugation at 5000g for 5 min at room temperature and washed three times using Hepes (40 mM) (pH 7.4). The bacteria were resuspended in Hepes [40 mM (pH 7.4) and 1 ml], and compounds of interest at different concentrations were added into the bacteria suspension. The mixtures were incubated at 37°C for 2 hours, followed by standing for 1 hour. Zeta potentials were measured on a Malvern Zetasizer Nano ZSP zetasizer.
For zeta potential measurements with RAW 264.7 or NIH/3T3 cells, 500,000 cells were seeded into a well of a cell culture dish. For each well, the final volume was 5 ml. Cells were incubated for 14 hours to attach to the plate and then were incubated with compounds of interest at different concentrations for 2 hours at 37°C in an incubator with a 5% CO2 atmosphere. Cells were washed with PBS three times, detached using trypsin-EDTA, and then pelleted down and resuspended with Hepes (40 mM) (pH 7.4) for immediate zeta potential measurements on a Malvern Zetasizer Nano ZSP zetasizer.
Cytoplasmic membrane depolarization assay
Mid-log bacterial cells were harvested by centrifugation at 5000g for 5 min at room temperature and resuspended using assay buffer [5 mM Hepes and 20 mM glucose (pH 7.4)]. The suspension was diluted 10-fold in assay buffer to reach a final concentration of 2 × 107 CFU/ml. DiSC3(5) (0.4 μM) was then added followed by a 1-hour incubation at room temperature to allow quenching of DiSC3(5). One hundred microliters of the above bacteria suspension with DiSC3(5) was added into each well of a 96-well white microplate, and compounds of interest were added into the wells at different concentrations. The fluorescence intensity [Ex = 620 nm, Em = 670 nm for DiSC3(5) dye] for each well was monitored for 30 min using a plate reader (Tecan Spark 10M). Assay buffer was used as the negative control, and all assays were performed at least three times.
ROS generation assay
E. coli (ATCC 29425) was cultured overnight in 4-ml CAMHB at 37°C, and the bacteria were washed three times and resuspended in PBS. The suspension was diluted to OD600 = 0.1, mixed with DCFH-DA (5 μM), and used as the working solution. Bacteria were placed into a 96-well plate (200 μl per well) and treated with oligomer 3 at different concentrations (0, 2, 4, 8, 16, and 32 μg/ml) for 3 hours at 37°C in the dark. The ROS analysis was performed immediately by using flow cytometry (Ex = 488 nm, Em = 533 ± 30 nm).
Effects of 3 on bacterial membrane permeability
PI was used as the fluorescent dye to evaluate integrity of bacterial membranes (16). A stationary-state culture of E. coli was washed three times with PBS and then resuspended to a working concentration (OD600 = 0.1) containing PI (100 μM). Oligomer 3 or ciprofloxacin was added at the indicated concentrations. The samples were incubated at 37°C for 4 hours. Cells were then analyzed via flow cytometry (Accuri C6 Plus, Becton Dickinson) using a 488-nm laser source. Red fluorescence emitted by PI was detected on FL2-PE. In all cases, 10,000 events were counted. For the membrane permeability of M. smegmatis, the bacteria were treated with 3 (10 μg/ml) or kanamycin for 15 min and then stained with 100 μM PI for 15 min. Then, the samples were transferred to a glass slide for confocal imaging.
Effects of 3 on mammalian cell membrane permeability
NIH/3T3 cells (200,000) were seeded into wells of a cell culture dish. For each well, the final volume was 5 ml. Cells were incubated for 14 hours to attach to the plate. The cells were then incubated with compounds for 24 hours at 37°C in an incubator with a 5% CO2 atmosphere. After incubation, 100 μM PI was used to stain the cells for 30 min. Cells were detached using trypsin-EDTA, washed three times using PBS, and resuspended into 500 μl of PBS. Cells were analyzed using a flow cytometer (Accuri C6 Plus, Becton Dickinson) equipped with a 488-nm laser. Red fluorescence emitted by PI was detected on FL2-PE. Cells were gated on forward scatter/side scatter (FSC/SSC) dot plots to exclude cell debris, and the FL2-PE histograms were derived from the intact cells. In all cases, 10,000 events were counted.
Study on the internalization pathway of oligomer 3
To explore the internalization mechanism of oligomer 3, RAW 264.7 cells were treated with RhB-3 under different conditions for observation of possible inhibitory effects. For the low temperature condition, cells were precooled at 4°C for 30 min, and RhB-3 (20 μg/ml) was added for cellular uptake for 2 hours at 4°C. For conditions with endocytic inhibitors, cells were preincubated with the corresponding medium with genistein (100 μg/ml), chlorpromazine (10 μg/ml), wortmannin (10 μg/ml), or mβCD (5 mM) for 30 min at 37°C, and RhB-3 (20 μg/ml) was added for cellular uptake for 2 hours at 37°C. The uptake levels of 3 under different conditions were quantified by flow cytometry.
Inhibition effect of LPS and DNA on oligomer activity
An overnight starter culture in CAMHB of A. b -1 (for DNA effect) or A. b (for LPS effect) was diluted 1000-fold in fresh CAMHB and then grown at 37°C to an OD600 value of ~0.3. The working solution was prepared by diluting this log-phase culture into fresh CAMHB to a final concentration of 5 × 105 CFU/ml and transferred into every well in a flat-bottom 96-well plate. Oligomer 3 and DNA or LPS were added to the wells via serial dilution, and the final volume in each well was 100 μl. Plates were incubated at 37°C, shaking at 220 rpm for 16 to 24 hours. The OD600 values were measured to determine bacterial growth inhibition effects in each well.
DNA binding study using DLS
Genomic DNA was extracted from A. baumannii with a Genomic DNA Mini Preparation Kit with a Spin Column (D0063, Beyotime Biotechnology, Shanghai), and its concentration was determined by NanoDrop 2000 (Thermo Fisher Scientific, Shanghai). The plasmid and linear DNA used were pCDFDuet-GFP and the corresponding digestion product of Nde I/Bam HI, respectively. In a representative experiment, DNA (final concentration = 0.05 or 0.25 pmol/ml for genomic DNA and 0.5 or 2.5 pmol/ml for plasmid and linear DNA) was mixed with oligomer 3 (final concentration = 50 μg/ml) in PBS, and the mixture was incubated for 60 to 120 min. The sample was measured on a Malvern Zetasizer Nano ZSP zetasizer.
Hoechst displacement study
Investigations on 3’s binding mode was carried out using Hoechst 33342 (a groove-binding molecule) and 3, and a non–DNA binding antibiotic kanamycin was used as the control. Binding was performed in PBS (pH 7.4) at 25°C. Plasmid pCold-ctx-m-15 (stored at 4°C) was extracted from E. coli (DH5α). pCold-ctx-m-15 (10 ng/μl) and Hoechst 33342 (4 μg/ml) were incubated for 30 min at 37°C, and the plasmid-dye mixture was titrated with 3 or kanamycin with a 30-min incubation at 37°C to reach equilibrium. The process was monitored by fluorescence spectroscopy. The experiments were performed at least in triplicate. The Kd (dissociation constant) value for Hoechst 33342 is 2.5 × 10−9 M (62).
Gel retardation assay
Plasmid pCold-ctx-m-15(40 μg/ml) was incubated with oligomer 3 of various concentrations (4, 8, 16, 32, and 64 μg/ml) for 30 min at 37°C. The mixture was run on a 1% agarose gel with Gel-Red (2 μg/ml) for 1.5 hours, and the DNA band shift was visualized under ultraviolet. The Kd value for Gel-Red is 5.6 × 10−8 M (50).
Oligomer-DNA docking study
We docked a fragment of oligomer 3 (containing four amidine groups) to (CGCGAATTCGCG)2 using the x-ray crystallographic structure (4U8A) (49). The receptor was prepared by setting the receptor docking grid center to the original ligand with a diameter of 10 to 15 Å in the crystal structure. Self-docking of the original ligand was carried out as a preliminary test of the ability of the receptor grid center to recover the crystallographic pose of the ligand. The protonation state of 3 was determined by using the “wash” function in Molecular Operating Environment (MOE) at pH = 7. 3 was prepared by using the “Prepare Ligands” function in Discovery Studio 2.55 using all possible tautomers and stereoisomers generated. Docking was carried out using the CDOCKER module in Discovery Studio 2.55.
Confocal microscopy of bacteria stained by oligomer 3
E. coli, B. subtilis, or M. smegmatis were suspended in DMEM containing 10% FBS (OD600 = 0.5). FITC-labeled oligomer 3 was added at a final concentration of 10 μg/ml. The bacteria were cultured at 37°C for 2 hours, followed by centrifugation at 5000g for 3 min. The supernatant was removed, and the cell pellet was resuspended in PBS with DAPI (10 μg/ml) and then stained for 15 min at room temperature. Bacteria were then washed with Hank’s balanced salt solution (HBSS), resuspended in HBSS with FM4-64 dye (5 μg/ml), and stained for 1 min on ice. The bacteria were washed again with PBS, twice, and transferred to a glass slide for confocal imaging. Samples were imaged with Nikon A1R MP confocal microscope with a 100× objective lens.
Confocal microscopy of mammalian cells stained by oligomer 3
NIH/3T3 cells were cultured in DMEM supplemented with 10% FBS at 37°C in an incubator with a 5% CO2 atmosphere. NIH/3T3 cells (2 × 105) in 1.5 ml of medium were seeded into each well, and the cells were incubated for 14 hours for attachment. RhB-3 was added to the culture (final concentration = 10 μg/ml) and was incubated for 2 hours. Medium was removed, and cells were washed again with PBS (1 ml), three times. The cells were incubated with DAPI (10 μg/ml) for 15 min for nuclear staining. For mitochondrial staining by MitoTracker Green (diluted to 50 nM in PBS), cells were fully immersed in 1 ml of staining solution and incubated at 37°C in an incubator with a 5% CO2 atmosphere for 30 min. Medium was removed and cells were washed again with PBS (1 ml), three times. Polyvinylpyrrolidone mounting medium (50 μl) was added, and the cells were imaged using a Nikon A1R MP confocal microscope with a 100× objective lens.
RT-PCR study
For each individual experiment, PBS or oligomer 3 (0 to 10 μg/ml) was added to different groups of samples (N = 3), and the samples were incubated as indicated vide supra. A. baumannii RNA and RNA of the NIH/3T3 cells or RAW 264.7 cells were extracted with the HiPure Bacterial RNA Kit (Magen, Shanghai) and the HiPure Total RNA Micro Kit (Magen, Shanghai), respectively, following the manufacturer’s instructions. The extracted RNA was converted to cDNA, and the qPCR was conducted in a real-time PCR system (QuantStudio 7 Flex, Thermo Fisher Scientific, USA) with the HiScript II One Step qRT-PCR SYBR Green Kit (Vazyme, Nanjing). Gene expression was normalized to the expression of the housekeeping gene (16S rRNA for A. baumannii and 18S rRNA for NIH/3T3 or 18S rRNA for RAW 264.7). The primer sequences for each gene are listed in data file S1.
Oligomer 3’s inhibition of bacterial protein expression
E. coli with pDW17-RFP were grown in CAMHB with chloramphenicol (50 μg/ml) at 37°C overnight, and the resulting bacterial suspension was transferred to the wells of a 96-well plate at 5 × 105 CFU/ml. Oligomer 3 was added into each well at different concentrations, and the final volume in each well was 100 μl. The plate was incubated for 24 hours at 37°C. The expression level of RFP was quantified by measuring the fluorescence intensity of RFP on a plate reader (Tecan Spark 10M).
SDS-PAGE and Western blot
Protein expression in NIH/3T3 cells treated with 3 for 24 hours was analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and Western blot. Briefly, all samples were prepared in radioimmunoprecipitation assay lysis buffer, normalized to equal protein concentrations by the Bradford method, heated at 100°C for 10 min, loaded onto a 10% SDS–polyacrylamide gel (Bio-Rad), and run at 120 V for 2 hours. After all samples were separated by SDS-PAGE, they were transferred to nitrocellulose membranes (PALL), and probed with antibodies specific for actin and tubulin. The bands were stained with horseradish peroxidase (HRP)–conjugated secondary antibodies. Last, the proteins were detected using the Immobilon Western Chemiluminescent HRP substrate. The bands were quantified using ImageJ.
Multistep resistance generation evaluation
The method for multistep resistance evolution was adapted from a reported procedure (63). Briefly, the broth microdilution method for MIC determination against E. coli K12 or M. smegmatis was repeated for 14 passages over a period of 8 to 15 days (for E. coli) or 42 days (for M. smegmatis). The initial inoculum was 5 × 105 CFU/ml in CAMHB. For each subsequent passage, the inoculum for MIC determination was adjusted to a final density of approximately 5 × 105 CFU/ml using the contents of a well containing 3 at a sub-inhibitory concentration (at which bacterial growth was observed from the previous passage). To measure the MIC of each passage, bacteria were transferred to a new 96-well microtiter plate. Compounds were added in triplicate to wells in the first row of the microtiter plate and then serially diluted. The plate was incubated at 37°C for a minimum of 24 hours (for E. coli) or 48 hours (for M. smegmatis) before the MIC was determined by reading OD600 values. The growth generations were calculated with a doubling time of 20 min or 3.5 hours for E. coli and M. smegmatis, respectively. Resistance was classified as a greater than a fourfold increase in the initial MIC.
RBC-bacterium coculture model
A clinical isolate of A. baumannii (A. b -1 in Table 1) was added to 4% sheep blood in PBS to a final concentration of 5 × 105 CFU/ml. Samples were then treated with 3 (final concentration = 8 μg/ml), meropenem (final concentration = 8 μg/ml), or PBS and incubated for 24 hours at 37°C. Bacteria loading was determined by plating an aliquot of sample (5 μl) with different serial dilutions onto an agar plate, and CFUs were counted after a 12-hour incubation at 37°C. To determine the percent hemolysis, samples were centrifuged, and the absorbance of the supernatant at 576 nm was measured using a Triton X-100–treated blood sample as the positive control (100% hemolysis) and a nontreated blood sample as the negative control (0% hemolysis).
NIH/3T3-bacterium coculture model
In a 12-well plate, NIH/3T3 cells (2 × 104 per ml) were cultured in DMEM supplemented with 10% FBS in a humidified atmosphere with 5% CO2 at 37°C. Cells were maintained for 12 hours to reach a confluent monolayer. Medium was removed from wells followed by addition of DMEM + 10% FBS containing ~108 CFU of A. baumannii (A. b -1 in Table 1), with or without 3 (10 μg/ml). Bacteria were not added to the control sample. The cell or cell-bacteria mixtures were then cultured at 37°C for 24 hours before the samples were imaged using an optical microscope (Nikon Ti-S, Nikon Co., Tokyo, Japan). The cultures were then plated onto agar plates, and CFUs were counted after incubation at 37°C overnight.
Evaluation on intracellular antimicrobial activity
RAW 264.7 cells were suspended in DMEM supplemented with 10% FBS and seeded into a T75 flask at 4 × 105 cells/ml. Cells were maintained for 12 to 24 hours to reach a confluent monolayer. Medium was then removed, and the cells were washed with 10 ml of PBS. M. smegmatis were passed through a 26-gauge needle five times to disperse clumps, and 4 ml of medium containing 109 CFU of M. smegmatis was added to infect the macrophage cells. Phagocytosis was permitted during a 2-hour incubation at 37°C in a humidified atmosphere with 5% CO2, after which macrophage cells were washed thoroughly with PBS to remove the nonphagocytosed M. smegmatis. The infection ratio was 10:1. The infected macrophage cells were then detached from the flask and reseeded on six-well plates at 4 × 105 cells/ml with 3 ml in each well, with or without compounds. After 1 hour, the macrophage cells were lysed with ice-cold water, and the intracellular M. smegmatis was released. The resulting cell lysate were serially diluted and plated onto agar plate for CFU determination after incubation at 37°C overnight. For confocal imaging, the infected cells were imaged after 20 hours to better visualize the replication of bacteria.
C. elegans model for antimicrobial efficacy evaluation
This animal study was performed in accordance with national regulations on animal studies. The whole animal model C. elegans (wild-type strain N2) was used to examine the efficacy of oligomer 3 in treating bacterial infection in vivo. C. elegans N2 were maintained at 20°C following standard procedures (64). Briefly, worms were grown for 3 days at 20°C on nematode growth medium (NGM) agar plates seeded with a lawn of E. coli OP50 and were allowed to lay eggs. The eggs were harvested by bleaching and maintained for 24 hours at room temperature with gentle agitation for hatching. Hatched larvae were transferred to a new NGM plate seeded with E. coli OP50 and were kept at room temperature for 2 to 3 days until they reached the adult stage (L4). Adult worms were collected, and the suspension was washed three times with PBS in a 1:10 ratio to remove E. coli.
S. aureus (Newman), A. baumannii (Bouvet and Grimont, ATCC 19606), P. aeruginosa (reference strain 14), and a clinical MDR A. baumannii (A. b -1 in Table 1) were used for C. elegans infection. S. aureus, P. aeruginosa, and A. baumannii were grown in LB medium to stationary phase. One hundred fifty microliters of overnight bacterial cultures was inoculated on modified NGM with 0.35% peptone to obtain a lawn of bacteria. Adult worms were then transferred to the NGM plates for infection. After 12 hours of infection, worms were collected and washed with PBS buffer, three times, before 10 worms were transferred to a centrifuge tube. Worms were incubated with compounds of interest (10 or 20 μg/ml) or PBS (negative control). After a 12-hour treatment, worms were washed three times with PBS buffer and examined for morphological changes and viability. The worms were then ground with a glass rod, and samples were serially diluted and plated onto LB plates containing different selection of antibiotics (5 μg/ml of nalidixic acid for S. aureus, 50 μg/ml of ampicillin for P. aeruginosa and A. baumannii, respectively). Plates were incubated at 37°C for 17 hours before CFUs were determined. The bacterial CFUs were then divided by the number of worms in each treatment, and the percent reduction in bacterial growth upon drug treatment relative to PBS-treated worms was then calculated.
Mice cutaneous abscess model for antimicrobial efficacy evaluation
This animal study was performed in accordance with national regulations on animal studies. Male Institute of Cancer Research (ICR) mice (6 to 8 weeks, each weighing 18 to 25 g) were used. Neutropenia was induced by injecting cyclophosphamide (J&K, Beijing, China) intraperitoneally at 4 days (150 mg/kg) and 1 day (100 mg/kg) before infection. Hair cream was used to remove hair. S. aureus (5.0 × 108 CFU/ml, 100 μl) was injected subcutaneously on the back of the mice. Successfully infected mice were picked as models, and 39 mice were used and divided into three groups: treated with 3 (N = 13), vancomycin (N = 13), and a control group (N = 13). Treatment started when the cutaneous abscess appeared (6 hours after infection). During the 7-day treatment, compound solution aliquots (5 mg/ml in 50 μl; final concentration of 10 mg/kg) were injected into the infected wound directly for 3 and the vancomycin group, whereas PBS (50 μl) was used for the control group. The survival rate of each group was recorded for 14 days. Statistical significance of differences of survival data was determined by the log-rank test.
To determine the in vivo toxicity of oligomer 3, male ICR mice (6 to 8 weeks, each weighing 27 to 30 g) were injected subcutaneously with a single dose of 3 (10 mg/kg) or PBS of the same volume each day for 7 days. Mice (N = 3) were weighed daily. After seven injections, observation was continued daily for another 8 days.
Antimicrobial efficacy evaluation on mice excision wound model
This animal study was performed in accordance with national regulations on animal studies. This protocol was based on previously reported protocols with modifications (65, 66). Thirty-five adult male ICR mice (6 to 8 weeks, each weighing 18 to 25 g) were anesthetized with an intraperitoneal injection of chloral hydrate (50 mg/kg), and one open excision wound (2 to 3 cm2) was created to the depth of loose subcutaneous tissue on the dorsal side skin for each mouse. P. aeruginosa (reference strain 14) suspensions (108 CFU) were inoculated on excision wound area to establish the wound infection model. Mice were separated into a PBS treatment group (N = 11), a 3 treatment group (20 mg/kg, N = 12), and a ciprofloxacin treatment group (20 mg/kg, N = 11). Treatments were started 24 hours after infection by applying 60 μl of compound solutions or PBS to the wound area and were repeated every 24 hours for 5 days. On day 2, the bacterial load on the wound surface of the survived mice was determined by wiping the full wound surface with a sterile cotton swipe to transfer the bacteria to PBS and plating the PBS on agar plate (supplemented with 20 μg/ml of ampicillin) for CFU determination. The wounds were observed every 24 hours, and the survival rate of each group was recorded for 14 days. Deceased mice (N = 4) from the PBS group were dissected, and organ bacterial burden was determined as described in the following session. On day 14, survived mice on day 14 from treatment groups (N = 3 for each group) were euthanized, and organ bacterial burden was determined as described in the following session. Statistical significance of differences of survival data and surface bacterial load was determined by the log-rank test and two-tail t test, respectively. R code used to generate the box plots was included in the Supplementary Materials.
Organ bacterial burden determination
To evaluate the efficacy of oligomer 3 on suppression of bacteria dissemination into organs, the excision wound model was applied with N = 4 for PBS group and N = 4 for oligomer 3 group (dose = 20 mg/kg). Treatment started 24 hours after infection by applying 60 μl of PBS or compound solutions to the wound area, and the mice were euthanized 24 hours after treatment. The organs (heart, liver, spleen, lung, and kidney) were collected and homogenized using a high-throughput tissue homogenizer Tissuelyser-24 (Jingxin, Shanghai), serially diluted in PBS and plated onto LB agar plates supplemented with ampicillin (20 μg/ml). Plates were incubated overnight at 37°C, and colonies were quantified to determine bacteria CFU. Statistical significance of differences of organ bacterial load was determined by two-tail t test.
Immunohistology
PBS buffer or oligomer 3 in PBS (40 mg/kg) was subcutaneously injected into mice. The mice were euthanized after 24 hours, and the major organs (heart, liver, spleen, lung, and kidney) were collected. Tissues were fixed in 10% neutrally buffered formalin for 24 hours, processed into paraffin, sectioned into approximately 3 μm, and stained with hematoxylin and eosin. The samples were evaluated by using a Pannoramic MIDI microscope.
Statistical analyses
Experimental replicates (N) were ≥ 3 unless otherwise noted in the corresponding figure captions or protocols. Error bars represent ±SD. Statistical analysis was performed using Student’s t test: *, **, and *** represents P ≤ 0.05, 0.01, and 0.001, respectively, and the difference was considered significant when P ≤ 0.05. Pearson’s R values were generated using the ImageJ software.
Acknowledgments
Y.B. and X.F. thank the support from the Institute of Chemical Biology and Nanomedicine, Hunan University. We thank C. A. Mirkin, D. R. Walt, M. Mrksich, and Xiaobing Zhang for helpful discussions and suggestions on this work. Funding: The funding support from the National Natural Science Foundation of China (grants 21877033 for Y.B. and 21807031 for X.F.), and the U.S. NIH (grants R01GM065307 and R01CA158191 for E.O. and R01AR069645 for S.C.Z.) are acknowledged. Author contributions: S.B., K.Y., Y.B., and X.F. designed the research. S.B., K.Y., J.W., C.Z., Y.X., J.S., Y.G., Z.C., M.W., C.S., B.A., M.C., Y.B., and X.F. performed the research. All authors contributed in data analysis. S.B., E.O., S.C.Z., Y.B., and X.F. wrote the paper. Competing interests: Y.B., X.F., S.B., J.W., and K.Y. are inventors on a pending patent application (no. 202010121259.7; filed 3 July 2020) for the oligomer reported in this manuscript. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The transcriptomic dataset has been deposited to NCBI Sequence Read Archive with identifier PRJNA609956.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc9917/DC1
REFERENCES AND NOTES
- 1.No time to wait: Securing the future from drug-resistant infections (Report to the Secretary-General of the United Nations, World Health Organization, Geneva, Switzerland, 2019).
- 2.O. B. Jonas, A. Irwin, F. C. J. Berthe, F. G. Le Gall, P. V. Marquez, Drug-resistant infections: A threat to our economic future (HNP/Agriculture Global Antimicrobial Resistance Initiative, World Bank Group, Washington D.C., 2017), vol. 2.
- 3.Silver L. L., Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oldfield E., Feng X., Resistance-resistant antibiotics. Trends Pharmacol. Sci. 35, 664–674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.L. L. Silver, The antibiotic future, in Antibacterials, J. F. Fisher, S. Mobashery, M. J. Miller, Eds. (Springer Berlin Heidelberg, 2017), vol. 1, pp. 31–67. [Google Scholar]
- 6.Blair J. M. A., Webber M. A., Baylay A. J., Ogbolu D. O., Piddock L. J. V., Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Silver L. L., Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6, 41–55 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Coates A. R. M., Halls G., Hu Y., Novel classes of antibiotics or more of the same? Brit. J. Pharmacol. 163, 184–194 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hurdle J. G., O’Neill A. J., Chopra I., Lee R. E., Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9, 62–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenawy E.-R., Worley S. D., Broughton R., The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 8, 1359–1384 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Carmona-Ribeiro A. M., Carrasco L. D. D., Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 14, 9906–9946 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y., Cai Z., Huang Z., Tang X., Zhang X., Antimicrobial cationic polymers: From structural design to functional control. Polym. J. 50, 33–44 (2018). [Google Scholar]
- 13.Lam S. J., Wong E. H. H., Boyer C., Qiao G. G., Antimicrobial polymeric nanoparticles. Prog. Polym. Sci. 76, 40–64 (2018). [Google Scholar]
- 14.Lam S. J., O’Brien-Simpson N. M., Pantarat N., Sulistio A., Wong E. H. H., Chen Y.-Y., Lenzo J. C., Holden J. A., Blencowe A., Reynolds E. C., Qiao G. G., Combating multidrug-resistant gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 1, 16162 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Chin W., Zhong G., Pu Q., Yang C., Lou W., De Sessions P. F., Periaswamy B., Lee A., Liang Z. C., Ding X., Gao S., Chu C. W., Bianco S., Bao C., Tong Y. W., Fan W., Wu M., Hedrick J. L., Yang Y. Y., A macromolecular approach to eradicate multidrug resistant bacterial infections while mitigating drug resistance onset. Nat. Commun. 9, 917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong M., Lee M. W., Mansbach R. A., Song Z., Bao Y., Peek R. M., Yao C., Chen L.-F., Ferguson A. L., Wong G. C. L., Cheng J. J., Helical antimicrobial polypeptides with radial amphiphilicity. Proc. Natl. Acad. Sci. U.S.A. 112, 13155–13160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuroda K., DeGrado W. F., Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 127, 4128–4129 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Kamaruzzaman N. F., Chong S. Q. Y., Edmondson-Brown K. M., Ntow-Boahene W., Bardiau M., Good L., Bactericidal and anti-biofilm effects of polyhexamethylene biguanide in models of intracellular and biofilm of Staphylococcus aureus isolated from bovine mastitis. Front. Microbiol. 8, 1518 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamaruzzaman N. F., Firdessa R., Good L., Bactericidal effects of polyhexamethylene biguanide against intracellular Staphylococcus aureus EMRSA-15 and USA 300. J. Antimicrob. Chemoth. 71, 1252–1259 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Chindera K., Mahato M., Sharma A. K., Horsley H., Kloc-Muniak K., Kamaruzzaman N. F., Kumar S., McFarlane A., Stach J., Bentin T., Good L., The antimicrobial polymer phmb enters cells and selectively condenses bacterial chromosomes. Sci. Rep. 6, 23121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu C.-H., Chen C. P., Jou M.-L., Lee A. Y.-L., Lin Y.-C., Yu Y.-P., Huang W.-T., Wu S.-H., Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 33, 4053–4064 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park C. B., Kim H. S., Kim S. C., Mechanism of action of the antimicrobial peptide buforin II: Buforin ii kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244, 253–257 (1998). [DOI] [PubMed] [Google Scholar]
- 23.Yan J., Wang K., Dang W., Chen R., Xie J., Zhang B., Song J., Wang R., Two hits are better than one: Membrane-active and DNA binding-related double-action mechanism of NK-18, a novel antimicrobial peptide derived from mammalian NK-lysin. Antimicrob. Agents. Chemother. 57, 220–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee M. W., Chakraborty S., Schmidt N. W., Murgai R., Gellman S. H., Wong G. C. L., Two interdependent mechanisms of antimicrobial activity allow for efficient killing in nylon-3-based polymeric mimics of innate immunity peptides. BBA-Biomembranes 1838, 2269–2279 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou M., Qian Y., Xie J., Zhang W., Jiang W., Xiao X., Chen S., Dai C., Cong Z., Ji Z., Shao N., Liu L., Wu Y., Liu R., Poly(2-Oxazoline)-based functional peptide mimics: Eradicating MRSA infections and persisters while alleviating antimicrobial resistance. Angew. Chem. Int. Ed. 59, 6412–6419 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Paine P. L., Moore L. C., Horowitz S. B., Nuclear envelope permeability. Nature 254, 109–114 (1975). [DOI] [PubMed] [Google Scholar]
- 27.Bhaduri S., Ranjan N., Arya D. P., An overview of recent advances in duplex DNA recognition by small molecules. Beilstein J. Org. Chem. 14, 1051–1086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahman A., O’Sullivan P., Rozas I., Recent developments in compounds acting in the DNA minor groove. MedChemComm 10, 26–40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alniss H. Y., Thermodynamics of DNA minor groove binders. J. Med. Chem. 62, 385–402 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Maher J. F., Nathans D., Multivalent DNA-binding properties of the hmg-1 proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 6716–6720 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barceló F., Capó D., Portugal J., Thermodynamic characterization of the multivalent binding of chartreusin to DNA. Nucleic Acids Res. 30, 4567–4573 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J., Bai Y., Chembazhi U. V., Peng S., Yum K., Luu L. M., Hagler L. D., Serrano J. F., Chan H. Y. E., Kalsotra A., Zimmerman S. C., Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U.S.A. 116, 8709–8714 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen B., Hamelberg D., Bailly C., Colson P., Stanek J., Brun R., Neidle S., Wilson W. D., Characterization of a novel DNA minor-groove complex. Biophys. J. 86, 1028–1041 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo J. N., Qin J., Ren Y. Y., Wang B., Cui H. Q., Ding Y. Y., Mao H. L., Yan F., Antibacterial activity of cationic polymers: Side-chain or main-chain type? Polym. Chem. 9, 4611–4616 (2018). [Google Scholar]
- 35.Liu L., Huang Y., Riduan S. N., Gao S., Yang Y., Fan W., Zhang Y., Main-chain imidazolium oligomer material as a selective biomimetic antimicrobial agent. Biomaterials 33, 8625–8631 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Patel M. M., Anchordoquy T. J., Contribution of hydrophobicity to thermodynamics of ligand-DNA binding and DNA collapse. Biophys. J. 88, 2089–2103 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haq I., Ladbury J. E., Chowdhry B. Z., Jenkins T. C., Chaires J. B., Specific binding of hoechst 33258 to the d(cgcaaatttgcg)2 duplex: Calorimetric and spectroscopic studies. J. Mol. Biol. 271, 244–257 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Stokes J. M., Gutierrez A., Lopatkin A. J., Andrews I. W., French S., Matic I., Brown E. D., Collins J. J., A multiplexable assay for screening antibiotic lethality against drug-tolerant bacteria. Nat. Methods 16, 303–306 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Stokes J. M., Lopatkin A. J., Lobritz M. A., Collins J. J., Bacterial metabolism and antibiotic efficacy. Cell Metab. 30, 251–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balaban N. Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D. I., Brynildsen M. P., Bumann D., Camilli A., Collins J. J., Dehio C., Fortune S., Ghigo J.-M., Hardt W.-D., Harms A., Heinemann M., Hung D. T., Jenal U., Levin B. R., Michiels J., Storz G., Tan M.-W., Tenson T., Van Melderen L., Zinkernagel A., Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17, 441–448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marcheschi R. J., Tonelli M., Kumar A., Butcher S. E., Structure of the hiv-1 frameshift site rna bound to a small molecule inhibitor of viral replication. ACS Chem. Bio. 6, 857–864 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohanski M. A., Dwyer D. J., Hayete B., Lawrence C. A., Collins J. J., A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Dwyer D. J., Belenky P. A., Yang J. H., MacDonald I. C., Martell J. D., Takahashi N., Chan C. T. Y., Lobritz M. A., Braff D., Schwarz E. G., Ye J. D., Pati M., Vercruysse M., Ralifo P. S., Allison K. R., Khalil A. S., Ting A. Y., Walker G. C., Collins J. J., Antibiotics induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A. 111, E2100–E2109 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stokes J. M., MacNair C. R., Ilyas B., French S., Cote J.-P., Bouwman C., Farha M. A., Sieron A. O., Whitfield C., Coombes B. K., Brown E. D., Pentamidine sensitizes gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2, 17028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halder S., Yadav K. K., Sarkar R., Mukherjee S., Saha P., Haldar S., Karmakar S., Sen T., Alteration of zeta potential and membrane permeability in bacteria: A study with cationic agents. SpringerPlus 4, 672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silhavy T. J., Kahne D., Walker S., The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2, a000414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herzog I. M., Fridman M., Design and synthesis of membrane-targeting antibiotics: From peptides- to aminosugar-based antimicrobial cationic amphiphiles. MedChemComm 5, 1014–1026 (2014). [Google Scholar]
- 48.Epand R. M., Walker C., Epand R. F., Magarvey N. A., Molecular mechanisms of membrane targeting antibiotics. BBA-Biomembranes 1858, 980–987 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Zhu W., Wang Y., Li K., Gao J., Huang C.-H., Chen C.-C., Ko T.-P., Zhang Y., Guo R.-T., Oldfield E., Antibacterial drug leads: DNA and enzyme multitargeting. J. Med. Chem. 58, 1215–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crisafuli F. A. P., Ramos E. B., Rocha M. S., Characterizing the interaction between DNA and GelRed fluorescent stain. Eur. Biophys. J. 44, 1–7 (2015). [DOI] [PubMed] [Google Scholar]
- 51.M. M. Cox, Progress in nucleic acid research and molecular biology, K. Moldave, Ed. (Elsevier Academic Press, 2000), vol. 63, pp. 311–366. [DOI] [PubMed] [Google Scholar]
- 52.Ogura T., Wilkinson A. J., AAA+ superfamily ATPases: Common structure–diverse function. Genes Cells 6, 575–597 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Snider J., Thibault G., Houry W. A., The AAA+ superfamily of functionally diverse proteins. Genome Biol. 9, 216 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. E., The phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajan R., Bell C. E., Crystal structure of reca from deinococcus radiodurans: Insights into the structural basis of extreme radioresistance. J. Mol. Biol. 344, 951–963 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. L., Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Peleg A. Y., Seifert H., Paterson D. L., Acinetobacter baumannii: Emergence of a successful pathogen. Clin. Microbiol. Rev. 21, 538–582 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Delcour A. H., Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta 1794, 808–816 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolcott R. D., Rhoads D. D., Bennett M. E., Wolcott B. M., Gogokhia L., Costerton J. W., Dowd S. E., Chronic wounds and the medical biofilm paradigm. J. Wound Care 19, 45–53 (2010). [DOI] [PubMed] [Google Scholar]
- 60.DeLeon S., Clinton A., Fowler H., Everett J., Horswill A. R., Rumbaugh K. P., Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro wound model. Infect. Immun. 82, 4718–4728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fazli M., Bjarnsholt T., Kirketerp-Møller K., Jørgensen B., Andersen A. S., Krogfelt K. A., Givskov M., Tolker-Nielsen T., Nonrandom distribution of Pseudomonas aeruginosa and Staphylococcus aureus in chronic wounds. J. Clin. Microbiol. 47, 4084–4089 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tawar U., Jain A. K., Chandra R., Singh Y., Dwarakanath B. S., Chaudhury N. K., Good L., Tandon V., Minor groove binding DNA ligands with expanded A/T sequence length recognition, selective binding to bent DNA regions and enhanced fluorescent properties. Biochemistry 42, 13339–13346 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Mwangi J., Yin Y., Wang G., Yang M., Li Y., Zhang Z., Lai R., The antimicrobial peptide ZY4 combats multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii infection. Proc. Natl. Acad. Sci. U.S.A. 116, 26516–26522 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stiernagle T., Maintenance of C. elegans. WormBook 2006, 1–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antonoplis A., Zang X. Y., Huttner M. A., Chong K. K. L., Lee Y. B., Co J. Y., Amieva M. R., Kline K. A., Wender P. A., Cegelski L., A dual-function antibiotic-transporter conjugate exhibits superior activity in sterilizing MRSA biofilms and killing persister cells. J. Am. Chem. Soc. 140, 16140–16151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stokes J. M., Yang K., Swanson K., Jin W., Cubillos-Ruiz A., Donghia N. M., MacNair C. R., French S., Carfrae L. A., Bloom-Ackerman Z., Tran V. M., Chiappino-Pepe A., Badran A. H., Andrews I. W., Chory E. J., Church G. M., Brown E. D., Jaakkola T. S., Barzilay R., Collins J. J., A deep learning approach to antibiotic discovery. Cell 180, 688–702.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc9917/DC1