

A CRISPR-Cas–driven positive feedback circuit with exponential dynamic enables ultrasensitive detection of genomic DNA.
Abstract
Artificial nucleic acid circuits with precisely controllable dynamic and function have shown great promise in biosensing, but their utility in molecular diagnostics is still restrained by the inability to process genomic DNA directly and moderate sensitivity. To address this limitation, we present a CRISPR-Cas–powered catalytic nucleic acid circuit, namely, CRISPR-Cas–only amplification network (CONAN), for isothermally amplified detection of genomic DNA. By integrating the stringent target recognition, helicase activity, and trans-cleavage activity of Cas12a, a Cas12a autocatalysis-driven artificial reaction network is programmed to construct a positive feedback circuit with exponential dynamic in CONAN. Consequently, CONAN achieves one-enzyme, one-step, real-time detection of genomic DNA with attomolar sensitivity. Moreover, CONAN increases the intrinsic single-base specificity of Cas12a, and enables the effective detection of hepatitis B virus infection and human bladder cancer–associated single-nucleotide mutation in clinical samples, highlighting its potential as a powerful tool for disease diagnostics.
INTRODUCTION
Artificial biochemical circuits could precisely control the dynamic and function of chemical reaction networks, which shows great promise in biosensing, bioregulation, and bioengineering (1, 2). Biochemical reaction networks/circuits based on nucleic acid chemistry have attracted great interest due to the intrinsically programmable interactions of DNA/RNA and their rich repertoire of various enzymatic transformations (3, 4). Among these circuits, the catalytic nucleic acid circuits integrate cascaded or cycling reactions to generate multiple outputs in response to a single input, providing valuable toolkits for signal amplification (2). Using nucleic acid–based molecular programming, various catalytic circuits, such as polymerase/exonuclease/nickase DNA circuit (5), self/cross-catalytic nucleic acids (6, 7), hybridization chain reaction (8), and catalytic hairpin assembly (3), have been developed. In addition, their dynamics are powered by DNA-manipulating enzymes, DNAzymes/RNAzymes, or strand displacement reactions. These circuits feature the autonomous amplification in an isothermal manner, which offers distinct and appealing alternatives to the gold-standard thermocycling-required polymerase chain reaction (PCR), exhibiting inherent merits compatible with in-field medical diagnosis and point-of-care tests (9). However, these catalytic circuits use single-stranded nucleic acids as inputs, yet direct inputting of untreated genomic double-stranded DNA (dsDNA) is currently unavailable. Moreover, although multilayer catalytic circuits have been assembled to further improve the amplifying efficiency, most of them remain moderately sensitive (picomolar to femtomolar levels). Therefore, artificial catalytic circuits applicable for ultrasensitive (attomolar sensitivity) response to genomic DNA input are highly desirable for clinical diagnostics, and new circuit modules and mechanisms are needed for this purpose.
Clustered regularly interspaced short palindromic repeats (CRISPR) system, a form of the prokaryotic adaptive immune system, has emerged as revolutionary tools for genome editing, transcript regulation, and molecular diagnostics (10). Recently, interests in genetic circuits adapted from CRISPR-mediated gene regulation have grown in synthetic biology (11–13), but a de novo design of the CRISPR system–based artificial chemical circuits is yet unexplored. Given that CRISPR-associated (Cas) effector proteins allow for precise sequence-based recognition and efficiently catalytic cleavage of nucleic acids, the CRISPR-Cas system is a potentially programmable platform for building catalytic nucleic acid circuits. The RNA-guided targeting and helicase activity of Cas proteins allow them to recognize genomic sequences under physiological conditions, suggesting the potential of Cas proteins as unique circuit modules capable of processing DNA/RNA dual inputs and directly inputting genomic dsDNA (11, 13). Moreover, some class 2 types V and VI Cas proteins, including Cas12a (14), Cas12b (15), Cas13a (16), and Cas14a (17), have the target loading–activated noncanonical trans-cleavage endonuclease activity toward single-stranded DNA (ssDNA) or RNA with high turnovers, which is adaptive for the biocatalytic signal amplifier.
To achieve ultrasensitive nucleic acid detection, several CRISPR-based diagnostics (CRISPR-Dx), such as SHERLOCK (18), DETECTR (14), HOLMES (19), and others (15, 20, 21), have been developed on the basis of sophisticated cascade amplification via coupling the Cas-catalyzed linear signal amplification and extra polymerase-based exponential amplification, e.g., recombinase polymerase amplification (14, 15, 18, 21), PCR (19), and loop-mediated isothermal amplification (20). Although approaching their attomolar sensitivity is still challenging for Cas protein itself or existing catalytic nucleic acid circuits, it is intriguing to see whether the integration of CRISPR-Cas system into catalytic nucleic acid circuits could provide simpler molecular mechanisms with comparable ultrasensitivity. Positive feedback circuits with exponential dynamics are a promising solution and are usually leveraged to achieve ultrasensitive response in biological regulation or artificial reaction networks (22–24). Considering that both inputs (DNA/RNA dual inputs) and outputs (cleaved nucleic acids) of Cas are nucleic acid–based, we envisioned that rational molecular programming of nucleic acid modules in the CRISPR-Cas system would wire up the inherent multifunctions of Cas to achieve the autocatalytic generation of active Cas proteins. This process could build a CRISPR-Cas–based positive feedback circuit with exponential amplification of catalysis. Thus, repurposing the CRISPR-Cas system for artificial reaction networks design will provide a novel paradigm for the development of nucleic acid circuits and CRISPR-Dx.
Here, we devise a CRISPR-Cas–powered catalytic nucleic acid circuit and deploy it as a CRISPR-Cas–only amplification network (termed CONAN) for ultrasensitive nucleic acid diagnostics. In CONAN, the stringent target recognition and efficient trans-cleavage activity of the CRISPR-Cas12a system are ingeniously integrated via a rationally designed switchable-caged guide RNA (scgRNA) with self-reporting capability. As a result, a Cas12a autocatalysis-driven chemical reaction network is programmed to construct a positive feedback circuit with exponential signal amplification. CONAN presents an unprecedented catalytic circuit-based CRISPR-Dx, which achieves one-enzyme (Cas12a), one-step real-time detection of genomic DNA with attomolar sensitivity, single-base specificity, and true isothermal amplification property at physiological temperature. Its versatility has been demonstrated by the sensitive detection of hepatitis B virus (HBV) infection and human bladder cancer–associated single-nucleotide mutation (SM) in clinical samples. Moreover, because of its positive feedback circuit–caused exponential dynamic, CONAN significantly improves the specificity of the Cas12a system in the discrimination of SMs, especially the protospacer adjacent motif (PAM)–distal mismatches. CONAN presents a powerful and promising tool in supersensitive and specific nucleic acid diagnostics.
RESULTS
Rational design of CONAN
In this work, CRISPR-Cas components are adapted for fabricating three signal processors, namely, transducer 1 (T1), transducer 2 (T2), and a signal amplifier (A), in which the parallel connection of A and T2 forms a positive feedback circuit (Fig. 1). To fabricate the CONAN, T1 and the positive feedback circuit are connected in series. Specifically, CRISPR-Cas12a, one of the widely used CRISPR-Cas systems with trans-cleavage activity (14), was chosen as a model. Cas12a and a gRNA for target dsDNA (gRNA-T) are preassembled as the T1, which converts the input of target dsDNA into active Cas12a protein (Cas12a/gRNA-T/DNA complex). A scgRNA with self-reporting capability functions as the A, which outputs amplified fluorescence signals and multiple active (decaged) gRNA molecules in response to each active Cas12a protein with trans-cleavage activity. T2 can transduce the resulting decaged gRNA into another active Cas12a protein, as T2 comprises Cas12a protein and assistant dsDNA probe (aDNA) that targeted by the decaged gRNA. In this scenario, each catalytic event of an active Cas12a releases a decaged gRNA via A and sequentially assembles a new active Cas12a protein via T2; thus, multiple turnovers, in turn, generate much more amounts of active Cas12a proteins and amplified fluorescence signals. In this reaction cycle, active Cas12a indirectly catalyzes its production, which forms an autocatalytic cycle of active Cas12a, thereby resulting in a positive feedback circuit between A and transducers T2 with a significantly amplified signal gain (Fig. 1). Therefore, each target nucleic acid molecule is converted into exponentially amplified fluorescence signals in CONAN, which enables ultrasensitive nucleic acid detection.
Fig. 1. Rational design of CONAN.
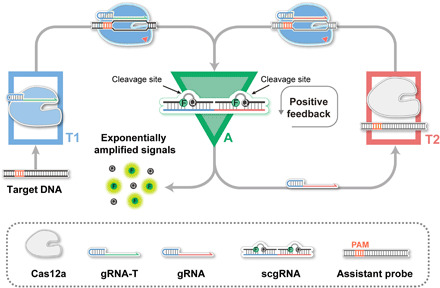
Principle of the CONAN for exponentially amplified detection of DNA using a Cas12a autocatalysis-driven positive feedback circuit.
Design and fabrication of scgRNA
In CONAN, the implementation of the positive feedback circuit relies on the ingenious coupling between signal amplifier A and transducer T2. Thus, the construction of a scgRNA that can be activated by the trans-cleavage of Cas12a is the priority. In this work, Cas12a from Lachnospiraceae bacterium ND2006 (LbCas12a; figs. S1and S2) was cloned and expressed (14). Previous studies have revealed that the conformation of gRNA plays a crucial role in the formation of Cas12a/gRNA/DNA complex (25, 26). Therefore, we sought to construct a caged gRNA (cgRNA) by hybridizing the gRNA with an inhibitor DNA (iDNA; Fig. 2A). A series of iDNAs that are complementary to the essential regions [21–nucleotide (nt) 5′-handle and 20-nt 3′-spacer; fig. S3] of gRNA were designed to generate gRNA/iDNA duplexes with different thermostabilities (Fig. 2B and fig. S4) (27, 28). To validate the inhibition effect of iDNA on gRNA, we tested the trans-cleavage activity of LbCas12a in the presence of different gRNA/iDNA duplexes individually. With the usage of a fluorescent probe as the reporter 6-carboxyfluorescein (FAM)-TTATT-black-hole-quencher 1 (BHQ1), denoted as FQ reporter), the inhibition efficiencies were determined. As shown in Fig. 2B, the hybridization of gRNA and iDNA resulted in a marked decline in the trans-cleavage activity of LbCas12a, and the inhibition efficiency exhibited a positive correlation with the thermostability of gRNA/iDNA duplexes. When a perfect complementary iDNA (iDNA VII) was adopted, almost complete inhibitions on both trans- and cis-cleavage activities of LbCas12a were observed (Fig. 2B and fig. S5), indicating the successful construction of cgRNA with high caging efficiency.
Fig. 2. Construction of cgRNA and scgRNA.
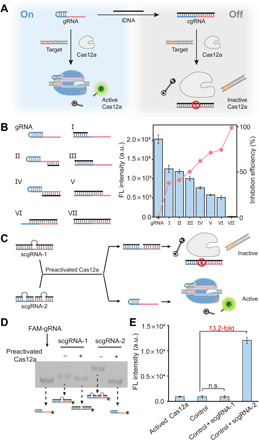
(A) Caging the activity of LbCas12a gRNA using an iDNA. (B) Inhibition on the trans-cleavage activity of LbCas12a by a variety of iDNAs. The 5′-handle and 3′-spacer regions of gRNA are shown in blue and red, respectively. LbCas12a, 50 nM; gRNA, 50 nM; iDNA, 50 nM; target dsDNA (aDNA), 50 nM; FQ reporter, 1.0 μM. FL, fluorescence; a.u., arbitrary units. (C) Design principle of scgRNAs and the release of active gRNA from scgRNA-2. The bulges in scgRNA are shown in gray. (D) Analysis of the cleavage of scgRNA (200 nM) by preactivated LbCas12a (50 nM) using native polyacrylamide gel electrophoresis (PAGE). The gRNA in each scgRNA is labeled by a 6-carboxyfluorescein (FAM-gRNA). (E) Evaluation of the activity of released gRNA from scgRNA-2. Each scgRNA (500 nM) was treated by preactivated LbCas12a (1.0 nM) and then mixed with LbCas12a protein (1.0 μM), aDNA (500 nM), and FQ reporter (1.0 μM). The control sample contains the same components except the scgRNA. Data represent means ± SD. n.s., not significant. Student’s t test.
Having verified the feasibility to cage the activity of gRNA by hybridization of iDNA, we next attempted to develop a scgRNA. Given the efficient trans-cleavage activity of Cas12a on ssDNA, we hypothesized that the introduction of unmatched ssDNA bulge structure into the gRNA/iDNA duplex might serve as substrates of active Cas12a and provide a feasible strategy to develop switchable cgRNA. To test this hypothesis, we treated the bulges with different lengths with active Cas12a protein and found that the bulges in duplex structure with 5 nt or above were effectively digested (fig. S6). Then, we designed and assembled two scgRNAs (scgRNA-1 and scgRNA-2; Fig. 2C), in which the gRNA was hybridized with one or two iDNAs (each iDNA in duplex contains a 7-nt bulge), respectively. According to the theoretical computation by DINAMelt (29), Ca12a-mediated digestion of the ssDNA bulges induced a sharp decrease in the melting temperature (Tm) value of scgRNA-2 (fig. S7). In contrast, scgRNA-1 showed only a small decrease in Tm after the cleavage of the bulge (fig. S8). Under the reaction temperature (37°C), active gRNA and ssDNA fragments were released from scgRNA-2, and the released ssDNA fragments were further digested by active Cas12a proteins. To verify this design, we incubated scgRNA-1 and scgRNA-2 with preactivated LbCas12a protein (LbCas12a/gRNA/aDNA complex), respectively, and analyzed the mixtures by gel shift assay. In the lane of active LbCas12a-treated scgRNA-2 sample, a band corresponding to that of gRNA appeared (lane 5 in Fig. 2D), suggesting the release (decaging) of gRNA from scgRNA-2. To verify the functionality of the released gRNA from scgRNA-2 to assemble active Cas12a effector, we collectively added LbCas12a apoprotein and aDNA into the mixture of active LbCas12a protein–treated scgRNA-2 and tested the fluorescence signal of trans-cleavage activity using the FQ reporter. An intensive fluorescence signal that is about 13.2-fold over the control sample of preactivated LbCas12a alone was detected (Fig. 2E), indicating the formation of additional active Cas12a proteins (LbCas12a/gRNA/aDNA complex). Together, by introducing iDNA with bulge structure, we have constructed a scgRNA-2 that can be effectively activated by the trans-cleavage of LbCas12a with ultralow background and high switching ratio.
Construction of CONAN
The two major functionalities of signal amplifier A in CONAN are to programmatically output multiple gRNA molecules and to produce amplified fluorescence signals in response to the input of each active LbCas12a protein. Because the former functionality has been achieved, we next sought to render the scgRNA-2 with the self-reporting ability to produce fluorescence signals. To this end, we modified a dye/quencher pair (FAM/BHQ1; fig. S9) at two deoxyribothymine (dT) sites with a 5-nt spacer in the bugle-forming region of each iDNA. As the formation of iDNA/gRNA duplexes (namely, the scgRNA in Fig. 1), the fluorescence of FAM is efficiently quenched by the BHQ1 through proximity-induced highly efficient fluorescence resonance energy transfer (FRET). In such a design, the FAM and BHQ1 remain close to each other even when partial strand dissociation occurs in scgRNA, contributing to a low background. Digestion of the ssDNA bulges in this scgRNA by active LbCas12a protein results in the release of gRNA, as well as the disruption of FRET between FAM and BHQ1, thereby generating appreciable fluorescence signals. With the usage of this scgRNA as signal amplifier A, CONAN for amplified detection of a dsDNA target is fabricated (Fig. 3A). Given the highly efficient trans-cleavage of ssDNA (~1250 turnover/s) by LbCas12a (14), signal amplifier A enables each target-activated LbCas12a protein to produce amplified fluorescence signals and numerous gRNA molecules. The released gRNA molecules are effectively converted into active LbCas12a proteins by transducer T2 due to the high affinity (Kd = 2.86 nM) between LbCas12a and its gRNA (25). These active LbCas12a proteins trigger the next round of cycle for active LbCas12a formation and signal gain, forming an autocatalytic cycle of Cas12a. As a consequence, a positive feedback circuit is developed, and the fluorescence signal increases exponentially in CONAN (Fig. 3B). On the contrary, the Cas12a gate without the positive feedback circuit, which contains the sole T1 transducer and the FQ probes without A and T2, was also constructed for comparison, and it could only accumulate fluorescence signals linearly (Fig. 3B).
Fig. 3. Highly sensitive and specific detection of DNA by CONAN.
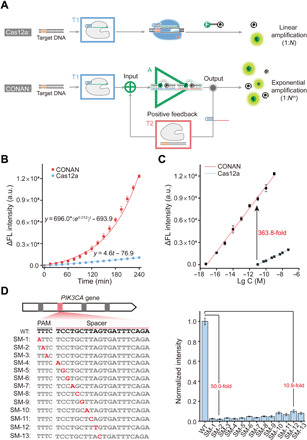
(A) Schematic illustration for the comparison of signal amplification patterns in CONAN and the Cas12a gate. (B) Experimentally determined (dots) and mathematically fitted (solid curves) time-dependent fluorescence signal changes in CONAN and the Cas12a gate. (C) Calibration curves of CONAN and the Cas12a gate in response to different concentrations of DNA target. Lg C, the logarithm of target concentration. (D) Sequence information (left) and the normalized fluorescence signals (right) of the wild-type (WT) target and SM strands. The negative strand is shown, and the mutation site is indicated in red. The concentration of each DNA strand is 1.0 nM. Data represent means ± SD.
Ultrasensitive and highly specific DNA detection by CONAN
To implement the catalytic nucleic acid circuit capable of directly inputting genomic dsDNA, we next investigated the analytical performance of CONAN in the detection of disease-associated genes. Somatic missense mutations in the PIK3CA gene, which encodes the class 1A phosphoinositide-3-kinase p110α catalytic subunit, have been reported to be frequent in several human tumors, such as breast, ovarian, colorectal, and bladder cancers (30). In particular, the E545K mutant (a single 1633G>A mutation) in PIK3CA was found to be highly correlated with the development of human bladder tumors (31). Here, the feasibility of CONAN in detecting the potential oncogene was examined using a synthetic dsDNA fragment (35 base pairs) of PIK3CA containing the 1633G>A mutation as the target/input. A gRNA-T targeting the positive strand of PIK3CA fragment was designed and used in CONAN and the Cas12a gate (nonfeedback control). Time-dependent fluorescence signals (enhanced intensity, ΔF) of CONAN and the Cas12a gate in response to 1.0 nM PIK3CA fragment were respectively monitored and compared. The signal in CONAN increased rapidly in an exponential growth pattern with a fitted equation of ΔF = 696.0 × (e0.012)t − 693.9, compared to the linear growth manner in the nonfeedback Cas12a gate (ΔF = 4.6t − 76.9) (Fig. 3B and fig. S10). Because of the exponential magnification effect of the positive feedback loop, the fluorescence signal of 10 pM PIK3CA from CONAN is much higher (363.8-fold) than that from the nonfeedback Cas12a gate, indicating the superior signal gain in CONAN caused by the autocatalysis-driven positive feedback circuit. Then, several key reaction parameters in CONAN, including the concentrations of LbCas12a and assistant probe, were optimized (figs. S11 and S12). Under the optimal condition (1.0 μM Cas12a and 0.5 μM aDNA), CONAN responded to PIK3CA in a broad linear concentration range from 5.0 aM to 1.0 nM, with the lowest detectable concentration of 5.0 aM (Fig. 3C and fig. S13), which is much lower than those of conventional catalytic nucleic acid circuits (4–8) and comparable to those of many CRISPR-Dx methods involving extra polymerase-based nucleic acid amplification (14, 15, 18–21). By contrast, the lowest detectable concentration of the nonfeedback Cas12a gate was 10 pM (Fig. 3C and fig. S14), which is similar to the values reported by other groups (19). These results suggested that the coupling of a positive feedback loop significantly enhanced the detection sensitivity of Cas12a by more than six orders of magnitude.
Aside from high sensitivity, good specificity is of great importance in molecular diagnostics. On account of the stringent target recognition of the CRISPR-Cas12a system (fig. S15), the specificity of CONAN was assessed by testing its capacity of discrimination toward SM. A series of targets with SM were analyzed by CONAN (Fig. 3D, left), and significant differences in fluorescence signal were observed between wild-type (WT) target and the SMs (Fig. 3D, right). In general, the signals of WT in CONAN were 10.9 to 50.0 times to those of SMs at the same concentration (1.0 nM), indicating the excellent specificity of CONAN in distinguishing SM. At the protospacer adjacent motif–distal sites (> 6 nt far from the 3′ terminus of PAM, namely, from SM-10 to SM-13), the ratios of WT to SM (ΔFWT/ΔFSM) in CONAN were 7.2 to 10.4 times as high as those in the nonfeedback Cas12a gate (fig. S16). Therefore, CONAN had improved specificity over Cas12a in discriminating SMs, especially at the PAM-distal sites, which might be attributed to the more efficient nonlinear accumulation of signal differences during the positive feedback processes (fig. S17). These results have demonstrated the unparalleled advantages of CONAN in both sensitivity and specificity compared with Cas12a itself (the Cas12a gate). Therefore, the target recognition, helicase, and self-signal amplification capacities of Cas12a protein are rationally amalgamated in CONAN, thereby enabling the isothermal detection of dsDNA with attomolar sensitivity, which addresses the limitations faced by conventional catalytic nucleic acid circuits. Moreover, CONAN enables a further improved specificity in discriminating SMs compared with the intrinsic specificity of Cas12a.
Analysis of virus infection by CONAN
To verify whether the CONAN is qualified in molecular diagnostics, we directly exploited the pathogenic virus genome as the input and further assessed the utility of CONAN on the detection of virus infection. Notably, sensitive and specific detection of HBV infection is critical for early diagnosis and treatment of HBV-related hepatitis, cirrhosis, and hepatocellular carcinoma (32). In the serum of patients with chronic hepatitis B, the HBV genome existed as partly double-stranded relaxed circular DNA (rcDNA), while it was found to form covalently closed circular DNA (cccDNA) in the nucleus of HBV-infected hepatocytes (33). To detect the HBV genome in both types, we designed a gRNA-T to target the positive strand of the core gene (2315-2334) of HBV and used in CONAN (fig. S18) (33). First, HBV-infected cultured liver cells (HLCZ01) were chosen as the model, and total DNA extracted from the cells was analyzed by CONAN directly. As shown in Fig. 4 (A and B), CONAN unambiguously distinguished HBV-infected cells from the uninfected cells. After the copies of HBV genomic DNA in the total DNA were quantified by quantitative real-time PCR (qRT-PCR), it was found that as low as 3 copies/μl of HBV genomic DNA (5.0 aM) from the infected cells could be detected by CONAN (fig. S19). However, 6 × 106 copies/μl of HBV genomic DNA (10 pM) is needed for the Cas12a gate without feedback to generate detectable signals, indicating the superior sensitivity of CONAN (Fig. 4C). To evaluate the feasibility of CONAN on the diagnosis of clinical samples, we carried out the analysis of the crude DNA extractions from the serums of nine healthy donors, and nine patients with chronic hepatitis B, respectively. As shown in Fig. 4 (D to F), CONAN could accurately detect HBV DNA in patient serum samples, and the signals in CONAN exhibited a positive correlation with the results of qRT-PCR (fig. S20). Together, these results demonstrate that CONAN is capable to directly detect HBV infection in clinical samples with excellent sensitivity and accuracy.
Fig. 4. Evaluation of CONAN for virus infection diagnosis.
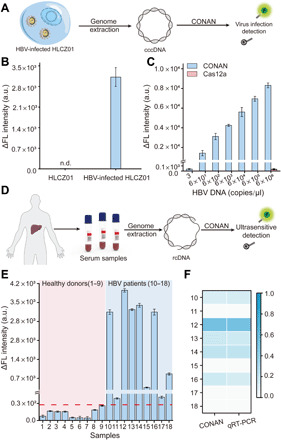
(A) Schematic illustration for the analysis of HBV infection in cultured liver cells (HLCZ01) by CONAN. (B) Fluorescence signals of HBV-infected and uninfected HLCZ01 cell samples in CONAN. The concentration of genomic DNA in each tested sample is 6 × 102 copies/μl. n.d., not detected. (C) Fluorescence signals of different amounts of genomic DNA from HBV-infected HLCZ01 in CONAN and the Cas12a gate. (D) Schematic of HBV infection detection in human serum samples by CONAN. (E) The fluorescence signals of 18 serum samples in CONAN. Serum samples from nine healthy donors (1 to 9) and nine patients with chronic hepatitis B (10 to 18) were tested, respectively. (The red dash line indicates the threshold of the highest response of healthy donor plus 3σ). (F) Heatmap of the normalized signal intensities of nine patient samples in CONAN and qRT-PCR. Data represent means ± SD.
Analysis of cancer-associated SM by CONAN
Many SMs in the human genome have been found to drive or facilitate the development of various cancers and thus have been regarded as prominent biomarkers for early diagnosis and targeted therapy (34). We next asked whether CONAN is applicable for clinical genomic DNA samples and effective in discriminating cancer-associated SM in human tissue samples. The PIK3CA_1633G>A mutation in tissue samples of human bladder cancer was tested as a model. Genomic DNAs of LO2 (a human hepatocyte cell line) and TCCSUP (a human bladder cancer cell line) cells were extracted and analyzed using a gRNA-T that targets the positive strand of PIK3CA locus containing the 1633G>A mutation (Fig. 5A). In the group of TCCSUP genomic DNA, a significant fluorescence signal that is 14.8 times as high as the LO2 group was observed, suggesting the existence of PIK3CA_1633G>A mutation in the TCCSUP genome (Fig. 5B). This result was further confirmed by DNA sequencing (Fig. 5C). Further studies validated that CONAN could detect 10 aM TCCSUP genomic DNA (Fig. 5D). For rapid detection, CONAN was able to detect 1.0 fM genomic DNA in 30 min (fig. S21). Moreover, mixture samples with different concentration ratios (0 to 5%) of TCCSUP genomic DNA to LO2 genomic DNA were prepared and tested by CONAN. The results revealed that CONAN enabled distinguishing the mutation of PIK3CA_1633G>A at a low rate of 0.1% (Fig. 5E). Next, genomic DNAs extracted from the tissue sections from the normal bladder and bladder cancer that were pathologically diagnosed (Fig. 5, F and G) were tested by CONAN, respectively. The signals from the bladder cancer tissues were 4.3- to 13.8-fold compared to those from the normal bladder tissues (Fig. 5H), which were comparable with the results of next-generation sequencing (NGS) (Fig. 5I). These results demonstrated that 1633G>A mutation of the PIK3CA gene occurs specifically in the cancer tissue. Therefore, these results clearly indicate the excellent sensitivity and specificity of CONAN in the diagnostic or prognostic detection of tumor-related SMs.
Fig. 5. Application of CONAN in cancer-associated SM diagnosis.
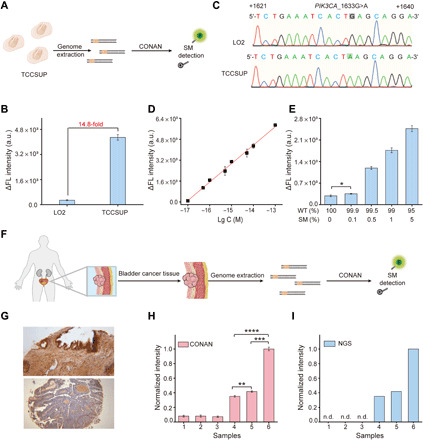
(A) Schematic illustration for the detection of SM in the genome of human bladder cells (TCCSUP) by CONAN. (B) Fluorescence signals of TCCSUP and LO2 cell samples in CONAN. The amount of genomic DNA in each tested sample is 10 fM. (C) Verification of PIK3CA_1633G>A in the genome of TCCSUP cells by DNA sequencing. The positive strand of PIK3CA from +1621 to +1640 is shown. (D) Calibration curve based on the plots of fluorescence signal versus the logarithm concentration of genomic DNA from TCCSUP cells. (E) Fluorescence signals of genomic DNA with different mutation rates. The total amount of genomic DNA in each sample is 10 fM. (F) Schematic illustration for the detection of bladder cancer–associated SM in human tissue samples by CONAN. (G) Immunohistochemical images of the tissue sections from the normal bladder (top) and bladder cancer (bottom). Original magnification, ×40. Response signals of the genomic DNA from different tissue samples in (H) CONAN and (I) NGS. Bladder tissue samples from three healthy donors (1 to 3) and three patients with bladder cancer (4 to 6) were tested, respectively. Data represent means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, Student’s t test.
DISCUSSION
Artificial nucleic acid circuits have attracted much attention in the fields of bioanalysis, bioregulation, and bioengineering due to the programmability and scalability of Watson-Crick base pairing and the diverse enzymatic or functional nucleic acid toolkits (1, 2, 5). Here, we verified that CRISPR-Cas system presented a promising artificial circuit–building module due to its gRNA-guided stringent nucleic acid recognition, RNA/dsDNA dual-inputting property, and cis/trans-cleavage nuclease activity enabling treating nucleic acids as outputs. These properties could be reprogrammed and connected to build artificial circuits by the rationally structural and functional design of nucleic acid modules, e.g., switchable gRNA modules integrated with self-reporting function in CONAN, unlike gene-regulating schemes generally used for CRISPR-Cas–based gene circuits. Given that Cas effectors could recognize, unwind, and bind any gene loci of interest under the guidance of gRNA (10, 25, 26), CRISPR-Cas systems provide a solution to address the limitation that conventional nucleic acid circuits lack recognition mechanisms for gene loci in genomic DNA. In CONAN, the RNA-guided target recognition and helicase activity of Cas12a are especially effective for target gene loci searching and binding at physiological temperature, which allows the nucleic acids circuits to directly process the input of genomic DNA, such as virus genomic DNA and human cancer–associated gene loci (Figs. 4E and 5F). Moreover, the trans-cleavage activity of Cas12a is highly efficient with an extraordinarily high turnover (14), and the exponential dynamic of Cas12a autocatalysis-driven positive feedback circuit further significantly increases the sensitivity of Cas12a in CONAN (Fig. 3, B and C). These properties render the Cas12a-based nuclei acid circuit with superior sensitivity (attomolar) and faster dynamics than the catalytic nucleic acid circuits powered by DNAzymes/RNAzymes or conventional nucleases. Therefore, taking CONAN as a proof of principle, we have shown that CRISPR-Cas system can be used to build artificial circuits capable of one-step genomic DNA detection with ultrasensitivity and single-base specificity, which provides a novel paradigm for the development of artificial nucleic acid circuits with promising potential in molecular diagnosis.
Compared with most of the CRISPR-Dx methods (14, 15, 18–21), CONAN not only avoids the extra target amplification step involving multiple enzymes and procedures but also generalizes the detection of different targets by simply fine-tuning the gRNA-T, hence exhibiting the advantages in simplicity, easy operation, and versatility. The positive feedback circuit driven by Cas12a autocatalytic cycle renders CONAN with several merits: (i) Although the sequence-based stringent recognition of Cas12a exhibits good intrinsic specificity in discriminating single-nucleotide mismatches, CONAN further improves the specificity of Cas12a by 7.2 to 10.4 times, especially the PAM-distal mismatches sites (>6 nt far from the 3′ terminus of PAM) where Cas12a shows relative unapparent discrimination ability (Fig. 3D and figs. S15 and S16). (ii) CONAN enables the DNA detection with a broad linear concentration range of above eight orders of magnitude (5.0 aM to 1.0 nM), which is better than those of conventional CRISPR-Dx methods (two to six orders of magnitude). This merit allows directly quantitative measurement of target DNA in clinical samples without the need for preanticipation of its measuring range and sample dilutions. (iii) Because of the synergistic effect of its excellent specificity and wide linear range, CONAN shows fantabulous performance in distinguishing different mutation levels of cancer tissue samples, implying the potency of CONAN in the accurate gene loci analysis of clinical samples. (iv) CONAN is a true isothermally amplified assay performed with a single step at physiological temperature, whereas most of the existing nucleic acid amplification techniques require the thermocycling or initial heating process to denature dsDNA. Hence, CONAN is highly suitable for in-field molecular diagnosis with limited resources.
In summary, CONAN has exploited the potential of the nucleic acid circuits in ultrasensitive genomic DNA detection by ingeniously designing a CRISPR-Cas12a autocatalysis-driven positive feedback circuit. The effectiveness of CONAN has demonstrated that the ingenious coupling of the CRISPR-Cas system and artificial reaction networks provides an alternative strategy for facilitating the practice application of nucleic acid circuits in molecular diagnosis. As a proof of concept, this system is adaptable and scalable to higher-order artificial circuits by further integrating functional modules: (i) other emerging Cas proteins with trans-cleavage activities, such as Cas12b, Cas13a, and Cas14a (15–17); (ii) other variants of Cas derivatives with different functionalities, for instances, Cas proteins or nuclease-dead Cas proteins with effector fusions (11, 35); and (iii) diverse structural and functional nucleic acid modules (2, 9). Therefore, this work opens up a new avenue for the development of high-performance artificial nucleic acid circuits, exhibiting promising potential in early diagnosis and disease treatment.
MATERIALS AND METHODS
Reagents and materials
Hydrochloric acid (HCl), sodium chloride (NaCl), magnesium chloride (MgCl2), and potassium chloride (KCl) were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Dithiothreitol (DTT), glycerol, tris, tetrasodium salt (EDTA), and heparin sodium (from porcine intestinal) were purchased from Sangon Biotech Co. Ltd. (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM), RPMI-1640, trypsin, and fetal bovine serum (FBS) were purchased from Invitrogen (Gibco, USA). Human bladder cancer cells (TCCSUP) and human normal hepatocyte cells (LO2) were obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). To create and maintain a ribonuclease-free environment, all the solutions containing RNAs were prepared by diethyl pyrocarbonate (DEPC)–treated water. The solutions for protein purification and other protocols were prepared using ultrapure water (resistivity of 18.2 megohm·cm).
Construction, expression, and purification of LbCas12a protein
The coding sequence of LbCas12a was derived from the plasmid pET-LbCpf1-2NLS (Addgene, #102566). The gene sequence of LbCas12a was codon-optimized and synthesized by Sangon Biotech Co. Ltd. (Shanghai, China) and then cloned into pET28a.The plasmid pET28a-LbCas12a was transformed into Escherichia coli BL21 Rosetta. For protein expression, a single clone was first cultured overnight in 5-ml liquid Luria-Bertani (LB) tubes and then 1% (v/v) inoculated into 3 liters of fresh liquid LB. Cells were grown with shaking at 220 rpm and 37°C until the optical density at 600 nm reached 0.8, and isopropyl-β-d-thiogalactopyranoside was then added to a final concentration of 0.2 mM, followed by further culture of the cells at 16°C for about 16 hours. After being harvested by centrifugation and resuspended in buffer [50 mM tris-HCl (pH 8.0) and 1.5 M NaCl], cells were lysed by sonication. Protein was purified by Ni-NTA agarose chromatography (ÄKTA, GE), and then the buffer was exchanged into desalination buffer [20 mM tris-HCl (pH 8.0), 600 mM NaCl, 2 mM DTT, and 10% glycerol] by desalination chromatography (ÄKTA, GE). The purified LbCas12a was quantified using the improved Bradford protein assay dye reagent kit with bovine serum albumin as the standard. Last, the LbCas12a was stored at −80°C until use.
RNA/DNA oligomers
All DNA strands were ordered from Sangon Biotech Co. Ltd. (Shanghai, China), and RNA strands were ordered separately from Sangon Biotech Co. Ltd. (Shanghai, China) and GenePharma (Shanghai, China). DNA and RNA strands were dissolved in DEPC-treated water and quantified to stock solutions with 100 μM by ultraviolet (UV)–vis absorption at 260 nm with an Agilent Cary 60 spectrophotometer (Agilent Technologies, Australia). All the dsDNA were prepared by hybridization of an equivalent molar amount of the two complementary oligomers at 95°C for 4 min in annealing buffer [20 mM tris-HCl (pH 7.5), 150 mM KCl, 1 mM EDTA, and 50 mM MgCl2], followed by gradient cooling to 25°C at a rate of 0.1°C/s. The cgRNA, scgRNA-1, and scgRNA-2 solutions were also generated by the hybridization of an equivalent molar amount gRNA and the customized iDNA using the same annealing procedure. For the fabrication of scgRNA in CONAN, iDNAs and gRNA were mixed at the molar ratio of 1.2:1 and hybridized using the same annealing procedure. The concentration of scgRNA in CONAN is regarded as the same as the concentration of the used gRNA. NUPACK software was used to model and design all cgRNAs and scgRNAs (36). The sequences of all oligonucleotides were listed in tables S1 to S4.
Fluorescence measurement in CONAN and the Cas12a gate
The fluorescence intensity tests were carried out on a 96-well reaction plate with the QuantStudio 7 Flex machine (Invitrogen Life Technologies, Carlsbad, CA, USA). FAM was excited at 495 nm, and its emission at 520 nm was recorded.
For Cas12a gate, the reaction was initiated by 50 nM LbCas12a and 50 nM gRNA with various concentrations of the target DNA and 1 μM FQ reporter substrates in 20-μl reaction volume [20 mM tris-HCl (pH 7.5), 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol, and heparin (50 μg/ml)]. The reactions were performed in the 96-well PCR plates (100 μl) at 37°C for 4 hours with fluorescence measurements taken every 30 s.
For CONAN, 1 μM LbCas12a with 50 nM gRNA-T, 500 nM aDNA, and 1 μM scgRNA was mixed with various concentrations of the target DNA that were added directly into the 20-μl reaction volume. The reactions were performed in the 96-well PCR plate (100 μl) at 37°C for 4 hours with fluorescence measurements taken every 30 s.
Gel electrophoresis studies
For SDS–polyacrylamide gel electrophoresis (PAGE), 10 μl of reaction solutions were analyzed by gel shift assay on 10% denaturing polyacrylamide gels at 110 V for 90 min. The theoretical molecular weight of LbCas12a was evaluated on the net (http://web.expasy.org/compute_pi/). For native PAGE, 10 μl of reactions (FAM-labeled DNA or gRNA) were analyzed by gel shift assay on 12 or 20% polyacrylamide gels (39:1 acrylamide/bisacrylamide) at 110 V for 30 min or 3 hours. After electrophoresis, the gels were scanned under UV light using a ChemiDoc MP system (Bio-Rad, USA) with its built-in FAM channel, and the intensities of the bands were quantified with ImageJ (National Institutes of Health) software.
Cell culture
The operation procedures for cell culture were performed as described previously (37). Briefly, two cell lines (TCCSUP and LO2) were cultured in a 100-mm dish containing 8 ml of DMEM or RPMI-1640 supplemented with 10% FBS and penicillin-streptomycin (100 U/ml) with a humidified atmosphere of 5% CO2 at 37°C. The hepatoma cell line (HLCZ01) was isolated and cultured according to the published literature (38). For HBV-infected HLCZ01 reactions, the HLCZ01 cells were inoculated with the different strains of sera from chronic patients diluted at 1:20 and were cultured for the indicated periods. Then, cell numbers were measured with a TC10 automated cell counter (Bio-Rad, USA). The human genomic DNAs of HLCZ01, TCCSUP, and LO2 cells (106 to 107 cells) were extracted using the TIANamp Genomic DNA Kit according to the manufacturer’s instruction. The concentration of the extracted genomic DNAs was routinely measured before incubation with CONAN using the Synergy Mx Multimode Microplate Reader Receives (BioTek, USA).
Human serum and bladder tissue sample collection
Institutional Review Board approval was obtained before study initiation (NCT03066310 and 2008S1108), and all patients signed informed consent. For 18 human serum samples, nine human health (four men and five women; mean age, 49 years; range, 21 to 67 years) and nine HBV (six men and three women; mean age, 30 years; range, 20 to 53 years) serum samples were collected between 2009 and 2011 in Xiangya Hospital of Central South University, China. For six bladder tissue samples, three healthy (two men and one woman; mean age, 54 years; range, 21 to 80 years) and three cancer (two men and one woman; mean age, 67 years; range, 51 to 84 years) tissue samples were also recruited from the Xiangya Hospital of Central South University, China. The study was approved by the Ethics Committee of Xiangya Hospital (Changsha, China). The human genomic DNAs were extracted from serum and bladder tissue using a TIANamp Genomic DNA Kit according to the manufacturer’s instruction.
qRT PCR, NGS, and immunohistochemical image
qRT-PCR experiment was performed on a 96-well reaction plate with the QuantStudio 7 Flex machine (Invitrogen Life Technologies, Carlsbad, CA, USA) using a 2× SYBR premix Ex TaqTM GC kit. First, prepare 1:2 serially diluted DNA as standard. Then, the PCR reaction mixture was prepared in a volume of 10 μl, consisting of 5 μl of 1× SYBR premix, 2 μl of genomic DNA with the specific amount, 0.2 μl of 10 μM primer F, 0.2 μl of 10 μM primer R, and 2.6 μl of deionized water. The primers pair HBV-F/R used for PCR to detect HBV DNA were shown in table S4. Analyze melting curves of cccDNA PCR products to check specificity and standard curve to determine the PCR efficiency.
For NGS, the genomic DNA from six bladder tissue samples was sequenced with Illumina NextSeq 500 (Illumina, San Diego, CA, USA). In addition, the NGS dates were provided by Genetalks Biotech Co. Ltd. (Changsha, China). The immunohistochemical staining of the tissue sections from the normal bladder and bladder cancer was imaged under an Olympus CX31-LV320 (Olympus, Tokyo, Japan) bright-field microscope. In addition, these data were provided by the Third Hospital of Hebei Medical University (Shijiazhuang, China).
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (21725503, 22034002, and 21974038). Author contributions: K.S., C.L., and Z.N. designed the study and drafted the manuscript. K.S., S.X., and S.W. performed the experiments and analyzed the data. C.L. and Z.N. directed the data analysis and interpretation. R.T. and H.Z. contributed to the construction of HLCZ01 cell line, collection of serum samples, and qRT-PCR experiment. Q.L. contributed to the NGS data. D.G. collected the tissue samples and performed the immunohistochemical experiment. Z.N. conceived and supervised the concept of the study. Competing interests: All authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc7802/DC1
REFERENCES AND NOTES
- 1.Zhang D. Y., Turberfield A. J., Yurke B., Winfree E., Engineering entropy-driven reactions and networks catalyzed by DNA. Science 318, 1121–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Li J., Green A. A., Yan H., Fan C., Engineering nucleic acid structures for programmable molecular circuitry and intracellular biocomputation. Nat. Chem. 9, 1056–1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin P., Choi H. M. T., Calvert C. R., Pierce N. A., Programming biomolecular self-assembly pathways. Nature 451, 318–322 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Zuo X., Xia F., Xiao Y., Plaxco K. W., Sensitive and selective amplified fluorescence DNA detection based on exonuclease III-aided target recycling. J. Am. Chem. Soc. 132, 1816–1818 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Gines G., Menezes R., Nara A., Kirstette A.-S., Taly V., Rondelez Y., Isothermal digital detection of microRNAs using background-free molecular circuit. Sci. Adv. 6, eaay5952 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F., Elbaz J., Teller C., Willner I., Amplified detection of DNA through an autocatalytic and catabolic DNAzyme-mediated process. Angew. Chem. Int. Ed. 50, 295–299 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Zou L., Wu Q., Zhou Y., Gong X., Liu X., Wang F., A DNAzyme-powered cross-catalytic circuit for amplified intracellular imaging. Chem. Commun. 55, 6519–6522 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Dirks R. M., Pierce N. A., Triggered amplification by hybridization chain reaction. Proc. Natl. Acad. Sci. U.S.A. 43, 15275–15278 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang F., Lu C.-H., Willner I., From cascaded catalytic nucleic acids to enzyme-DNA nanostructures: Controlling reactivity, sensing, logic operations, and assembly of complex structures. Chem. Rev. 114, 2881–2941 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Knott G. J., Doudna J. A., CRISPR-Cas guides the future of genetic engineering. Science 361, 866–869 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jusiak B., Cleto S., Perez-Piñera P., Lu T. K., Engineering synthetic gene circuits in living cells with CRISPR technology. Trends Biotechnol. 34, 535–547 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Nakamura M., Srinivasan P., Chavez M., Carter M. A., Dominguez A. A., La Russa M., Lau M. B., Abbott T. R., Xu X., Zhao D., Gao Y., Kipniss N. H., Smolke C. D., Bondy-Denomy J., Qi L. S., Anti-CRISPR-mediated control of gene editing and synthetic circuits in eukaryotic cells. Nat. Commun. 10, 194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kempton H. R., Goudy L. E., Love K. S., Qi L. S., Multiple input sensing and signal integration using a split Cas12a system. Mol. Cell 78, 184–191.e3 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Chen J. S., Ma E., Harrington L. B., Da Costa M., Tian X., Palefsky J. M., Doudna J. A., CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teng F., Guo L., Cui T., Wang X.-G., Xu K., Gao Q., Zhou Q., Li W., CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 20, 132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.East-Seletsky A., O’Connell M. R., Knight S. C., Burstein D., Cate J. H. D., Tjian R., Doudna J. A., Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington L. B., Burstein D., Chen J. S., Paez-Espino D., Ma E., Witte I. P., Cofsky J. C., Kyrpides N. C., Banfield J. F., Doudna J. A., Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gootenberg J. S., Abudayyeh O. O., Lee J. W., Essletzbichler P., Dy A. J., Joung J., Verdine V., Donghia N., Daringer N. M., Freije C. A., Myhrvold C., Bhattacharyya R. P., Livny J., Regev A., Koonin E. V., Hung D. T., Sabeti P. C., Collins J. J., Zhang F., Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S.-Y., Cheng Q.-X., Wang J.-M., Li X.-Y., Zhang Z.-L., Gao S., Gao R.-B., Zhao G.-P., Wang J., CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li L., Li S., Wu N., Wu J., Wang G., Zhao G., Wang J., HOLMESv2: A CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 8, 2228–2237 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Gootenberg J. S., Abudayyeh O. O., Kellner M. J., Joung J., Collins J. J., Zhang F., Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nistala G. J., Wu K., Rao C. V., Bhalerao K. D., A modular positive feedback-based gene amplifier. J. Biol. Eng. 4, 4 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu M., Zhang Q., Chang D., Gu J., Brennan J. D., Li Y., A DNAzyme feedback amplification strategy for biosensing. Angew. Chem. Int. Ed. 56, 6142–6146 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Patharkar O. R., Walker J. C., Floral organ abscission is regulated by a positive feedback loop. Proc. Natl. Acad. Sci. U.S.A. 112, 2906–2911 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong D., Ren K., Qiu X., Zheng J., Guo M., Guan X., Liu H., Li N., Zhang B., Yang D., Ma C., Wang S., Wu D., Ma Y., Fan S., Wang J., Gao N., Huang Z., The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Swarts D. C., Jinek M., Mechanistic insights into the cis- and trans-acting DNase activities of Cas12a. Mol. Cell 73, 589–600.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanewich-Hollatz M. H., Chen Z., Hochrein L. M., Huang J., Pierce N. A., Conditional guide RNAs: Programmable conditional regulation of CRISPR/Cas function in bacterial and mammalian cells via dynamic RNA nanotechnology. ACS Cent. Sci. 5, 1241–1249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oesinghaus L., Simmel F. C., Switching the activity of Cas12a using guide RNA strand displacement circuits. Nat. Commun. 10, 2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markham N. R., Zuker M., DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 33, W577–W581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henderson S., Chakravarthy A., Su X., Boshoff C., Fenton T. R., APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 4, 1833–1841 (2014). [DOI] [PubMed] [Google Scholar]
- 31.López-Knowles E., Hernández S., Malats N., Kogevinas M., Lloreta J., Carrato A., Tardón A., Serra C., Real X. F., PIK3CA mutations are an early genetic alteration associated with FGFR3 mutations in superficial papillary bladder tumors. Cancer Res. 66, 7401–7404 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kannan P., Subramanian P., Maiyalagan T., Jiang Z., Cobalt oxide porous nanocubes-based electrochemical immunobiosensing of hepatitis B virus DNA in blood serum and urine samples. Anal. Chem. 91, 5824–5833 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Li X., Zhao J., Yuan Q., Xia N., Detection of HBV covalently closed circular DNA. Viruses 9, 139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loeb L. A., Human cancers express mutator phenotypes: Origin, consequences and targeting. Nat. Rev. Cancer 11, 450–457 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makarova K. S., Wolf Y. I., Alkhnbashi O. S., Costa F., Shah S. A., Saunders S. J., Barrangou R., Brouns S. J. J., Charpentier E., Haft D. H., Horvath P., Moineau S., Mojica F. J. M., Terns R. M., Terns M. P., White M. F., Yakunin A. F., Garrett R. A., van der Oost J., Backofen R., Koonin E. V., An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 13, 722–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfe B. R., Porubsky N. J., Zadeh J. N., Dirks R. M., Pierce N. A., Constrained multistate sequence design for nucleic acid reaction pathway engineering. J. Am. Chem. Soc. 139, 3134–3144 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Luo X., Xue B., Feng G., Zhang J., Lin B., Zeng P., Li H., Yi H., Zhang X.-L., Zhu H., Nie Z., Lighting up the native viral RNA genome with a fluorogenic probe for the live-cell visualization of virus infection. J. Am. Chem. Soc. 141, 5182–5191 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Yang D., Zuo C., Wang X., Meng X., Xue B., Liu N., Yu R., Qin Y., Gao Y., Wang Q., Hu J., Wang L., Zhou Z., Liu B., Tan D., Guan Y., Zhu H., Complete replication of hepatitis B virus and hepatitis C virus in a newly developed hepatoma cell line. Proc. Natl. Acad. Sci. U.S.A. 13, E1264–E1273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swarts D. C., van der Oost J., Jinek M., Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tiollais P., Pourcel C., Dejean A., The hepatitis B virus. Nature 317, 489–495 (1985). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc7802/DC1