

KLF11 is a negative regulator of CSC stemness in sarcoma by repressing YAP/TEAD-dependent transcription.
Abstract
Cancer stem cells (CSCs) are involved in tumorigenesis, recurrence, and therapy resistance. To identify critical regulators of sarcoma CSCs, we performed a reporter-based genome-wide CRISPR-Cas9 screen and uncovered Kruppel-like factor 11 (KLF11) as top candidate. In vitro and in vivo functional annotation defined a negative role of KLF11 in CSCs. Mechanistically, KLF11 and YAP/TEAD bound to adjacent DNA sites along with direct interaction. KLF11 recruited SIN3A/HDAC to suppress the transcriptional output of YAP/TEAD, which, in turn, promoted KLF11 transcription, forming a negative feedback loop. However, in CSCs, this negative feedback was lost because of epigenetic silence of KLF11, causing sustained YAP activation. Low KLF11 was associated with poor prognosis and chemotherapy response in patients with sarcoma. Pharmacological activation of KLF11 by thiazolidinedione effectively restored chemotherapy response. Collectively, our study identifies KLF11 as a negative regulator in sarcoma CSCs and potential therapeutic target.
INTRODUCTION
Osteosarcoma is the most common type of bone cancer and the second leading cause of cancer-related deaths in children and adolescents (1). Osteosarcoma often displays highly aggressive behavior and, accordingly, a poor outcome with overall 5-year survival rate of only 20% in patients with metastatic disease (2). The multidisciplinary treatments, including surgery and chemotherapy, have remained essentially unchanged over the past three decades (2). However, the expected benefits of these therapies are tempered by poor response and high rate of recurrence (3). Therefore, osteosarcoma presents a substantial clinical challenge, and it is critical to identify potential therapeutic targets (4).
The high recurrence rate and treatment failure of osteosarcoma prompt us to focus attention on a distinct subpopulation of cells called cancer stem cells (CSCs) (5). CSCs exhibit sustained self-renewal potential and tumor-initiating ability, thereby contributing to tumorigenesis, therapy resistance, recurrence, and, in some cases, metastasis (6). The identification of underlying mechanisms governing CSC propagation is an active area of research, fueled by the promise that a combination of conventional chemotherapy with specific CSC inhibitors will increase therapeutic success and even eradicate cancer (7). Genetic screens are powerful tools for identifying causal genes in various hallmarks of cancer (8). In recent years, the CRISPR-Cas9 system has been widely applied to identify genes involved in cancer cell survival and drug resistance (9, 10). However, CRISPR screen in CSCs has rarely been reported.
In this study, by a genome-wide CRISPR screen and in-depth mechanistic studies, we identify Kruppel-like factor 11 (KLF11) as a key negative regulator of osteosarcoma CSCs and demonstrate the mechanism by which down-regulation of KLF11 contributes to CSCs stemness, focusing on its involvement in regulating the transcription activity of Yes Associated Protein (YAP). Pharmacological activation of KLF11 synergizes with chemotherapy for osteosarcoma treatment. More generally, we suggest that KLF11 may serve as a common regulator for sarcoma CSCs.
RESULTS
A reporter-based CRISPR screen for regulators of osteosarcoma CSCs
To enable a pooled CRISPR screen for regulators of osteosarcoma CSCs, we used a reporter to monitor the expansion of CSC population. Evaluation of the levels of stemness genes in osteosarcoma spheres revealed that Oct4 (pou5f1) had the most significant increase (fig. S1, A and B). Loss-of-function and gain-of-function experiments in osteosarcoma cells with high (143B and SJSA-1) or low (MG63 and U2-OS) Oct4 basal levels proved the critical role of Oct4 for CSC-like properties, demonstrated by self-renewal capacity, tumor-initiating ability, and metastasis (fig. S1, C to J). To examine whether an Oct4-expressing reporter could be suited to detect osteosarcoma CSCs as previously reported (11, 12), we lentivirally introduced enhanced green fluorescent protein (EGFP) reporter driven by human Oct4 promoter (pOct4-EGFP) or trimer of Oct4 enhancers (eOct4-EGFP) (13) into osteosarcoma cells. eOct4-EGFP+ cells exhibited closer characteristics of CSC than pOct4-EGFP+ cells (fig. S2, A to G) (13). The frequency of eOct4-EGFP+ cells ranged from 3 to 22% in different osteosarcoma cells, and the EGFP signal remained constant after 14 days of adherent culture and increased under suspended culture (fig. S2H).
We used eOct4-EGFP reporter–containing MG63 osteosarcoma cells for genome-wide CRISPR screen. eOct4-EGFP− cells were selected and transduced with the human lentivirus–based genome-scale CRISPR knockout (GeCKO v2) library (14) in lentiCRISPRv2 vector. After 14 days of culturing, EGFPhigh (the highest 50% of EGFP fluorescence peak) and EGFP− cell populations were sorted out for deep sequencing of single-guide RNA (sgRNA) representation (Fig. 1A). The read distributions of two biologically independent samples showed a high level of correlation (fig. S3A). Our genetic screen effectively recovered functional genes known to modulate CSCs, as sgRNAs targeting positive CSC regulators (e.g., Sox2 and Pou5f1) being deleted and those targeting negative regulators (e.g., Gsk3β, Arid3a, and Gli3) being highly enriched in EGFPhigh cells (Fig. 1B; fig. S3, B and C; and table S1). The asymmetric nature of the volcano plot showed that the statistical power to detect depleted sgRNAs was lower than the power to detect enriched sgRNAs (Fig. 1B) (15). Hence, our further analysis was focused on the enriched sgRNAs. First, we used small interfering RNAs (siRNAs) targeting the enriched hits with false discovery rate (FDR) < 0.05 and evaluated their effect on eOct4-luc reporter (fig. S3D). The residual 14 candidates were further validated on Oct4 expression and self-renewal ability (fig. S3, E and F). Using this approach, we identified KLF11, a member of the Sp1/KLF family, as the top candidate.
Fig. 1. A reporter-based CRISPR screen identifies KLF11 as a negative regulator of osteosarcoma CSCs.
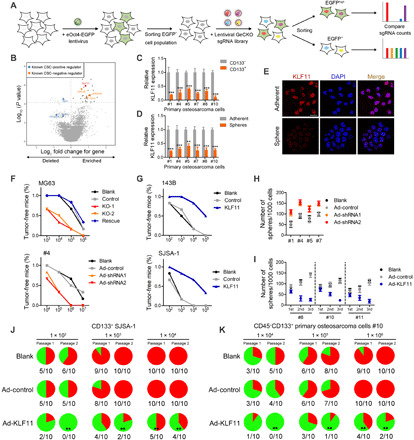
(A) Experimental design of an eOct4-EGFP reporter–based genome-wide CRISPR screen. (B) Volcano plot displaying the log2 fold change and adjusted P value for all sgRNAs identified in the screen. Negative regulators with a threshold of FDR < 0.05 are labeled as small dark gray dots, selected genes with known function as large blue and orange dots, and KLF11 as large red dot. (C) qRT-PCR analysis of KLF11 expression in CD133+ and CD133− cells from primary osteosarcoma cells (n = 3). (D) qRT-PCR analysis of KLF11 expression in spheres and adherent cells from primary osteosarcoma cells (n = 3). (E) Immunofluorescence detection of KLF11 (red) in primary osteosarcoma #4 spheres and adherent cells. The nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 25 μm. (F) In vivo limiting dilution assay of KLF11-deleted and ectopic KLF11 rescue MG63 cells, and KLF11-knockdown primary osteosarcoma cells (sample #4). KO, knockout. n = 6 for each group. (G) In vivo limiting dilution assay of KLF11-overexpressing and control osteosarcoma spheres (143B and SJSA-1). n = 6 for each group. (H) Sphere formation assay of KLF11-knockdown primary osteosarcoma cell. (I) Sphere formation assay of KLF11-overexpressing primary osteosarcoma cells (n = 3). (J) Incidences of tumorigenesis of CD133+ SJSA-1 cells infected with indicated adenovirus in serial transplantation models. **P < 0.01 compared with untreated CD133+ SJSA-1 cells in the first inoculation by Fisher’s exact test. (K) Incidences of tumorigenesis of CD45−CD133+ primary osteosarcoma cells (sample #10) infected with indicated adenovirus in serial transplantation models. **P < 0.01 compared with untreated primary osteosarcoma cells in the first inoculation by Fisher’s exact test.
KLF11 negatively regulates osteosarcoma CSCs
We first determined the expression of KLF11 in osteosarcoma CSCs. CD133 is a well-known marker for osteosarcoma CSCs (16). Pearson correlation analysis revealed that KLF11 levels were negatively correlated with the expression of CD133 in osteosarcoma tissues (fig. S4A). As shown in Fig. 1 (C to E), KLF11 levels were down-regulated in sorted CD133+ primary osteosarcoma cells and in spheres derived from primary osteosarcoma cells. Similar results were observed in osteosarcoma cell lines (fig. S4, B and C).
To explore the regulatory role of KLF11 on CSCs, we transduced MG63 and U2-OS osteosarcoma cells with two distinct sgRNAs targeting KLF11 (fig. S4D). KLF11-deleted cells exhibited enhanced CSC-like properties, including proportion of CD133+ cells (fig. S4E), stemness gene expression (fig. S4, D and F), self-renewal ability (fig. S4G), tumorigenicity (Fig. 1F), and metastasis (fig. S4H), as well as chemotherapy tolerance (fig. S4, I and J). These effects could be rescued by the sgRNA-resistant KLF11 complementary DNA (cDNA) (fig. S4, D to J). In line with these results, yielded KLF11 overexpression impaired the stemness characteristics of 143B and SJSA-1 osteosarcoma cells (Fig. 1G and fig. S5). Similar results were obtained in eOct4-EGFP + and eOct4-EGFP− cells (fig. S6).
We further validated the role of KLF11 in primary osteosarcoma CSCs by using KLF11 short hairpin–mediated RNA (shRNA) or KLF11-expressing adenovirus. Flow cytometry analysis showed that knockdown of KLF11 increased the proportion of primary CD133+ cells (fig. S4E). Primary osteosarcoma cells with KLF11 knockdown exhibited enhanced self-renewal capacity and increased expression of stemness genes (Fig. 1H and fig. S4, D and F). Overexpression of KLF11 in primary osteosarcoma cells had the opposite effects (Fig. 1I and fig. S5, A and B).
Furthermore, serial transplantation was performed by subcutaneous injection of the CD133+ cells isolated from SJSA-1 cells (Fig. 1J) or primary osteosarcoma cells (Fig. 1K) into a second and subsequently third batch of mice. Adenovirus-delivered KLF11 substantially restrained the tumorigenicity upon serial passage in vivo of sorted CD133+ cells, suggesting that KLF11 impaired the tumor formation ability of osteosarcoma CSCs. Similar results were obtained upon KLF11 deletion in CD133− SJSA-1 and MG63 cells (fig. S6, F and G). Collectively, these results support an inhibitory role of KLF11 in osteosarcoma CSCs.
KLF11 restrains YAP/TEAD transcriptional output
To study the molecular mechanism of KLF11 in the regulation of osteosarcoma CSCs, we performed RNA sequencing (RNA-seq) in control and KLF11-knockdown osteosarcoma cells (Fig. 2A, fig. S7A, and table S2). Gene set enrichment analysis (GSEA) demonstrated that the stemness-related signature was awakened upon KLF11 reduction (fig. S7B). We noticed that YAP signature was the most significantly enriched upon KLF11 knockdown among several stemness-related pathway signatures (Fig. 2B). Moreover, KLF11 knockdown selectively increased the transcript abundance of YAP target genes compared to non-YAP targets (Fig. 2C and fig. S7C), which was verified by quantitative reverse transcriptase polymerase chain reaction (qRT-qPCR) (fig. S7D). These observations indicated that KLF11 knockdown facilitated the transcriptional output of YAP.
Fig. 2. KLF11 restrains YAP/TEAD transcriptional output.
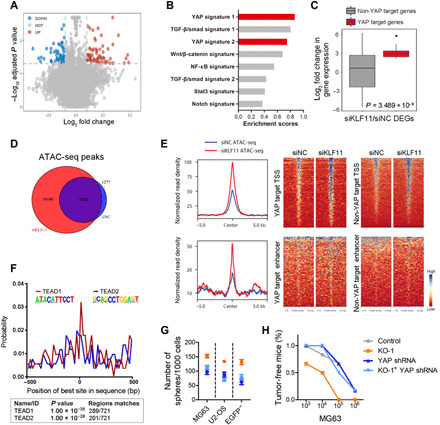
(A) Volcano plots compare KLF11-knockdown to control group. Absolute log2 fold change beyond 1.0 with adjusted P < 0.05 were considered as significant difference. Red circles represent up-regulated genes in the KLF11-knockdown group. Blue circles represent down-regulated genes in the KLF11-knockdown group. (B) GSEA using indicated signatures in RNA-seq from KLF11-knockdown MG63 cells. TGF-β, transforming growth factor–β; NF-κB, nuclear factor κB. (C) Fold change in deregulated genes [121 differentially expressed genes (DEGs)] expression of non-YAP target genes versus YAP target genes in MG63 cells upon KLF11 knockdown. siNC, negative control siRNA. Data are presented as box-and-whiskers plots. Box includes values within the 25th and 75th percentiles (with the median highlighted by the line in the middle), and whiskers extend from the 5th to the 95th percentile. (D) Venn diagram for the numbers of ATAC-seq peaks in KLF11-knockdown and control MG63 cells. (E) Density profiles (left) and heatmaps (right) of ATAC-seq data showing the read density in a ±5-kb window surrounding the center of YAP target TSS and enhancer peaks as well as non-YAP target TSS and enhancer peaks in KLF11-knockdown and control MG63 cells. (F) Centrimo analysis for TEAD1 and TEAD2 binding motifs at KLF11-bound enhancer sites in a 1-kb window. The reads are centered on the respective KLF11 peak. bp, base pairs. (G) Sphere formation assay of KLF11-deleted osteosarcoma cells (MG63, U2-OS and eOct4-EGFP− MG63) transfected with indicated plasmids (n = 3). (H) In vivo limiting dilution assay of KLF11-deleted MG63 cells transfected with indicated plasmids. Tumors were observed over 2 months; n = 6 for each group. See also figs. S7 and S8.
We hypothesized that KLF11 might locally impair chromatin configurations to repress YAP target genes. To test this possibility, we performed assay for transposase-accessible chromatin with sequencing (ATAC-seq) to evaluate open chromatin regions. KLF11 knockdown led to more significant ATAC-seq peaks (additional 19,190) compared to control cells (Fig. 2D and fig. S7, E and F), including 5700 KLF11 bound sites, suggesting that KLF11 may serve as a transcription repressor at these sites. The increased chromatin accessibility at the upstream of genes upon KLF11 knockdown was more pronounced at YAP targets compared to non-YAP targets (Fig. 2E), and these sites were centrally enriched for TEA Domain Transcription Factor (TEAD)–binding motifs (Fig. 2F), implying that chromatin becomes more accessible at TEAD-binding sites (TBSs) after KLF11 knockdown. In line with this, knockdown of YAP eliminated the discrepant stemness properties triggered by KLF11 depletion (Fig. 2, G and H, and fig. S8), indicating that YAP is required for KLF11-mediated stemness regulation.
KLF11 and YAP/TEAD bind to joint sites in a cooperative manner
To gain further insights into the mechanism, we performed chromatin immunoprecipitation sequencing (ChIP-seq) to compare the binding sites of KLF11 and YAP/TEAD (Fig. 3A). The expression of genes containing a KLF11 peak within a 50-kb window of their promoter was notably induced upon KLF11 knockdown (fig. S9A), indicating that KLF11 acts mainly as a transcription repressor. Analysis of the distribution of binding sites revealed that most peaks were located farther than 10 kb from the closest transcription start site (TSS) (fig. S9B). KLF11 and YAP/TEAD preferentially bound to enhancers as previously reported (fig. S9, C and D) (17, 18). The overlap of KLF11 and YAP/TBS at enhancers was significantly better compared to promoters (Fig. 3B). Furthermore, the TEAD1 signal demonstrated a very narrow spatial distribution around KLF11 peaks at enhancers and a bimodal signal for H3K27ac surrounding the signal for KLF11 and TEAD1 (Fig. 3C). Similar binding patterns were observed for KLF11/YAP/TEAD occupancy on representative gene loci, and KLF11 loss led to increased DNA accessibility at these gene loci (Fig. 3D and fig. S9E). Further analysis for KLF- and TEAD-binding motifs within a 500–base pair (bp) window surrounding the top 500 KLF11 peaks revealed that both KLF- and TEAD-binding motifs are significantly enriched in the vicinity of KLF11 peaks (Fig. 3E). This finding suggests that specific sites for KLF11 and TEAD1 have coevolved so that these genomic regions can be bound by both transcription factors at the same time. We also found that YAP knockdown induced a preferential loss of KLF11 from YAP target promoter and enhancer compared to those of non-YAP targets (fig. S9, F and G), suggesting that the localization of KLF11 to those sites may be dependent on YAP binding.
Fig. 3. KLF11 and YAP/TEAD bind to joint sites in a cooperative manner.
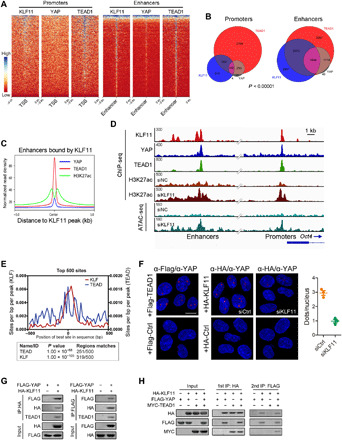
(A) Heatmaps of ChIP-seq data showing the occupancy of KLF11, YAP, and TEAD1 in a ±5-kb window surrounding the center of all RefSeq transcriptional start sites (TSSs) and all enhancer regions in MG63 cells. (B) Venn diagram showing the numbers of promoters and enhancers, respectively, bound by KLF11, YAP and TEAD1in MG63 cells. (C) Density profiles of ChIP-seq data showing the occupancy of YAP, TEAD1, and H3K27ac in a ±5-kb window surrounding the center of KLF11 peaks at enhancer sites. (D) Genome browser view of ChIP-seq and ATAC-seq signals at Oct4 promoters and enhancers as indicated locus in MG63 cells. (E) Centrimo analysis for KLF and TEAD motifs to identify enriched binding motifs surrounding the top 500 KLF11 ChIP-seq peaks. (F) Left: Representative microscopic images from PLA experiments in MG63 sphere-derived cells. PLA signals (red dots) were detected for interaction of endogenous YAP and exogenous FLAG-TEAD (positive control, left) or HA-KLF11 after treatment with siRNAs targeting KLF11 (siKLF11) or control siRNAs (siCtrl). The nuclei were stained with DAPI (blue). Scale bar, 10 μm. Right: Quantification of PLA signals for the YAP1-KLF11 interaction in MG63 sphere-derived cells treated with siKLF11 or siCtrl. (G) Co-IP of exogenous KLF11 and YAP in lysates from human embryonic kidney (HEK) 293T cells overexpressing HA-KLF11 and FLAG-YAP. (H) Two-step co-IP to test for ternary complex formation of KLF11, YAP, and TEAD1. HEK293T cells were transfected with HA-KLF11, FLAG-YAP, and Myc-TEAD1. The first immunoprecipitation was performed with an anti-HA antibody. The complex was eluted with the HA peptide, followed by the second step of immunoprecipitation with an anti-FLAG antibody. The resulting precipitates were analyzed by Western blot analysis with the indicated antibodies. See also figs. S9 and S10.
We next analyzed the potential physical interaction between KLF11 and YAP. The colocalization of KLF11 and YAP was observed by immunofluorescence (Pearson’s correlation R = 0.629) (fig. S10A) and proximity ligation assays (PLAs) (Fig. 3F). Coimmunoprecipitation (co-IP) validated the interaction between KLF11 and YAP (Fig. 3G and fig. S10B). In vitro pull-down assay using purified proteins verified the direct interaction between KLF11 and YAP (fig. S10C). Moreover, re-IP assay demonstrated that KLF11, YAP, and TEAD1 were in a ternary complex (Fig. 3H). Re-ChIP assay demonstrated that KLF11, YAP, and TEAD1 existed as a transcription complex on Oct4 enhancer region (fig. S10D). Subsequent domain mapping showed that the R3 domain of KLF11 interacted specifically with the WW1/WW2 region of YAP (fig. S10, E and F). Together, we propose that KLF11 and YAP/TEAD bind to joint DNA sites stabilized with direct protein-protein interaction.
KLF11 recruits SIN3A/HDAC corepressor to YAP/TEAD complex
We next further deciphered the precise mechanism by which KLF11 represses YAP/TEAD transcriptional activity. By using a TEAD-YAP fusion protein, we found that the KLF11-mediated repressive effects were not dependent on the interaction between YAP and TEAD (fig. S11A). KLF11 is known to repress gene expression by recruiting chromatin partners, such as SIN3 transcription regulator family member A/histone deacetylase (SIN3A/HDAC) complex, WD40 proteins, and heterochromatin protein 1-H3K9 histone methyltransferase (HP1-HMT) system (19). Therefore, we used KLF11 mutants that could specifically uncouple KLF11 from each of these chromatin remodeling proteins, namely, KLF11 mutant wherein amino acids E29 and A30 were mutagenized to proline residues (EAPP) (SIN3A/HDAC), A347S (WD40), and Δ486 (HP1α/HMT) (20). Only EAPP failed to repress the expression of YAP targets (Fig. 4A), suggesting that KLF11 may facilitate SIN3A/HDAC complex recruitment to the YAP/TEAD complex. Co-IP assays revealed that SIN3A and HDAC2 interacted with YAP, and KLF11 deletion blocked the interaction (Fig. 4B). Moreover, re-ChIP assay demonstrated that KLF11, YAP/TEAD, and SIN3A/HDAC formed a transcription complex on Oct4 enhancer region (fig. S11B).
Fig. 4. KLF11 recruits SIN3A/HDAC corepressor to YAP/TEAD complex.
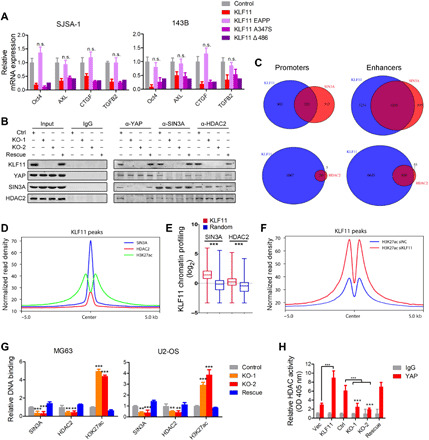
(A) qRT-PCR analysis of indicated stemness genes in osteosarcoma sphere-derived cells (SJSA-1 and 143B) transfected with indicated plasmids after 48 hours (n = 3). n.s., not significant. (B) Co-IP of endogenous YAP and SIN3A/HDAC2 complex in lysates from MG63 cells upon KLF11 deletion and ectopic KLF11 rescue. (C) Venn diagram showing the numbers of promoters and enhancers, respectively, bound by KLF11 and SIN3A (top), KLF11 and HDAC2 (bottom) in MG63 cells. (D) Density profiles of ChIP-seq data showing the occupancy of SIN3A, HDAC2, and H3K27ac in a ±5-kb window surrounding the center of KLF11 peaks. (E) Box plot of the SIN3A and HDAC2 ChIP-seq signal (log2) overlapping with KLF11 bound regions. Data are presented as box-and-whiskers plots (whiskers extend from the 5th to the 95th percentile; the box extends from the 25th to the 75th percentile; the line within the box represents the median). ***P < 10−10 (one-tailed Mann-Whitney U test). (F) Density profiles of ChIP-seq data showing the occupancy of H3K27ac in a ± 5-kb window surrounding the center of KLF11 peaks regions in KLF11-knockdown and control MG63 cells. (G) ChIP assay of the enrichment of SIN3A/HDAC2 and H3K27ac on Oct4 enhancer relative to immunoglobulin G (IgG) in KLF11-deleted and ectopic KLF11 rescue osteosarcoma cells (MG63 and U2-OS) (n = 3). Eluted DNAs were subjected to conventional qPCR. **P < 0.01 and ***P < 0.001. (H) HDAC activity assay of YAP pull-down immunoprecipitates in indicated MG63 cells (n = 3). See also fig. S11. OD, optical density.
For further validation, we performed ChIP-seq for SIN3A and HDAC2 in MG63 cells. Similar to KLF11 and YAP/TEAD, both factors preferentially bound to enhancers compared to promoters (figs. S9C and S11C). At enhancer sites, SIN3A and HDAC2 peaks exhibited a highly significant overlap with the KLF11 peaks (P < 0.001) (Fig. 4C and fig. S11D). The signals of SIN3A and HDAC2 demonstrated a very narrow spatial distribution around KLF11 peaks (Fig. 4D). In line, SIN3A and HDAC2 were enriched at KLF11-bound regions compared to 6000 random sequences (Fig. 4E).
As SIN3A/HDAC was implicated in the regulation of enhancers by deacetylating H3K27ac (21, 22), we performed ChIP-seq to examine the global effects of KLF11 on H3K27ac. KLF11 knockdown led to more H3K27ac peaks compared to control cells (fig. S11E), coupled with a substantial increase of H3K27ac signal at the KLF11 target sites (Fig. 4F). Furthermore, ChIP-quantitative PCR (qPCR) assay confirmed that deletion of KLF11 impaired the recruitment of SIN3A/HDAC2 complex to target genes, along with increased H3K27ac status (Fig. 4G), suggesting that recruitment of SIN3A/HDAC2 to active enhancer is KLF11 dependent. HDAC activity assay revealed that the enzymatic activity of recruited HDAC2 is functionally involved in KLF11-mediated gene repression by using YAP-pulled immunoprecipitates (Fig. 4H). Last, the repressive effect by KLF11 was fully relieved by HDAC inhibitor treatment (fig. S11, F and G). Collectively, these results support a scenario in which KLF11 recruits the SIN3A/HDAC complex to restrain YAP/TEAD-dependent transactivation.
Hypermethylation of KLF11 promoter contributes to its reduced expression and loss of response to YAP in osteosarcoma CSCs
We observed evident location of YAP and TEAD1 peaks at the promoter of KLF11 in our ChIP-seq data (Fig. 5A), suggesting that YAP/TEAD may regulate KLF11 expression. As shown in fig. S12A, KLF11 levels were positively regulated by YAP. However, overexpression of a YAP mutant without TEAD-binding capacity (YAP-5SA-S94A) or verteporfin treatment, a drug that selectively disrupts YAP/TEAD binding, failed to promote KLF11 expression (fig. S12B). Bioinformatics analysis predicted three consensus TBSs in KLF11 promoter region. As shown in fig. S12C, introduction of constitutively activated YAP (YAP-5SA) promoted KLF11 luciferase reporter activity but not the reporter with TBS mutation. ChIP-qPCR assay demonstrated the enrichment of YAP and TEAD1 on the TBSs of KLF11 promoter (Fig. 5B). Furthermore, re-ChIP assay confirmed the cooccupancy of YAP and TEAD1 on the KLF11 promoter (fig. S12, D and E). These data indicated that YAP/TEAD complex bound to the KLF11 promoter and promoted KLF11 transcription, therefore forming a negative feedback circuit.
Fig. 5. Hypermethylation-mediated low KLF11 expression predicts poor prognosis and response to chemotherapy.
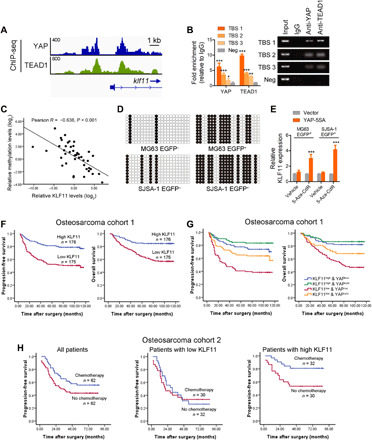
(A) Genome browser view of ChIP-seq signals at KLF11 promoters as indicated locus in MG63 cells. (B) ChIP assay of the enrichment of YAP and TEAD1 on KLF11 promoter relative to IgG in MG63 cells (n = 3). Eluted DNAs were subjected to conventional qPCR. A random region without TEAD1-binding sites (TBSs) acted as a negative control (Neg). *P < 0.05, **P < 0.01, and ***P < 0.001. (C) The correlation between KLF11 methylation levels and KLF11 expression in human osteosarcoma tissues (n = 42) was analyzed by Pearson’s correlation. (D) Bisulfite sequencing analysis of the methylation status of the KLF11 promoter in eOct4-EGFP+ and eOct4-EGFP− osteosarcoma cells (MG63 and SJSA-1). Open and closed circles indicate the unmethylated and methylated CpG dinucleotides, respectively. (E) qRT-PCR analysis of KLF11 expression in 5-Aza-CdR–treated eOct4-EGFP+ MG63 and SJSA-1 cells transfected with indicated plasmids. ***P < 0.001. (F) Kaplan-Meier analysis of progression-free survival (left; P < 0.001, log-rank test) and overall survival (right; P < 0.001, log-rank test) of patients with osteosarcoma in low (red line) and high (blue line) KLF11 groups from our cohorts. (G) Kaplan-Meier analysis of progression-free survival (P < 0.001) and overall survival (P < 0.001) of patients with osteosarcoma with high or low KLF11 and cytoplasmic or nuclear YAP from our cohorts. (H) Kaplan-Meier analysis of progression-free survival in patients with osteosarcoma with and without doxorubicin-based chemotherapy. Left: All patients (P = 0.059). Middle: Patients with low KLF11 expression (P = 0.771). Right: Patients with high KLF11 expression (P = 0.016). The median KLF11 expression was used as a cutoff. See also figs. S12 to S14 and tables S3 to S6.
Notably, the above experiments were performed in osteosarcoma cells, whereas the regulation of KLF11 by YAP was not observed in sorted osteosarcoma CSCs that express low levels of KLF11 (fig. S12, F and G), leading us to speculate that KLF11 expression is silenced in CSCs. Epigenetic modification, especially DNA methylation, has been proposed to be responsible for the down-regulation of KLF11 (23). Methylation-specific PCR in 42 cases of osteosarcoma samples showed a strong negative correlation between KLF11 promoter methylation and KLF11 expression (Fig. 5C and fig. S13A). As shown in fig. S13B, three distinct CpG islands within 2000 bp upstream of the KLF11 gene were found by MethPrimer (24). The bisulfite sequencing of this region indicated that the CpG sites were hypermethylated in osteosarcoma CSCs but not in non-CSCs (Fig. 5D). In addition, treatment with 5-Aza-dC, an inhibitor of DNA methyltransferase, resulted in significantly increased KLF11 expression following YAP-5SA overexpression, while YAP-5SA overexpression alone had no notable effect on KLF11 expression (Fig. 5E). These results demonstrate that hypermethylation of KLF11 promoter contributes to its reduced expression and loss of response to YAP in osteosarcoma CSCs.
Low KLF11 predicts poor prognosis and response to chemotherapy
We next investigated the clinical significance of KLF11. The average levels of KLF11 were lower in osteosarcoma tissues than adjacent nontumor tissues by immunohistochemistry (IHC) (fig. S14A), even lower in patients with metastasis (fig. S14B). In osteosarcoma tissues, KLF11 levels were negatively correlated with the expression of stemness gene Oct4 (fig. S14C). Kaplan-Meier analysis showed that patients with lower KLF11 levels exhibited shorter time to recurrence and worse overall survival in our patient cohort and online datasets (Fig. 5F; fig. S14, D and E; and tables S3 and S4), indicating that low expression of KLF11 correlates with poor prognosis in patients with osteosarcoma. In addition, patients with both repressed KLF11 level and increased Oct4 expression displayed even worse prognosis (fig. S14F). To further support the role of the KLF11/YAP circuit in osteosarcoma progression, we performed immunochemistry staining of YAP and KLF11 in tissue microarray (fig. S14G). Although low KLF11 predicts a poor prognosis, patients with osteosarcoma with both repressed KLF11 and nuclear YAP displayed even worse prognosis (Fig. 5G), indicating a better prognostic value of combining the two factors.
Chemotherapy might have unrealized clinical benefits for patients with osteosarcoma who are masked because of a lack of reliable biomarkers in the selection of responders (25). As KLF11 affected the chemotherapy tolerance of osteosarcoma cells (figs. S4I and S5, E and F), and the average levels of KLF11 were higher in the good-response group than poor-response patients (fig. S14H), we explored whether KLF11 expression could predict the clinical response to chemotherapy in patients with osteosarcoma. As shown in Fig. 5H, chemotherapy provided limited benefit to the progression-free survival (PFS) of overall patients with osteosarcoma, whereas patients with high KLF11 expression in tumors exhibited a superior PFS after receiving chemotherapy compared with those in the control group. In contrast, patients with low expression of KLF11 had a poor response to chemotherapy. There was no significant difference in clinical characteristics between the control group and the chemotherapy treatment group (table S5). In both univariate and multivariate analyses, chemotherapy correlated with improved PFS of patients with osteosarcoma with high KLF11 expression (table S6). In addition, a significant interaction was observed between KLF11 and therapy with respect to the effect on the PFS of patients with osteosarcoma (p = 0.005 for interaction). Thus, KLF11 could serve as an independent predictor for chemotherapy response in patients with osteosarcoma.
Pharmacological activation of KLF11 by thiazolidinedione restores chemotherapy response in osteosarcoma
In the stem cell hypothesis, relapse after treatment results from the inability to eradicate CSCs. With the above results, we expect that KLF11 activation is able to improve the efficacy of chemotherapy by eroding CSCs. Peroxisome proliferator–activated receptor gamma (PPARγ) has been reported to induce KLF11 (17, 26); thus, we used synthetic ligands of PPARγ, the thiazolidinediones (TZDs), to assess the treatment potential of pharmacologic activation of KLF11 in osteosarcoma. Pioglitazone (Pio) and third-generation TZD Efatutazone (Efat) are clinically used TZD and were confirmed for the induction of PPARγ transcription activity as well as induction of KLF11 (Fig. 6A). As shown in fig. S15A, TZD treatment led to increased expression of KLF11 and decreased expression of Oct4 and connective tissue growth factor (CTGF), which is PPARγ dependent. We found that Pio and Efat treatment induced demethylation of KLF11 promoter (fig. S15B). It has been reported that PPARγ has the ability to direct local demethylation around its binding sites by recruiting methylcytosine hydroxylation enzyme ten-eleven translocation (TET) proteins (27). Among the TET family proteins (TET1, TET2, and TET3) (28), knockdown of TET1 significantly impaired the TZD-mediated KLF11 activation (fig. S15C). Moreover, Pio and Efat treatment enhanced the interaction between PPARγ and TET1 (Fig. 6B) and their binding on the KLF11 promoter (fig. S15D), accompanied with increased 5-hydroxymethylcytosine (5hmC) production (fig. S15D), suggesting that a PPARγ agonist promotes the demethylation of KLF11 promoter by PPARγ-mediated TET1 recruitment.
Fig. 6. Pharmacological activation of KLF11 by TZD restores chemotherapy response.
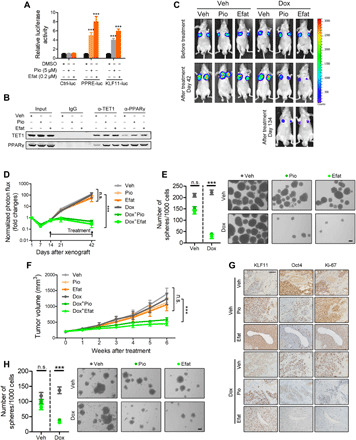
(A) Luciferase activity of the peroxisome proliferator–responsive element (PPRE) containing reporter (PPRE-luc) or KLF11-luc in eOct4-EGFP+ SJSA-1 cells transfected with indicated plasmids and treated with Pio (left) or Efat (right) (n = 3). Data were normalized against Renilla luciferase activity. DMSO, dimethyl sulfoxide. (B) Co-IP of endogenous PPARγ and TET1 in lysates from eOct4-EGFP+ SJSA-1 cells treated with Pio (5 μM) or Efat (0.2 μM). (C) Representative bioluminescent images of nude mice xenografted with MG63R-luc cells (1 × 106 cells) were intraperitoneally injected with doxorubicin (3 mg/kg, twice a week for 3 weeks), or orally administrated with Pio (25 mg/kg per day for 4 weeks) or Efat (1 mg/kg per day for 4 weeks) (n = 5 per group). (D) Quantification of bioluminescent imaging signal intensities of nude mice xenografted with MG63R-luc cells (1 × 106 cells) were intraperitoneally injected with doxorubicin (Dox) or orally administrated with Pio or Efat (n = 5 per group). Veh, vehicle. ***P < 0.001. (E) Sphere formation assay of MG63R-luc xenograft-derived cells from indicated groups (n = 3). ***P < 0.001. (F) Tumor volumes of PDX subcutaneously xenografted in immunodeficient nonobese diabetic/severe combined immunodeficiency mice intraperitoneally injected with doxorubicin or orally administrated with Pio or Efat (n = 5 per group). ***P < 0.001. (G) Representative images of indicated IHC in consecutive tumor sections from aforementioned indicated groups (F). Scale bar, 100 μm. (H) Sphere formation assay of PDX-derived cells from indicated groups. ***P < 0.001. See also figs. S15 and S16.
Both Pio and Efat significantly recovered the chemotherapy sensitivity of resistant MG63 cells (MG63R) (fig. S15, E and F). A similar trend was also observed by introducing the constitutively active form of PPARγ (M3-PPARγ). These effects were attenuated by KLF11 deletion (fig. S15, G and H), indicating that the synergistic effect was KLF11 dependent. Furthermore, both Pio and Efat restored the sensitivity of MG63R-derived xenografts to concurrent doxorubicin treatment (Fig. 6, C and D). The treatment was discontinued after 28 days, but no tumor regrowth was observed in the combined treatment group even after 3 months. This was associated with significantly prolonged survival of mice (fig. S16A) and decreased self-renewal ability (Fig. 6E). No notable weight loss was observed upon treatment (fig. S16B). Xenografts from TZD-treatment group showed increased KLF11 expression and decreased Oct4 expression (fig. S16C). More intriguingly, the combination of TZD plus doxorubicin treatment prevented the emergence of resistance, indicating a potential strategy for suppressing chemotherapy resistance (fig. S16D).
Furthermore, we evaluated the therapeutic potential of Pio and Efat in a patient-derived xenograft (PDX) model derived from a patient with osteosarcoma who achieved partial response to doxorubicin but relapsed after 13 months with acquired resistance. Tumor burden remained unchanged in the mice after monotherapy with either TZD or doxorubicin. In contrast, we observed significant tumor shrinkage in mice treated with these drugs in combination (Fig. 6F), accompanied by increased levels of KLF11, decreased levels of Oct4 and proliferation (Fig. 6G and fig. S16E), and impaired self-renewal potential in the posttreatment tumors (Fig. 6H). No notable weight loss of mice was observed during treatment (fig. S16F). Collectively, these data demonstrate that activation of KLF11 with a PPARγ agonist restores the sensitivity of osteosarcoma cells to chemotherapy.
DISCUSSION
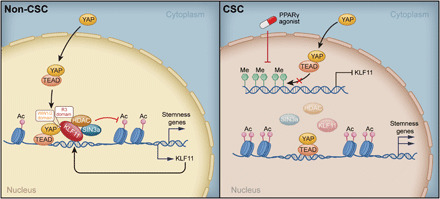
Here, we performed a genome-wide CRISPR-Cas9 screen to identify important regulators of osteosarcoma CSCs using an eOct4-EGFP reporter. Our results demonstrated that KLF11 substantially suppressed CSC stemness of osteosarcoma by restraining YAP/TEAD transcription activity via recruiting SIN3A/HDAC (Fig. 7). In turn, YAP/TEAD promoted KLF11 transcription to form a negative feedback loop in osteosarcoma non-CSCs. Intriguingly, hypermethylation of KLF11 promoter led to its reduced expression and loss of response to YAP/TEAD in osteosarcoma CSCs, facilitating sustained transcriptional output of YAP, including stemness gene expression (Fig. 7). Our work further indicated that the PPARγ agonist TZD, which activates KLF11, might serve as an effective strategy to overcome chemoresistance through eradicating CSCs in osteosarcoma.
Fig. 7. Schematic diagram of KLF11-based signaling circuit in osteosarcoma CSCs.
CRISPR-Cas9 screen is a powerful tool for identifying the causal relationship between genotype and phenotype (8). In this study, we performed genome-wide loss-of-function screen to identify essential regulators of osteosarcoma CSCs basing on an eOct4-EGFP reporter. Although the use of a single marker to measure CSCs introduces caveats and there are likely to be some “hits” unrelated to CSCs in reporter-based screen, our screen identified most of the known positive and negative osteosarcoma CSC regulators, as well as potential novel regulators, for Oct4 is a well-defined marker to faithfully reflect osteosarcoma CSCs (11, 12). To date, the application of CRISPR screen in CSCs was rare because of the difficulty to obtain large number of CSCs. Our strategy provides a paradigm for dissecting CSC regulators and may be broadly applied to other type of CSCs that can be monitored by a fluorescence-based reporter.
DNA-facilitated transcription factor interaction/pairing is common between cotranscription factors (29). Here, we found that KLF- and TEAD-binding motifs were closely located in the genome, especially in the enhancer region, suggesting cooperation between these two transcription factors. Apart from TEAD-binding motifs, we also found the presence of GATA- and FOX-binding motifs in the vicinity of KLF-binding motifs, suggesting potential cooperation between KLF11 and these transcription factors. Intriguingly, our study showed that KLF11 and YAP bound to each other directly, which might further stabilize their interaction. Thus, the complex formation of KLF11 and YAP/TEAD is probably dictated by both adjacent DNA motifs and protein-protein interaction. Our results broaden the scope of the precise modulation among interacted transcription factors.
Hippo pathway has been reported to be involved in the regulation of CSC stemness and tumorigenesis in various sarcomas (30, 31), including osteosarcoma (32). However, given the broad physiological functions of YAP such as tissue homeostasis, general targeting strategy may cause cellular toxicity and side effects. In our study, we found that KLF11 repressed the expression of a proportion of YAP targets sharing joint binding sites, among which include genes related to CSCs, while those with individual YAP recognition motifs are affected to a lesser extent or even spared from this repressive effect. In this regard, our study represents a previously unrecognized type of mechanism for the regulation of Hippo signaling and suggests that the manipulation of KLF11 would be an attractive alternative for anti-YAP strategy to specifically target CSCs.
Maintenance of DNA methylation has been reported to be critical for CSC properties (33), which might suppress the expression of differentiation- and apoptosis-related genes. Here, we observed that preexisting methylation of KLF11 promoter in osteosarcoma CSCs presents a roadblock to impede its activation by YAP/TEAD and thus blocked the negative feedback loop between KLF11 and YAP, leading to sustained YAP transcription activity in osteosarcoma CSCs. Previous studies have shown that Oct4 and Nanog could up-regulate DNMT1 to maintain DNA methylation and undifferentiated state in stem cells (34), but the exact mechanism underlying the methylation of KLF11 promoter in osteosarcoma CSCs remains to be further determined.
Chemotherapy might have unreliable clinical benefits for patients with osteosarcoma due to the lack of reliable biomarkers in the selection of responders during clinical routine. Here, we showed that low expression of KLF11 in pretherapy osteosarcoma tissues significantly correlated with a poor chemotherapy response, whereas patients with high KLF11 expression exhibited a marked improvement in prognosis after receiving treatment. Therefore, it is advisable to evaluate KLF11 expression in osteosarcoma tumors or circulating tumor cells to select patients who might benefit from a more specific and tailored treatment (35). Although TZDs as PPARγ agonists have been reported to induce differentiation of CSCs (36–38), the mechanisms remain elusive. Our study revealed that activation of KLF11 was responsible for the suppressive effect on osteosarcoma CSCs by TZDs in both cell line–derived xenografts and PDX models, which restored the chemosensitivity of resistant osteosarcoma cells. Thus, we propose that CSC-targeted therapy via TZD treatment may be a potential adjuvant therapy along with chemotherapy for patients with osteosarcoma. However, prospective, large-scale, randomized clinical trials are necessary to confirm our results before this strategy can be applied in the clinic.
Last, we explored the role of KLF11 in other types of sarcoma. Similar to osteosarcoma, KLF11 suppressed the CSC properties of sarcoma cells via YAP (fig. S17, A to C). KLF11 expression was down-regulated in the tumors of patients with sarcoma (fig. S17, D and E) and correlated with prognosis in sarcoma patient cohorts including kidney sarcoma (fig. S18 and tables S7 and S8). The common dependence of osteosarcoma cells and other types of sarcoma cells on KLF11/YAP to regulate CSC stemness, together with the prognostic significance of KLF11 and YAP in clinical patients, indicates that the found pathway is probably a conserved mechanism in the regulation of sarcoma CSCs, which needs further exploration.
MATERIALS AND METHODS
Cell lines
The osteosarcoma cell lines MG63, U2-OS, 143B, and SJSA-1; Ewing sarcoma cell line SK-ES-1; and human embryonic kidney (HEK) 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (Gibco, USA). The MG63R drug-resistance cell line was generated from MG63 cells with doxorubicin treatment at increasing concentrations until cells were resistant to treatment.
In vivo xenograft and treatment experiments
All animal experiments were undertaken in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals with the approval of the Institutional Animal Care and Use Committee at Nanjing University. Four- to 6-week-old female athymic BALB/c nude mice and immunodeficient nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice (GemPharmatech Co. Ltd., China) were housed and fed in standard pathogen-free conditions.
PDX models were established by transplanting fragments of fresh human osteosarcoma tissues subcutaneously into the flanks of female NOD/SCID mice. When tumors have successfully engrafted, xenografts were passage into next generation of NOD/SCID mice for the following studies. When the sizes of PDXs were matched, the mice were randomized for intraperitoneal injection of doxorubicin (3 mg/kg, twice a week for 3 weeks), oral administration of Pio (25 mg/kg per day for 4 weeks) or Efat (1 mg/kg per day for 4 weeks), or combination treatment. Mice xenografted with luciferase-labeled MG63 cells were intraperitoneally injected with doxorubicin (3 mg/kg per week), orally administrated with Pio (25 mg/kg per day) or Efat (1 mg/kg per day), or combination treatment for 16 weeks.
For in vivo serial passaging, xenografts were mechanically disaggregated and digested with type II/IV collagenase (1 mg/ml) and deoxyribonuclease (DNase) (1 mg/ml) at 37°C for 1 hour. The cell suspension was filtered through an 80-μm filter and centrifuged at 300g for 5 min at 4°C. The pellets were suspended and sorted for CD45−CD133+ cells followed with indicated adenovirus treatment as previously described (39). Then, serial concentrations of CD45−CD133+ osteosarcoma cells were injected subcutaneously into the flanks of NOD/SCID mice.
For in vivo limiting dilution assay, serial concentrations of cells were injected subcutaneously into the flanks of NOD/SCID mice. Kinetic of tumor formation was evaluated weekly for 8 weeks. The frequency of T-ICs was determined using ELDA software (http://bioinf.wehi.edu.au/software/elda/index.html).
For in vivo lung metastasis model, 1 × 106 single cells were injected into the tail vein of nude mice. Mice were euthanized 12 weeks after inoculation, and consecutive sections of the whole lung were subjected to hematoxylin-eosin staining.
All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering. Mice body weight and xenograft size were measured weekly. Xenograft volumes were evaluated by caliper measurements of two perpendicular diameters and calculated individually with the following formula: Volume = a × b2/2 (a represents length and b represents width) or by bioluminescent imaging technology (Xenogen IVIS 100 Imaging System).
Patients and clinical samples
This study was approved by all the institutional review boards with participating sites providing the necessary institutional data-sharing agreements before initiation. All clinical samples and clinical data were obtained under institutional review board approval and informed consent.
Fresh tumor tissues used for PDXs and primary cell cultures were obtained from patients who had undergone a surgical osteosarcoma excision or radical nephrectomy at Nanjing General Hospital. The detailed clinical characteristics of these patients are provided in table S9.
For evaluating the correlation between KLF11 expression and clinical characteristics, we incorporated 351 patients with osteosarcoma as osteosarcoma cohort 1 and 62 patients with kidney sarcoma as kidney sarcoma cohort. The collection of tissue samples was carried out between 2004 and 2015 consecutively. The detailed clinical characteristics of these patients were provided in tables S3 and S7.
For evaluating the correlation between KLF11 expression and chemotherapy response, we incorporated 124 patients with osteosarcoma between 2004 and 2015 as osteosarcoma cohort 2. Patients in the chemotherapy group (n = 62) received at least two cycles of doxorubicin-based chemotherapy, and patients in the control group (n = 62), who did not receive any therapy or received less than one cycle of chemotherapy because of unacceptable adverse effects, were matched to chemotherapy-receiving patients on baseline clinicopathologic characteristics. We assessed therapeutic response according to Response Evaluation Criteria in Solid Tumors. The detailed clinical characteristics of these patients are listed in table S5.
All patients were followed up until December 2018. If the primary end point is PFS (for prognostic power evaluation, refer to time from initial surgery to the occurrence of local recurrence or distant metastasis; for therapeutic response predictor evaluation, refer to time from the initiation of chemotherapy to the date of first radiological evidence of progressed disease), and the secondary is overall survival (time from initial surgery to death). Progression was defined as the development of a local or regional nodal recurrence on computed tomography (CT) or magnetic resonance imaging (MRI); distant metastasis was seen on CT or MRI; death from osteosarcoma. Progressed disease was defined by radiologic assessment of ≥25% tumor regrowth, or appearance of new lesions, or symptomatic deterioration during chemotherapy. Patients who were alive or progression free at the time of analysis were censored using the time from initial surgery or treatment to the last follow-up.
Online data cohorts
The differential expression of KLF11 in various sarcoma tissues was assessed in National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) databases (GSE2712, GSE2719, and GSE49972). In the correlation between KLF11 expression and other gene expression, clinical characteristics were evaluated in NCBI GEO databases (GSE21257, GSE30929, GSE40021, GSE42352, and GSE63155). The correlation between KLF11 expression and chemotherapy response was assessed in NCBI GEO databases (GSE39055 and GSE87437).
Cell transfection and real-time reverse transcription PCR analysis
Transfection of plasmids was performed using jetPEI (PolyPlus Transfection, France). Transfection of siRNA was performed using Lipofectamine RNAiMAX (Invitrogen, USA) at a final concentration of 100 nM. Sequences of siRNA (GenePharma, China) against specific targets were listed in table S10.
Total RNAs of cells were extracted by TRIzol (Invitrogen, USA). qRT-PCR was performed in a reaction mixture of SYBR Green (Takara, China) with ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, USA). The expression of indicated genes was normalized to endogenous reference control β-actin by using a 2-ΔΔCt method. Sequences of primers used for qRT-PCR in this study were listed in table S11.
IHC, immunocytochemistry, and PLA
For IHC, paraffin-embedded sections were deparaffinized, rehydrated, and followed by antigen retrieval. After primary and secondary antibody incubation, slides were finally incubated with diaminobenzidine (Dako, USA), and counterstained with hematoxylin (Sigma Chemical Co., USA).
For immunocytochemistry, osteosarcoma cells or spheres were plated in 12-well plates and fixed with 10% paraformaldehyde solution for 15 min at room temperature, permeabilized with 0.4% Triton X-100 in phosphate-buffered saline (PBS) for 5 min, and then blocked with 1% bovine serum albumin in PBS for 1 hour at 37°C. The blocked cells were incubated with primary antibody overnight at 4°C, followed by incubation with Alexa Fluor 488–conjugated anti-mouse immunoglobulin G (IgG) antibody and Alexa Fluor 555–conjugated anti-rabbit IgG antibody (Invitrogen, Carlsbad, CA) for 2 hours. Nuclear staining of cells was conducted using 4,6-diamidino-2-phenylindole (DAPI). Representative images were acquired using a confocal microscope.
For PLA, osteosarcoma cells were seeded on fibronectin-coated glass chamber slides and transfected with HA-KLF11 or Flag-TEAD1, which serves as a positive control. After 24 hours after transfection, cells were fixed in 4% formaldehyde for 10 min at room temperature and blocked with the blocking buffer supplied by the manufacturer for 1 hour. In situ PLA was performed with the DuoLink In Situ Red Starter Kit (mouse/rabbit) (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Briefly, after incubating with primary antibodies overnight at 4°C, (i) cells were incubated with plus and minus probes for 1 hour, (ii) DNA strands were subjected to ligase treatment for 30 min at 37°C, (iii) DNA was amplified for 100 min at 37°C with the addition of polymerase, and (iv) slides were mounted and PLA dots were detected via confocal microscopy. When the proximity of two PLA probes is less than 40 nm, the red fluorescent emissions would be detected.
Assessment of the IHC staining was based on the staining intensity and the percentage of positively stained cells using Image-Pro Plus 6.0 software (Media Cybernetics Inc., USA). All samples were reviewed by three pathologists (H.C., X.W., and X.D.Z.). IHC of YAP or KLF11 was scored by determining the percentage of positive tumor cells (<25% focal, 25 to 50% moderate, and 51 to 100% diffuse), and their staining intensity (0, negative; 1+, weak; 2+, moderate; and 3+, strong; see fig. S14G). Nuclear YAP samples were defined by at least moderate (2+) nuclear staining in ≥25% of tumor cells. Cytoplasmic YAP samples were defined by no visible nuclear staining or weak (1+) nuclear staining.
Western blot
Cell lysates were electrophoresed by 10% SDS–polyacrylamide gel electrophoresis and transferred into a nitrocellulose membrane. Membrane was incubated with primary specific antibodies, followed by IRDye 800CW–conjugated secondary antibody (Rockland Immunochemicals, USA). The intensity of the fluorescence was scanned by the Odyssey fluorescence scanner system (LI-COR Biosciences, USA) and quantified by Quantity One software (Bio-Rad, USA). Primary antibodies used in this study are listed in table S12.
Immunoprecipitation
Osteosarcoma cells under indicated treatments were harvested in IP lysis buffer supplemented with a complete protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The cell lysates were immunoprecipitated using specific primary antibody or control IgG preloaded with protein A/G agarose beads.
For re-IP, the first round of immunoprecipitants was eluted with IP lysis buffer containing 250 mM NaCl and HA peptide (1 mg/ml). The second immunoprecipitation was performed using the eluate from the first immunoprecipitation and anti-FLAG antibody, followed by the addition of protein A/G agarose beads.
For in vitro IP, reaction mixtures containing 1 μM purified human KLF11 protein (Abnova, NBP258081PEP) and 1 μM human YAP protein (Abnova, NBP262669PEP) in the binding buffer of 50 mM tris-HCl (pH 7.5) and 1 mM dithiothreitol (DTT) (Sangon) were incubated at 37°C for 90 min. Then, co-IP was performed with protein A/G Sepharose. After extensive washing with the lysis buffer, the immunoprecipitants were resolved by loading buffer, followed by Western blot analysis.
Plasmid construction
The wild-type (WT) and mutant forms of KLF11 plasmids were provided by G. Lomberk (Department of Surgery, Medical College of Wisconsin, Milwaukee, WI, USA) (40), and cloned into pcDNA3.1-HA. The WT and mutant forms of YAP plasmids were constructed previously by our group (41). TEAD-YAP fusion plasmid was provided by K. Guan (Department of Pharmacology and Moores Cancer Center, University of California, San Diego, La Jolla, USA) (42), which was generated by cloning YAP-S94A (inactive for TEAD binding) into pQCXIH-TEAD1 and in frame with the TEAD1 coding region. PPARγ luciferase reporter was provided by X. Han (Key Laboratory of Human Functional Genomics of Jiangsu Province, Jiangsu Diabetes Center, Nanjing Medical University, Nanjing, Jiangsu, China). Constitutively active mutant of PPARγ plasmid (M3-PPARγ) were provided by D. Gong (Division of Endocrinology, Diabetes and Nutrition, Department of Medicine, University of Maryland School of Medicine at Baltimore, USA) (43).
KLF11-deficient osteosarcoma cells were established according to the previously reported protocol (14). KLF11 sgRNAs (sgRNA-1, 5′-TGAGGGGTCTTATCCGCAAC-3′; sgRNA-2, 5′-AAGGTCTACTTGCAGCATCT-3′) were designed through online software (http://crispr.mit.edu/) and cloned into lentiCRISPRv2. To rescue the KLF11 deletion, a KLF11 cDNA (GAGATCCACCTGTTCCATAT) resistant to sgRNA-2 was constructed by site-directed mutagenesis (Agilent) of the bases in the seed sequence corresponding to KLF11 sgRNA-2. shRNA oligos (Oct4 shRNA-1, 5′-GCTTCAAGAACATGTGTAA-3′; Oct4 shRNA-2, 5′-GGAGGAAGCTGACAACAAT-3′; KLF11 shRNA-1, 5′-CCAAGTAACAGATTCCAAA-3′; KLF11 shRNA-2, 5′-GACAGTTTACTCAGCACTA-3′; PPARγ shRNA, 5′- GCCCTTCACTACTGTTGAC-3′) were synthesized by Sangon Co. Ltd. After annealing, double-strand oligos were inserted to lentiviral pLKO.1-Puro vector or AdMax Adenovirus Vector (Microbix Inc., Ontario, Canada). All constructs were confirmed by DNA sequencing.
Luciferase reporter assay
Some of the following methods are similar to those previously published (41). Cells were cotransfected with luciferase reporter and indicated plasmids using Lipofectamine 2000 (Invitrogen, USA). Each group was run in triplicate in 48-well plates. The luciferase activity was detected by the Synergy 2 Multidetection Microplate Reader (BioTek Instruments Inc.) 48 hours after transfection. Renilla luciferase activity was normalized against firefly luciferase activity.
HDAC activity assay
Cell lysates were incubated with an anti-YAP antibody precoupled with protein A/G agarose beads for 4 hours. The beads were washed three times with NP-40 lysis buffer and then fixed in 85 μl of double-distilled H2O. Then, 10 μl of 10× HDAC Assay Buffer and 5 μl of HDAC colorimetric substrate were added to each well (Colorimetric HDAC Activity Assay Kit, BioVision Incorporated). The plates were incubated at 37°C overnight. The reaction was stopped by adding 10 μl of lysine developer and mixed well at 37°C for 1 hour. The samples were analyzed in an enzyme-linked immunosorbent assay plate reader at 405 nm.
Sphere formation assay and in vitro limiting dilution assay
One thousand single cells were seeded into 96-well Ultra-Low Attachment Microplates (Corning, USA) in serum-free DMEM/F12 medium (Invitrogen, USA) supplemented with B27 (1:50, Invitrogen), epidermal growth factor (EGF) (20 ng/ml) (PeproTech), basic fibroblast growth factor (bFGF) (10 ng/ml) (Invitrogen), and insulin (4 μg/ml) (Sigma-Aldrich). Spheres were counted 7 days after seeding (primary spheres). To propagate spheres in vitro, spheres were collected by centrifugation and trypsinized with 0.25% trypsin to obtain single cells. Equal numbers of cells were then seeded into an ultralow attachment plate (secondary spheres).
Osteosarcoma cells were seeded into 96-well Ultra-Low Attachment Microplates (Corning, USA) at various cell doses and incubated under sphere forming conditions for 7 days. On the basis of the frequency of wells with sphere forming, the proportion of tumor-initiating cells was determined using Poisson distribution statistics and the LCalc Version 1.1 software program (Stem Cell Technologies Inc. Vancouver, Canada).
CRISPR-Cas9 screen and data analysis
The human genome-scale CRISPR knockout library (GeCKO v2, Addgene #1000000048) in the lentiCRISPR v2 vector (Addgene #52961) consists of 123,411 sgRNAs that target 19,050 protein-coding genes (6 sgRNAs per gene) and 1864 microRNAs (4 sgRNAs per microRNA) and also includes around 1000 nontargeting control sgRNAs (9, 14). We transduced the human GeCKO v2 library into MG63 cells by lentivirus at a multiplicity of infection of 0.4. Our data showed high coverage of the sgRNA library with <0.25% missing sgRNAs across samples. Cells were selected with puromycin and kept in culture in serum-free DMEM/F12 medium supplemented with B27, EGF, bFGF, and insulin for 10 days followed by fluorescence-activated cell sorting based on their EGFP expression. An “unsorted” sample was used to assess presort sgRNA library coverage, while sorted EGFPhigh and EGFP− populations were subjected to genomic DNA extraction, and the inserted sgRNA library was amplified by two steps of PCR for next-generation sequencing. Each screen was performed twice.
For data analysis, reads from the fastq files generated by sequencing were tallied for each guide by taking the first 20 bp from each read and mapping that sequence to the identical short gRNA sequence. For each screen, a table of reads per guide that includes the counts from the EGFPhigh and EGFP− populations of both replicates was generated and loaded into MAGeCK (44) by comparing the EGFPhigh and EGFP− populations. Top genes were determined on the basis of mean log2 fold change, FDR, and robust ranking aggregation (RRA) score (table S1). Figures were generated using the normalized read counts in R and RStudio (R project, Revolution Analytics).
RNA-seq and bioinformatic analysis
Some of the following methods are similar to those previously published (41). Total RNA was extracted using the RNeasy Mini Kit (Qiagen), and contaminant DNA was removed by the RNase-Free DNase Set (Qiagen). RNA integrity was verified with the Agilent 2100 Bioanalyzer automated electrophoresis system. mRNA was isolated using the Poly(A) mRNA Magnetic Isolation Module (NEB), and library preparation was conducted with the Ultra RNA Library Prep Kit for Illumina with Dual Index Primers (Multiplex Oligos for Illumina, NEB). Libraries were quantified with the Agilent 2100 Bioanalyzer automated electrophoresis system and subjected to Illumina Sequencing (HiSeq 2500) by DIATRE Biotechnology, Shanghai, China.
After removing the adapter sequences and reads with poor quality by Trim Galore (www.bioinformatics.babraham.ac.uk/projects/trim_galore), the qualified reads were mapped to the reference genome (hg19) and the expression level of genes were measured by RSEM (v1.3.1) with the reference annotation file obtained from Ensemble GRCh37. The expression level of genes was calculated in the form of fragments per kilobase of transcript per million mapped reads (FPKM). Genes whose expression level was not detected in all samples (FPKM < 1) were excluded in the following analysis. Differentially expressed genes (DEGs) were defined as genes with fold change >2 or <0.5 and Benjamini and Hochberg–adjusted P < 0.05. DEGs were used for Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis by using the clusterProfiler package and GSEA (GSEA v2.0). Gene sets were obtained from published gene signatures (18, 45–51). Statistical significance was assessed by comparing the enrichment score to enrichment results generated from 1000 random permutations of gene set to obtain P values.
ChIP and ChIP-seq
ChIP assays were conducted as previously described (41). Briefly, cells were cross-linked with 1% formaldehyde, washed with PBS, and lysed in ChIP lysis buffer [20 mM tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, and 1 mM EDTA]. Chromatin was sonified to obtain fragments of 200 to 800 bp. Equilibration of protein A Dynabeads (Invitrogen, 5-μl Dynabeads/1-μg antibody) with antibodies (2 μg for ChIP and 10 μg for ChIP-seq) was performed overnight, following immunoprecipitation of chromatin (100 μg for ChIP and 200 μg for ChIP-seq) for 6 hours. After extensive washing, bound chromatin was eluted using ChIP elution buffer [50 M tris (pH 8.0), 1 mM EDTA, 1% SDS, and 50 mM NaHCO3] and decross-linked and ribonuclease A/proteinase K digested overnight. The DNA was purified by the QIAquick PCR Purification Kit (Qiagen). Re-ChIP assays were conducted as previously described (41). Briefly, bead eluates from the first round of immunoprecipitation were incubated with 10 mM DTT at 37°C for 30 min and diluted 1:50 in dilution buffer [1% Triton X-100, 2 mM EDTA, 150 mM NaCl, and 20 mM tris-HCl (pH 8.1)] followed by reimmunoprecipitation with the second antibodies. The final elution step was performed using 1% SDS solution in tris-EDTA buffer (pH 8.0). ChIP samples were analyzed by PCR using specific primers or subjected to in-depth whole-genome DNA sequencing by DIATRE Biotechnology, Shanghai, China. The libraries were sequenced on an Illumina HiSeq 2500 platform. Sequences of primers used for ChIP-qPCR in this study were listed in table S13.
ChIP-seq data were analyzed using a publicly available pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline2) based on The Encyclopedia of DNA Elements (ENCODE) transcription factor and histone ChIP-seq pipeline specifications. Briefly, the raw sequencing reads from ChIP-seq were aligned to the human reference genome (UCSC GRCh37, hg19) using Bowtie 2 and further subjected to peak calling analyzed by model-based analysis for ChIP-seq (MACS) with default settings. Only peaks with significance at an irreproducible discovery rate (IDR) of 0.1 were considered in the downstream analysis.
Heatmaps of read density and average binding profiles around TSSs or enhancer or peak center were produced using the program Deeptools. Each row in the heatmaps represents a genomic region around a peak summit or TSS, and rows are ranked according to signal intensity. The genomic location of the peaks and their distance to the TSS of annotated genes were calculated using the Homer annotatePeaks.pl tool. The overlap of peaks from different ChIP-seq experiments was determined using the Intervene tool. Normalized read density [reads per kilobase of transcript per million mapped reads (RPKM)] was calculated from pooled replicates using the bamCoverage function in Deeptools and displayed using Integrative Genomics Viewer. Normalized read density was calculated with a resolution of 10 bp.
As coordinates for the promoters, 2-kb-wide regions centered on each TSS of all RefSeqs were used. The enhancer sites in mesodermal cells were retrieved from the UCSC ENCODE database where enhancers are defined by the enhancer-specific chromatin mark acetylated lysine 27 on histone H3 (H3K27ac) and by hypersensitivity to DNAse I. The BEDTools suite was used to infer overlaps of ChIP-Seq summits with enhancer or promoter regions. Read count quantitation was obtained using SeqMonk (Babraham Institute, www.bioinformatics.babraham.ac.uk/projects/seqmonk/). The regions of interest were loaded into SeqMonk as an Annotation set and quantitated by the read count quantitation method, with identical reads removed and correction for RPKM.
Transcription factor–bound enhancers were defined as enhancers overlapping with peaks. Similarly, transcription factor–bound promoters were defined as promoter regions (as defined above) overlapping with peaks. YAP target genes were defined as previous reported (18).
ATAC-seq and bioinformatics analysis
ATAC-seq were performed with two biological replicates per condition. Briefly, 50,000 cells were used per reaction. Nuclei were isolated after resuspension and centrifugation in lysis buffer. The 50-μl transposase reaction with isolated nuclei was incubated at 37°C for 30 min. DNA was purified using a MinElute PCR purification column (Qiagen). The transposed DNA fragments were preamplified by a first PCR reaction with five cycles containing barcoded Nextera PCR primers. The optimal number of cycles was determined by a SYBR Green qPCR reaction containing a 5-μl aliquot from the first PCR. The second PCR was then carried out with eight cycles, and the libraries were first purified by MinElute PCR purification column (Qiagen) and then further size-selected by AMPure XP beads to obtain libraries with a size distribution between 150 and 1000 bp. We used a publicly available ATAC-seq pipeline based on the official pipeline specification of ENCODE (https://github.com/kundajelab/atac-seq-pipeline). For calling ATAC-seq peaks, only peaks with an IDR < 0.1 were considered significant.
Motif discovery
Known motif and de novo motif discovery were performed with the findMotifsGenome.pl function from Homer. Motifs were searched within a 500-bp window around the peak summit.
Statistical analysis
All statistical analyses in this study were performed with SPSS 16.0 software (SPSS Inc., USA). Data were presented as means ± SD. The significance of mean values between two groups was analyzed by two-tailed Student’s t test. Spearman’s correlation analysis was performed to determine the correlation between two variables. Pearson chi-square test acted to analyze the clinical variables. In Kaplan-Meier survival analysis, patients were divided into low or high group based on the median gene expression, and the survival distributions were compared with the log-rank test. Cox proportional hazards regression analysis was used to analyze the effect of clinical variables on patient survival. A P < 0.05 was considered significant.
Acknowledgments
We thank the members of the oncology laboratory for input throughout the course of the project; X. Li, S. Y. Wang, and J. Hou for valuable comments; and all the patients as well as families who made this study possible. Funding: This study was supported by grants from the National Natural Science Foundation of China (nos. 81772740 to L.Q., 81972333 to C.C., 81872074 to L.W., 81902600 to Y.L., and 81702175 to Y.W.), Initiative Postdocs Supporting Program (no. BX201700105 to L.Q.), and Foundation for Youths of Jiangsu Province (no. BK20170621 to L.Q.). Author contributions: L.Q. designed the experiments. C.C., Y.W., J.W., H.C., C.S., K.Z., X.Y., and H.H. conducted the experiments; Y.Y., C.C., C.X., Z.W., and L.Q. performed the data analysis; J.Z., G.Z., J.G., W.Z., Xuhui Zhou, and L.W. provided clinical samples; X.C., Q.R., X.W., Xiaodie Zhou, B.L., and G.L. provided technical assistance; L.Q., C.C., Y.Y., Y.L., and K.G. drafted the manuscript; all authors discussed and approved the manuscript; L.Q. was responsible for research supervision, coordination, and strategy. Competing interests: K.G. is a cofounder of and has equity interest in Vivace Therapeutics. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The RNA-seq, ATAC-seq, and ChIP-seq data generated in this study have been deposited in the GEO database under accession GSE133312. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabe3445/DC1
REFERENCES AND NOTES
- 1.Casali P. G., Bielack S., Abecassis N., Aro H. T., Bauer S., Biagini R., Bonvalot S., Boukovinas I., Bovee J., Brennan B., Brodowicz T., Broto J. M., Brugieres L., Buonadonna A., De Alava E., Tos A. P. D., Del Muro X. G., Dileo P., Dhooge C., Eriksson M., Fagioli F., Fedenko A., Ferraresi V., Ferrari A., Ferrari S., Frezza A. M., Gaspar N., Gasperoni S., Gelderblom H., Gil T., Grignani G., Gronchi A., Haas R. L., Hassan B., Hecker-Nolting S., Hohenberger P., Issels R., Joensuu H., Jones R. L., Judson I., Jutte P., Kaal S., Kager L., Kasper B., Kopeckova K., Krakorova D. A., Ladenstein R., Le Cesne A., Lugowska I., Merimsky O., Montemurro M., Morland B., Pantaleo M. A., Piana R., Picci P., Piperno-Neumann S., Pousa A. L., Reichardt P., Robinson M. H., Rutkowski P., Safwat A. A., Schoffski P., Sleijfer S., Stacchiotti S., Strauss S. J., Hall K. S., Unk M., Van Coevorden F., van der Graaf W. T. A., Whelan J., Wardelmann E., Zaikova O., Blay J. Y.; Esmo Guidelines Committee, PaedCan and ERN EURACAN , Bone sarcomas: ESMO–PaedCan–EURACAN clinical practice guidelines for diagnosis, treatment and follow-up. Annals Oncol. 29, IV79–IV95 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Anderson M. E., Update on survival in osteosarcoma. Orthop. Clin. 47, 283–292 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Isakoff M. S., Bielack S. S., Meltzer P., Gorlick R., Osteosarcoma: Current treatment and a collaborative pathway to success. J. Clin. Oncol. 33, 3029–3035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kansara M., Teng M. W., Smyth M. J., Thomas D. M., Translational biology of osteosarcoma. Nat. Rev. Cancer 14, 722–735 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batlle E., Clevers H., Cancer stem cells revisited. Nat. Med. 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Clarke M. F., Clinical and therapeutic implications of cancer stem cells. N. Eng. J. Med. 380, 2237–2245 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Sánchez-Rivera F. J., Jacks T., Applications of the CRISPR–Cas9 system in cancer biology. Nat. Rev. Cancer 15, 387–393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shalem O., Sanjana N. E., Hartenian E., Shi X., Scott D. A., Mikkelson T., Heckl D., Ebert B. L., Root D. E., Doench J. G., Zhang F., Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen S., Sanjana N. E., Zheng K., Shalem O., Lee K., Shi X., Scott D. A., Song J., Pan J. Q., Weissleder R., Lee H., Zhang F., Sharp P. A., Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gibbs C. P., Kukekov V. G., Reith J. D., Tchigrinova O., Suslov O. N., Scott E. W., Ghivizzani S. C., Ignatova T. N., Steindler D. A., Stem-like cells in bone sarcomas: Implications for tumorigenesis. Neoplasia 7, 967–976 (2005); published online EpubNov (10.1593/neo.05394). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levings P. P., McGarry S. V., Currie T. P., Nickerson D. M., McClellan S., Ghivizzani S. C., Steindler D. A., Gibbs C. P., Expression of an exogenous human oct-4 promoter identifies tumor-initiating cells in osteosarcoma. Cancer Res. 69, 5648–5655 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotta A., Cheung A. Y., Farra N., Vijayaragavan K., Seguin C. A., Draper J. S., Pasceri P., Maksakova I. A., Mager D. L., Rossant J., Bhatia M., Ellis J., Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 6, 370–376 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pusapati G. V., Kong J. H., Patel B. B., Krishnan A., Sagner A., Kinnebrew M., Briscoe J., Aravind L., Rohatgi R., CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell 44, 113–129.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tirino V., Desiderio V., Paino F., De Rosa A., Papaccio F., Fazioli F., Pirozzi G., Papaccio G., Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J. 25, 2022–2030 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Loft A., Forss I., Siersbaek M. S., Schmidt S. F., Larsen A. S., Madsen J. G., Pisani D. F., Nielsen R., Aagaard M. M., Mathison A., Neville M. J., Urrutia R., Karpe F., Amri E. Z., Mandrup S., Browning of human adipocytes requires KLF11 and reprogramming of PPARγ superenhancers. Genes Dev. 29, 7–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zanconato F., Forcato M., Battilana G., Azzolin L., Quaranta E., Bodega B., Rosato A., Bicciato S., Cordenonsi M., Piccolo S., Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 17, 1218–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tetreault M.-P., Yang Y., Katz J. P., Krüppel-like factors in cancer. Nat. Rev. Cancer 13, 701–713 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Calvo E., Grzenda A., Lomberk G., Mathison A., Iovanna J., Urrutia R., Single and combinatorial chromatin coupling events underlies the function of transcript factor krüppel-like factor 11 in the regulation of gene networks. BMC Mol. Biol. 15, 10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whyte W. A., Bilodeau S., Orlando D. A., Hoke H. A., Frampton G. M., Foster C. T., Cowley S. M., Young R. A., Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482, 221–225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solaimani P., Wang F., Hankinson O., SIN3A, generally regarded as a transcriptional repressor, is required for induction of gene transcription by the aryl hydrocarbon receptor. J. Biol. Chem. 289, 33655–33662 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potapova A., Hasemeier B., Römermann D., Metzig K., Göhring G., Schlegelberger B., Länger F., Kreipe H., Lehmann U., Epigenetic inactivation of tumour suppressor gene KLF11 in myelodysplastic syndromes. Eur. J. Haematol. 84, 298–303 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Li L.-C., Dahiya R., MethPrimer: Designing primers for methylation PCRs. Bioinformatics 18, 1427–1431 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Linch M., Miah A. B., Thway K., Judson I. R., Benson C., Systemic treatment of soft-tissue sarcoma—gold standard and novel therapies. Nat. Rev. Clin. Oncol. 11, 187–202 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Yin K.-J., Fan Y., Hamblin M., Zhang J., Zhu T., Li S., Hawse J. R., Subramaniam M., Song C.-Z., Urrutia R., KLF11 mediates PPARγ cerebrovascular protection in ischaemic stroke. Brain 136, 1274–1287 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujiki K., Shinoda A., Kano F., Sato R., Shirahige K., Murata M., PPARγ-induced PARylation promotes local DNA demethylation by production of 5-hydroxymethylcytosine. Nat. Commun. 4, 2262 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Huang Y., Rao A., Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 30, 464–474 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jolma A., Yin Y., Nitta K. R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J., DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Eisinger-Mathason T. K., Mucaj V., Biju K. M., Nakazawa M. S., Gohil M., Cash T. P., Yoon S. S., Skuli N., Park K. M., Gerecht S., Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 112, E3402–E3411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye S., Lawlor M. A., Rivera-Reyes A., Egolf S., Chor S., Pak K., Ciotti G. E., Lee A. C., Marino G. E., Shah J., YAP1-mediated suppression of USP31 enhances NFκB activity to promote sarcomagenesis. Cancer Res. 78, 2705–2720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Basu-Roy U., Bayin N. S., Rattanakorn K., Han E., Placantonakis D. G., Mansukhani A., Basilico C., Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 6, 6411 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bröske A.-M., Vockentanz L., Kharazi S., Huska M. R., Mancini E., Scheller M., Kuhl C., Enns A., Prinz M., Jaenisch R., DNA methylation protects hematopoietic stem cell multipotency from myeloerythroid restriction. Nat. Genet. 41, 1207–1215 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Tsai C.-C., Su P.-F., Huang Y.-F., Yew T.-L., Hung S.-C., Oct4 and Nanog directly regulate Dnmt1 to maintain self-renewal and undifferentiated state in mesenchymal stem cells. Mol. Cell 47, 169–182 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Pantel K., Alix-Panabières C., Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 16, 409–424 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Mueller E., Sarraf P., Tontonoz P., Evans R. M., Martin K. J., Zhang M., Fletcher C., Singer S., Spiegelman B. M., Terminal differentiation of human breast cancer through PPARγ. Mol. Cell 1, 465–470 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Demetri G. D., Fletcher C. D., Mueller E., Sarraf P., Naujoks R., Campbell N., Spiegelman B. M., Singer S., Induction of solid tumor differentiation by the peroxisome proliferator-activated receptor-γ ligand troglitazone in patients with liposarcoma. Proc. Natl. Acad. Sci. U.S.A. 96, 3951–3956 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prost S., Relouzat F., Spentchian M., Ouzegdouh Y., Saliba J., Massonnet G., Beressi J.-P., Verhoeyen E., Raggueneau V., Maneglier B., Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 525, 380–383 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Zhang H., Chen Q., Yang M., Zhu B., Cui Y., Xue Y., Gong N., Cui A., Wang M., Shen L., Zhang S., Fang F., Chang Y., Mouse KLF11 regulates hepatic lipid metabolism. J. Hepatol. 58, 763–770 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Lomberk G., Mathison A. J., Grzenda A., Seo S., DeMars C. J., Rizvi S., Bonilla-Velez J., Calvo E., Fernandez-Zapico M. E., Iovanna J., Sequence-specific recruitment of heterochromatin protein 1 via interaction with Krüppel-like factor 11, a human transcription factor involved in tumor suppression and metabolic diseases. J. Biol. Chem. 287, 13026–13039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu L., Wu Z., Li Y., Xu Z., Liu B., Liu F., Bao Y., Wu D., Liu J., Wang A., A feed-forward loop between lncARSR and YAP activity promotes expansion of renal tumour-initiating cells. Nat. Commun. 7, 12692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao B., Ye X., Yu J., Li L., Li W., Li S., Yu J., Lin J. D., Wang C. Y., Chinnaiyan A. M., Lai Z. C., Guan K. L., TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y., Lazar M. A., Differential gene regulation by PPARγ agonist and constitutively active PPARγ2. Mol. Endocrinol. 16, 1040–1048 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Li W., Xu H., Xiao T., Cong L., Love M. I., Zhang F., Irizarry R. A., Liu J. S., Brown M., Liu X. S., MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker S., Wang C., Walradt T., Hong B. S., Tanner J. R., Levinsohn J. L., Goh G., Subtil A., Lessin S. R., Heymann W. R., Vonderheid E. C., King B. A., Lifton R. P., Choi J., Identification of a gain-of-function STAT3 mutation (p.Y640F) in lymphocytic variant hypereosinophilic syndrome. Blood 127, 948–951 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cordenonsi M., Zanconato F., Azzolin L., Forcato M., Rosato A., Frasson C., Inui M., Montagner M., Parenti A. R., Poletti A., Daidone M. G., Dupont S., Basso G., Biciato S., Piccolo S., The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Adorno M., Cordenonsi M., Montagner M., Dupont S., Wong C., Hann B., Solari A., Bobisse S., Rondina M. B., Guzzardo V., A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Padua D., Zhang X. H.-F., Wang Q., Nadal C., Gerald W. L., Gomis R. R., Massagué J., TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H., Zhu F., Yong J., Zhang P., Hou P., Li H., Jiang W., Cai J., Liu M., Cui K., Qu X., Xiang T., Lu D., Chi X., Gao G., Ji W., Ding M., Deng H., Generation of induced pluripotent stem cells from adult rhesus monkey fibroblasts. Cell Stem Cell 3, 587–590 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Bild A. H., Yao G., Chang J. T., Wang Q., Potti A., Chasse D., Joshi M.-B., Harpole D., Lancaster J. M., Berchuck A., Olson J. A. Jr., Marks J. R., Dressman H. K., West M., Nevins J. R., Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439, 353–357 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Boyer L. A., Lee T. I., Cole M. F., Johnstone S. E., Levine S. S., Zucker J. P., Guenther M. G., Kumar R. M., Murray H. L., Jenner R. G., Gifford D. K., Melton D. A., Jaenisch R., Young R. A., Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabe3445/DC1