

CXCL1/2, elevated in GBM to promote tumor progression, hold potential as clinical targets in GBM treatment.
Abstract
The invasiveness and high immune suppression of glioblastoma multiforme (GBM) produce poor survival of afflicted patients. Unfortunately, in the past decades, no therapeutic approach has remarkably improved the survival time of patients with GBM. Our analysis of the TCGA database and brain tumor tissue arrays indicated that CXCL1 and CXCL2 overexpression is closely associated with GBM’s aggressiveness. Our results showed that elevation of CXCL1 or CXCL2 facilitated myeloid cell migration and simultaneously disrupted CD8+ T cell accumulation at tumor sites, causing accelerated tumor progression. Yet, blockade of CXCL1/2 significantly prevented myeloid-derived suppressor cell migration and thereby increased CD8+ T cell accumulation in vitro and in vivo. CXCL1/2 also promoted the paracrine factor S100A9 and further activated Erk1/2 and p70S60k, whereas blocking CXCL1/2 down-regulated these prosurvival factors. The combination of targeting CXCL1/2 and standard temozolomide chemotherapy improved upon the antitumor efficacy of chemotherapy alone, extending the overall survival time in GBM.
INTRODUCTION
Glioblastoma multiforme (GBM) is the most common and fatal brain malignancy, with a poor 5-year survival rate of less than 5% (1). The life expectancy of patients with GBM is very limited, with a median survival time of approximately 15 months (2). In the past decades, no new therapeutic strategies have demonstrated a significant prolongation of the survival of patients with GBM beyond that afforded by standard-of-care chemotherapy and radiotherapy.
GBM is known for its highly suppressive tumor immunity, which is a critical hurdle for immunotherapy. A number of mechanisms modulate the molecular pathways and cellular functions of tumor immunity and account for the tumor-associated immunosuppression produced by GBM. GBM tumor cells produce potent immune-suppressive molecules, including transforming growth factor–β, interleukin-10, and Indoleamine-pyrrole 2,3-dioxygenase (IDO) (3, 4). GBM can also express immune checkpoint ligands that markedly inhibit immune effector responses (5, 6). Moreover, abundant accumulation of suppressive regulatory T cells (Tregs), M2-like tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment has been reported to correlate with poor overall survival of patients with GBM (7). Multiple clinical trials of checkpoint inhibition for GBM are ongoing with the goal of reinvigorating immunity directed against GBM (4). But despite the promising antitumor immune responses in preclinical studies (8–13), blockade of checkpoint regulators in GBM clinical trials has shown only limited improvement of therapeutic efficacy.
Emerging evidence reveals that high expression levels of chemokines (C-X-C motif) ligand 1 [CXCL1, or keratinocytes-derived chemokine (KC), growth-regulated oncogene-alpha (GROα)] and (C-X-C motif) ligand 2 (CXCL2 or macrophage inflammatory protein 2 and GROβ) in tumors are associated with advanced cancer stage, larger tumor size, tumor aggressiveness, and poor prognosis and overall survival in different types of cancer (14–16). CXCL1 and CXCL2 are typically overexpressed in tumors, signaling through G protein–coupled chemokine receptor CXCR2 (the sole receptor of CXCL1 and CXCL2) to recruit tumor-associated MDSCs. CXCL1 and CXCL2 share high homology in protein sequence and are generally overexpressed in tumor and stromal cells in a variety of cancers (17), including breast carcinoma, colorectal cancer, prostate cancer, pancreatic ductal adenocarcinoma, bladder cancer, and others (15, 18–22). CXCR2 is expressed mainly on myeloid populations (neutrophils, monocytes, and macrophages). This receptor directs the egress of myeloid-derived cells from the bone marrow and their migration to CXCL1- and CXCL2-overexpressing tumor sites, where they facilitate tumor immune evasion (23, 24) by suppressing effector T cell proliferation, activation, and motility and stimulating Treg numerical expansion (25).
In addition to their immune-suppressive role, CXCL1 and CXCL2 recruit myeloid cells to produce paracrine factors, such as S100A9, that promote cancer cell survival (18, 19). Calcium-binding small-molecule S100A9 can be secreted by abundant MDSCs and TAMs and stimulates diverse mitogen signaling pathways (26). Upon interaction with Toll-like receptor 4 (TLR4) and receptor for advanced glycation end products (RAGE) on the tumor cell surface, S100A9 triggers the activation of prosurvival factors, including extracellular signal‑regulated protein kinase 1/2 (Erk1/2), nuclear factor κB (NFκB), and p70S6K. Hiratsuka et al. (27) indicated that S100A9 facilitates the migration of myeloid suppressor cells to form premetastatic niches. In a recent study, Alafate et al. (28) found that silencing CXCL1 inhibited human glioma xenograft progression by down-regulating NFκB and mesenchymal transition. Moreover, the CXCL1/2-S100A9 axis has been shown to play an essential role in chemoresistance and metastasis in breast cancer and ovarian cancer (18, 29). However, the role of the CXCL1/2-S100A9 paracrine network in promoting tumorigenesis and poor prognosis in GBM remains undefined.
In this study, our hypothesis was that the CXCL1/2-S1009 axis promoted GBM progression, serving as a treatment target along with the standard-of-care chemotherapy for GBM. Analysis of human GBMs in The Cancer Genome Atlas (TCGA) database showed that the expression levels of CXCL1 and CXCL2 were positively correlated with glioma grade and survival time. Our experiments showed that overexpression of CXCL1/2 attracted abundant MDSCs and TAMs but inhibited effector immune infiltrates. Moreover, exogenous CXCL1/2 elevated S100A9-mediated activation of prosurvival signaling in GBM tumors. By contrast, blockade of CXCL1/2 limited the migration of immune suppressor cells, allowing the accumulation of effector T cells in tumors, suppressing S100A9-induced cancer cell survival pathways, and prolonging survival in GBM-bearing mice. Together, our results suggest that CXCL1 and CXCL2 are previously unknown targets for improving the efficacy of treatment in GBM.
RESULTS
High levels of CXCL1/2 expression are associated with high-grade and recurrent GBM with poor survival.
To define the clinical importance of CXCL1 and CXCL2 in brain tumors, we analyzed the mRNA expression levels of CXCL1 and CXCL2 in tumors from patients with low-grade glioma or high-grade GBM obtained from the RNA sequencing (RNA-seq) results in the TCGA database. Both CXCL1 and CXCL2 expression levels were significantly higher in GBMs than in low-grade gliomas (CXCL1 means ± SD: 116.7 ± 17.69 versus 20.32 ± 5.024, P = 0.0002; CXCL2 means ± SD: 677.8 ± 128.5 versus 96.02 ± 12.03, P < 0.0001) (Fig. 1A). Moreover, CXCL1 and CXCL2 expression levels were significantly higher in recurrent GBM tumors than primary GBM tumors (P = 0.0445 and 0.036, respectively) (Fig. 1B). Given that high levels of CXCL1/2 often correlate with poor prognosis in patients with cancer, we determined whether CXCL1 and CXCL2 expression had an impact on the disease-free survival (DFS) of these patients with GBM by ranking the patients by the CXCL1 and CXCL2 mRNA expression levels and comparing their DFS in the TCGA dataset. The patients with expression levels of both CXCL1/2 above the median levels were classified in the CXCL1/2-high group, and the patients with expression levels of both CXCL1/2 below the median levels were classified in the CXCL1/2-low group. Log rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests both showed that patients in the CXCL1/2-high group had significantly shorter DFS times (P = 0.0072 and 0.0268, respectively) than patients in the CXCL1/2-low group (Fig. 1C). The median DFS times in the CXCL1/2-low and CXCL1/2-high groups were 8.647 and 5.753 months, respectively. These results, summarized from a clinical database, suggested that CXCL1 and CXCL2 are substantially induced in high-grade and recurrent GBM, leading to the poor DFS observed in patients with GBM.
Fig. 1. Expression of CXCL1 and CXCL2 genes is associated with prognosis in GBM progression in patients and mouse models.

(A) Data on gene expression (mRNA) of CXCL1 (left) and CXCL2 (right) in patients with low-grade glioma or GBM were obtained from the TCGA glioma database. (B) Waterfall graph shows gene expression (mRNA) of CXCL1 and CXCL2 in patients with primary or recurrent GBM (www.oncomine.org). (C) The DFS durations of patients with GBM were stratified by CXCL1 and CXCL2 gene expression levels downloaded and retrieved from the TCGA data portal (www.cbioportal.org/public-portal/). Kaplan-Meier analysis for DFS of the patients with GBM is shown. The patients with both CXCL1 and CXCL2 expression levels above median levels were classified into the CXCL1/2hi group, and those with both expression levels below median levels were classified into the CXCL1/2low group. The log rank test was used to compare overall survival among groups. (D) Tissue arrays of human brain tissues and brain tumor tissues of different stages were purchased from US Biomax and were subjected to immunohistochemical staining of CXCL1 and CXCL2. The dot graph represents the intensity (means ± SEM) of CXCL1 or CXCL2 staining. Scale bars, 100 μm. NS, not significant. (E) The correlation of CXCL1 and CXCL2 expression (mRNA) and MDSC signature gene index (HLA−DR−CD33+) in patients with GBM, obtained from the TCGA database, was determined by the Pearson product test. (F) The experimental schema used for measuring the effects of CXCL1 and CXCL2 overexpression in GBM orthotopic (n = 4) tumor models. (G) Representative bioluminescence images (BLI) and tumor growth curves of FFluc-DBT transfected with control vector, CXCL1, CXCL2, or CXCL1 + CXCL2 in the GBM orthotopic model. (H) DBT tumor–bearing brains from (F) were harvested and subjected to lysis; CXCL1 and CXCL2 levels were determined by ELISA. Bar graphs show the concentrations of CXCL1 and CXCL2 (means ± SEM). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
CXCL1/2 promote GBM tumor progression
Because the mRNA transcription levels of CXCL1 and CXCL2 are higher in GBM than in low-grade gliomas, we assessed whether the production of these chemokines correlates with tumor grade. We stained normal cerebrum tissue and different grades of astrocytomas, including high-grade GBM, with the antihuman CXCL1 and CXCL2 antibodies. Immunohistochemistry staining demonstrated no expression of CXCL1 and CXCL2 in normal cerebrum and grade 1 astrocytomas, increased staining intensity in grade 2 and 3 astrocytomas, and the highest staining intensity in high-grade GBM tissues (grade 4) (Fig. 1D). To determine how these chemokines regulate GBM tumor immunity, we also analyzed the RNA-seq dataset from the human GBM TCGA provisional database and found that the expression levels of human CXCL1 and CXCL2 were highly associated with the MDSC index (HLA−DR+CD33+) in GBM (Fig. 1E) To clarify the role of CXCL1 and CXCL2 in glioma growth in vivo, DBT cells that stably overexpress CXCL1, CXCL2, or CXCL1 plus CXCL2 were labeled with FFluc and used to establish orthotopic GBM tumors in mouse brains (Fig. 1F). The tumor development was markedly faster in CXCL1 and CXCL2 double-overexpressing DBT tumors (Fig. 1G). CXCL1 and CXCL2 levels in brains were verified via enzyme-linked immunosorbent assay (ELISA) (Fig. 1H). GL261 and DBT tumors were also established subcutaneously. Systemic hydrodynamic delivery of control, CXCL1, CXCL2, or CXCL1 plus CXCL2 DNA was initiated 1 week before tumor inoculation and repeated weekly three times (fig. S1A). Tumor growth in both models was promoted by CXCL1 plus CXCL2 codelivery (fig. S1, B and C). CXCL1 and CXCL2 levels in the GL261 and DBT tumors after cognate gene delivery were validated via ELISA (fig. S1, D and E). These data suggested that up-regulation of CXCL1 and CXCL2 is associated with the rapid progression of advanced-stage GBMs.
Overexpression of CXCL1 and CXCL2 recruits MDSCs to tumors, thereby suppressing CD8+ T cell accumulation
CXCL1 and CXCL2 are potent chemoattractants for MDSCs through the CXCL1/2-CXCR2 axis, which is required for homing of MDSCs to tumors from the general circulation. Our TCGA database analysis also showed that the MDSC profile was associated with CXCL1/2 expression in GBM tumors (Fig. 1E). MDSCs are composed of a variety of immature myeloid cells and have two subtypes, polymorphonuclear MDSCs (PMN-MDSC: CD11b+Ly6G+Ly6Clo) and monocytic MDSCs (M-MDSC: CD11b+Ly6G−Ly6Chi). In the context of mouse GBM tumors, CXCR2 is located mainly on MDSCs and is found at lower levels on M2 macrophages (CD11b+F4/80+CD206+) (Fig. 2A). We therefore postulated that CXCL1/2 account for the suppressive immunity of GBM tumors by modulating the mobilization of myeloid cells and inflammatory infiltrates.
Fig. 2. CXCL1 and CXCL2 recruit MDSCs and, in turn, reduce CD8+ T cells in GBM tumors.

(A) DBT tumor–bearing brains generated as shown in Fig. 1F (n = 4) were dissociated and stained with M2 macrophages or MDSC markers as well as CXCR2, the receptor of CXCL1 and CXCL2. Flow cytometry was performed to assess CXCR2 expression on the macrophage population (left; CD11b+F4/80+CD206+) and the MDSC population (right; CD11b+Ly-6G+ and CD11b+Ly-6C+). (B to E) DBT orthotopic tumors were established as described in Fig. 1F. Tumor-bearing mice were euthanized 10 days after inoculation. M2 macrophage (B), M-MDSC (C), PMN-MDSC (D), and CD8+ T cell (E) accumulation was assessed in dissociated tumor cells via flow cytometry. Bar graphs represent the frequency (means ± SEM) of the indicated cell populations in brains. The results represent three repeated experiments. FITC, fluorescein isothiocyanate. *P < 0.05; **P < 0.01; ****P < 0.0001.
To test this hypothesis, a Boyden chamber transwell system was established to simulate the physiological conditions of effector immune cell migration into tumor sites. GBM tumor cells were seeded in the lower chambers, while sham control, carboxyfluorescein diacetate succinimidyl ester–labeled CD8+ T cells, or CD8+ T cells plus bone marrow–derived myeloid cells were placed in the upper chambers in the presence or absence of recombinant CXCL1 and CXCL2 for 24 hours. Migration of CD8+ T cells to the tumor cells was disrupted by myeloid cells and further inhibited by the CXCL1 and CXCL2 recombinant proteins (fig. S2, A and B), which, in contrast, enhanced the migration of myeloid cells to tumor cells (fig. S2, C and D). In agreement with this in vitro observation, overexpression of CXCL1 and CXCL2 by DBT tumor cells synergistically increased the density of M2 macrophages and also boosted PMN-MDSCs and M-MDSCs in orthotopic tumors (Fig. 2, B to E). Such recruitment of suppressive immune cells into DBT tumors resulted in marked reduction of effector CD8+ T cells (Fig. 2E). In the GL261 and DBT subcutaneous tumor models described in fig. S1A, the impact of overexpressed CXCL1 and CXCL2 in GBM tumor immunity suppression was consistent with our findings in orthotopic tumors, showing synergistic increase of MDSCs and M2 macrophages and diminished effector T cells (fig. S3). These data suggested that abundant CXCL1 and CXCL2 may protect GBM tumor cells against tumor immune surveillance.
Blocking CXCL1/2 in orthotopic GBM models can overcome immune suppression
Our results showing that CXCL1/2 play a critical role in protumorigenic tumor immune suppression in GBM cells suggested that inhibiting CXCL1/2 would overcome immune suppression in GBM tumors. In the Boyden chamber in vitro system, blocking CXCL1/2 by using antibodies significantly prevented migration of bone marrow–derived myeloid cells and, at the same time, facilitated CD8+ T cell migration to GBM cells (Fig. 3, A and B, and fig. S2). To evaluate the antitumor efficacy of targeting CXCL1 and CXCL2 in vivo, Balb/c mice were implanted with FFluc-DBT cells to establish orthotopic GBM tumors and treated with control immunoglobulin G (IgG), anti-CXCL1, anti-CXCL2, or anti-CXCL1 plus anti-CXCL2 antibodies twice weekly (Fig. 3C). The bioluminescence intensity of GBM tumors showed that anti-CXCL1 or anti-CXCL2 alone may delay DBT tumor progression compared with the control IgG. While the combination therapy totally eradicated the aggressive brain tumors in two of four mice as early as day 14, other groups of mice had to be euthanized due to advanced tumor burden or neurologic issues (Fig. 3, D and E). The survival time of DBT tumor–bearing mice was markedly prolonged by anti-CXCL1 plus anti-CXCL2 treatment (Fig. 3F), and no adverse effects were observed in these mice, compared to the naïve tumor–bearing littermates (fig. S4, A and B). In vivo analysis using three independent intracranial GBM tumor models, GL261, DBT, and CT2A, found that blocking CXCL1/2 remarkably reduced the density of PMN-MDSCs and M-MDSCs in tumor-bearing brains and tended to suppress an immune-suppressive tumor-supportive phenotype (i.e., M2 skewing as detected by CD206 expression) among the TAMs (Fig. 3, G to I, and fig. S5, A, B, and D). The decrease of myeloid cells upon CXCL1/2 blockade was consistent with the earlier finding that CXCR2, the receptor of CXCL1/2, is expressed mainly on myeloid cells in GBM (Fig. 2A).
Fig. 3. Blockade of CXCL2 and CXCL2 stimulates GBM tumor immunity.

(A and B) A transwell assay was used to determine the extent of migration of CD8+ T cells (A) and MDSCs (B) to tumor cells in the presence or absence of anti-CXCL1 (5 μg/ml) and anti-CXCL2 (5 μg/ml) blocking antibodies. Bar graphs represent the frequency (means ± SEM) of CD8+ T cells (A) or MDSCs (B) in tumors. rCXCL1/2, recombinant CXCL1(2 μg/ml) and CXCL2 proteins (2 μg/ml). (C) This experimental schema was used to test the effects of CXCL1 and CXCL2 blockade in GBM intracranial tumor models. i.p., intraperitoneal; i.c., intracranial. (D) Representative bioluminescence images of FFluc-DBT tumor growth in the DBT orthotopic model. (E) Bioluminescence kinetics of FFluc-DBT tumor growth (three to four mice per group) in the DBT orthotopic models. (F) Survival curves were generated by the Kaplan-Meier method for DBT tumor–bearing mice (three to four mice per group) treated with control IgG or anti-CXCL1/2 antibodies. (G to I and K) DBT tumor–bearing mice were subjected to treatments described in (C). Tumor-bearing mice were euthanized 14 days after inoculation. M2 macrophages (G), MDSCs (H and I), and CD8+ T cell populations (K) in brains were assessed using flow cytometry. Bar graphs represent the frequency (means ± SEM) of the indicated immune cell populations in brains. The results represent three repeated experiments. (J) DBT orthotopic tumor-bearing mice were treated as described in (C). Tumor-bearing mice were euthanized 14 days after inoculation. Tregs were identified using anti-FOXP3 immunohistochemical staining. Scale bar, 50 μm. Bar graphs represent numbers of FOXP3+ cells per high-power field (HPF; means ± SEM). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Given that MDSCs may facilitate the expansion and activation of Tregs (25), we quantified Tregs in sections of brain tissue bearing DBT, GL261, or CT2A GBM via immunohistochemistry and found that blocking CXCL1/2 substantially reduced the density of Tregs in these tumors (Fig. 3J and fig. S6). Because MDSCs, M2-skewed TAMs, and Tregs are major inducers of tumor immune suppression, hampering T cell recruitment and activation in tumors, we evaluated the effect of chemokine blockade on the density of effector CD8+ T cells in brain tissues bearing GBM tumors. Notably, inhibition of CXCL1 and CXCL2 synergistically boosted the accumulation of CD8+ T cells in these GBM tumor models, as determined via flow cytometry analysis (Fig. 3K and fig. S5, C and D). Together, these results suggested that CXCL1/2 blockade significantly diminished the recruitment of MDSCs, TAMs, and Tregs and, in turn, promoted CD8+ T cell accumulation in GBM tumors.
CXCL1/2 promote GBM cell survival through the myeloid cell–derived S100A9 cascade
Tumor immune-suppressing myeloid cells recruited by redundant CXCL1/2 in GBM may also produce paracrine factors to enhance cancer cell survival (18). Coexpression analysis of TCGA GBM RNA-seq datasets suggested that CXCL1/2 expression is strongly associated with expression of S100A9 (Pearson correlation coefficients r = 0.55 and 0.38, respectively) (Fig. 4A), which encodes a small molecule that triggers RAGE and TLR4 to activate the downstream prosurvival signaling pathways [such as Mitogen-Activated Protein Kinase (MAPK) and NFκB] in cancer cells (18, 26, 30, 31). We validated that overexpression of CXCL1 and CXCL2 elevated the production of S100A9 in DBT orthotopic tumors as well as GL261 and DBT subcutaneous tumors, as determined by immunohistochemistry and immunoblots. Further, as reported in other cancers, the CXCL1/2-S100A9 loop triggered the phosphorylation of NFκBp65, Erk1/2, and p70S6K, suggesting that overexpressed CXCL1/2 stimulated S100A9-mediated prosurvival effects through activation of protein translation and NFκB and MAPK cascades (Fig. 4, B to D, and figs. S7 and S8).
Fig. 4. CXCL1 and CXCL2 regulate tumor cell survival through S100A9 signaling.

(A) Correlation of CXCL1 and CXCL2 expression (mRNA) and S100A9 in GBM data obtained from the TCGA. (B-D) Orthotopic DBT tumor–bearing mice (n = 4) described in Fig. 1F were euthanized 10 days after inoculation. DBT tumor sections were subjected to anti–phosphor-Erk (B), anti-S100A9 (C), or anti–phopshor-p60S6 kinase (D) antibody staining. Bar graphs represent the numbers of indicated marker-expressing cells per high-power field (means ± SEM of five fields per slide and three slides per sample) in tumor sections. The results represent three repeated experiments. Scale bars, 100 μm in 20× images (top rows) and 50 μm in 40× images (bottom rows). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To address whether inhibition of the CXCL1/2-S100A9 survival loop may disrupt prosurvival signaling in cancer cells, DBT, GL261, and CT2A orthotopic tumor-bearing mice were treated with control IgG, αCXCL1 antibody, αCXCL2 antibody, or a combination of αCXCL1 plus αCXCL2 antibodies. Immunohistochemical staining results showed that S100A9, phosphor-p70S6K, and phosphor-Erk were reduced by treatment with αCXCL1 plus αCXCL2 (Fig. 5 and fig. S9). These results highlight the potential of targeting CXCL1 and CXCL2 to rescue stimulatory tumor immunity and attenuate prosurvival signaling in GBM tumors.
Fig. 5. Blockade of CXCL1 and CXCL2 inhibits GBM tumor cell survival signaling in orthotopic models.
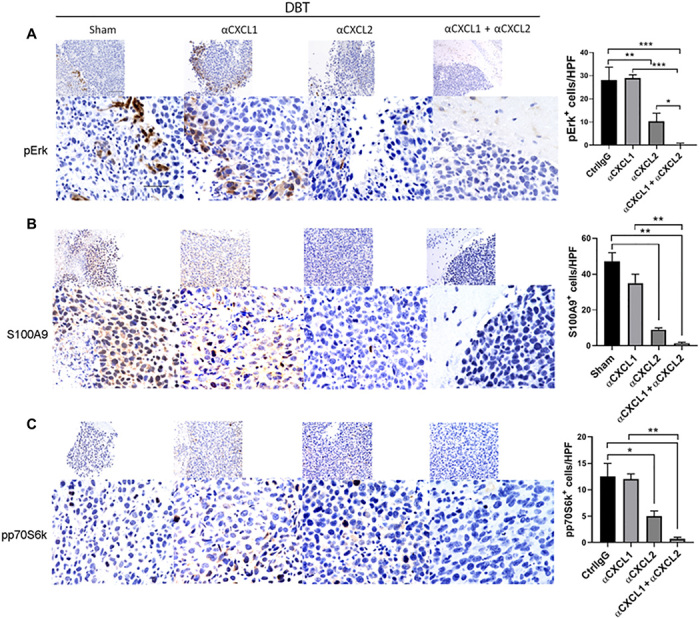
(A-C )DBT tumor–bearing mice (n = 3 to 4) were euthanized 14 days after inoculation. DBT tumor–bearing brain sections from mice treated with control IgG (CtrlIgG), anti-CXCL1, anti-CXCL2, or anti-CXCL1 plus anti-CXCL2 antibodies were subjected to anti–phosphor-Erk (A), anti-S100A9 (B), or anti–phosphor-p60S6 kinase (C) antibody staining. Bar graphs represent the numbers of indicated marker-expressing cells per high-power field (means ± SEM of five fields per slide and three slides per sample) in tumor sections. The results represent three repeated experiments. Scale bars, 100 μm in 20× images (top rows) and 50 μm in 40× images (bottom rows). *P < 0.05; **P < 0.01; ***P < 0.001.
Blockade of CXCL1 and CXCL2 endowed a survival advantage in orthotopic GBM models
To test this, we postulated that CXCL1 and CXCL2 blockade could inhibit GBM tumor progression and thereby extend overall survival. Notably, αCXCL1 plus αCXCL2 significantly prolonged the survival time of mice bearing CT2A or GL261 orthotopic tumors and eradicated the GL261 tumor in one of six mice (Fig. 6, A and B).
Fig. 6. Targeting CXCL1 and CXCL2 can improve the therapeutic efficacy of chemotherapy in orthotopic GBM.

(A and B) CT2A (A) and GL261 (B) orthotopic GBM tumor-bearing mice were subjected to treatments as described in Fig. 3C. Survival curves were generated by the Kaplan-Meier method for tumor-bearing mice treated with control IgG or anti-CXCL1/2 antibodies. (C) This experimental schema was used to determine the effects of treatment with temozolomide (TMZ) with or without CXCL1 and CXCL2 blockade in the DBT orthotopic GBM model. (D) Survival curves were generated for DBT tumor–bearing mice subjected to treatments as described in (C). *P < 0.05; **P < 0.01.
During standard cancer treatment, a number of chemotherapeutic agents may stimulate tumor cells to release CXCL1/2, further triggering the CXCL1/2-S100A9 axis and compromising antitumor efficacy (32, 33). In agreement with these reports, we observed that temozolomide, the standard chemotherapeutic for patients with GBM, up-regulated CXCL1 and CXCL2 in orthotopic GBM tumors determined by ELISA (fig. S10). We therefore hypothesized that combined CXCL1/2 blockade would sensitize GBMs to temozolomide. In the DBT orthotopic GBM model, blockade of CXCL1/2 plus temozolomide administration showed a significant survival advantage over temozolomide administration alone (Fig. 6, C and D). These promising preclinical results raised the potential of targeting CXCL1/2 along with providing the standard-of-care chemotherapy to improve the therapeutic efficacy of chemotherapy in GBM.
DISCUSSION
A number of immune-suppressive mechanisms regulate immune escape of GBM, protecting tumorigenesis. One possible mechanism is infiltration of substantial numbers of MDSCs, M2 macrophages, and Tregs promoted by soluble factors secreted from GBM tumors, a widely observed phenomenon (34–36). These suppressive immune populations constitute a large portion of the glioma tumor mass (37–39), impeding the success of immune therapies. The importance of the tumor immune environment and the promise of targeting suppressive immunity in GBM warrant identification of its key regulators. To address this challenge, we analyzed the expression of CXCL1 and CXCL2 in the GBM TCGA database, and our findings implicate overexpression of these genes in GBM aggressiveness.
Elevation of CXCL1/2 in tumors has been shown to play a fundamental role in tumor malignancy and metastasis in many cancers (40, 41). Chemokine receptor CXCR2, which exclusively binds CXCL1/2, is mainly expressed on myeloid-derived neutrophils and macrophages to recruit MDSCs and TAMs to tumors. There is ample evidence that MDSCs and TAMs in tumors suppress the expansion and effector functions of T cells by inducing checkpoint regulator and exhaustion markers, thereby protecting tumor cell survival and metastasis (42, 43). MDSC and TAM infiltration may, in turn, recruit Tregs to contribute to the highly suppressive tumor immunity exhibited by these tumors (25). In the context of GBM, overexpression of CXCL1 and CXCL2 synergistically increased the infiltration of MDSCs in orthotopic DBT and subcutaneous GL261 and DBT tumor models. By contrast, and consistent with observations in human GBM patients, mouse orthotopic GBM tumors naturally expressed high levels of CXCL1 and CXCL2, and blocking CXCL1/2 diminished the migration of MDSCs, TAMs, and Tregs into tumors. This great alleviation of suppressive immunity in GBM tumors by blockade of CXCL1/2 led to high accumulation of CD8+ T cells and thereby reinvigorated the antitumor immune response against GBM.
Immunity against tumors is suppressed not only by myeloid cell migration into tumors but also by small soluble factor S100A9, which is produced by myeloid cells at markedly elevated levels in GBM. S100A9 not only serves as an inflammatory hallmark (44, 45) but also, more importantly, is up-regulated in a number of cancers to promote cancer progression (30, 46). Ichikawa et al. demonstrated that S100A9 enhanced MDSC migration to tumors by inducing the autocrine feedback loop (26, 47). Studies from other groups indicated that S100A9 activated the TLR4- and RAGE-mediated signaling pathways to promote tumor growth and premetastatic niches (48–50). Acharyya et al. (18) showed that, in mice engrafted with S100A9−/− bone marrow, breast cancer growth and metastasis occurrence were significantly reduced compared with that in littermates transplanted with S100A9+/+ bone marrow; furthermore, inhibition of Erk, p70S6K, or p38 completely or partially abrogated S100A9-mediated cancer cell survival. Upon stimulation with S100A9, colon tumor cells secreted higher levels of CXCL1 as positive feedback (26). Our results argue that both orthotopic GBM tumors that were stably transfected with CXCL1 and CXCL2 and subcutaneous GBM tumors that were loaded with CXCL1 and CXCL2 via multiple hydrodynamic deliveries attract large numbers of MDSCs and TAMs into tumors, which, in turn, produces the soluble molecule S100A9 in the tumor environment. In response to S100A9 stimulation, prosurvival signaling pathways, including NFκB, Erk1/2, and p70S6K, are activated to promote tumor growth. Orthotopic GBM tumors naturally produce high levels of CXCL1 and CXCL2, which recruit myeloid cells and create a suppressive tumor immunity to protect tumor cells from being attacked by immune surveillance. Blockade of CXCL1/2 in GBM limited the source of S100A9, resulting in down-regulation of cancer cell survival signaling, further confirming that CXCL1/2 are not only targets for rescuing immune suppression in GBM but also function as drivers of cancer cell survival through S100A9-mediated signaling.
The highly proliferative character and acquired resistance to chemotherapy of GBM often leads to early tumor recurrence after surgery. In particular, stem-like cells within GBMs are highly resistant to chemotherapy and radiotherapy, which underlies the poor prognosis of patients with this tumor. A combination of intrinsic tumor cell– and extrinsic tumor environment–mediated pathways are involved in the development of drug resistance. Many lines of evidence show that chemotherapy leads to cytokine and chemokine storm in tumors (32, 33). Our results support this discovery by showing that treatment with temozolomide substantially induced CXCL1 and CXCL2 expression in the GBM brain environment, and blockade of CXCL1/2 greatly extended the overall survival time of temozolomide-treated mice in our preclinical orthotropic GBM model. These results suggest that CXCL1/2 induction may regulate chemoresistance during GBM treatment, which requires further in-depth investigation in the future.
In conclusion, our study reveals overexpression of CXCL1 and CXCL2 in GBM tumors and provides insights into these chemokines’ roles in increasing suppressive immune cell chemotaxis and activating the S100A9-mediated cancer cell survival axis. These chemokines, along with inflammatory modulator S100A9, facilitated the escape of GBM tumor cells from antitumor immune responses and, moreover, led to a survival advantage for these cancer cells that promoted rapid tumor progression. Our discovery suggests the potential for clinical targeting of these chemokines to limit tumor development and improve the therapeutic efficacy of chemotherapy in GBM.
MATERIALS AND METHODS
cBioPortal for cancer genomics
CXCL1/2 gene expression data and DFS data were obtained from TCGA portal (www.cbioportal.org/public-portal/). Since the z-scores of gene expression show a normalized mRNA expression relative to non-neoplastic samples (51), the patients with both CXCL1 and CXCL2 z-scores above the median levels were classified as CXCL1highCXCL2high group, and the patients with both values below the median levels were classified as CXCL1lowCXCL2low group (13, 52). The comparison of CXCL2 expression between primary and recurrent GBM tumors was obtained from the Oncomine database (www.oncomine.org/). To identify relationships between these variables, Pearson correlation coefficients were calculated for the chemokines CXCL1/2 and MDSC or S100A9 gene expression using the R statistical computing environment. A P value < 0.05 was considered statistically significant.
Animal studies
Six- to eight-week-old Balb/c or C57bl/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The mouse care and handling procedures were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center.
The orthotopic GBM brain tumor models were generated by mixing tumor cells (GL261, DBT, or CT2A) with an equal volume of 3% methylcellulose in phosphate-buffered saline (PBS) solution. Tumor cells in a total volume of 5 μl were injected intracerebrally into mice (5 × 104 cells per mouse for GL261 and CT2A models and 1 × 104 or 5 × 103 cells per mouse for DBT tumor model) as described in a previous publication (53). The subcutaneous GBM models (GL261, DBT, or CT2A) were generated by inoculating tumor cells suspended in 30 μl of PBS into the mouse flank (2 × 105 cells per mouse).
CXCL1- and/or CXCL2-encoding and control plasmid DNA (5 μg per mouse in 1.5 ml of 0.45% saline solution) were injected hydrodynamically through the tail vein of mice. Mice received the treatment indicated once per week starting 1 week before tumor cell inoculation.
Mice were treated with either 250 μg of isotype control antibody or rabbit antihuman/mouse CXCL1 and/or CXCL2 antibody (Alpha Diagnostic International, San Antonio, TX) intraperitoneally, twice per week, starting 3 days after tumor inoculation. Rabbit anti-CXCL1 antibody was generated using a 15–amino acid peptide corresponding to residues 71 to 85 of mouse CXCL1 for immunization, and rabbit anti-CXCL2 antibody was generated using an 18–amino acid peptide corresponding to residues 70 to 88 of mouse CXCL2 for immunization; both antibodies were purified on a protein G column (see Table 1 for a complete list of antibodies).
Table 1. Antibody information.
HRP, horseradish peroxidase.
| Antibody | Source | Cat. no. |
| h/m CD11B-BV605 | BioLegend | 101237 |
| Mouse Ly-6g/Ly-6C-PE | BioLegend | 108408 |
| Mouse CXCR2-FITC | BioLegend | 149309 |
| Mouse CD8a-PE | Tonbo Biosciences | 50-0081 |
| h/m CD11B-FITC | BioLegend | 101206 |
| Mouse CD8a-PE/CY7 | BioLegend | 100721 |
| Mouse F4/80-EF450 | eBioscience | 48-4801 |
| Mouse CD206-PE/CY7 | BioLegend | 141720 |
| Mouse CD45-EF450 | Tonbo Biosciences | 75-0451 |
| Mouse S100A9 | Bioss | bs-2697r |
| h/m pErk | Cell Signaling Technology | 4370 |
| h/m pp70S6 kinase | Cell Signaling Technology | 9028 |
| Mouse Foxp3 | eBioscience | 14-4771 |
| h/m NFκB p65 | Cell Signaling Technology | 8242 |
| h/m NFκB p65-phosphorylated | Cell Signaling Technology | 3033 |
| h/m p70S6 kinase | Cell signaling Technology | 9202 |
| h/m Erk | Santa Cruz Biotechnology |
sc-93 |
| h/m β-actin | Thermo Fisher Scientific | PA1-183 |
| Goat anti-mouse IgG AF405 | Thermo Fisher Scientific | A31553 |
| Goat anti-rat IgG AF488 | Thermo Fisher Scientific | A11006 |
| Goat anti-rabbit IgG AF488 | Thermo Fisher Scientific | A11008 |
| Goat anti-mouse IgG AF546 | Thermo Fisher Scientific | A10036 |
| HRP anti-rabbit IgG | Thermo Fisher Scientific | PI314600 |
| Biotin anti-rabbit IgG | Thermo Fisher Scientific | SA5-10230 |
| Rabbit anti-mouse CXCL1 (neutralization) |
Alpha Diagnostic International |
Custom made from peptide: TQTEVIATLKNGREAC |
| Rabbit anti-mouse CXCL2 (neutralization) |
Alpha Diagnostic International |
Custom made from peptide: TLKGGQKVCLDPEAPLVQK |
Tumor volumes of subcutaneous tumors were calculated using the following formula: π(length g × width2)/8. The length represents the longest axis, and the width is at right angles to the length. DBT orthotopic tumor growth was monitored by bioluminescence imaging via IVIS Spectrum imaging system (PerkinElmer).
Cells
Mouse GBM cell lines GL261 and CT2A were obtained from the U.S. National Cancer Institute. The DBT cell line was provided by L. Metelitsa (Baylor College of Medicine, Houston, TX) (54). Tumor cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. All tumor cell lines were characterized by DNA fingerprinting at MD Anderson’s Characterized Cell Line Core Facility within 6 months of initiating the experiments. All cells were treated with mycoplasma removal agent (Bio-Rad, Hercules, CA) to ensure that cells were mycoplasma-negative before inoculation.
Enzyme-linked immunosorbent assay
Tumors were collected from tumor-bearing mice that underwent the indicated treatments and subjected to lysis. The levels of CXCL1 and CXCL2 in the tumor lysates were measured by using ELISA kits (R&D Systems, Minneapolis, MN).
Immunoblotting
Frozen tissue samples were smashed before being homogenized using a minibead beater with five to eight silicone beads (BioSpec Products, Bartlesville, OK) in 0.4 ml of ice-cold radioimmunoprecipitation assay lysis buffer. The homogenized tumor cells were then subjected to lysis with this buffer. The protein extracts were separated from the tissue residues by centrifugation at the maximum speed for 20 min at 4°C. Forty-microgram samples of the total protein were fractionated by 10% SDS–polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes using a Trans-Blot Turbo transfer system (Bio-Rad). The membranes were blotted with different primary and secondary antibodies to detect the proteins of interest.
Flow cytometry
Cells were sequentially incubated with primary and secondary antibodies for 30 min each at 4°C. Stained cells were analyzed using an Attune acoustic focusing cytometer (Applied Biosystems, Foster City, CA) or BD LSR Fortessa (BD Biosciences, San Jose, CA). Flow cytometry data were analyzed using the FlowJo software program (BD Biosciences).
Immunofluorescence and immunohistochemistry
Frozen tumor sections were sequentially fixed with cold acetone, acetone plus chloroform (1:1), and acetone. Paraffin-embedded sections were deparaffinized and heated in antigen retrieval buffer. Tissue sections were blocked with 3% hydrogen peroxide in distilled water for 20 min and then in blocking buffer (5% normal horse serum and 1% normal goat serum in PBS). Slides were incubated with primary antibody overnight at 4°C and secondary antibody for 1 hour at room temperature. For immunohistochemical staining, the secondary antibody was biotin-conjugated, the sections were treated with avidin–biotin complex (ABC) reagent, and the nuclei were counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO). Tumor sections were mounted with Cytoseal mounting medium (Life Technologies, Carlsbad, CA). Immunohistochemical staining was quantified in three randomly selected low-power fields (20×) per slide. For immunofluorescence staining, tumor sections were mounted in antifade with 4′,6-diamidino-2-phenylindole fluorescence mounting medium. Slides were visualized under a Nikon Eclipse Ti fluorescence microscope.
Migration assay
The capacity of myeloid cells and T cells to migrate to tumor cells in vitro was assessed using a Boyden chamber transwell system. DBT, CT2A, or GL261 GBM cells were seeded in the lower chambers (1 × 105 cells per chamber), while sham control, CellTracker-labeled CD8+ T cells, or CD8+ T cells plus bone marrow–derived myeloid cells were placed in the upper chambers (5 × 105 cells per chamber) in the presence or absence of recombinant CXCL1 plus CXCL2 proteins or combined CXCL1/2 antibodies for 24 hours. The cells in the lower chambers were collected, and proportions of CD8+ T cells or MDSCs were identified and quantified via flow cytometry.
Statistical analysis
The directly measured outcomes were compared using a two-sided Student’s t test (two treatment groups) or one-way analysis of variance (more than two treatment groups). The statistical significance of each comparison was determined using the Prism software program (GraphPad Software, San Diego, CA). All data values represent replicates shown as median ± SEM in figure legends. Significance was defined as *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Survival curves were generated and analyzed using Kaplan-Meier estimates with log rank test.
Supplementary Material
Acknowledgments
We greatly appreciate K. Hale and A. Ninetto from MD Anderson’s Editing Services, Research Medical Library, and D. M. Wildrick from MD Anderson’s Department of Neurosurgery for editing this manuscript. Funding This research was supported by grants from the U.S. NIH (CA203493) to S.L. This study was also supported by the Cancer Center Support Grant P30 CA016672. The following MD Anderson Cancer Center Support Grant core resources were used: Genetically Engineered Mouse Facility and Veterinary Pathology Services. Author contributions: J.H. designed and conducted most of the experiments, data analysis, and interpretation and wrote and edited the manuscript. Q.Z. conducted animal studies. L.-Y.K. inoculated GBM cells to prepare orthotopic models. J.W. conducted all the statistical analyses. J.Y. helped with some animal work. X.X. prepared the DNA constructs. Z.J. performed histology sectioning and staining. A.B.H. developed the study concept, provided materials, and reviewed the manuscript. S.L. directed the experimental design, oversaw the development of the study concept, and reviewed and edited the manuscript. All authors reviewed the manuscript and approved the content. Competing interests: A.B.H. serves on the advisory board of Caris Life Sciences, WCG Oncology, receives royalties from Celldex Therapeutics and DNAtrix, and research funding from Merck. The other authors declare no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The TCGA datasets are publicly accessible at www.cbioportal.org/public-portal/. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc2511/DC1
REFERENCES AND NOTES
- 1.Carlsson S. K., Brothers S. P., Wahlestedt C., Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 6, 1359–1370 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J. B., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O.; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Tran T.-T., Uhl M., Ma J. Y., Janssen L., Sriram V., Aulwurm S., Kerr I., Lam A., Webb H. K., Kapoun A. M., Kizer D. E., McEnroe G., Hart B., Axon J., Murphy A., Chakravarty S., Dugar S., Protter A. A., Higgins L. S., Wick W., Weller M., Wong D. H., Inhibiting TGF-β signaling restores immune surveillance in the SMA-560 glioma model. Neuro Oncol. 9, 259–270 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Preusser M., Lim M., Hafler D. A., Reardon D. A., Sampson J. H., Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 11, 504–514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hodges T. R., Ott M., Xiu J., Gatalica Z., Swensen J., Zhou S., Huse J. T., de Groot J., Li S., Overwijk W. W., Spetzler D., Heimberger A. B., Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 19, 1047–1057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garber S. T., Hashimoto Y., Weathers S.-P., Xiu J., Gatalica Z., Verhaak R. G. W., Zhou S., Fuller G. N., Khasraw M., de Groot J., Reddy S. K., Spetzler D., Heimberger A. B., Immune checkpoint blockade as a potential therapeutic target: Surveying CNS malignancies. Neuro Oncol. 18, 1357–1366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yue Q., Zhang X., Ye H.-X., Wang Y., Du Z.-G., Yao Y., Mao Y., The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J. Neurooncol 116, 251–259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladomersky E., Zhai L., Lenzen A., Lauing K. L., Qian J., Scholtens D. M., Gritsina G., Sun X., Liu Y., Yu F., Gong W., Liu Y., Jiang B., Tang T., Patel R., Platanias L. C., James C. D., Stupp R., Lukas R. V., Binder D. C., Wainwright D. A., IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin. Cancer Res. 24, 2559–2573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speranza M.-C., Passaro C., Ricklefs F., Kasai K., Klein S. R., Nakashima H., Kaufmann J. K., Ahmed A.-K., Nowicki M. O., Obi P., Bronisz A., Aguilar-Cordova E., Aguilar L. K., Guzik B. W., Breakefield X., Weissleder R., Freeman G. J., Reardon D. A., Wen P. Y., Chiocca E. A., Lawler S. E., Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro Oncol. 20, 225–235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J. E., Patel M. A., Mangraviti A., Kim E. S., Theodros D., Velarde E., Liu A., Sankey E. W., Tam A., Xu H., Mathios D., Jackson C. M., Harris-Bookman S., Garzon-Muvdi T., Sheu M., Martin A. M., Tyler B. M., Tran P. T., Ye X., Olivi A., Taube J. M., Burger P. C., Drake C. G., Brem H., Pardoll D. M., Lim M., Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin. Cancer Res. 23, 124–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reardon D. A., Gokhale P. C., Klein S. R., Ligon K. L., Rodig S. J., Ramkissoon S. H., Jones K. L., Conway A. S., Liao X., Zhou J., Wen P. Y., Van Den Abbeele A. D., Hodi F. S., Qin L., Kohl N. E., Sharpe A. H., Dranoff G., Freeman G. J., Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol. Res. 4, 124–135 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Hu J., Yan J., Rao G., Latha K., Overwijk W. W., Heimberger A. B., Li S., The duality of Fgl2 - secreted immune checkpoint regulator versus membrane-associated procoagulant: Therapeutic potential and implications. Int. Rev. Immunol. 35, 325–339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J., Kong L. Y., Hu J., Gabrusiewicz K., Dibra D., Xia X., Heimberger A. B., Li S., FGL2 as a multimodality regulator of tumor-mediated immune suppression and therapeutic target in gliomas. J. Natl. Cancer Inst. 107, djv137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang D., Dubois R. N., Richmond A., The role of chemokines in intestinal inflammation and cancer. Curr. Opin. Pharmacol. 9, 688–696 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyake M., Lawton A., Goodison S., Urquidi V., Rosser C. J., Chemokine (C-X-C motif) ligand 1 (CXCL1) protein expression is increased in high-grade prostate cancer. Pathol. Res. Pract. 210, 74–78 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Murphy C., McGurk M., Pettigrew J., Santinelli A., Mazzucchelli R., Johnston P. G., Montironi R., Waugh D. J. J., Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin. Cancer Res. 11, 4117–4127 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Balkwill F., Cancer and the chemokine network. Nat. Rev. Cancer 4, 540–550 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Acharyya S., Oskarsson T., Vanharanta S., Malladi S., Kim J., Morris P. G., Manova-Todorova K., Leversha M., Hogg N., Seshan V. E., Norton L., Brogi E., Massagué J., A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 150, 165–178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., Sun H., Wei J., Cen B., DuBois R. N., CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res. 77, 3655–3665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steele C. W., Karim S. A., Leach J. D. G., Bailey P., Upstill-Goddard R., Rishi L., Foth M., Bryson S., McDaid K., Wilson Z., Eberlein C., Candido J. B., Clarke M., Nixon C., Connelly J., Jamieson N., Carter C. R., Balkwill F., Chang D. K., Evans T. R. J., Strathdee D., Biankin A. V., Nibbs R. J. B., Barry S. T., Sansom O. J., Morton J. P., CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 29, 832–845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seifert L., Werba G., Tiwari S., Giao Ly N. N., Alothman S., Alqunaibit D., Avanzi A., Barilla R., Daley D., Greco S. H., Torres-Hernandez A., Pergamo M., Ochi A., Zambirinis C. P., Pansari M., Rendon M., Tippens D., Hundeyin M., Mani V. R., Hajdu C., Engle D., Miller G., The necrosome promotes pancreatic oncogenesis via CXCL1 and mincle-induced immune suppression. Nature 532, 245–249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H., Ye Y.-L., Li M.-X., Ye S.-B., Huang W.-R., Cai T.-T., He J., Peng J.-Y., Duan T.-H., Cui J., Zhang X.-S., Zhou F.-J., Wang R.-F., Li J., CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 36, 2095–2104 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Oppenheim J. J., Zachariae C. O. C., Mukaida N., Matsushima K., Properties of the novel proinflammatory supergene “intercrine” cytokine family. Annu. Rev. Immunol. 9, 617–648 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Talmadge J. E., Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin. Cancer Res. 13, 5243–5248 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Gabrilovich D. I., Ostrand-Rosenberg S., Bronte V., Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichikawa M., Williams R., Wang L., Vogl T., Srikrishna G., S100A8/A9 activate key genes and pathways in colon tumor progression. Mol. Cancer Res. 9, 133–148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hiratsuka S., Watanabe A., Aburatani H., Maru Y., Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 8, 1369–1375 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Alafate W., Li X., Zuo J., Zhang H., Xiang J., Wu W., Xie W., Bai X., Wang M., Wang J., Elevation of CXCL1 indicates poor prognosis and radioresistance by inducing mesenchymal transition in glioblastoma. CNS Neurosci. Ther. 26, 475–485 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taki M., Abiko K., Baba T., Hamanishi J., Yamaguchi K., Murakami R., Yamanoi K., Horikawa N., Hosoe Y., Nakamura E., Sugiyama A., Mandai M., Konishi I., Matsumura N., Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat. Commun. 9, 1685 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebhardt C., Németh J., Angel P., Hess J., S100A8 and S100A9 in inflammation and cancer. Biochem. Pharmacol. 72, 1622–1631 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Vogl T., Tenbrock K., Ludwig S., Leukert N., Ehrhardt C., van Zoelen M. A. D., Nacken W., Foell D., van der Poll T., Sorg C., Roth J., Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 13, 1042–1049 (2007). [DOI] [PubMed] [Google Scholar]
- 32.DeNardo D. G., Brennan D. J., Rexhepaj E., Ruffell B., Shiao S. L., Madden S. F., Gallagher W. M., Wadhwani N., Keil S. D., Junaid S. A., Rugo H. S., Hwang E. S., Jirstrom K., West B. L., Coussens L. M., Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilbert L. A., Hemann M. T., DNA damage-mediated induction of a chemoresistant niche. Cell 143, 355–366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kikuchi K., Neuwelt E. A., Presence of immunosuppressive factors in brain-tumor cyst fluid. J. Neurosurg. 59, 790–799 (1983). [DOI] [PubMed] [Google Scholar]
- 35.Jordan J. T., Sun W., Hussain S. F., DeAngulo G., Prabhu S. S., Heimberger A. B., Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol. Immunother. 57, 123–131 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komohara Y., Ohnishi K., Kuratsu J., Takeya M., Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 216, 15–24 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Wainwright D. A., Sengupta S., Han Y., Lesniak M. S., Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 13, 1308–1323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badie B., Schartner J. M., Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 46, 957–961; discussion 961-2 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Almand B., Clark J. I., Nikitina E., van Beynen J., English N. R., Knight S. C., Carbone D. P., Gabrilovich D. I., Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 166, 678–689 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Jamieson T., Clarke M., Steele C. W., Samuel M. S., Neumann J., Jung A., Huels D., Olson M. F., Das S., Nibbs R. J. B., Sansom O. J., Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 122, 3127–3144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saintigny P., Massarelli E., Lin S., Ahn Y.-H., Chen Y., Goswami S., Erez B., O’Reilly M. S., Liu D., Lee J. J., Zhang L., Ping Y., Behrens C., Solis Soto L. M., Heymach J. V., Kim E. S., Herbst R. S., Lippman S. M., Wistuba I. I., Hong W. K., Kurie J. M., Koo J. S., CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 73, 571–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Talmadge J. E., Gabrilovich D. I., History of myeloid-derived suppressor cells. Nat. Rev. Cancer 13, 739–752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuang D.-M., Zhao Q., Peng C., Xu J., Zhang J.-P., Wu C., Zheng L., Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roth J., Vogl T., Sorg C., Sunderkötter C., Phagocyte-specific S100 proteins: A novel group of proinflammatory molecules. Trends Immunol. 24, 155–158 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Roth J., Goebeler M., Sorg C., S100A8 and S100A9 in inflammatory diseases. Lancet 357, 1041 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Salama I., Malone P. S., Mihaimeed F., Jones J. L., A review of the S100 proteins in cancer. Eur. J. Surg. Oncol. 34, 357–364 (2008). [DOI] [PubMed] [Google Scholar]
- 47.Ostrand-Rosenberg S., Cancer and complement. Nat. Biotechnol. 26, 1348–1349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghavami S., Rashedi I., Dattilo B. M., Eshraghi M., Chazin W. J., Hashemi M., Wesselborg S., Kerkhoff C., Los M., S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 83, 1484–1492 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hiratsuka S., Watanabe A., Sakurai Y., Akashi-Takamura S., Ishibashi S., Miyake K., Shibuya M., Akira S., Aburatani H., Maru Y., The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 10, 1349–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Gebhardt C., Riehl A., Durchdewald M., Németh J., Fürstenberger G., Müller-Decker K., Enk A., Arnold B., Bierhaus A., Nawroth P. P., Hess J., Angel P., RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 205, 275–285 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cancer Genome Atlas Research Network , Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan J., Zhao Q., Gabrusiewicz K., Kong L.-Y., Xia X., Wang J., Ott M., Xu J., Davis R. E., Huo L., Rao G., Sun S.-C., Watowich S. S., Heimberger A. B., Li S., FGL2 promotes tumor progression in the CNS by suppressing CD103+ dendritic cell differentiation. Nat. Commun. 10, 448 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei J., Wang F., Kong L.-Y., Xu S., Doucette T., Ferguson S. D., Yang Y., McEnery K., Jethwa K., Gjyshi O., Qiao W., Levine N. B., Lang F. F., Rao G., Fuller G. N., Calin G. A., Heimberger A. B., miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 73, 3913–3926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiong G., Husseiny M. I., Song L., Erdreich-Epstein A., Shackleford G. M., Seeger R. C., Jäckel D., Hensel M., Metelitsa L. S., Novel cancer vaccine based on genes of salmonella pathogenicity island 2. Int. J. Cancer 126, 2622–2634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc2511/DC1