

Spatially resolved metabolic profiling and isotope-tagged protein analysis revealed unique lipid distributions in tissues.
Abstract
Spatially resolved RNA and protein molecular analyses have revealed unexpected heterogeneity of cells. Metabolic analysis of individual cells complements these single-cell studies. Here, we present a three-dimensional spatially resolved metabolomic profiling framework (3D-SMF) to map out the spatial organization of metabolic fragments and protein signatures in immune cells of human tonsils. In this method, 3D metabolic profiles were acquired by time-of-flight secondary ion mass spectrometry to profile up to 189 compounds. Ion beams were used to measure sub–5-nanometer layers of tissue across 150 sections of a tonsil. To incorporate cell specificity, tonsil tissues were labeled by an isotope-tagged antibody library. To explore relations of metabolic and cellular features, we carried out data reduction, 3D spatial correlations and classifications, unsupervised K-means clustering, and network analyses. Immune cells exhibited spatially distinct lipidomic fragment distributions in lymphatic tissue. The 3D-SMF pipeline affects studying the immune cells in health and disease.
INTRODUCTION
Molecular profiles of RNAs, proteins, and metabolites in cells and tissues are informative about health and disease state at the single-cell level (1, 2). Spatially resolved analysis of immune organs is critical to map out tissue organization (3). Chemical compounds are altered in spatially distinct locations in tissues to regulate cell-cell interactions and intracellular decision-making. In particular, a human tonsil is a secondary lymphoid organ that exhibits a heterogeneous distribution of immune cells, such as T cells and B cells, among other subtypes of immune and structural cells (4). Tonsils play a large role in accepting and rejecting foreign organisms that enter the human airway (5). The tonsils lie at the beginning of the alimentary tract, where they continually monitor and capture pathogens such as viruses and bacteria to perform immune surveillance. This network of cellular interactions is guided by immune responses, secreted antibodies, and metabolic regulators in the tonsil (6). Immune tissue function and architecture in tonsils has been previously investigated by histological analysis and immunofluorescence to examine various cell types and molecular distributions. However, these techniques have been limited to a relatively small number of markers (7), leading to an incomplete map of chemical compositions in immune networks.
Tissue architecture, cellular identification, subcellular localization, and metabolic regulation in immune organs require simultaneous measurements of many chemical fingerprints. Regenerative medicine and immunoengineering applications require a deeper understanding of native tissue functionality and the role of metabolites with high spatial detail (8). To resolve these cell features, multiplexed and highly spatially localized metabolic profiles of immune cells in human tissues are crucial. Thus, technological developments are needed to deliver spatially localized metabolic profiles of individual immune cells in human tissues.
Here, we interrogate metabolites that are spatially distributed in specific anatomical regions of tonsil samples using time-of-flight secondary ion mass spectrometry (TOF-SIMS) (9). The TOF-SIMS system raster scans ion beams across the tissue sample to profile atomic masses ranging from 1 to 10,000 atomic mass units (amu). The system can detect elements down to concentrations of a few parts per million of a monolayer, making it a technique capable of dopant-level surface and depth profile characterization. It can produce chemical maps and three-dimensional (3D) chemical reconstructions from both positive and negative ions with single-nanometer depth resolution and submicrometer lateral resolution (10, 11). While implementing 3D depth profile results in the destruction of the sample, the damage is localized to a small volume of material. If no depth information is needed, then TOF-SIMS is quasi-nondestructive, meaning that damage depth is less than one monolayer.
Before destructive TOF-SIMS analysis, optical imaging (12) allows for nondestructive and highly accurate spatial location of cellular boundaries and stained cells in volume across multiple micrometers in-depth to define an anatomical region in tonsils. Once the regions of interest (ROIs) such as germinal centers (GCs) have been identified from the optical microscopy experiments, TOF-SIMS is used to examine the areas of interest. After recording the spatial coordinates of the GCs using optical inspection, metabolic profiles were recorded inside, outside, and at the border of GCs in healthy tonsil tissue.
Another important advance is to incorporate cell-specific enrichment in spatial metabolic profiling in tissues. The labeling method was adapted from cytometry by time of flight (CyTOF) (13) and imaging mass cytometry (IMC) (14) technologies that routinely use up to 35-plex isotope tags to profile and spatially localize individual cell types and structural components of cell suspensions and tissues. The toolkits (15) are readily available to conjugate these isotope tags onto the antibodies using MaxPar labeling, and the efficiency of antibody detection was validated by IMC measurements in tonsil tissues without metabolic distributions. In this platform, we deployed these isotope-tagged antibody libraries to stain the tonsil tissues, followed by TOF-SIMS measurements around GCs of the labeled tonsil section. While unlabeled tonsil sections were initially analyzed, a library of 20-plex isotope-tagged antibodies was labeled onto tonsil slices to correlate cell-specific features to metabolites of interest. This paper presents 3D spatially resolved metabolomic profiling framework (3D-SMF) as a method to profile volumetric metabolic fragment maps and cell-specific features of immune cells in spatially distinct anatomical regions of a tonsil architecture (Fig. 1).
Fig. 1. 3D-SMF.
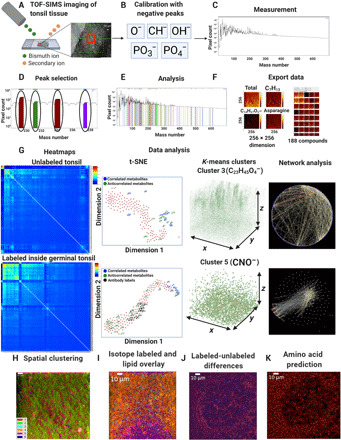
(A) The schematics of 3D metabolic profiling over 50 to 150 depth layers in a tonsil specimen is illustrated. (B) Calibration of the mass peak lists was performed using the negatively charged ions. (C) Postanalysis of the acquired TOF-SIMS data included extraction of the mass channels, their corresponding intensity counts, and duration of data collection. (D) To determine the exact value of a mass channel, we identified the peaks of the relevant mass channels during the image acquisition or after the measurements. (E) Mass channels were carefully chosen on the basis of the intensity and width of mass channels. (F) The acquired images from the mass channels were analyzed using log transforms and filtering, followed by the data export into an American Standard Code for Information Interchange (ASCII) text. (G) 3D metabolic distributions in the acquired TOF-SIMS datasets were processed by postanalysis methods. (H) The spatial clustering of all TOF-SIMS datasets into six distinct clusters was performed using six unique colors. (I) Overlaid images of isotope-labeled mass channels with those containing distinct 10 lipid fragments were shown. (J) TOF-SIMS signal difference between isotope-labeled and unlabeled dataset of the same GC was illustrated. (K) Prediction of isotope-labeled dataset amino acid mass channel was calculated from mass image differences of labeled and unlabeled channels.
First, the experimental procedures to record 3D metabolic data cubes were demonstrated and validated after calibration of abundant mass channels across serial TOF-SIMS imaging of 256 pixel by 256 pixel regions of tonsil tissue. This step was followed by digital quantification of up to 189 metabolic channels using dimensionality reduction, 3D spatial correlation, and unsupervised 3D clustering analysis. Second, isotope-tagged antibody labeling of a tonsil section was introduced in TOF-SIMS experiments to associate cell-specific features such as surface receptor distributions for immune cells and functional markers to correlate phenotypic signatures to metabolic distributions.
Last, spatially distinct regions that contain or exclude GCs were mapped in labeled tonsil sections and analyzed to determine unique lipidomic profiles that are correlated or anticorrelated across the 3D volume distributions. The 3D-SMF provides high-dimensional maps of immune cell distributions in immune organs and can be broadly applicable to other tissues from diseased samples such as tumors.
RESULTS
3D spatially resolved metabolic profiling and data analysis
In the 3D-SMF approach, formalin-fixed and paraffin-embedded (FFPE) tonsil tissue slices were subjected to repeated sputtering cycles with cesium (Cs) ion gun to remove the oxidized/intermediate layer and subsequent analysis with bismuth (Bi) ion gun producing secondary ions to measure a series of 50 to 150 depth images from the same field of view that were stacked to generate a 3D volumetric reconstruction of the chemistry of tonsil tissues (16). The overall procedure to handle the measurement datasets acquired from TOF-SIMS imaging was outlined (Fig. 1A). To extract prominent mass channels, we used peaks of lower masses from negatively charged ions (e.g., abundant phosphorus, sulfur, and oxygen ions) for calibration of the list of peaks over a large range (Fig. 1B). The measurement window was used to monitor and quantify the mass number and the corresponding intensity count (Fig. 1C). The selection of peaks could be performed either before or after the measurement process (Fig. 1D). Analysis of mass channels was carried out using the SurfaceLab (version 6) software to export data that are designed to be compatible with Python pipelines (Fig. 1, E and F). Spatially resolved clustering of the resultant mass spectra was performed to create pseudo-colored images of cell-specific features and metabolic fragments (Fig. 1, H and I). For validation, the same GC was also imaged from the serial tissue sections, wherein the difference analysis was performed between the two similar biological targets that were labeled with the isotype-antibody pairs, and another one was left unlabeled (Fig. 1J). A predictive analysis was developed to calculate contributions of antibodies used in labels to the amino acid fragments (Fig. 1K), providing a computational solution to measure multimass spectra components of labels in the 3D-SMF platform.
To identify each compound in the dataset, we used two models to assign chemical targets to the potential mass fragments. First, mass numbers that were compiled from the previous TOF-SIMS literature (9, 17–23) were assigned to the lipidomic fragment signatures in the 3D-SMF datasets (data file S1). Upon collection of mass identification information corresponding to relevant chemical compositions from the TOF-SIMS datasets, the number of occurrences for each identified mass numbers was statistically analyzed. The most repeatable and probably mass signatures were then assigned to each of the mass numbers. This is akin to the community-based platform provided by METASPACE 2020 (24) for the metabolic annotation from the matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) datasets that were created from a public repository, which inspired our mass target identification from the TOF-SIMS literature. Second, the TOF-SIMS library contains a repository of empirically identified mass spectra from previous calibration experiments using relatively pure targets (data file S2). TOF-SIMS uses the elemental and molecular isotope ratios that were previously extracted from the large library of samples to predict the compound names corresponding to mass numbers (25, 26). Here, the SurfaceLab program searches for the peak in the spectrum that is closest to the mass number in question, after which the center of the mass peak is determined for target identification. For the compounds closest in mass to the peak center, the database lists them in order of the difference of their library mass from the selected peak’s centroid. For each compound listed, a match is calculated at the positions of possible isotopic permutations. If a signal is detected at these positions, then the “explained” value for that compound is 100%. If there is no detected signal at some predicted location(s), then the explained value is correspondingly lower than 100%. These values statistically match chemical compounds such as amino acids, lipids, and other metabolic fragments. While these two methods are the major mass identification strategies, correlation of the TOF-SIMS data to other MS techniques such as MALDI (27)) and desorption electrospray ionization (28), as well as other neural network-based training methods of known samples to identify unknown targets, can, in principle, be used in a large-scale sample library (29). Target identification that is presented in this platform is still a proof-of-principle model, which can directly benefit from emerging target identification approaches in the MS field.
The volumetric postprocessing steps were performed in custom-developed Python scripts to quantify 3D metabolite distributions in the unlabeled and labeled tonsil data using the t-distributed stochastic neighbor embedding (t-SNE) maps, heatmaps, K-means clustering algorithm, and network analysis (Fig. 1G). Data reduction by the t-SNE maps suggested metabolites that have high-degree colocalization of their spatial distributions. Hierarchical clusters denoted heatmaps from 3D correlation analysis of 10 binned slices that combined the average of 10 subsequent depth images using Pearson’s coefficients that spanned a range of −1 to 1 values, providing correlated and anticorrelated metabolic mass channels across volumetric data. The effect of 5 to 20 slices binning on the heatmaps was illustrated (fig. S1). These correlation values were then visualized as a network of metabolic channels using nodes and edges, wherein edge thickness referred to a higher correlation and the dark red color corresponds to positive correlations. To classify metabolic voxels, we used an unsupervised K-means clustering method to determine the organization of metabolic differences in spatially distinct regions of tonsil samples. For instance, of the six clusters obtained in K-means analysis from an unlabeled tonsil tissue sample, cluster 3 provided mass compounds that were enriched in 385 mass/charge ratio (m/z) (C23H45O4−, Cholesterol) mass channel, and cluster 5 yielded 42.2 m/z compounds. In spatial distributions, the first compound in cluster 5 showed circular cellular features, whereas the latter one was more distributed around the peripheral regions of the cells. While these round metabolic distributions are similar to those single-cell nuclear images in the 4′,6-diamidino-2-phenylindole channel obtained from optical images, we will broadly refer to cellular features to avoid any miscalling. The resultant t-SNE maps, heatmaps with clustergrams, and K-means clusters of 3D metabolic distributions provided spatially resolved 3D chemical fingerprints that contain largely distinct lipidomic signatures in tonsils.
The 3D metabolomic fragment measurements were performed in a 5-μm-thick unlabeled tonsil slice (Fig. 2A). The peak list for 189 compounds was identified, and the measurement data were exported for postanalysis (Fig. 2B). To categorize metabolic compounds into spatially resolved groups, we computed 3D Pearson’s correlations across the volumetric 189 mass channels to define “correlated” and “anticorrelated” metabolite groups across 20 binned slices (tables S1 and S2). Correlated compounds exhibited highly colocalized spatial distributions, while the anticorrelated ones showed different spatial cellular maps. A ranking of correlation coefficients was carried out on the basis of the highest spatial similarity and dissimilarity, wherein the highest correlation ranking for 20 to 50 correlated compounds and lowest ranking for 20 to 50 anticorrelated metabolites were determined. To allow comparisons across datasets, we displayed the compounds that commonly occur in the rankings in the form of bar graphs. In the unlabeled tissue dataset, the highest correlated compounds included m/z mass channels 16, 17, 50, 60, 61, 63, 66, 74, 76, 79, 98, 109, 118, 179, 185, 201, and 190 m/z (C7H13NPO3+, lipid) and 385 m/z (C23H45O4−, cholesterol); and the lowest correlated (anticorrelated) metabolites were mass channels 50, 60, 72, 73, 74, 76, 96, 97, and 98 m/z (C5H12NO+, lipid), 179 and 190 m/z (C7H13NPO3+, lipid), and 385 m/z (C23H45O4−, cholesterol) (Fig. 2C). Notably, some metabolite identities (e.g., 50, 60 m/z, etc.) appeared in both lists due to spatial correlations with one subset of metabolites and simultaneous noncolocalized distributions for a different set of metabolic signatures. The pairwise correlations among the 189 metabolites were visualized using heatmaps.
Fig. 2. Anticorrelated and correlated spatially resolved metabolic distributions in tissues.
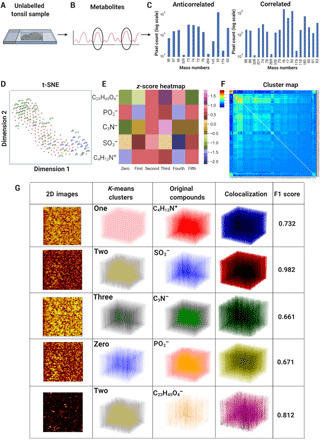
(A) A 5-μm-thick unlabeled tonsil slice was imaged by a TOF-SIMS device, providing 3D metabolite images over 138 depth slices. (B) The calibration and identification of the 189 mass channels were carried out, and the resultant measurement data were exported into an ASCII text file. (C) The bar graphs for total ion 3D signals were used for the highest 50 correlated and anticorrelated metabolite pairs. (D) The t-SNE plot for the unlabeled tonsil dataset demonstrated the highest (blue) and lowest correlated (anticorrelated) metabolites (green). (E) Six clusters were obtained from a 3D K-means algorithm to visualize selected five metabolites of 50, 63, 74, 79, and 385 m/z (C23H45O4−, cholesterol) on a heatmap of normalized z-scores. (F) Pairwise correlations of the 189 metabolites of the unlabeled tonsil tissue dataset were computed on a clustered heatmap. (G) Five metabolites of 50, 63, 74, 79, and 385 m/z (C23H45O4−, cholesterol) were visualized for six clusters. The first column contains pixel-binned 2D images, the second column shows the 3D clusters, and the third column displays the 3D voxels of the five metabolites. The clusters and the original metabolites were quantified by F1 scores (0.661 to 0.982).
The t-SNE plot validated anticorrelated and correlated compounds (Fig. 2D). The resultant t-SNE maps were generated through optimization of nonlinearity parameters such as perplexity, learning rate, and exaggeration. These data reduction maps provided a decent separation of metabolic compounds. Unlike proteomic datasets to separate cell types such as T cells and B cells, the presented t-SNE maps cover a mixture of metabolites without distinctive separation due to heterogeneous distribution of metabolic markers in any cell subset of immune cells. As a proof of concept, a selected list consisting of m/z mass channels 50, 63, 74, 79, and 385 m/z (C23H45O4−, cholesterol) that exhibit spatially distinct distributions was used in the 3D K-means algorithm to quantify and visualize volumetric metabolite differences across six K-means clusters. From 3D data cubes, z-score row-normalized values of these compounds were plotted in the heatmap, where unique clusters (1 to 6) capture the spatial structural 3D localization of each compound (Fig. 2E). For pairwise analysis, hierarchical clustering from 3D metabolites showed correlated and anticorrelated compounds in the distinct clustergram groups of a heatmap (Fig. 2F).
For the same selected five metabolic compounds, the quantification of the overlap between the calculated clusters and the original metabolomic compounds was validated using colocalization and F1 scores (30). The first column showed the 2D images from metabolic images. The second column denotes the results of one cluster of six that separates 3D spatial metabolite patterns. The third column demonstrates the original voxel values, and the fourth column shows colocalization of the clusters and voxels as visual representations of the 3D overlap, whereas the F1 score was a quantification metric that corresponds to the similarity of the clusters that are colocalized with the original metabolic voxel distributions in the specific mass channels. The F1 metric obtained ranges from 0.661 to 0.982, providing a high-degree overlap for all the five metabolic compounds (Fig. 2G). The lower F1 score between the mass channels 50 and 63 might be attributed to the similarity in their spatial distributions as the density-based K-means analysis would not be able to efficiently separate them. Alternatively, the higher F1 scores correspond to the successful classification of anticorrelated and spatially different compounds.
3D spatial metabolomic profiling with cell-specific labels
Metabolomic fragment signatures are widely distributed across many cell types. The specificity of a metabolite of interest to a well-defined cell identity is rather limited. For instance, lipidomic markers are distributed across the cell membranes and cytosol, and most immune cell types heterogeneously express these features. To introduce specificity to a metabolomic profiling pipeline, 3D-SMF used isotope-tagged antibody libraries that were implemented from IMC experiments (31, 32). These isotope-tagged markers (Table 1) corresponded to the specific cell features that included structural tissue [169 collagen I and 141 smooth muscle actin (SMA)], epithelial cells (148 PanKeratin), T cells (170 CD3, 156 CD4, 162 CD8A, 155 Foxp3, and 173 CD45RO), B cells (161 CD20), checkpoints [165 programmed cell death protein 1 (PD1) and 150 Programmed Cell Death Ligand 1 (PDL1)], nuclear enriched signatures (171 histone 3 and 191/193 intercalators), and other functional markers (143 vimentin, 158 E-cadherin, 168 Ki-67, and 167 granzyme B), and together with unlabeled or background channels (127, 131, 138, 152, 190, and 208). In 3D-SMF, a key criterion is that cells and tissues should have a minimal natural background (33) for these mass channels such that the labeling will provide the enriched contribution of labels for cell-specific ion signals but not from native metabolic distributions. The extensive CyTOF and IMC literature have validated minimal cell background in these channels, while 3D-SMF may still have a small contribution from metabolites of interest. Thus, the decoupling of relatively low metabolic signals and cell-specific ions will not be at the center of this current pipeline. 3D correlative analysis of cell-type features and metabolic compounds provides data-driven analysis of enhanced specificity in the 3D-SMF results. Notably, the sensitivity of secondary ion detection per isotope channel for IMC and 3D-SMF might also be different due to the source differences (bismuth and oxygen) and the dimensions of the imaging volume, which are intrinsically controlled by the device settings (34). However, 3D-SMF is a correlation-based and spatially resolved clustering solution to the image-based metabolic dissection of tissue architectures that should, in principle, relate cell-feature association to compounds at a high confidence level.
Table 1. Library of isotope-tagged antibodies for cell labeling in TOF-SIMS.
| Number | Mass | Label | Description |
| 1 | 141 | Pr | SMA |
| 2 | 143 | Nd | Vimentin |
| 3 | 148 | Nd | PanKeratin |
| 4 | 149 | Sm | H3K9me3 |
| 5 | 150 | Nd | PDL1 |
| 6 | 155 | Gd | Foxp3 |
| 7 | 156 | Gd | CD4 |
| 8 | 158 | Gd | E-cadherin |
| 9 | 159 | Tb | CD68 |
| 10 | 161 | Dy | CD20 |
| 11 | 162 | Dy | CD8A |
| 12 | 165 | Ho | PD1 |
| 13 | 167 | Er | Granzyme B |
| 14 | 168 | Er | Ki-67 |
| 15 | 169 | TM | Collagen I |
| 16 | 170 | Er | CD3 |
| 17 | 171 | Yb | Histone 3 |
| 18 | 173 | Yb | CD45RO |
| 19 | 191 | Ir | Intercalator (DNA) |
| 20 | 193 | Ir | Intercalator (DNA) |
Cell-specific labeling of an unlabeled tonsil was carried out using an IMC-optimized isotope-tagged antibody library (Fig. 3A). As described, tonsil tissue slices were stained with 20-plex functional and structural cellular labels to obtain specificity in the 3D-SMF platform (Fig. 3, B and C). 3D metabolic data were captured across a 150 μm by 150 μm area of the labeled tonsil tissue at 256 pixel by 256 pixel sampling over 150 slices. To demonstrate the separation of mass labels and metabolic compounds on the t-SNE map, each isotope label was displayed in black, the highest correlated compounds were shown in blue, and lowest correlated (anticorrelated) metabolites were denoted in green colors (Fig. 3D). Isotope-tagged antibody library for 20 targets and metabolic compounds for another 169 peaks were extracted from the calibrated mass spectra (Fig. 3E). These two distinct classes are correlated in 3D to associate cell features to metabolites of interest, providing cell type enrichment of a subset of compounds in tonsil tissues.
Fig. 3. Isotope-conjugated antibodies were labeled on tissue sections.
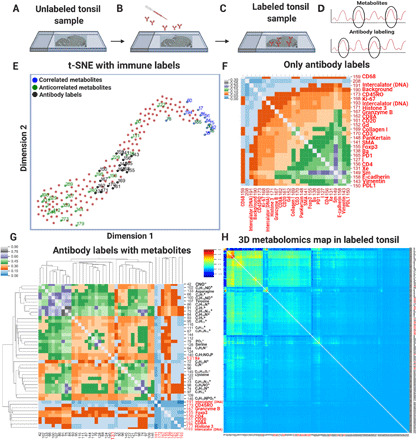
(A) Cell-specific labeling of an unlabeled tonsil-sliced sample using an isotope-tagged antibody library. (B) Specific cellular features and metabolic compounds were identified. (C) After labeling, the tonsil tissue was analyzed by TOF-SIMS imaging to acquire a 3D dataset. (D) The t-SNE plot exhibited the highest correlated metabolites (blue), anticorrelated metabolites (green), and isotope-tagged antibody labels (black) in the labeled tissue dataset. (E) The calibrated mass spectra consisted of 189 compounds that contained 20 peaks for cell features and 169 peaks for metabolites. (F) Pairwise 3D correlations for only the antibody-labeled masses of the isotope antibody–labeled tonsil dataset across 20 binned slices were shown on a hierarchically clustered heatmap. (G) A clustered heatmap was used to represent the pairwise 3D correlations between a subset of metabolic compounds and cell-specific labels. (H) The hierarchically clustered heatmap showed pairwise 3D correlations of the five binned slices of the metabolite compounds and antibody labels. The dominant red/green colors in the heatmaps included a colormap for blue (low correlations), green (medium correlations), and red (high correlations).
The 3D correlation heatmap of the inside isotope antibody–labeled tonsil dataset is displayed for metabolic compounds and isotope-tagged mass channels. To validate cell-specific features, we presented 3D correlations across 20 binned slices from only antibody channels (Fig. 3F). In this analysis, several correlations correspond to cellular features that were expected from labeling schemes. For instance, 171 histone 3 proteins were correlated with 193 intercalator (DNA) channels, providing good agreement for voxel distributions across the nuclear regions. Because of the sensitivity differences of each isotope channel, these correlations would not provide perfect correlations. A subset of the metabolic compounds was uniquely correlated with the cell-specific labels. For instance, the CD4 cells (155 and 156 masses) were associated with 42, 64, 79, 87, 95, 102, 112, 124, and 144 mass channels, whereas CD20 cells (161 mass) were linked to 66, 75, 84, 86, 91, 100, and 107 metabolites (Fig. 3G). The entire 3D correlation map across five binned slices of the ion data cube that contains antibody labels and metabolite compounds is displayed (Fig. 3H). The large heatmap showed that the immune labels correlated with a considerable number of other metabolomic compounds, which are denoted as a dominant green/red cluster in a colormap of blue (low correlations), green (medium correlations), and red (high correlations).
3D metabolomic profiling in spatially distinct tonsil anatomical regions
Tonsil tissue architecture comprises spatially unique anatomical regions. One biologically interesting region to regulate immune responses is GCs that regulate antibody production through B cell and T cell interactions (35). The communication between these cells is regulated by the lipid structures (36) that are functionally linked to the ligand and receptor pairs for enabling cell-cell recognition. To determine the relations of cell-specific features to metabolomic fragments and particularly lipidomic fragment maps, we performed multiple 3D-SMF measurements in spatially distinct regions. To identify ROIs, we used a separate optical imaging step to observe the inside and outside regions of a GC that is close to one of the tissue edges in the labeled dataset (Fig. 4A and figs. S2 and S3). To align ROIs in the TOF-SIMS, we also used an internal optical camera to exactly match the inside, outside, and at the border of the same GC while starting the mass acquisition for the 3D-SMF datasets (Fig. 4, B to D). Spatially unique lipidomic signatures (37) around the GC region will be useful to identify the key chemical fingerprints that control immune-cell functionality in tonsils.
Fig. 4. Cell type–specific and metabolic correlations for the GC in tonsil tissues.
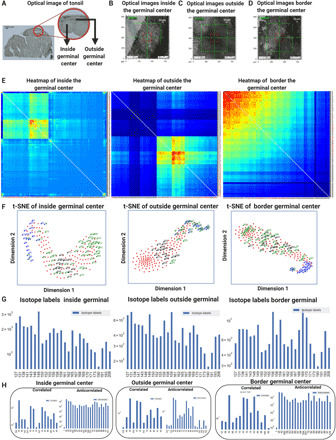
(A) An optical imaging technique was used to observe the 5-μm sliced tonsil section before TOF-SIMS. (B) The selected ROI was identified consistently between the optical imaging platform and the TOF-SIMS platform. In the TOF-SIMS device, an internal optical camera was used to align the ROIs. (C) The image showed the outside of the GC captured by the internal optical camera before the TOF-SIMS measurement process. (D) The images were shown for the border of the GC captured by the internal optical camera image before the TOF-SIMS measurement process started. (E) Comparative hierarchically clustered heatmaps for the 3D correlations were implemented for inside, outside, and at the border of the GC in the labeled tonsil datasets, yielding unique lipidomic features in each dataset. (F) Comparative t-SNE maps of the same GC of the labeled tonsil tissues was displayed. The t-SNE contained correlated (blue), anticorrelated (green), and antibody-labeled (black) channels. (G) Bar plots of the secondary ion signal levels were computed for the selected antibody labels and the metabolite compounds. (H) The bar graphs showed the 50 highest correlated and anticorrelated rankings of metabolic pairs from the 3D Pearson correlations.
The hierarchically clustered heatmaps for the 3D correlations of 20 binned slices inside, outside, and at the border of GC were demonstrated for comparisons (Fig. 4E and figs. S4 to S6). While correlation clustering inside and outside the GC exhibit unique lipidomic features, the border regions of the GC showed a transition state from the clusters with varying heatmaps. In the t-SNE maps, the correlated (blue), anticorrelated (green), and antibody-labeled (black) channels showed distinct clusters for each category (Fig. 4F).
The extracted secondary ion signal levels from the selected cell-specific and metabolic compounds were then compared across these three spatial regions of the GCs in the form of bar plots (tables S3 to S8). In the total signal level across the 3D datasets, mass channels of 127, 131, and 149 m/z were the most distinguished for inside the GC; 131, 149, and 159 m/z were the most prominent for outside the GC; and the cell-specific markers for T cell markers of 156 and 170 m/z were the most enriched compounds in the border of the GC (Fig. 4G). The enrichment of B cells was higher inside the GC at the 161 m/z mass channel, which was expected from B cell distributions in the GCs of a tonsil sample. The correlated and anticorrelated metabolite and cell-specific compounds that were the most commonly found in the highest correlations for each of the three labeled datasets were displayed (Fig. 4H). The correlated and anticorrelated list were uniquely distributed inside, outside, and at the border of GCs, providing spatially distinct compositions of tonsil anatomical regions (Table 2).
Table 2. Unique compound distributions inside, outside, and at the center of GCs in tonsil tissues.
| Masses (m/z) | ||
| Inside germinal | Common correlated compounds |
194, 35, 66, 100, 201, 42, 75, 76, 107, 16, 84, 88, 185, 91, and 60 |
| Common anticorrelated compounds |
1, 35, 42, 50, 185, 58, 57, 63, 64, 194, 66, 196, 201, 74, 75, 84, 85, 86, 88, 91, and 107 |
|
| Top correlation compounds pairs |
75-42, 91-42, 75-91, 84-91, and 84-42 |
|
| Top anticorrelation compounds pairs |
50-16, 50-179, 50-76, 16-122, and 359-431 |
|
| Outside germinal | Common correlated compounds |
128, 66, 35, 100, 42, 75, 107, 79, 84, 86, 91, and 63 |
| Common anticorrelated compounds |
1, 128, 133, 35, 42, 64, 66, 72, 73, 75, 84, 86, 91, 92, 95, 100, 102, 107, and 124 |
|
| Top correlation compounds pairs |
75-84, 60-76, 75-91, 60-16, and 16-17 |
|
| Top anticorrelation compounds pairs |
73-75, 73-42, 73-91, 73-84, and 73-100 |
|
| Border germinal | Common correlated compounds |
97, 98, 35, 66, 72, 73, 42, 75, 74, 50, 84, 86, 185, and 60 |
| Common anticorrelated compounds |
1, 35, 42, 185, 58, 63, 64, 66, 67, 68, 194, 201, 75, 82, 84, 85, 86, 87, 88, 91, and 124 |
|
| Top correlation compounds pairs |
75-42, 50-185, 50-74, 75-91, and 50-73 |
|
| Top anticorrelation compounds pairs |
1-158, 1-155, 1-98, 208-1, and 267-1 |
|
To visualize 3D distributions of metabolic compounds, we used unsupervised K-means clustering for a combination of anticorrelated and correlated metabolites. To select several mass channels for 3D visualization, we used the 3D correlation strengths, commonly occurring compounds, and 3D ion signal levels from bar graphs as a classification strategy. For 3D clustering analysis, the dominant compounds in the inside GC datasets were 50 and 385 m/z (C23H45O4−, cholesterol) and 122 and 98 m/z (C5H12NO+, lipid), providing colocalization values using F1 score in the range of 0.841 to 0.999 (Fig. 5A). The noteworthy mass channels outside GC datasets were 75, 80, 229, and 42, yielding F1 scores of 0.889 to 0.999 (Fig. 5B). Similarly, important compounds for GC border were 75, 42, 50, and 144, exhibiting F1 scores of 0.683 to 0.995 (Fig. 5C).
Fig. 5. 3D clustering of metabolic fragments in spatially distinct cell distributions in tissues.
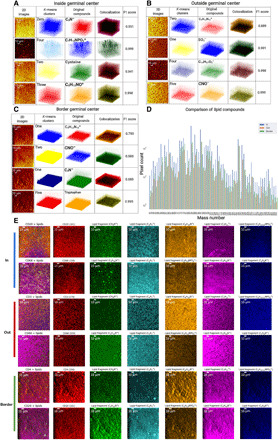
(A) The prominent mass channels were identified in the inside GC dataset that contained 50 and 385 m/z (C23H45O4, cholesterol) and 122 and 98 m/z (C5H12NO+, lipid). The first column contains pixel-binned images, the second column shows the 3D clusters, and the third column displays the 3D visualization of voxels of the four metabolic channels. The 3D overlaps between metabolites and clusters were represented using colocalization in the fourth column and an F1 score in the range of 0.661 to 0.982 in the fifth column. (B) The prominent mass channels in the outside GC are 75, 80, 229, and 42 m/z. The F1 scores were 0.889 to 0.999. (C) The prominent mass channels in the border GC are 75, 42, 50, and 144 m/z. The F1 scores were 0.683 to 0.995. (D) A comparison of the detected metabolic compounds was shown in the form of a bar graph. Distinct distributions of lipidomics were obtained for inside, outside, and at the border of the same GC. (E) A comparison of TOF-SIMS mass channel images that were overlaid using the identified immune labels and lipid fragments. In each row, immune markers and lipid fragment metabolic images were assigned to unique colors.
A comparison in the form of a bar graph of the detected metabolic fragments was presented (Fig. 5D). Distinct subsets of lipidomic compounds were identified inside, outside, and at the border of GCs. This trend can be observed across the hierarchically clustered heatmaps (Fig. 4E and figs. S7 to S11), wherein high concentrations of B cells inside the GC and enrichment of T cells outside the GCs yielded cell type to lipidomic correlations. The distinct distribution of the lipidomic signatures correlated with higher concentrations of the B cells inside the GCs and higher concentrations of T cells outside the GCs (Fig. 5D). Several anticorrelated compounds have been displayed in the form of original 2D images for comparisons (figs. S12 to S15). These dominant correlated and anticorrelated metabolites and the mass channels corresponding to the cell-specific labels were also presented as networks of nodes (figs. S16 and S17) (38). The 3D spatial correlations among these mass channels were also denoted with connecting lines, where the dark red lines show high correlations and fainter gray lines demonstrated lower correlations.
In addition to the molecular correlation and network analyses, spatially resolved clustering of mass channels was performed on all of the mass channels (n = 189) and only in lipid fragment channels (n = 47), yielding six distinct spatial distributions of 3D-SMF measurements that are provided as uniquely pseudo-colored mass channel images (figs. S18 to S24). Using an analytical formalism (see Eq. 1 in Materials and Methods) for K-means analysis of mass channel images, these clusters showed spatial similarity or differences of each metabolic fragment and other potential distribution by the isotope antibody labeling. To visualize the results of clustered mass images, the original TOF-SIMS data were binned across four pixels in x and y dimensions of 2D images and colored by unique assignments. Among 189 mass channels, spatially distinct 10 mass channels for lipid fragments showed substantial localization variability around the GCs (in, out, and border), wherein visual representations of the overlaid images of isotope-specific channels for immune cells and other 10 mass images from lipid fragments in the form of two row sets were shown in the first column, single pseudo-colored cell-specific channels were presented in the second column, and colored metabolic images from major lipid fragments were included in the third to seventh columns (Fig. 5E). This coloring demonstration is a proof-of-principle analysis for 12 mass channels and can be extended to the entire set of 17 isotope-labeled channels and 47 lipid fragments.
To further validate the presented 3D-SMF results, we obtained another set of TOF-SIMS data in the same GC from serial tissue sections of the tonsil samples. The first tissue sample was labeled by seven-plex isotope-enrichment targets (171Yb, histone 3; 149Sm, histone H3 lysine 9 trimethylation (H3K9me3); 153Eu, CD44; 143Nd, vimentin; 158Gd, E-cadherin; 168Er, Ki-67; 192Ir/193Ir, intercalator for DNAs), and the second tissue sample with the same anatomical structure was left unlabeled to serve as a control. The imaging settings were kept the same between the labeled and the unlabeled GC targets. To perform histological comparisons, we obtained the optical images of labeled and unlabeled tonsil tissues using the internal optical microscope in the TOF-SIMS device (fig. S25). To quantify the similarities in the 2D distributions of TOF-SIMS signals, we performed spatial clustering analyses of all mass channels and separately of only lipid fragment channels from both unlabeled and labeled distinct tissues in the same GC (fig. S26 to S29). These spatially clustered mass channels corresponding to unique lipidomic compounds from the labeled and unlabeled GC datasets were presented as pseudo-colored and composite images (Fig. 6A). To link morphological features to TOF-SIMS signals, we overlaid histological optical images of the labeled and the unlabeled tonsil tissue slices with the same 10 lipid fragment datasets (Fig. 6B). Compared to the wet-form imaging of the tonsils using a separate optical microscope, the dry-form tonsil imaging inside the TOF-SIMS provides better matching cell patterns. Notably, the surface of imaging may be separated by a few micrometers in the TOF-SIMS data acquisition from the labeled and unlabeled GC due to the sectioning process of tissues despite analyzing the same GC, potentially exhibiting biological variations of lipid fragments and cell-specific targets.
Fig. 6. Spatially resolved clustering and coloring of isotope-labeled and unlabeled GC in serial tonsil sections.

(A) A comparison of TOF-SIMS mass channel images overlaid on microscopy images with lipid fragments from the labeled and unlabeled GC was shown. Two rows of lipid fragment metabolic images were assigned a unique color and then overlaid together with the morphological image. (B) A comparison of TOF-SIMS mass channel images overlaid from the isotope-labeled mass data of immune targeted channels and lipid fragments for labeled and unlabeled GC was shown. Each row immune marker and lipid fragment metabolic image were assigned a unique color and then overlaid together.
The 3D-SMF platform may help explain the role of unique lipid signatures for major pathways in immune regulation. To identify functions of distinct lipid fragments in our GC datasets, we used the library from the lipid maps consortium to classify the 47 lipid fragment types mainly as fatty acids (FAs), among major eight lipid classification categories (tables S9 to S11) (39). FAs in distinct channels showed enhancement or reduction inside the GCs in the average signals maps of lipids in the labeled and unlabeled GCs, yielding unique spatial lipid codes per channel (fig. S30). These differential changes in FA concentrations agree well with the findings that support the mechanism of B cells in GCs that selectively oxidized FAs for energy (40). Vitamin E (205 m/z) and cholesterol (385 m/z) may also contribute to the membrane compositions to regulate cell-to-cell recognition (41, 42).
3D-SMF analyses of the same GCs from serial tissue slices enable a quantitative framework to quantify signals from other mass fragments in the labeling assembly that contains isotopes, antibodies, and polymers. After labeling, the sum of pixel intensities across 138 z-scans from these datasets for the GC showed intensity enhancement in the total signal from all mass channels (fig. S31A). The total signal in the TOF-SIMS measurements can be modeled as multiple mass spectral analyses, in which the distinct chemical compounds can be numerically computed in different ranges of mass channels (see Eq. 2 in Materials and Methods). In the case of seven isotopes used for the labeling, the total signal of 3D TOF-SIMS per isotope-targeting channels showed an increased summed intensity, while the other mass channels either showed enhancement or reduction in the total signal per channel (fig. S31, B and C). The antibodies are expected to get fragmented into amino acids based on prior TOF-SIMS analysis of pure antibodies (43). Thus, we assigned 11 mass channels to amino acid fragments and calculated the signal enhancement due to labeling in each amino acid channel of the labeled and unlabeled GC data, providing a consistent enhancement in all the mass channels for amino acid fragments and a spatial patterning in the labeled data for the visual representation of mass images (fig. S32, A to C). For further quantification factor of the total signal levels per channel, signals in the labeled samples were subtracted by those in the unlabeled channels (fig. S33) that were then divided by the total labeled signal, yielding positive factors for amino acid channels due to antibodies, positive factors for isotopes, and heterogeneous positive and negative factors for lipids that might be due to the biological variability (fig. S34). In the case of the polymers used to conjugate antibodies to the isotopes, the mass spectra should be in the range of 525 (only polymer) that is added by the isotope’s mass number (e.g., 85 amu), demonstrating a mass peak at 610 m/z and beyond, which was previously experimentally verified by single quadrupole electrospray ionization MS measurements of the same maleimido-mono-amide–1,4,7,10-tetraazacyclododecane-1, 4,7,10-tetraacetic acid (DOTA) (mal-DOTA) polymer complex (15). This fairly large mal-DOTA complex should, in principle, be detected by the TOF-SIMS in the 400 to 600 m/z range based on prior polymer studies in the TOF-SIMS literature (44). Therefore, the spectral decomposition of total TOF-SIMS signal and prior mass analysis on antibodies and polymers allowed isolation of signal contributions that are arising from a labeling complex, an important framework for the 3D-SMF platform to decode chemical species, cell-specific features, and natural compounds such as lipid fragments.
In addition to signal-level analysis, a spatial cross-correlation–based quantification was performed across the labeled and unlabeled datasets per mass channels that correspond to amino acids, lipids, and isotopes. Here, the labeled image of a single mass channel was spatially cross-correlated with the unlabeled image of the same mass number, providing a Gaussian peak and the height of this peak indicating a higher similarity of spatial distributions (fig. S35A). Antibody fragmentation in amino acid channels may show higher spatial similarity, while the isotope-specific channels should be more spatially unique. This expectation was recapitulated in the heatmap that contained the cross-correlation amplitudes of mass channel pairs from 189 compounds (fig. S35B), where all of the amino acids and several other mass compounds showed substantial spatial similarity and some of the lipid channels also showed spatial similarity that might be due to their related functionality in cell regulation.
To further test out the spatial enrichments of antibodies in the amino acid channels, we developed an inverse model from the difference of labeled and unlabeled data (see Eqs. 3 and 4 in Materials and Methods). Because antibody fragments may arise from the same locations around the GCs, the inverse model computes these common distributions by taking the average of the differences between labeled and unlabeled samples in the amino acid mass channels. Another amino acid channel (e.g., 82 m/z) that was not used for calculating this mean difference can then be modified by this common spatial pattern, providing a predicted labeled image without the use of original labeled data (fig. S36, A to D). This inverse predictive model was then used to predict experimental images in major 11 amino acid channels (fig. S37), wherein the comparison of the original experimental labeled image of this predicted amino acid channel and the predicted image of a labeled image provided a decent agreement with a structural similarity of 0.2 to 0.43. These predictive solutions for the mass spectral dependent analysis of the same biological targets with and without isotope labels will be shedding light on the underlying isotope-enriched biological signatures while separating the contributions of antibodies or polymers used in the 3D-SMF technology.
DISCUSSIONS
3D metabolomic profiling platforms were previously implemented in cells and tissues based on various MS technologies. Using TOF-SIMS devices, 3D metabolomic profiling datasets lacked cell type specificity with a comparable nanoscale resolution to the presented 3D-SMF platform (17, 18, 20). On the other hand, another metabolomic profiling measurement was limited to low-imaging resolution in the order of 10 to 100 μm and large sample volumes using MALDI (21). Additional 3D biological systems used metabolomic flux analyzers and shotgun metabolomic profilers to measure chemical fingerprints in 3D cultures (19, 22). An OrbiSIMS platform (23) was also used to profile up to 100 metabolites with custom machine engineering at 300-nm resolutions. Another NanoSIMS device was leveraged to map out up to seven targets at 100-nm resolution (45, 46). Recently, cell specificity was also introduced in NanoSIMS with antibody-based oligo-tagging with seven mass channels (47). To simultaneously achieve multiplexing and specificity, 3D-SMF offers both cell labeling and metabolic analysis up to 200 markers in a single-imaging MS measurement at sub–100-nm resolutions.
In addition to the lipidomic measurements in the 3D-SMF platform, antibodies that were made by the B cells inside the GC region would have been interesting targets to profile in the same experiment, but the secondary ions from this experiment would not be sufficient to call the type of the proteins [e.g, immunoglobulin A (IgA) and IgG] (48) definitively. These experiments can be achieved by a combined analysis of MS and imaging mass analysis, but conventional MS would not reach a nanoscale spatial resolution that renders its use insufficient in the 3D-SMF platform. An ultimate imaging MS platform should combine protein and metabolite identification at nanoscale measurements, but the device trade-offs make it challenging either to map higher-resolution profiles with the loss of protein identity or to map lower-resolution complete profiling in tissues. To address these issues, 3D-SMF will be an alternative route to retain protein specificity with metabolite calling with high-resolution capabilities, wherein antibody-labeled additional isotopes should also be included to cover IgA and IgG molecular detection.
Last, alternative routes are emerging to perform 3D metabolomic profiling measurements with high spatial resolution up to 50 markers. The first direction is to use label-free Raman imaging to visualize chemical fingerprints in the vibrational spectra. Recent approach supermultiplex vibrational imaging (49) allows up to 24-plex measurements in even live specimens. In combination with computational debarcoding of optical spectra, the multiplex vibrational analysis should, in principle, reach highly multiplex metabolomic profiling measurements. Using 3D scanning of optical paths or sample stages, these metabolomic profiling technologies should achieve exciting 3D spatial metabolomic profiling platforms. The latter direction is to profile RNA or protein signatures of metabolites of interest using readily available multiplexed transcriptional and proteomic mapping technologies. Along these lines, a recent demonstration included antibody-based detection of protein markers for enzymes to study T cell biology using CyTOF and multiplexed ion beam imaging (50). Multiplex RNA profiling of metabolic targets by fluorescent in situ hybridization (FISH)–based technologies such as sequential FISH (51, 52) and multiplexed error-robust FISH (53, 54) should provide metabolic regulators in tissues. A complementary RNA profiling approach illustrated the use of spatial transcriptomics data to reveal metabolic alterations in prostate cancers, albeit being limited to the 2D images of tissue sections (55).
Summary
In summary, spatially resolved metabolomic profiling analysis framework, 3D-SMF, demonstrated cell-specific labeling and metabolomic compound detection for up to 189 markers using a TOF-SIMS device. To quantify interactions of cell features and metabolites, we performed 3D spatial correlation analysis to reveal correlated and anticorrelated compounds in tonsil tissues. These associations were presented in the network graphs with nodes and edges. To visualize 3D distributions, we used an unsupervised clustering method in the 3D metabolite and cell-labeled mass channels. F1 score was the validation method of the colocalization measure in the 3D visuals. To explore spatially distinct regions of tonsil tissues, we analyzed 3D-SMF results inside, outside, and at the border of a GC region after defining an ROI by prior optical imaging. Unique lipidomic profiles obtained for each spatial region were linked to specific immune subsets. The presented 3D-SMF results may affect the understanding of the spatial organization within a tonsil architecture by highlighting metabolic cues that play key roles in cell-cell interactions toward deciphering the spatial dynamics of immune responses.
MATERIALS AND METHODS
The presented 3D-SMF method uses experimental and computational procedures to profile metabolic and cell-specific compounds in tissues as detailed in the following sections.
Tissue
The human tonsil tissues were obtained from an FFPE tonsil block (ID 1320.06.3D, BioMax). This FFPE tissue block was cut into sections at a thickness of 5 μm and mounted on Superfrost Plus Gold Slides (catalog number FT4981GLPLUS, Thermo Fisher Scientific). The tissue samples on microscope slides were then deparaffinized by immersing them in xylene and then rehydrated through sequential immersing steps in descending concentrations of ethanol (100, 95, 80, 70, and 50%) and a final wash step in deionized water. After dehydrating the tissue, unlabeled tonsil sections were then analyzed by optical microscopy and TOF-SIMS imaging under dry conditions.
Tissue labeling
After performing the tissue deparaffinization step as described in the previous section, the heat-induced epitope retrieval process was then achieved by immersing the tissue slides in a target retrieval solution with pH 9 (catalog number S2367, Agilent Dako) and using a pressure cooker on a high-pressure setting for 20 min. Protein-blocking step was performed by immersing the tissue slides in a serum-free, ready-to-use blocking buffer (catalog number X090930-2, Agilent Dako) for 30 min at room temperature. After three washes of Maxpar phosphate-buffered saline (PBS) (catalog number 201058, Fluidigm), the tonsil tissues were then stained with metal-conjugated antibody cocktail mix for immune markers overnight at 4°C following Fluidigm-labeling protocol. The tissue slides were then washed with three changes of Maxpar PBS, and the nuclear staining was performed using an iridium-conjugated intercalator (catalog number 201192A, Fluidigm) for 10 min at room temperature. Last, slides were washed with three changes of Maxpar PBS and left to dry overnight.
Optical microscopy
To define anatomical regions of the tonsil tissue, tonsil tissues were initially scanned and imaged by an optical microscope (Keyence IX810) using 20× and 60× objective lenses in the bright-field and ultraviolet channel with autoexposure settings. The microscopic images corresponded to the light microscopy patterns or autofluorescence images of cells in tissues. Landmark patterns were identified and the coordinates of which were saved in a grid view. The optical imaging was carried out before the TOF-SIMS experiments because the sample retained its original shape and structure only before TOF-SIMS. Once the coordinates of the ROI are obtained from the optical images and the TOF-SIMS optical camera, spatial distinct regions (e.g., GCs) were aligned properly for 3D metabolic imaging (fig. S2). Notably, after metabolic measurements, the cells in tissues are ablated destructively, and thus, optical registrations would not be accurate.
TOF-SIMS imaging
The TOF-SIMS (IONTOF 5 GmbH, Münster, Germany) instrument uses a bismuth liquid metal ion gun as the primary ion source to generate secondary ions from the sample surface, followed by identification of m/z of secondary ions by a TOF analyzer. The bismuth source can be used in different modes: Singly charged 25-kV monoatomic (Bi+), singly charged 25-kV three-atom cluster (Bi3+), or doubly charged 50-kV cluster (Bi3++) mode. Distinct modes exhibit extra cluster mass and energy that can increase the yield (i.e., the number of secondary ions per primary ion) of heavier molecular weight secondary ion species. In this device, the secondary ions were collected and then accelerated across a voltage gap and directed into an ultrahigh-vacuum flight tube toward the detector (a combination channel plate and photomultiplier tube). The flight time of secondary ions is proportional to the ion m/z; thus, lighter ions (lower m/z) arrive at the detector more quickly, and the heavier the ion, the longer the TOF. For depth profiling, another cesium ion gun (Cs+ ions, 2-kV energy, and microampere current) was used to iteratively sputter away very thin layers, followed by bismuth (Bi) bombardment to generate secondary ions from the tissue sample before and after Cs sputtering cycles.
Mass channel calibrations
TOF-SIMS spectra simultaneously record thousands of peaks. However, a peak for a metabolite of interest may shift fractionally because of device tuning settings. The uncertainty in mass for a peak increases with the mass of the compound, so when measuring high molecular weight compounds, accurate and large range calibrations are needed to minimize the errors in the peak identification (56). Thus, abundant lower mass channels (atomic ions, hydrogen, phosphate, and sulfur-containing channels) were used to define the “offset” in the experimental spectra for each peak, and a larger range of mass channels was included to achieve calibration accuracy for large molecules.
Mass imaging modes
To achieve optimal calibration performance, different imaging settings are available for TOF-SIMS imaging that includes burst alignment (BA), BA-burst, and high-current bunch (HCBU) modes (57). While the HCBU mode provides high mass and low spatial resolution with high ion counts, the BA-burst mode achieves high mass and high spatial resolution but with low counts. On the other hand, BA mode yields high spatial resolution (in submicrometers) and medium mass resolution with counts higher than that of BA-burst mode and lower than that of HCBU (5 μm). Each mode has unique advantages, and thus, a combined procedure was developed to optimize both modes at the same time. First, the HCBU mode is used to choose the metabolite of interest. Second, BA-burst mode is used to refine the peaks of each compound. Last, BA mode is performed to capture higher spatial resolution and higher count rate.
Imaging mode polarity
In this 3D-SMF platform, negative and positive modes were experimentally tested. As the metabolomic profiling measurements in unlabeled samples provided more noteworthy spatial distributions in the negative mode, the rest of the experiments were performed to detect the negatively charged compounds as the selection of polarity. The critical question is whether a positive ion and a negative ion of the same compound have the same mass. The opposite polarity ions differ in mass by two electrons. Thus, if neutral C is exactly 12.0000 Da, then C- is ~12.0005 Da, and C+ is ~ 11.9995 Da. Even with the high mass resolution in the HCBU mode (resolution = m/Δm ~ 5000), those masses would overlap almost completely. As long as the m/z axis scaling is carefully performed in the mass calibration, the peak position should be accurate in either polarity. Thus, the list of compounds from various literatures should be generally applicable to the identification of the detected mass channels.
Mass image acquisition
Negatively charged ion mass spectra were collected on 150 μm by 150 μm areas with 256 pixel by 256 pixel density. After extensive experimentation at different settings, the optimal configuration was this pixel size of 580 nm by 580 nm in the BA mode. The peak masses were initially calibrated using the HCBU mode and then fine-tuned with the BA mode, followed by image acquisition in the BA-burst mode in labeled tonsil tissues. Additional factors that include ion beam instability, sample roughness, and analyzer settings slightly shift the detected masses. Biological tissues have high concentrations of anions (e.g., phosphates and chlorides) that can be identified with a high degree of certainty for a given set of samples, providing accurate calibration settings for the mass channel (m/z) axis of the overall spectra. After calibration, peaks of interest can be selected either before or after the acquisition of the spectral information. The criteria for selection of the peaks include the signal-to-noise ratio, the width of peaks, and the degree of spectral overlap. Once optimizations are complete, mass spectra is saved to identify any metabolite of interest after the measurements using postanalysis.
Postacquisition data analysis
After the data acquisition, the IONTOF SurfaceLab software (version 6) was used to perform basic image processing operations on the acquired spatial mass spectra. The spatial distribution for each selected peak was visualized in 2D/3D, and the files containing the coordinates and pixel intensity values were generated. A total of 189 peaks were selected, and 256 pixel by 256 pixel images for each peak were obtained. The selected peaks were each normalized to the total pixel intensities available, and each mass peak was normalized to the total intensity of the remaining peaks in the mean-centered spectra. The data were then exported in American Standard Code for Information Interchange (ASCII) format into a text file. Open-sourced codes in Python and R were used for the postanalysis of the exported mass channel files. The computational analysis included data dimensionality reduction technique (t-SNE), hierarchically clustered 3D correlation heatmaps, 3D spatially resolved clustering algorithms, and 3D network maps.
3D clustering method
K-means is a robust clustering algorithm that splits the data into k different clusters, wherein each datum is assigned to the cluster with the mean nearest to the spatial density distributions. The k value needs to be defined before the clustering to generate many clusters accordingly. In the 3D classification algorithm, the cluster means and the cluster associated with each data point are iteratively updated until the algorithm reaches the upper limit on the number of iterations. Compared to most other algorithms, K-means has the advantage of being computationally efficient as it only requires computing means of the ion voxels. F1 score is used to check the validity of the computed clusters. F1 score is defined as the harmonic mean of precision and recall. Precision captures how much the method is prone to produce false positives, wherein high precision implies that the method does not produce a lot of false positives. The recall is a measure of the ability of the method to correctly identify all the true-positive cases. In this pipeline, the predicted clusters are compared with the original compounds to quantify the spatial colocalization between the two, yielding typically a range of 0.6 to 0.9 values.
Network analysis of compounds
Network analysis methods visualize the bipartite association structure between the correlated and anticorrelated metabolites and cell-specific compounds. A bipartite network is a particular class of graph that separates the nodes into two sets and has a specific property of only allowing connections (edges) between two nodes in different sets. The network nodes are the metabolites and cell-specific compounds with the weights represented by the 3D spatial correlation between two compounds that are extracted from a voxel level of the TOF-SIMS images. Two types of networks are obtained from the bipartite network: circular layout and force atlas. The networks were plotted using Gephi, a network visualization software used in various disciplines that include social network analysis, biology, and genomics. The circular layout places the nodes of the graph on a circle, and the nodes are evenly spaced. On the other hand, ForceAtlas2 is a force-directed layout simulating a physical system to spatialize the network by giving repulsion forces to the nodes and attraction force to the edges.
Spatial clustering and coloring
Spatial maps are generated on the basis of K-means clustering of the mass peak images. From pixel intensity, the acquired mass images are used for unsupervised clustering. Each cluster is assigned to a unique colormap. The mean image of each cluster is used to construct a spatial map showing spatial structures. Each single mass channel image is colored in the associated cluster color to show the similarity between mass channels within a single cluster. Here, the K-means clustering divides the set of 189 images into six disjoint clusters C, each described by the mean μj of the images in the cluster (Eq. 1). Each image is represented by a unique vector xi obtained by reshaping a 256 pixel by 256 pixel image to a 65,536 size vector. The K-means algorithm aims to find the centroids that minimize the within-cluster sum of squares of distances as follow with n = 189
| (1) |
Multiple mass spectra contributions of labeling
The spectral dependent analysis separates the contributions of the total signal into amino acid channels, isotope-specific channels, lipid channels, and polymer channels. The pixel intensity at position (i, j) for the total signal is split into multiple spectra by the following Eq. 2
| (2) |
with M as the total signal, Am as the signal split into the amino acid channel at mass m, Im as the signal split into the isotope-labeled channel at mass m, Lm as the signal split into lipid channel at mass m, Pm as the signal split into polymer channel at mass m, and Rm as the rest of the mass channel at mass for m ∈ δ with δ of all the mass channels.
δ= AA + L + I + P + R, with mass channel identified with amino acids (AA), lipids (L), isotope-labeled (I), polymer (P), and the rest (R).
1) P = (610).
2) AA = (42, 44, 56, 61, 70, 72, 73, 74, 82, 84, and 86).
3) I = (141, 143, 148, 149, 150, 152, 155, 156, 158, 159, 161, 162, 165, 167, 168, 169, 170, 171, 173, 191, and 193).
4) L = (30, 34, 55, 57, 58, 59, 60, 66, 67, 68, 69, 71, 81, 83, 85, 88, 91, 93, 95, 97, 98, 100, 102, 104, 105, 111, 146, 166, 182, 184, 185, 190, 194, 196, 198, 206, 210, 212, 224, 226, 238, 240, 246, 252, 254, 256, and 282).
5) R = (50, 200, 201, 202, 203, 204, 205, 207, 208, 209, 214, 216, 217, 220, 221, 222, 223, 225, 229, 232, 236, 243, 248, 249, 250, 253, 259, 261, 263, 265, 267, 190, 273, 275, 276, 279, 289, 301, 307, 308, 310, 318, 385, 332, 339, 344, 353, 359, 369, 380, 385, 397, 412, 424, 431, 436, 448, 457, 468, 474, 479, 498, 63, 501, 510, 514, 518, 520, 529, 544, 552, 570, 586, 64, 593, 610, 1, 75, 16, 76, 78, 79, 80, 17, 87, 96, 107, 109, 112, 115, 116, 117, 118, 121, 122, 124, 125, 127, 128, 131, 133, 134, 138, 35, 140, 144, 151, 164, and 179).
Isotope-labeled amino acid mass channel image prediction
Let IL, m(i, j) be the pixel intensity at position (i, j) for the labeled mass channel m and IU, m(i, j) for the unlabeled mass channel m with 0 ≤ i ≤ w and 0 ≤ j ≤ h, w as the width and h as the height of the image (here, 256 by 256) (Eq. 3). The difference between labeled and unlabeled pixel can be expressed as
| (3) |
Therefore, using a linear model, the predicted labeled mass channel image of amino acid can be obtained by the following Eq. 4
| (4) |
With AA as the set of the mass channel corresponding to amino acid (42, 44, 56, 61, 70, 72, 73, 74, 82, 84, and 86) and n as the size of AA minus 1.
Spatial cross-correlation analysis
2D cross-correlation between the isotope-labeled and unlabeled TOF-SIMS signals was computed for each pair of mass images. The signal differences of each mass channel at each pixel Dm(i, j) location are shown in Eq. 3. Each signal difference is represented by a unique vector xk obtained by reshaping 256 pixel by 256 pixel to a 65,536 size vector. The pairwise correlation between mass channel k and l is computed by the Pearson correlation
| (5) |
with n = 65536, xk, i and xl, i as the individual signal point in xk and xl, respectively, indexed with i, and as the mean value of xk and analogously for .
Acknowledgments
Funding: A.F.C. holds a Career Award at the Scientific Interface from Burroughs Wellcome Fund, National Institute of Health K25 Career Development Award (K25AI140783), and a Bernie-Marcus Early-Career Professorship. A.F.C. was supported by start-up funds from the Georgia Institute of Technology and Emory University. This work was performed in part at the Materials Characterization Facility (MCF) at Georgia Tech. The MCF is jointly supported by the GT Institute for Materials (IMat) and the Institute for Electronics and Nanotechnology (IEN), which is a member of the National Nanotechnology Coordinated Infrastructure supported by the NSF (grant ECCS-2025462). Authors contributions: S.G., T.H., and E.W. conducted the data acquisition and analysis. M.A. and S.C. performed the tissue handling and labeling. W.H. contributed to the manuscript writing and editing. A.F.C. supervised the project and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. 3D-SMF datasets are available at https://doi.org/10.5281/zenodo.4087670. Custom Python (Jupyter Notebook) codes are available at https://github.com/coskunlab/Spatially-resolved-3D-metabolomic-profiling. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd0957/DC1
REFERENCES AND NOTES
- 1.Regev A., Teichmann S. A., Lander E. S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M., Clevers H., Deplancke B., Dunham I., Eberwine J., Eils R., Enard W., Farmer A., Fugger L., Göttgens B., Hacohen N., Haniffa M., Hemberg M., Kim S., Klenerman P., Kriegstein A., Lein E., Linnarsson S., Lundberg E., Lundeberg J., Majumder P., Marioni J. C., Merad M., Mhlanga M., Nawijn M., Netea M., Nolan G., Pe’er D., Phillipakis A., Ponting C. P., Quake S., Reik W., Rozenblatt-Rosen O., Sanes J., Satija R., Schumacher T. N., Shalek A., Shapiro E., Sharma P., Shin J. W., Stegle O., Stratton M., Stubbington M. J. T., Theis F. J., Uhlen M., van Oudenaarden A., Wagner A., Watt F., Weissman J., Wold B., Xavier R., Yosef N.; Human Cell Atlas Meeting Participants , Science forum: The human cell atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rozenblatt-Rosen O., Regev A., Oberdoerffer P., Nawy T., Hupalowska A., Rood J. E., Ashenberg O., Cerami E., Coffey R. J., Demir E., Ding L., Esplin E. D., Ford J. M., Goecks J., Ghosh S., Gray J. W., Guinney J., Hanlon S. E., Hughes S. K., Hwang E. S., Iacobuzio-Donahue C. A., Jané-Valbuena J., Johnson B. E., Lau K. S., Lively T., Mazzilli S. A., Pe’er D., Santagata S., Shalek A. K., Schapiro D., Snyder M. P., Sorger P. K., Spira A. E., Srivastava S., Tan K., West R. B., Williams E. H.; Human Tumor Atlas Network , The Human Tumor Atlas Network: Charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park J.-E., Botting R. A., Conde C. D., Popescu D.-M., Lavaert M., Kunz D. J., Goh I., Stephenson E., Ragazzini R., Tuck E., Wilbrey-Clark A., Roberts K., Kedlian V. R., Ferdinand J. R., He X., Webb S., Maunder D., Vandamme N., Mahbubani K. T., Polanski K., Mamanova L., Bolt L., Crossland D., de Rita F., Fuller A., Filby A., Reynolds G., Dixon D., Saeb-Parsy K., Lisgo S., Henderson D., Vento-Tormo R., Bayraktar O. A., Barker R. A., Meyer K. B., Saeys Y., Bonfanti P., Behjati S., Clatworthy M. R., Taghon T., Haniffa M., Teichmann S. A., A cell atlas of human thymic development defines T cell repertoire formation. Science 367, eaay3224 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richter F. C., Obba S., Simon A. K., Local exchange of metabolites shapes immunity. Immunology 155, 309–319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scadding G. K., Immunology of the tonsil: A review. J. R. Soc. Med. 83, 104–107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.J.-M. Anaya, Y. Shoenfeld, A. Rojas-Villarraga, R. A. Levy, R. Cervera, Autoimmunity: From Bench to Bedside (El Rosario University Press, 2013); www.ncbi.nlm.nih.gov/books/NBK459447/. [PubMed]
- 7.Lin J.-R., Izar B., Wang S., Yapp C., Mei S., Shah P. M., Santagata S., Sorger P. K., Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alexandrov T., Spatial metabolomics and imaging mass spectrometry in the age of artificial intelligence. Annu. Rev. Biomed. Data Sci. 3, 61–87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passarelli M. K., Winograd N., Lipid imaging with time-of-flight secondary ion mass spectrometry (ToF-SIMS). Biochim. Biophys. Acta 1811, 976–990 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brison J., Robinson M. A., Benoit D. S. W., Muramoto S., Stayton P. S., Castner D. G., TOF-SIMS 3D imaging of native and non-native species within HeLa cells. Anal. Chem. 85, 10869–10877 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fletcher J. S., Latest applications of 3D ToF-SIMS bio-imaging. Biointerphases 10, 018902 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Itzkan I., Qiu L., Fang H., Zaman M. M., Vitkin E., Ghiran I. C., Salahuddin S., Modell M., Andersson C., Kimerer L. M., Cipolloni P. B., Lim K.-H., Freedman S. D., Bigio I., Sachs B. P., Hanlon E. B., Perelman L. T., Confocal light absorption and scattering spectroscopic microscopy monitors organelles in live cells with no exogenous labels. Proc. Natl. Acad. Sci. U.S.A. 104, 17255–17260 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bendall S. C., Simonds E. F., Qiu P., Amir E.-a. D., Krutzik P. O., Finck R., Bruggner R. V., Melamed R., Trejo A., Ornatsky O. I., Balderas R. S., Plevritis S. K., Sachs K., Pe’er D., Tanner S. D., Nolan G. P., Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687–696 (2011).21551058 [Google Scholar]
- 14.Giesen C., Wang H. A. O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P. J., Grolimund D., Buhmann J. M., Brandt S., Varga Z., Wild P. J., Günther D., Bodenmiller B., Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Han G., Spitzer M. H., Bendall S. C., Fantl W. J., Nolan G. P., Metal-isotope-tagged monoclonal antibodies for high-dimensional mass cytometry. Nat. Protoc. 13, 2121–2148 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noël C., Busby Y., Mine N., Houssiau L., ToF-SIMS depth profiling of organic delta layers with low-energy cesium ions: Depth resolution assessment. J. Am. Soc. Mass Spectrom. 30, 1537–1544 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Singh A. V., Jungnickel H., Leibrock L., Tentschert J., Reichardt P., Katz A., Laux P., Luch A., ToF-SIMS 3D imaging unveils important insights on the cellular microenvironment during biomineralization of gold nanostructures. Sci. Rep. 10, 261 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breitenstein D., Rommel C. E., Stolwijk J., Wegener J., Hagenhoff B., The chemical composition of animal cells reconstructed from 2D and 3D ToF-SIMS analysis. Appl. Surf. Sci. 255, 1249–1256 (2008). [Google Scholar]
- 19.Lu X., Lodi A., Tiziani S., FASEB J. 33, 639.6–639.6 (2019). [Google Scholar]
- 20.Robinson M. A., Graham D. J., Castner D. G., ToF-SIMS depth profiling of cells: z-Correction, 3D imaging, and sputter rate of individual NIH/3T3 fibroblasts. Anal. Chem. 84, 4880–4885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dueñas M. E., Essner J. J., Lee Y. J., 3D MALDI mass spectrometry imaging of a single cell: Spatial mapping of lipids in the embryonic development of zebrafish. Sci. Rep. 7, 14946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell S., Wojtkowiak J., Neilson A., Gillies R. J., Metabolic profiling of healthy and cancerous tissues in 2D and 3D. Sci. Rep. 7, 15285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Passarelli M. K., Pirkl A., Moellers R., Grinfeld D., Kollmer F., Havelund R., Newman C. F., Marshall P. S., Arlinghaus H., Alexander M. R., West A., Horning S., Niehuis E., Makarov A., Dollery C. T., Gilmore I. S., The 3D OrbiSIMS—Label-free metabolic imaging with subcellular lateral resolution and high mass-resolving power. Nat. Methods 14, 1175–1183 (2017). [DOI] [PubMed] [Google Scholar]
- 24.T. Alexandrov, K. Ovchinnikova, A. Palmer, V. Kovalev, A. Tarasov, L. Stuart, R. Nigmetzianov, D. Fay, Key METASPACE contributors, M. Gaudin, C. G. Lopez, M. Vetter, J. Swales, M. Bokhart, M. Kompauer, J. McKenzie, L. Rappez, D. Velickovic, R. Lavigne, G. Zhang, D. Thinagaran, E. Ruhland, M. Sans, S. Triana, D. A. Sammour, S. Aboulmagd, C. Bagger, N. Strittmatter, A. Rigopoulos, E. Gemperline, A. M. Joensen, B. Geier, C. Quiason, E. Weaver, M. Prasad, B. Balluff, K. Nagornov, L. Li, M. Linscheid, C. Hopf, D. Heintz, M. Liebeke, B. Spengler, B. Boughton, C. Janfelt, K. Sharma, C. Pineau, C. Anderton, S. Ellis, M. Becker, J. Pánczél, G. D. Violante, D. Muddiman, R. Goodwin, L. Eberlin, Z. Takats, S. Shahidi-Latham, METASPACE: A community-populated knowledge base of spatial metabolomes in health and disease. bioRxiv 10.1101/539478 [Preprint]. 3 February 2019. 10.1101/539478. [DOI]
- 25.Madiona R. M. T., Welch N. G., Scoble J. A., Muir B. W., Pigram P. J., Determining the limit of detection of surface bound antibody. Biointerphases 12, 031007 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Welch N. G., Madiona R. M. T., Payten T. B., Easton C. D., Pontes-Braz L., Brack N., Scoble J. A., Muir B. W., Pigram P. J., Surface immobilized antibody orientation determined using ToF-SIMS and multivariate analysis. Acta Biomater. 55, 172–182 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Walch A., Rauser S., Deininger S.-O., Höfler H., MALDI imaging mass spectrometry for direct tissue analysis: A new frontier for molecular histology. Histochem. Cell Biol. 130, 421–434 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiseman J. M., Ifa D. R., Zhu Y., Kissinger C. B., Manicke N. E., Kissinger P. T., Cooks R. G., Desorption electrospray ionization mass spectrometry: Imaging drugs and metabolites in tissues. Proc. Natl. Acad. Sci. U.S.A. 105, 18120–18125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta S., Aires-de-Sousa J., Comparing the chemical spaces of metabolites and available chemicals: Models of metabolite-likeness. Mol. Divers. 11, 23–36 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Wiwie C., Baumbach J., Röttger R., Comparing the performance of biomedical clustering methods. Nat. Methods 12, 1033–1038 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Ali H. R., Jackson H. W., Zanotelli V. R. T., Danenberg E., Fischer J. R., Bardwell H., Provenzano E.; CRUK IMAXT Grand Challenge Team, Rueda O. M., Chin S.-F., Aparicio S., Caldas C., Bodenmiller B., Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 1, 163–175 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Hartmann F. J., Bendall S. C., Immune monitoring using mass cytometry and related high-dimensional imaging approaches. Nat. Rev. Rheumatol. 16, 87–99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao Y., Liu R., Shin M. S., Trentalange M., Allore H., Nassar A., Kang I., Pober J. S., Montgomery R. R., CyTOF supports efficient detection of immune cell subsets from small samples. J. Immunol. Methods 415, 1–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comi T. J., Do T. D., Rubakhin S. S., Sweedler J. V., Categorizing cells on the basis of their chemical profiles: Progress in single-cell mass spectrometry. J. Am. Chem. Soc. 139, 3920–3929 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen C. D. C., Okada T., Cyster J. G., Germinal-center organization and cellular dynamics. Immunity 27, 190–202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.K. Kwak, H. Sohn, J. Brzostowski, J. Manzella-Lapeira, S. K. Pierce, J. Immunol. 196, 195.8 (2016).
- 37.Yin L., Zhang Z., Liu Y., Gao Y., Gu J., Recent advances in single-cell analysis by mass spectrometry. Analyst 144, 824–845 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Jacomy M., Venturini T., Heymann S., Bastian M., ForceAtlas2, a continuous graph layout algorithm for handy network visualization designed for the gephi software. PLOS ONE 9, e98679 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fahy E., Subramaniam S., Murphy R. C., Nishijima M., Raetz C. R. H., Shimizu T., Spener F., van Meer G., Wakelam M. J. O., Dennis E. A., Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50, S9–S14 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisel F. J., Mullett S. J., Elsner R. A., Menk A. V., Trivedi N., Luo W., Wikenheiser D., Hawse W. F., Chikina M., Smita S., Conter L. J., Joachim S. M., Wendell S. G., Jurczak M. J., Winkler T. H., Delgoffe G. M., Shlomchik M. J., Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat. Immunol. 21, 331–342 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monroe E. B., Jurchen J. C., Lee J., Rubakhin S. S., Sweedler J. V., Vitamin E imaging and localization in the neuronal membrane. J. Am. Chem. Soc. 127, 12152–12153 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Kurczy M. E., Kozole J., Parry S. A., Piehowski P. D., Winograd N., Ewing A. G., Relative quantification of cellular sections with molecular depth profiling ToF-SIMS imaging. Appl. Surf. Sci. 255, 1158–1161 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu F., Dubey M., Takahashi H., Castner D. G., Grainger D. W., Immobilized antibody orientation analysis using secondary ion mass spectrometry and fluorescence imaging of affinity-generated patterns. Anal. Chem. 82, 2947–2958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore C., McKeown P., LCMS/MS and TOF-SIMS identification of the color bodies on the surface of a polymer. J. Am. Soc. Mass Spectrom. 16, 295–301 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Steinhauser M. L., Bailey A. P., Senyo S. E., Guillermier C., Perlstein T. S., Gould A. P., Lee R. T., Lechene C. P., Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature 481, 516–519 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saka S. K., Vogts A., Kröhnert K., Hillion F., Rizzoli S. O., Wessels J. T., Correlated optical and isotopic nanoscopy. Nat. Commun. 5, 3664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.X. Rovira-Clave, S. Jiang, Y. Bai, G. Barlow, S. Bhate, A. F. Coskun, G. Han, B. Zhu, C.-M. K. Ho, C. Hitzman, S.-Y. Chen, F.-A. Bava, G. P. Nolan, Subcellular localization of drug distribution by super-resolution ion beam imaging. bioRxiv 10.1101/557603 [Preprint]. 22 February 2019. 10.1101/557603. [DOI]
- 48.Curran R. C., Jones E. L., Immunoglobulin-containing cells in human tonsils as demonstrated by immunohistochemistry. Clin. Exp. Immunol. 28, 103–115 (1977). [PMC free article] [PubMed] [Google Scholar]
- 49.Wei L., Chen Z., Shi L., Long R., Anzalone A. V., Zhang L., Hu F., Yuste R., Cornish V. W., Min W., Super-multiplex vibrational imaging. Nature 544, 465–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartmann F. J., Mrdjen D., McCaffrey E., Glass D. R., Greenwald N. F., Bharadwaj A., Khair Z., Verberk S. G. S., Baranski A., Baskar R., Graf W., Van Valen D., Van den Bossche J., Angelo M., Bendall S. C., Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol. (2020); doi:10.1038/s41587-020-0651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lubeck E., Coskun A. F., Zhiyentayev T., Ahmad M., Cai L., Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 11, 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eng C.-H. L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.-C., Cai L., Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia C., Fan J., Emanuel G., Hao J., Zhuang X., Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc. Natl. Acad. Sci. U.S.A. 116, 19490–19499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen K. H., Boettiger A. N., Moffitt J. R., Wang S., Zhuang X., Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y., Ma S., Ruzzo W. L., Spatial modeling of prostate cancer metabolic gene expression reveals extensive heterogeneity and selective vulnerabilities. Sci. Rep. 10, 3490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Green F. M., Gilmore I. S., Seah M. P., TOF-SIMS: Accurate mass scale calibration. J. Am. Soc. Mass Spectrom. 17, 514–523 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Holzlechner G., Kubicek M., Hutter H., Fleig J., A novel ToF-SIMS operation mode for improved accuracy and lateral resolution of oxygen isotope measurements on oxides. J. Anal. At. Spectrom 28, 1080 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabd0957/DC1