

Abstract
Emerging evidence suggests comprehensive immune profiling represents a highly promising, yet insufficiently tapped approach to identify potentially prognostic signatures for periodontitis. In this report, we agnostically identified a periodontitis-associated inflammatory expression network with multiple biomarkers identified within gingival crevicular fluid samples from study participants by applying principal component analysis. We identified an IL-17–dominated trait that is associated with periodontal disease and is inversely modified by the level of IL-10. IL-10 mitigated chemokine CXCL5 and CXCL1 expressions in IL-17–stimulated peripheral blood monocytic cells and peripheral blood monocytic cell–derived macrophages. Il10-deficient mice presented more bone loss, which was associated with more Il17 and IL-17–mediated chemokine and cytokine expression at the transcriptional levels in comparison with control wild-type mice in both the Porphyromonas gingivalis–induced experimental murine periodontitis and ligature-induced alveolar bone-loss models. The dampening effect of IL-10 on the excessive signaling of IL-17 appeared to be mediated by innate immune cells populations rather than by gingival epithelial cells, which are the major cell target for IL-17 signaling. Additionally, elevated IL-17 response in Il10-deficient mice specifically elicited an M1-skewing macrophage phenotype in the gingiva that was associated with the advanced bone loss in the ligature model. In summary, IL-17 dominated an inflammatory network characteristic of periodontitis, and IL-10 dampens this excessive IL-17–mediated periodontitis trait.
Periodontitis is a dysbiosis-initiated inflammatory disease that clinically presents with irreversible loss of tooth supporting structures and eventually leads to tooth loss. This irreversible form of periodontal disease affects an estimated 42% of adult Americans and is associated with other systemic diseases such as diabetes (1, 2). Because of the heterogeneity of etiology, inflammation, and clinical presentation, periodontitis can be classified into different traits through clustering analyses. For example, based on the microbial burden in plaque, the level of inflammatory marker IL-1β in the gingival crevicular fluid (GCF), and the disease severity obtained by clinical measurements, we defined several periodontitis traits or phenotypes such as “Socransky trait” or “Porphyromonas gingivalis trait” through the principal component analysis (PCA) in a well-defined, community-dwelling subject pool (3).
Both longitudinal and cross-sectional studies have reported that the protein levels of an array of GCF inflammatory mediators, including cytokines, chemokines, growth factors, and enzymes, are elevated in periodontal disease (4, 5). A portion of those GCF inflammatory mediators are effector molecules closely associated with the innate immune response and directly exert their biological activities on the responding cells. For example, both CXCL1 and IL-8 chemokines that are produced by periodontal resident cells, especially gingival epithelial cells, are strong chemoattractants that recruit neutrophils to the local gingival tissues upon pathogen invasion (6). However, studies investigating GCF inflammatory mediators that modulate the cytokine/chemokine production network are lacking. An unbiased examination of the inflammatory network characteristics of periodontitis would provide critical insight into more robust stratification strategies of different periodontitis traits.
IL-17 is a signature Th17 cytokine that bridges the innate and adaptive immune responses at mucosal barriers. Although homeostatic IL-17 activity is critical to maintain mucosal barrier integrity and defense, excessive IL-17 activity plays a destructive role in several mucocutaneous inflammatory diseases (7-9). IL-17–mediated inflammatory pathways in chronic periodontitis have not been fully defined. Furthermore, the specific effect of IL-17 on gingival epithelium, which constitutes the first line of defense, and gingival immune populations such as macrophages have remained largely unknown. In an independent, gene-centric, genome-wide association study, we previously identified that potential loss-offunction loci present in the IL-17R adaptor protein gene TRAF3IP2 (or ACT1), which nonredundantly participates in IL-17 signaling pathway, is significantly associated with periodontitis (3). IL-10 is another immune-regulatory cytokine that actively suppresses the secretion of a wide range of proinflammatory mediators in LPS- or IFN-γ– stimulated immune cells (10). This anti-inflammatory activity of IL-10 reduces alveolar bone loss in a ligature-induced murine model (11-13). However, the specific modulatory effect of IL-10 on IL-17–mediated immune responses in immune and nonimmune cell populations present in periodontal tissue has not been well defined.
In this study, we applied a PCA, an unbiased clustering approach, to identify the inflammatory immune networks and key regulators assayed by multiplex GCF analyses. We agnostically identified a periodontitis-associated inflammatory expression trait in the study participants. This inflammatory network highlights the importance of IL-17 as a dominant cytokine for periodontitis. The biological activity of IL-17, which can be modified by IL-10 expression, mimics the critical role of IL-17 that has been reported in other inflammatory diseases (14). We further evaluated the IL-17–mediated inflammatory responses in gingival tissues in both pathogen-induced and ligature-induced alveolar bone-loss models in Il10 knockout (KO) mice and studied the mechanism of IL-17–associated innate immune response in alveolar bone loss in this report. We concluded that IL-10 plays a protective role in periodontitis by dampening an excessive IL-17–mediated inflammatory response majorly through innate immune cells.
Materials and Methods
Participants and samples collections
One hundred and seven white subjects enrolled in the Dental Atherosclerosis Risk in Communities (ARIC) study were included for clinical measurements and multiplex GCF-mediator analyses (15). All subjects provided informed written consent to an approved institutional review board on research in which human subjects are involved at the University of North Carolina at Chapel Hill (UNC-CH) and at each ARIC examination center. The collection of periodontal data, including probing depth, attachment level, gingival index, plaque index, and bleeding upon probing, were described in detail elsewhere (15). Trained and calibrated examiners with >90% agreement performed the periodontal measurements on all sites of full mouth, including third molars (3). GCF samples were collected with PerioPaper Strips from the mesiobuccal region of each first molar or, if missing, an alternate site, as previously described from those 107 participants (16). Those paper strips were flash-frozen chairside and stored in liquid nitrogen until use. Four GCF strips were eluted and analyzed individually for each participant. GCF-mediator concentration data were pooled to calculate a patient mean value in picograms per milliliter.
An additional patient pool of 43 participants provided informed consent and were recruited into this study (17). Eighteen gingival biopsies were obtained from different participants with chronic periodontitis, whereas 25 gingival control samples were harvested from subjects free of periodontitis. The diagnosis criteria of periodontitis were based on 1999 Workshop definition (18). Control samples were collected from either periodontally healthy subjects or the healthy sites that did not present bleeding upon probing of gingivitis patients. The inclusion and exclusion criteria for participation of the clinical study and clinical periodontal measurements were described previously (17). Biopsied gingival tissues were incubated within RNAlater (catalog no. AM7020; Thermo Fisher Scientific, Grand island, NY) overnight at 4°C and then stored at − 80°C until RNA isolation.
Mice
Il10-deficient (Il10−/−) and age- and gender-matched wild-type (WT) C57BL6 mice were generously provided by Dr. R. Balfour Sartor from UNC-CH School of Medicine. Act1(Traf3ip2)−/− and age- and gender-matched WT on C57BL6 background were generous gifts from Dr. U. Sienbenlist at National Institute of Allergy and Infectious Diseases. All components of animal studies were approved by the Institutional Animal Care and Use Committee at the UNC-CH and at the University of Iowa.
Cell culture, peripheral blood monocytic cell isolation, and in vitro differentiation into macrophages
Primary human gingival epithelial cells (pHGEs) were purchased from CELLnTEC (catalog no. HGEPp; CELLnTEC, Research Triangle Park, NC) and maintained in the fully supplemented culture medium (catalog no. CnT-PR; CELLnTEC). Freshly prepared buffy coats were isolated from healthy donors by density gradient centrifugation as described previously (19, 20). Briefly, mononuclear cells from 50 ml peripheral blood were purified with Ficoll Paque PLUS (catalog no. 17-1440-03; GE Healthcare, Pittsburgh, PA)–based density centrifugation. Purified cells were then incubated with magnetic-labeled CD14 beads (catalog no. 130-050-201; Miltenyi Biotec, San Diego, CA) to isolate monocytic cells according to manufacturer’s instruction. Purified monocytic cells from healthy donors were then plated at a density of 0.25 × 105 per well onto a 96-well plate and cultured in RPMI 1640 supplemented with penicillin–streptomycin and 10% FBS. For monocyte-derived macrophage differentiation, M-CSF (catalog no. 300-25-10UG; PeproTech, Rocky Hill, NJ) was added to the culture medium DMEM to achieve a final concentration of 100 ng/ml after monocytes were plated. Culture media supplemented with M-CSF was replenished every 72 h.
Lentiviral short hairpin RNA–mediated gene knockdown in pHGEs
Several bacterial glycerol stocks containing relevant clones targeting human ACT1 (TRAF3IP2) (gene accession no. NM_147200; https://www.ncbi.nlm.nih.gov/nuccore/NM_147200) and a stock containing a scramble short hairpin RNA (shRNA) clone were obtained from the University of North Carolina Lenti-shRNA Core Facility. shRNAs targeting human ACT1 (TRAF3IP2) or scramble clones were prepared with an EndoFree Plasmid Maxi Kit (catalog no. 12362; QIAGEN) and packaged into separate lentiviral vector particles by the Lenti-shRNA Core Facility staff using 293T cells as the host. The titers of those viral vector particles in the supernatant were typically ranging from 1 × 105 to 1 × 108 IU/ml. A total of 1–2 × 106 pHGEs were transduced with those shRNA target or scramble vector particles for 48 h. Two micrograms per milliliter puromycin was used to select successfully transduced cells. Puromycin contained cell culture medium was changed every 48 h. The TRAF3IP2 knockdown (KD) efficiency was evaluated with the immunoblot with a primary anti–ACT1/TRAF3IP2 Ab (catalog no. sc-398161; Santa Cruz Biotechnology, Dallas, TX). Cells exhibiting best KD efficiency were then seeded into 6-well or 24-well plates with downstream cytokine challenge experiments.
Cytokine treatment in cell cultures
PBMCs or PBMC-derived macrophages were seeded onto a 96-well plate with a density of 0.25 × 105 per well. pHGEs, scramble shRNA KD pHGEs, or ACT1 (TRAF3IP2) KD pHGEs were seeded into 6-well or 24-well plates in CnT-PR fully supplemented medium.
Cells were either treated with recombinant human IL-17(A) cytokine (catalog no. 7955-IL-025/CF; R&D Systems, Minneapolis, MN) at a final concentration of 100 ng/ml in the presence or absence of 10 ng/ml recombinant human IL-10 (catalog no. 217-IL-005/CF; R&D Systems) or treated with PBS as a control. After 3 h of treatment, cells were collected for mRNA analysis, whereas the supernatant of the remaining cells after 16 h treatment was collected for protein analysis.
Immunohistochemistry and immunofluorescence
Gingival tissues were fixed with 4% paraformaldehyde overnight. Paraffin-imbedded tissues were section into 5-μm slides, followed by deparaffin and rehydration before heat-induced epitope retrieval was performed with 1× Lab Vision HIER Buffer L (catalog no. TA-135-HBL; Thermo Fisher Scientific). After incubation with either Protein Block Serum-Free (catalog no. X0909; Dako, Carpinteria, CA) solution for 10 min or Thermo Diluent (catalog no. TA-125-ADQ; Thermo Fisher Scientific) for 1 h at room temperature, those tissue slides were then incubated overnight at 4°C with Abs specific for humans. The following primary Abs were used: rabbit anti–IL-17R with a dilution 1:300 (catalog no. SC-30175; Santa Cruz Biotechnology), rabbit anti–IL-17 with a dilution 1:250 (catalog no. SC-7927; Santa Cruz Biotechnology), or mouse anti–ROR-γδ with a dilution 1:200 (catalog no. MABF81; MilliporeSigma, Billerica, MA) in Thermo Diluent. After washing with 1× Tris (pH 7.6) buffer, slides were incubated for 1 h at room temperature with a secondary Ab. The secondary Abs used for coimmunoflorescence include Cy5 donkey anti-rabbit IgG (catalog no. 711-175-152; Jackson ImmunoResearch Laboratories, West Grove, PA) and Cy3 goat anti-mouse IgG (catalog no. 115-165-146; Jackson ImmunoResearch Laboratories). Sections were mounted in a DAPI-containing mounting medium (catalog no. 17985-50; Electron Microscopy Sciences, Hatfield, PA) before being coverslipped. Sections were observed and analyzed by an Olympus BX61 Widefield microscope for immunohistochemistry and LSM 700 Confocal Laser Scanning Microscope (ZEISS, Jena, Thuringia, Germany) for immunofluorescence. No primary Ab incubation was used as a negative control.
Analyses of inflammatory mediator in GCF, cell culture, and serum
GCF samples were assayed for a total of 16 inflammatory mediators, including IL-10, IL-1β, IL-4, IL-5 IL-6, IL-8, TNF-α, G-CSF, CCL3 (MIP1α), CCL4 (MIP1β), CCL5 (RANTES), MCP1, IFN-γ, IL-17, CXCL5 (ENA78), and IL-RA, using a Bio-Plex 200 multiplex format (Bio-Rad Laboratories, Hercules, CA). All reagents for GCF mediator analyses were purchased from R&D Systems (Minneapolis, MN). The measurement procedure that was used for GCF mediator analyses in this study was elaborated elsewhere (21). GCF levels of mediator were log transformed to be carried forward for PCA.
Similarly, the supernatant levels of several mediators in cell cultures were also quantified by a customized detection Ab mixture using the Bio-Plex 200 multiplex system. All reagents, magnetic capture beads, and Ab mixture used for the Bio-Plex were purchased from R&D Systems. Serum level of murine CXCL1 was assessed by ELISA using a Mouse CXCL1 DuoSet ELISA Kit (catalog no. DY453-05; R&D Systems).
RNA isolation, real-time quantitative PCR, and RT2 Profiler PCR Arrays
Total RNA was extracted from human gingival biopsy tissues, mouse gingiva, and cells with an RNeasy Mini Kit (catalog no. 74104; QIAGEN, Valencia, CA). cDNA from 300 ng RNA isolated from human or murine gingival tissues was synthesized by a SuperScript Vilo cDNA Synthesis Kit (catalog no. 11754050; Thermo Fisher Scientific). The following TaqMan quantitative real-time PCR probes were used: IL17(A) (Hs00174383_m1), IL1B (Hs00174097_m1), IL23 (Hs00900828_g1), IL6 (Hs00985639_m1), CCL5 (Hs00982282_m1), IL10 (Hs00961622_m1), CXCL5 (Hs01099660-g1), and GAPDH (Hs03929097_g1) for humans, and Ccl5 (Mm01302427_m1), Il17 (Mm00439618_m1), Csf3 (Mm00438334_m1), Cxcl5 (Mm00436451-g1), Cxcl1 (Mm04207460_m1), and Gapdh (Mm99999915_g1) were assessed for mice. The quantitative real-time PCR was performed in a 7000 Sequence Detection System (ABI Prism; Thermo Fisher Scientific) according to the manufacturer’s instruction. The fold change was calculated using the 2−ΔΔCT method (22). Six hundred-nanogram RNA samples isolated from pHGEs for PCR arrays were reverse transcribed, and the transcriptional level of mediators was determined by a Cytokine and Chemokines RT2 Profiler PCR Array (catalog no. PAHS-150Z; QIAGEN).
P. gingivalis culture and oral challenge regimen
P. gingivalis A7436 grown on Brucella Blood Agar (catalog no. AS-141; Anaerobe Systems, Morgan Hill, CA) under the anaerobic condition at 37°C were harvested and resuspended at 0.5 × 1010 CFU/ml in sterile PBS containing 2% carboxymethylcellulose (CMC) (catalog no. 419273; Sigma-Aldrich, St. Louis, MO). Murine oral challenge was conducted according to the regimen previously described with modifications (23, 24). Briefly, 11- to 12-wk-old WT mice and globally Il10−/− mice, both of which were on C57/B6 background, received antibiotic treatment, with 0.87 mg/ml sulfamethoxazole and 0.17 mg/ml of trimethoprim in drinking water ad libitum, for 10 d to reduce the initial oral bacterial burden, followed by a 3-d antibiotic-free period before oral challenge. WT or Il10−/− mice were orally inoculated with 0.5 × 109 CFU P. gingivalis A7436 daily for 14 consecutive d. Mock inoculated with 2% CMC in PBS were included as controls. A portion of mice were euthanized 2 d after the last dose of oral challenge for gingival mRNA assessment or short-term serum chemokine analysis, whereas the remaining mice were euthanized 42 d after the last P. gingivalis oral challenge for alveolar bone loss analysis and long-term serum mediator analysis.
Murine gingival sample preparation and laser capture microdissection
Laser capture microdissection for dissecting murine gingival epithelium was performed with a PALM MicroBeam system at the University of North Carolina Translational Pathology Laboratory Core Facility under an RNase-free condition. The whole maxilla was fixed with 4% paraformaldehyde overnight before decalcification in 10% EDTA (pH 7.4) for 6 d. All reagents or solutions were prepared under RNase-free condition. The paraffinembedded maxillae were sectioned into 5-μm serial slices that were mounted onto a MembraneSlide NF 1.0 PEN (catalog no. 415190-9081000; ZEISS, Oberkochen, Germany). Epithelial layers of the sliced samples were captured with the PALM MicroBeam system. The captured samples were acquired within an AdhesiveCap (catalog no. 415190-9201-000; ZEISS), and RNA was isolated by a RecoverAll Total Nucleic Acid Isolation Kit (AM1975; Thermo Fisher Scientific) following the manufacturer’s instruction.
Ligature-induced alveolar bone loss model in mice
We followed a well-described method to create the ligature-induced alveolar bone-loss model in mice. The detailed procedure of ligature placement was described elsewhere (25). Eleven days after ligature placement, mice were euthanized by CO2 inhalation. Gingival tissues approximate to where the ligature was placed were harvested and stored in RNAlater, and mouse maxillas were fixed with 4% paraformaldehyde for micro computed tomography analysis.
Gene expression assessment by NanoString
Total RNA that was isolated in the gingival tissues from the age- and gender-matched mice in the ligature model was hybridized with reporter and capture probes for a customized CodeSet of murine IL-17 cytokine pathway (NanoString Technologies, Seattle, WA) as per manufacturer’s instructions. Data were normalized to several housekeeping genes and spiked positive controls. Transcript counts fewer than the mean of the negative control transcripts plus 1 SD for each sample were considered as background. Hybridization from all RNA samples passed the quality control. Normalization and quality control were performed by the nSolver Analysis Software (NanoString Technologies). The transcriptional expression of CodeSet target genes is the direct count of mRNA hybridized to the probes without reverse transcription.
Alveolar bone loss measurement
Fixed mouse maxillas obtained from 42 d after the last P. gingivalis oral inoculation or 11 d after ligature placement were subjected to micro computed tomography scanning for image analysis by a SCANCO microCT40 system (Scanco Medical, Bruttisellen, Switzerland). The distance between cementoenamel junction (CEJ) to the alveolar bone crest (ABC) was measured at 12 predetermined sites, including the mesial and distal buccal root region from the first to the third maxillary molars at both sides, by the MicroView software (Parallax Innovations, Ilderton, Canada) for P. gingivalis-induced bone loss measurements. The measurement was performed as described previously (26). The distance between CEJ to ABC at the distal buccal root region of the mouse first molar (m1) and the mesial buccal root region of the mouse second molar (m2) was measured for ligature-induced bone loss measurements.
Preparation single-cell suspension and flow cytometry analysis of immune cells
Spleen tissues and cervical lymph nodes were collected from Il102/2 and WT mice. Single-cell suspension was prepared by mashing tissues with a syringe plunger through a cell strainer with a pore size of 40 mm. Cells were collected in RPMI media and were stimulated for 3.5 h with 50 ng/ml PMA (catalog no. P8139; Sigma-Aldrich) and 2.5 μg/ml ionomycin (catalog no. 13909; Sigma-Aldrich) in the presence of GolgiPlug (catalog no. 555029; BD Biosciences). After incubation, cells were stained with anti-mouse CD45–Alexa Fluor 700 (catalog no. 56–0451-80; Invitrogen), CD3-PerCP Abs (catalog no. 561089; BD Pharmingen), and LIVE/DEAD Fixable Green Dead Cell Stain Kit (catalog no. L23101; Thermo Fisher Scientific). After fixation, the cells were further stained with anti-mouse IL-17–BV510 (catalog no. 564168; BD Horizon) and ROR-γt–PE (catalog no. 12-6981-80; Invitrogen) Abs.
We followed a published method with modifications to prepare single-cell suspension from murine gingival tissues for profiling innate immune cells (27). Seven days after ligature placement, gingival tissues at the ligature side or contralateral control side without ligature were excised separately from either Il10−/− mice or WT mice. Gingival tissues from the ligature or nonligature side were pooled from three separate animals for each strain of animals for each experiment. Samples were digested in a mixture of 3.2 mg/ml type IV collagenase (catalog no. 17104–019; Life Technologies) and 0.15 μg/ml DNase (catalog no. DN25–100MG; Sigma-Aldrich) for 30 min at 37°C with agitation. Then, a single-cell suspension was further prepared by mashing enzyme-digested gingival tissues with a syringe plunger through a cell strainer with a pore size of 70 μm. Isolated single cells from gingiva were directly stained with anti-mouse CD45-Alexa Fluor 700 (catalog no. 103127; BioLegend), CD11b-VioBright FITC (catalog no. 130–113-805; MACS Miltenyi), F4/80-APC (catalog no. 123115; BioLegend), and Ly-6G–PerCP (catalog no. 127653; BioLegend) Abs and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (catalog no. L-34965; Thermo Fisher Scientific). After fixation, cells were further stained with anti-mouse inducible NO synthase (iNOS)–PE (catalog no. 12-5920-80; Invitrogen) and CD206-BV605 (catalog no. 141721; BioLegend) Abs. Ab-staining panel without iNOS-PE was applied as fluorescence minus one control to facilitate gating. After staining, all samples were run on an LSR II Violet flow cytometer (BD Biosciences) within 16 h.
Anti-IL-17-neutralizing Ab injection in mouse gingiva
A neutralizing mAb to murine IL-17A (catalog no. BE0173; BioCell) or an mAb IgG1antibody with unknown specificity (catalog no. BE0083; BioCell) serving as isotype control was locally injected to separate Il10−/− mice with a 33-gauge removeable needle (catalog no. 7803–05; Hamilton) attached to a microliter syringe (catalog no. 7635-01; Hamilton). Two micrograms of either anti–IL-17 mAb or IgG1 mAb was injected into the palatal gingiva approximate to the m1 and m2 where the ligature was placed. The injection was initiated 1 d before ligature placement and was repeated every other day until 11 d after ligature placement.
Statistical analyses
PCA by the PROC PRINCOMP procedure with SAS v9.4 (SAS Institute, Cary, NC) was used to create GCF periodontal inflammatory traits (PITs). Correlation matrices of inflammatory traits with clinical measurements were created using a SAS PROC CORR procedure.
GCF biomarker levels were dichotomized into high or low categories depending on whether each individual mediator’s measurements were above or below the mean for those 107 subjects. ANOVA was used to compare the extent of probing depth ≥4-mm sites (EPDGE4) or the extent of clinical attachment loss ≥3 mm at the interproximal sites (EALGE3i) among stratified groups. ANOVA was also applied to comparing supernatant inflammatory mediator levels, murine alveolar bone loss, and the serum CXCL1 level in mice. A Student t test was applied to comparing variables between two groups whenever applicable. Linear regression was performed to model the relationship between cytokine ratios and clinical parameters.
We used the Mann–Whitney–Wilcoxon two-sample test to analyze the human gingival mediator transcriptional level, whereas a Kruskal–Wallis test by ranks was performed to compare the transcriptional level of mediators in murine gingiva. The significance threshold was set at p = 0.05 without adjusting for multiple comparisons.
Results
PIT4 is a periodontitis-associated trait
To ascertain a periodontal disease–associating inflammatory phenotype or trait, we first performed a PCA using the multiplex proteomic data of a panel of 16 GCF inflammatory biomarkers in 107 Dental ARIC subjects. The mediators assayed in those subjects include chemokines, common inflammatory cytokines (IL-17, IL-1β, IL-6, and TNF-α), classical Th1 and Th2 cytokines, anti-inflammatory cytokines (IL-10 and IL-1RA), and G-CSF. The loadings of the first six PITs defined by PCA are presented in Table I, and each of them accounts for at least 5% of the overall variance in the aforementioned analytes. PIT1, in which most of the mediators are evenly loaded, explains the highest (~31%) overall mediator variance. The correlations of principle component profile with the constitutive inflammatory mediators are shown in a graphic representation in Supplemental Fig. 1.
Table I.
Component loadings of GCF inflammatory mediators for PITs determined by PCA
| GCF Mediatorsa | PIT1 | PIT2 | PIT3 | PIT4 | PIT5 | PIT6 |
|---|---|---|---|---|---|---|
| IL-10 | 0.159 | 0.172 | 0.456 | −0.196 | −0.015 | −0.067 |
| IL-1β | 0.280 | 0.229 | −0.087 | 0.159 | 0.189 | −0.425 |
| IL-4 | 0.139 | 0.451 | −0.225 | 0.055 | 0.307 | 0.284 |
| IL-5 | 0.137 | −0.384 | −0.004 | −0.252 | 0.546 | 0.082 |
| IL-6 | 0.283 | −0.308 | −0.004 | 0.041 | 0.231 | −0.014 |
| IL-8 | 0.338 | 0.124 | −0.119 | −0.062 | −0.261 | −0.288 |
| TNF-α | 0.335 | −0.097 | −0.330 | −0.087 | 0.097 | −0.015 |
| G-CSF | 0.307 | 0.292 | −0.003 | 0.149 | 0.3462 | −0.200 |
| CCL3 | 0.343 | −0.097 | −0.046 | 0.114 | −0.322 | −0.078 |
| CCL4 | 0.352 | −0.210 | −0.017 | 0.007 | −0.343 | 0.082 |
| CCL5 | 0.139 | −0.110 | 0.115 | 0.599 | 0.047 | 0.454 |
| CCL2 | 0.313 | −0.245 | −0.0008 | −0.154 | −0.017 | 0.350 |
| IFN-γ | 0.073 | 0.395 | −0.359 | −0.099 | −0.201 | 0.447 |
| IL-17 | 0.119 | 0.104 | 0.488 | 0.436 | 0.006 | 0.034 |
| CXCL5 | 0.094 | 0.252 | 0.386 | −0.391 | 0.135 | 0.244 |
| IL-1RA | 0.264 | 0.079 | 0.280 | −0.294 | −0.199 | 0.049 |
| Portion of variance explained | 0.310 | 0.138 | 0.104 | 0.078 | 0.062 | 0.060 |
PCA was applied to create PITs by using the multiplex proteomic data containing 16 inflammatory mediators from the GCF samples of 107 ARIC subjects. Only first six PITs are shown, because each of the trait explains at least 5% of the overall mediator variance.
By performing a correlation matrix analysis, we identified that only PIT4 is positively and significantly associated with almost all clinical disease parameters reflected by the EPDGE4 (p = 0.0004), EALGE3i (p = 0.004), the extent of bleeding upon probing (p = 0.0009), or the extent of gingival index ≥1 (p = 0.014; Table II). We also observed that PIT4 is characterized by a high positive loading of IL-17 and CCL5 and high negative loadings of IL-10, CXCL5, IL-5, and IL-1RA with the loading of each of the mediators close to or above 0.2 (Table I). To delineate the diseaseassociating cytokine signature, we compared the cytokine loadings between PIT4 and PIT3, a trait that is not associated with periodontitis. We found that the loadings of CXCL5, IL-1RA, and IL-10 are still high but positively correlated to PIT3, whereas the loading of IL-17 remains almost the same. It suggests that the inverse association between IL-17 and CXCL5, or anti-inflammatory cytokines IL-10 or IL-1RA determines the clinical periodontitis phenotype.
Table II.
Correlation matrix of PITs with clinical measurements of disease
| Clinical Measuresa | PIT1 | PIT2 | PIT3 | PIT4 | PIT5 | PIT6 |
|---|---|---|---|---|---|---|
| EPDGE4 | 0.011 | 0.066 | −0.116 | 0.335 | 0.188 | −0.128 |
| p value | (0.909) | (0.500) | (0.237) | (0.0004) | (0.053) | (0.189) |
| EALGE3i | −0.010 | 0.0187 | −0.008 | 0.336 | 0.1947 | −0.12921 |
| p value | (0.915) | (0.849) | (0.936) | (0.0004) | (0.0445) | (0.1847) |
| EBL | 0.091 | 0.121 | −0.048 | 0.316 | 0.219 | −0.008 |
| p value | (0.354) | (0.216) | (0.623) | (0.0009) | (0.023) | (0.935) |
| EPQGE1 | 0.061 | −0.076 | 0.033 | 0.186 | 0.166 | −0.003 |
| p value | (0.556) | (0.459) | (0.750) | (0.069) | (0.105) | (0.974) |
| EGIGE1 | 0.087 | −0.136 | −0.035 | 0.260 | 0.204 | −0.009 |
| p value | (0.420) | (0.205) | (0.747) | (0.014) | (0.055) | (0.931) |
Correlation coefficients and p values are shown in the table; boldface indicates statistically significant (p < 0.05) correlations and PIT4.
EBL, extent bleeding score; EPQGE1, extent plaque score ≥1; EGIGE1, extent gingival index score ≥1.
IL-10 antagonizes IL-17 in determining clinical disease phenotype of periodontitis
Next, we evaluated whether mediator IL-17, a cytokine that was positively correlated with PIT4, or cytokines that were negatively correlated with PIT4 including CXCL5, IL-10, orIL-1RA had an impact on clinical disease activity alone or in combination. We found that although subjects with high (above mean) GCF IL-17 levels tended to have more sites with deep pockets or advanced attachment loss than subjects with low (below mean) IL-17 level, the difference was not statistically significant (Fig. 1A, 1B). Similarly, the GCF IL-10 level was not associated with the extent of clinical disease activities as reflected by EPDGE4 (p = 0.14; Fig. 1C) and EALGE3i (p = 0.412; Fig. 1D). Neither the dichotomized CXCL5 nor IL-1RA levels alone were associated with the clinical measurements (data not shown). Then, we interrogated the interactions between high or low IL-17 level and the levels of other three mediators in determining clinic disease severity. In this study, we show that only subjects with high IL-17 and low IL-10 levels presented with significantly more severe clinical disease, as reflected by EPDGE4, than subjects with low or high levels of both cytokines (p = 0.011 for low IL-17 and low IL-10; p = 0.014 for high IL-17 and high IL-10) or subjects with high IL-10, but low IL-17 levels (p = 0.016; Fig. 1E). Similarly, only subjects with the high IL-17 and low IL-10 combination had significantly higher EALGE3i scores, as compared with other groups (Fig. 1F). The GCF IL-17/IL-10 ratio was positively and significantly correlated with EPDGE4 (R2 = 0.052, p = 0.018; Fig. 1G) and with EALGE3i (R2 = 0.074, p = 0.005; Fig. 1H). The interactions between high or low IL-17 and CXCL5 were not significantly associated with the clinical disease (Supplemental Fig. 2A, 2B). However, the interactions of a high IL-17 and low IL-1RA levels were associated with significantly more disease sites (Supplemental Fig. 2C, 2D). These data suggest that IL-10 or IL-1RA may act as a key negative regulator in attenuating the excessive IL-17 activity to reduce clinical expression of periodontitis. Because the anti-inflammatory activity of IL-1RA has been described to be mostly passive in blocking IL-1R, we decided to focus on the modulatory effect of IL-10 on IL-17 signaling (28).
FIGURE 1.
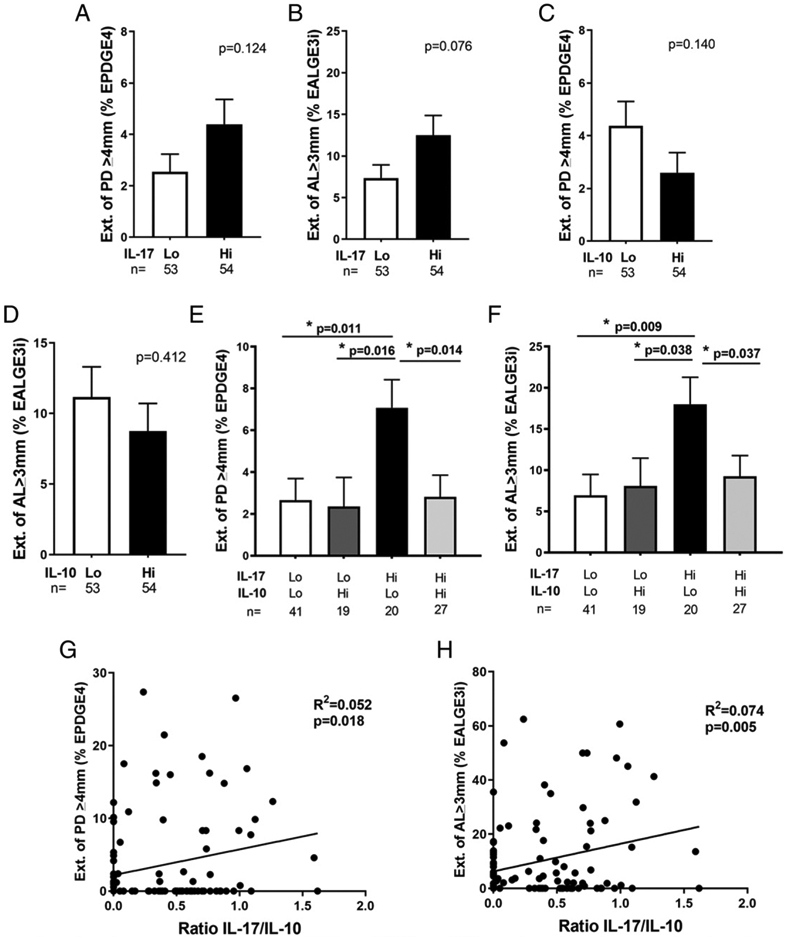
GCF levels of IL-17 interacted with IL-10 levels to determine the periodontal disease severity. The mediator levels of 107 clinical participants were dichotomized into low (Lo) or high (Hi) groups based on whether the log10-transformed GCF IL-17 or IL-10 levels exceeded the mean level of either cytokine of those participants. Extent of periodontal sites with probing depth ≥4 mm (A) or with attachment level ≥3 mm at interproximal sites was compared in Lo IL-17 to Hi IL-17 groups (B). The same comparisons of probing depth and attachment level were also compared in Lo IL-10 to Hi IL-10 groups (C and D). Clinical disease activity reflected by EPDGE4 (E) or attachment level at the interproximal sites ≥3 mm was compared among groups determined by the interactions between Lo or Hi IL-17 and IL-10 levels (F). GCF IL-17/IL-10 ratio was correlated to EPDGE4 (G) or of attachment level at the interproximal sites ≥3 mm (H). Data are shown as mean ± SE and were analyzed by either Student t test for two group comparisons or ANOVA for multigroup comparisons. Linear regression was applied for the correlation analysis. *p < 0.05.
IL-17 and its upstream signal cytokines are elevated in gingival tissues with periodontitis
We further assessed the transcript levels of IL-17 and IL-10 in gingival biopsies in an independent patient pool to validate our findings with the ARIC dataset. The transcript levels of IL-17 were significantly higher in periodontitis gingival tissues than in samples free of periodontitis (p = 0.042; Fig. 2A). The levels of IL-1B and IL-6, both of which are essential for Th17 differentiation, were also significantly higher in periodontitis patients (p = 0.046 and p = 0.0073, respectively; Fig. 2B, 2C). The transcript levels of IL-23 were not different (p = 0.477; Fig. 2D). Then, we examined the transcript levels of chemokine CCL5 and CXCL5 (Fig. 2E, 2F), which are downstream chemokines of the IL-17 signaling axis. Higher levels of CCL5 were observed in periodontitis samples (Fig. 2E). We also found that the IL10 mRNA levels in periodontitis samples were higher than the periodontitis-free group with a marginal p value (Fig. 2G). Notably, the ratio of IL-17/IL-10 was elevated in periodontitis gingival tissues than in control specimens (Fig. 2H).
FIGURE 2.

Comparisons of the transcriptional levels of IL-17 and IL-17–associated cytokines and chemokines in clinical gingival biopsies between periodontitis patients and subjects free of periodontitis. qRTPCR was performed to compare the mRNA levels of IL17 (A), Th17-differentiation upstream cytokines, IL-1B (B), IL-6 (C), and IL-23 (D) and IL-17–regulated chemokines, CCL5 (E) and CXCL5 (F), from periodontitis gingival biopsies (n = 18) to gingival tissues harvested from periodontitis-free subjects (n = 25). mRNA level of IL10 was also compared between two groups of gingival samples (G). The mRNA ratio of IL-17/IL-10 was compared between periodontitis gingival samples and periodontitis-free tissues (H). Expression of IL-17R in human gingival tissues is shown in periodontal health (I) and periodontitis lesion (J) by immunohistochemistry staining. The transcription factor specific for IL-17–secreting cells, ROR-γt (red), and IL-17 (green) expression stained by coimmunofluorescence are shown in the gingival tissues from periodontitis-free (K) and periodontitis lesion (L). Box plots show the median, interquartile range, maximal, and minimal values except (H), which shows mean ± SE. Nuclei were stained with DAPI (blue). Mann–Whitney U test was applied for comparisons of mediator at the mRNA level, whereas Student t test was used to compare the ratio of IL-17/IL-10 between periodontitis and disease-free tissues. *p < 0.05.
Using immunohistochemistry, we found that IL-17R was positively stained in several cell populations, including gingival epithelium from both periodontitis and periodontitis-free tissues (Fig. 2I, 2J). Immune cell infiltrates in the connective tissue compartment, especially from periodontitis biopsies, were also positive for IL-17R staining. We observed that IL-17+ROR-γt+ cells are more abundantly present in the inflammatory infiltrates from the gingival tissue with chronic periodontitis (Fig. 2K, 2L).
Effects of IL-17–induced chemokine expression in epithelial and monocytic cells
Because gingival epithelium appears to be a major target for IL-17, we next evaluated the biological effects of IL-17 on gingival epithelial cells. We profiled the transcriptional activation of IL-17– stimulated chemokine production in pHGEs. We used shRNA-expressing vectors to KD ACT1 expression and, therefore, to disrupt IL-17 signaling (Fig. 3A). We compared the difference in fold induction of chemokine transcription in IL-17–stimulated control scramble shRNA-transduced pHGEs to that of ACT1-KD pHGEs. We found that the transcription of several chemokine genes, including CCL7, IL8, IL23A, CXCL5, CXCL1, and CCL2, were reduced ~50% in ACT1-KD pHGEs as compared with scramble transduced control cells (Fig. 3A). We further confirmed that IL-17–stimulated pHGEs significantly increased CXCL1 (Fig. 3B) and CXCL5 secretion (Fig. 3C), whereas the supernatant IL-1RA level was not affected by IL-17 treatment (Fig. 3D). We also confirmed that inhibition of ACT1 expression by shRNA in pHGEs indeed drastically reduced supernatant level of chemokine CXCL1 (Fig. 3E). These data indicate that human gingival epithelial cells respond to IL-17 treatment by increasing chemokine mediator production in vitro.
FIGURE 3.

Inflammatory responses of the IL-17 signaling in pHGEs. The mRNA induction fold upon IL-17 stimulation was compared between ACT1 KD and mock KD pHGEs (A). Expression of ACT1 was knocked down by shRNA-mediated lentivirus transduction. Cells with ACT1 KD were challenged by IL-17 (100 ng/ml) for 3 h. The fold changes upon IL-17 stimulation of a panel of chemokine and cytokine mRNA levels in relation to PBS-treated, scramble shRNA–transduced pHGEs cells were determined by RT2 Profiler and indicated in blue bars, whereas mRNA inductions of IL-17–challenged pHGEs that were transduced by ACT1-specific shRNA lentivirus in relation to PBS-treated cells were indicated by orange bars. ACT1 KD efficiency in pHGEs mediated by two shRNA species is shown by Western blotting (lower panel). Expression levels of CXCL1 (B), CXCL5 (C), and IL-1RA (D) in the supernatant of pHGEs upon IL-17 treatment were determined by multiplex and compared with mock treatment (PBS) without IL-17. CXCL1 expression levels in the presence or absence of IL-17 stimulation were also measured in the supernatant of pHGEs in which ACT1 was knocked down by shRNA1 and compared with cells transduced with scramble shRNA (E). Data of multiplex protein expression in pHGEs were from one representative of three independent experiments. Student t test was applied for mediator comparison among different treatments. Mean ± SE was shown for multiplex data. **p < 0.01.
Because innate immune cells are also a potential target of IL-17 (Fig. 2J), we further assessed the inflammatory response of PBMCs and PBMC-derived macrophages to IL-17 stimulation with or without IL-10. IL-17 treatment alone significantly enhanced the production of CXCL5 in monocytes or macrophages whereas the addition of IL-10 significantly attenuated the CXCL5 secretion in the supernatant that was stimulated by IL-17 in both cell types (Fig. 4A, 4D). The same trend was also observed in monocytes or PBMC-derived macrophages for CXCL1, which is a classical IL-17 downstream effector chemokine (Fig. 4B, 4E). Supernatant levels of IL-1RA were not affected by IL-17 alone or cotreatment with IL-10 in both cell types (Fig. 4C-F).
FIGURE 4.

Inflammatory responses of the IL-10–modulated IL-17 signaling in PBMCs and PBMC-derived macrophages. Supernatant levels of CXCL5 (A), CXCL1 (B), and IL-RA (C) from peripheral blood monocytic cells challenged by IL-17 (100 ng/ml), IL-10 (10 ng/ml), or IL-17, and IL-10 cotreatment were measured by multiplex and compared among different treatments. Similarly, levels of those mediators from cytokine-treated, PBMC-derived macrophages were also compared among different treatments (D–F). Two independent experiments of stimulating peripheral blood monocytic cells that were isolated from two donors were performed. Data from one representative experiment were shown. ANOVA was applied for mediator comparison among different treatments. Mean ± SE was shown for multiplex data. **p < 0.01.
IL-17–mediated inflammation response is elevated in a P. gingivalis–induced alveolar bone-loss model in Il10 KO mice
We isolated immune cells from cervical lymph nodes from Il10−/− and WT mice to compare the steady-state local IL-17 response by flow cytometry. We found that more IL-17–secreting T cells (CD45+CD3+ROR-γt+IL-17+) were present in Il10−/− mice than WT mice (2.89% versus 0.45%, Fig. 5A). Similarly, more IL-17–secreting T cells were found in Il10−/− mouse spleen than in WT mouse spleen (Supplemental Fig. 3). These results indicate that the baseline IL-17 activity is elevated in Il10−/− mice.
FIGURE 5.

Transcription of IL-17–mediated inflammatory genes in gingival tissues in a P. gingivalis–induced murine alveolar bone-loss model. Flow cytometry plots show the percentages of IL-17–secreting cells (ROR-γt+IL-17+) on previously gated T cells (CD45+CD3+) isolated from WT (A, left panel) and Il10−/− mouse cervical draining lymph nodes (A, right panel) at baseline. Data were from one representative of three separate experiments. Alveolar bone loss from WT and Il10−/− mice that were orally challenged with either sham (CMC) or P. gingivalis is illustrated by the distance between CEJ (outlined by dashed blue line) and ABC (outlined by red line) in representative μCT scans (B, left panel) and compared among different groups (B, right panel). mRNA levels of Il17 (C) and IL-17–associated inflammatory genes, Cxcl1 (D), Ccl5 (E), Csf3 (F), and Cxcl5 (G) in gingival tissues were compared among different groups. Serum levels of CXCL1 from mice 2 d after the last P. gingivalis challenge (H) and 42 d after last challenge (I) were determined by ELISA. mRNA levels of Cxcl1, Cxcl5, and Csf3 from the laser-captured murine gingival epithelia were compared among different groups (J–L). Each experiment was repeated at least twice. Data were pooled for analysis. Each dot represents one sample for mRNA analysis. ANOVA was used to analyze bone loss and serum level of CXCL1, which was shown as the means ± SD. mRNA data were analyzed by Kruskal–Wallis test. *p < 0.05, **p < 0.01.
To delineate the in vivo role of the IL-10-modulated IL-17 inflammatory responses, we used a well-described, pathogen-induced periodontitis murine model to compare the alveolar bone loss and the IL-17–mediated local inflammatory response of Il10-null mice to WT controls. The alveolar bone loss was significantly more in the P. gingivalis A7436–challenged Il10−/− mice than in the P. gingivalis–challenged WT mice (Fig. 5B). Interestingly, Il10−/− mice showed significantly more bone loss than WT controls even under the mock challenge condition by CMC. Il10−/− mice were more prone to P. gingivalis–induced periodontitis than were the WT mice (Fig. 5B). In Il10 KO mouse gingival tissues, mRNA levels of Il17, Cxcl1 and Ccl5, and Csf3 were all significantly higher in Il10 KO mice upon P. gingivalis challenge than in WT mice under the same treatment (Fig. 5C-F). Cxcl5 was also increased in Il10−/− mouse gingiva upon P. gingivalis challenge but did not reach statistical significance (Fig. 5G). In addition, the transcription of Il17 was also significantly higher in the mock-challenged Il10−/− mouse gingiva than in the WT mice, which accompanied a phenotype of significantly more bone loss in Il10−/− mice than the WT mice under CMC treatment (Fig. 5B). Systemically, Il10−/− mice had a significantly highly serum level of CXCL1, a surrogate marker of IL-17, than did WT mice in both a short period (2 d) or a long period (42 d) after P. gingivalis challenge (Fig. 5H, 5I). These data suggest that IL-10 thwarts an excessive IL-17–mediated hyperinflammatory network that is pathogenic for the P. gingivalis–induced periodontal bone loss.
Because our ex vivo data clearly indicate the gingival epithelial cells were among the target of IL-17 and IL-10 may regulate inflammatory responses in nonimmune cells such as epithelial cells (29, 30), we also investigated the in vivo modulatory role of IL-10 in IL-17 response in gingival epithelium. There was no difference in Cxcl1, Cxcl5, or Csf3 at the transcriptional level in the laser-captured gingival epithelium among those groups (Fig. 5J-L). These data suggest that the role of IL-10 in vivo to prevent an excessive IL-17–associated inflammatory damage in the periodontal tissue is not modulated through gingival epithelial cells.
IL-17 is elevated in a ligature-induced alveolar bone-loss model in Il10 KO mice
We next determined the role of IL-10 in modulating IL-17–mediated inflammatory response in a Iigature-induced bone-Ioss modeI, which was more rapidIy progressing and destructive than the pathogen-induced bone Ioss. The bone Ioss in the mesiaI of the m2 from the Il10−/− mice was significantly more than in that of the WT control mice (Fig. 6A, 6B). The ligature-induced bone loss in the m2 from Act1−/− mice was significantly less than Il10−/− mice (p = 0.015), whereas a trend of more bone loss in Act1−/− mice than in WT controls was present (p = 0.066; Fig. 6B). These data indicate that IL-10 plays a role in preventing the Iigature-induced aIveoIar bone Ioss. The data also suggest that homeostatic, but not excessive, IL-17 activity contributes to the periodontal health. Using a customized IL-17 NanoString panel, we identified that Il17 transcripts from the Il10 KO mouse gingiva were significantIy more than those of WT mice (Fig. 6C). Similarly, the transcriptional levels of Ccr2, a receptor gene for a monocyte chemotaxis-associated chemokine Ccl2, and Rank, an osteocIast differentiation and activator marker, were also significantly more in Il10−/− mouse gingiva than in WT gingival tissues in the ligature model (Fig. 6D, 6E). The fold changes with p values of Il10 KO mouse gingiva of all 53 CodeSet target genes in relation to WT controls are shown in a volcano plot (Supplemental Fig. 4).
FIGURE 6.
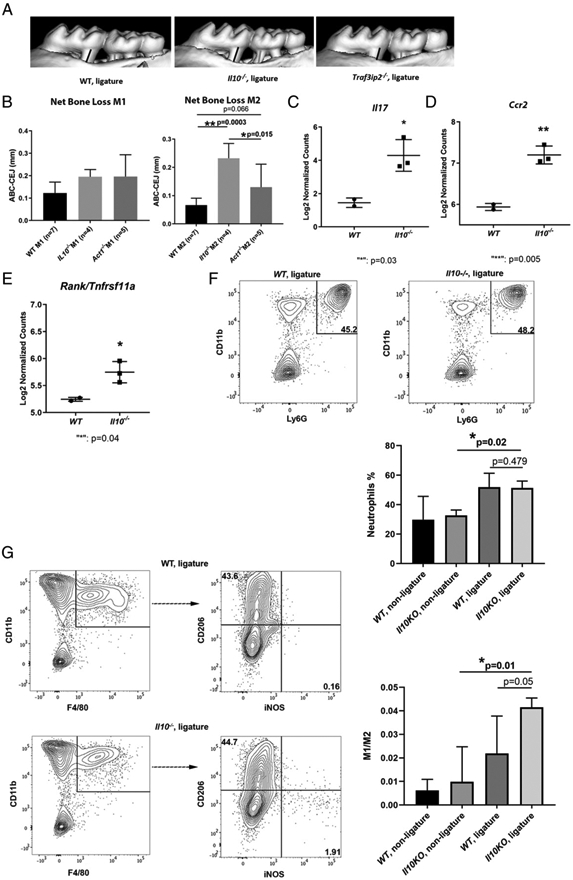
Alveolar bone loss and IL-17–mediated inflammatory response in gingival tissues in a ligature-enabled plaque accumulation-induced murine periodontitis model. A ligature was placed between the m1 and m2 on the right side and kept for 11 d. mCT measurements were performed between the CEJ to ABC from the distal root of the m1 and the mesial root of the m2 on the right side of maxilla after ligature placement. Representative μCT scans are shown (A). The black bar indicates the distance between CEJ of the distal root of m1 to ABC, whereas the gray bar indicates the distance between CEJ of the mesial root of m2 to ABC. The bone level of the distal root of m1 and mesial root of m2 was compared among WT, Il10 KO, and Act1 KO mice (B). Transcriptional expressions of Il17 (C), chemokine receptor Ccr2 (D), and bone resorption signal Rank/Tnfrsf11a (E) were compared between WT and Il10 KO mouse gingiva from the ligatured site by NanoString gene expression analysis. Direct mRNA counts were normalized first by nSolver software and then log2 transformed for comparison. Representative flow cytometry plots show the percentage of neutrophils (CD11b+F4/80−Ly-6G+) that were previously gated on CD45+cells from the ligature side of WT gingiva (F, left panel) and the ligature side of Il10−/− mouse gingiva (F, right panel). The comparisons of neutrophil infiltration in murine gingiva are also shown (F, lower panel). Representative flow cytometry plots show the percentage of M1-skewed (CD45+CD11b+Ly-6G−F4/80+iNOS+CD206−) and M2-skewed (CD45+CD11b+Ly-6G−F4/80+iNOS−CD206+) macrophages from the ligature side of WT mouse gingiva (G, upper panel) and the ligature side of Il10−/− mouse gingiva (G, lower panel). The ratios of M1/M2 were compared among different groups of gingival tissues (G, right panel). ANOVA test was applied for bone level comparisons and flow cytometry data, whereas Student t test was applied for mediator expression. Bone loss was shown as the means ± SD. Samples were pooled from two independent experiments of the ligature-induced periodontitis model for bone level analysis. Each dot or square indicates one sample for mediator transcriptional expression analysis. For flow cytometry analysis, gingival tissues from the ligature side or the nonligature side were pooled from three animals with the same genotype for each experiment. Three independent experiments were included for statistical analysis. Gingival tissues from the contralateral side of the maxilla where no ligature was placed were used as controls. *p < 0.05, **p < 0.01.
Elevated M1/M2 gingival macrophage phenotype in the ligature model of Il10−/− mice
We further investigated the mechanism of how the excessive IL-17 signals in Il10 KO mouse gingiva are associated with more bone Ioss. We compared the neutrophiIs and the polarization of macrophage differentiation reflected by M1/M2 in the ligature model of Il10 KO mice to WT mice. The flow cytometry anaIysis of ceIIs isoIated from mouse gingiva showed that the percentage of neutrophiIs, which were gated by CD45+ CD11b+F4/80− Ly-6G+, in the ligature side of Il10−/− mice was not significantly different from the neutrophil recruitment in WT mouse gingiva (Fig. 6F). CeIIs isolated from the contraIateraI gingiva, where no Iigature was pIaced, were incIuded as controIs. We then compared the ratio of M1 (CD45+CD11b+Ly-6G−F4/80+iNOS+CD206−) and M2 (CD45+CD11b+Ly-6G−F4/80+iNOS−CD206+) macrophage phenotypes between Il10−/− and WT mouse gingivae. We found that a higher M1/M2 ratio in the ligature side of Il10−/− mouse gingiva than the ratio from the Iigature side of WT gingivaI tissue with a borderline significance (p = 0.05; Fig. 6G). In summary, alveolar bone loss in the ligature model from Il10−/− mice was associated with a more pronounced M1-skewing proinflammatory macrophage phenotype promoted by a hyper–IL-17 response in comparison with the WT mice.
Inhibition of IL-17 alleviates the ligature-induced alveolar bone loss in Il10−/− mice
We injected neutralizing IL-17 Ab in the palatal gingiva where the ligature was placed in Il10−/− mice and compared the bone loss level to Il10−/− mice that received isotype control Ab injection. We found that the ligature-induced bone loss at the m2 mesial root of mice that received anti–IL-17 Ab injection was significantly less as compared with mice that received control Ab injection (Fig. 7).
FIGURE 7.
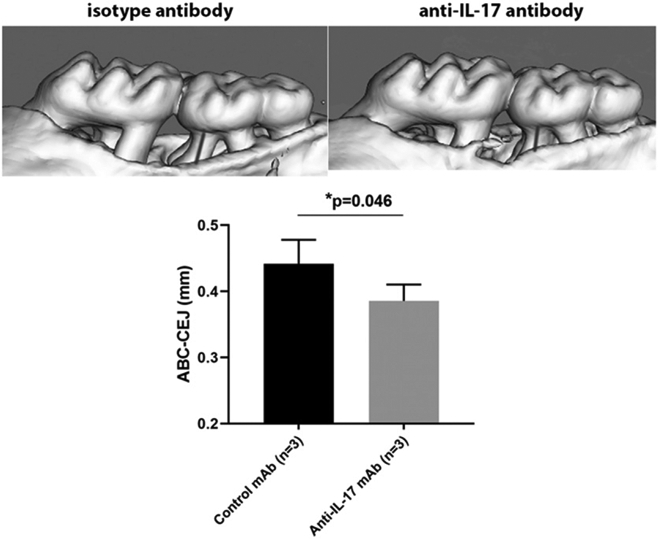
Local effect of anti–IL-17 mAb in the alveolar bone loss in a ligature-induced periodontitis model. Two micrograms of anti–IL-17 mAb or isotype IgG1 Ab was locally injected into the palatal gingival of Il10−/− mice. The injection was repeated every other day until the end of the ligature model. Representative mCT scans are shown. The gray bar indicates the distance between the CEJ to ABC in the mesial root of the m2 (upper panel). The bone level of the mesial root of m2 was compared between Il10−/− mice received isotype control Ab and anti-IL-17 Ab injections (lower panel). Bone loss was shown as the mean ± SD. Student t test was applied. *p < 0.05.
Discussion
In this study, we identified a periodontitis-associated inflammatory network, PIT4, using a PCA approach, in a well-defined clinical population and found that the interaction between IL-10 and IL-17 within the PIT4 network determines clinical disease phenotypes. Using two alveolar-bone-loss murine periodontitis models, we report that IL-10 dampens an excessive IL-17–mediated inflammatory response in preventing periodontitis. We further found that such an inhibitory anti-inflammatory activity of IL-10 appears to be mediated through an innate immune cell population rather than through gingival epithelial cells. Bone loss associated with an excessive IL-17 signaling is mediated through a proinflammatoryskewing gingival macrophage phenotype, as reflected by an increased M1/M2 ratio, in the absence of IL-10.
Studies associating single inflammatory mediators with periodontitis have generated conflicting findings (31). Our data also suggest that neither IL-17 nor IL-10 alone was associated with clinical disease. Homeostatic secretions of cytokines, chemokines, growth factor, and anti-inflammatory mediators are orchestrated in networks to protect periodontal health. Disrupting the harmony of those mediators may contribute to disease initiation or progression. We hypothesize that the clinical expression of periodontitis is associated with a definable signature cytokine/chemokine secretion network. Most chemokines and G-CSF included in the GCF mediator panel can be upregulated by IL-17 signaling in epithelial cells. Compared with traits that are not associated with clinical disease, PIT4 is characterized by the inverse loadings of inflammatory cytokine IL-17 and anti-inflammatory cytokines IL-10 and IL-1RA. We prioritized the modulatory effect of IL-10 on IL-17–mediated inflammatory response in this study. The anti-inflammatory activity of IL-10 usually involves de novo synthesis of molecules that actively participate in the anti-inflammatory processes (32). In addition, IL-10 can stimulate IL-1RA production (33). Therefore, the anti-inflammatory effect of IL-1RA could be mediated through IL-10 signaling.
The data that a high GCF IL-17 level combined with a low IL-10 production was significantly associated with a more prevalent disease phenotype echo previous studies suggesting that a high Th17/regulatory T cell ratio was associated with periodontitis (34). In our cytokine transcriptome analysis of clinical samples, we also found that the IL-17/IL-10 ratio was nonsignificantly increased in periodontitis patients. The mRNA level of IL-10 was also nonsignificantly increased in periodontitis tissues. In the GCF proteomic analysis, the clinical phenotype was defined by the prevalence or extent of periodontitis, which are continuous variables, whereas in the transcriptome analysis, the clinical phenotype was dichotomized as either periodontitis or nonperiodontitis. However, the increased IL17/Il10 ratio in diseased gingiva supports the proteomic findings in the ARIC subject pool (Fig. 1G, 1H). In addition, an increased Th17 response and a dampened IL-10–producing regulatory T cell response were associated with rheumatoid arthritis, another common inflammation disease that is mechanistically associated with periodontitis (35).
The main target cell populations of IL-17 are nonimmune cells. For example, IL-17 stimulates the expression of chemokines molecules CXCL1/2/5, IL-8/CXCL8, CCL2, granulopoietic growth factor G-CSF, and antimicrobial peptides in epithelial cells and keratinocytes (29, 30). However, very few studies have documented the activity of IL-17 in innate immune cells (36). Our ex vivo data indicated that innate immune cells can readily respond to IL-17 stimulation by producing chemokines. Although homeostatic physiological IL-17 production protects the mucosal barrier by walling off pathogens through engaging neutrophils, excessive secretion of this cytokine plays a destructive role in several mucocutaneous inflammatory diseases such as psoriasis and systemic lupus erythematosus (37, 38). Excessive IL-17 production has also been reported in periodontitis gingival tissues and such a hyper–IL-17 response is triggered by a dysbiotic microbiome (39). Dutzan et al. (40) identified an expansion of Th17 cells in the gingival tissue in periodontitis patients and mechanistically associated with periodontal disease. Our data also suggested that more IL-17–producing cells are present in the immune infiltrate of periodontitis-involved gingiva than periodontitis-free tissues. On the contrary, IL-10 possesses a potent anti-inflammatory activity to suppress the expression of several proinflammatory mediators such as TNF-α, IL-6, or IL-1 (41). Our data indicate that IL-10 can directly antagonize the IL-17–stimulated chemokine production in monocytes and macrophages. The mechanism of how IL-10 attenuated IL-17–mediated inflammatory response in PBMCs or macrophages was not explored in this study. Several studies have shown that SOCS3-mediated NF-κB deactivation is used by IL-10 as a mechanism to suppress TLR4-mediated inflammatory mediator synthesis (42).
More IL-17–secreting T cells present in Il10 KO spleen and draining cervical lymph nodes than WT mice at the steady-state level echo the human subject data that IL-10 prevents an exaggerated IL-17 response. To further delineate the modulatory effect of IL-10 on an IL-17–mediated inflammatory network, we analyzed the local transcriptional expression of Il17 and its downstream effector mediators in Il10 KO gingiva upon P gingivalis oral challenge. Significantly higher transcriptional levels of Il17 and its downstream molecules in Il10−/− mice support the hypothesis that IL-10 at the tissue level antagonizes IL-17–mediated inflammatory responses and plays a suppressive role in pathogen-induced periodontal inflammation. Previous studies have reported that IL-10 reduces alveolar bone loss in murine models and such a protective role of IL-10 for bone loss is IL-1 independent (13, 24). Our pathogen-induced bone loss data are supported by a previous report by Moretti et al. (43) demonstrating that the expression level of IL-17(A) was elevated in Il10−/− mice throughout the course of P. gingivalis infection. However, they observed that another IL-17 cytokine family member, IL-17F, possesses a protective role against bone loss in the P. gingival oral infection model. Although we did not assess the role of IL-17F in our two animal models, the signaling of IL-17F in gingival tissue may explain why the bone loss was not protected in Act1 KO mice in our ligature-induced alveolar bone loss in comparison with WT animals (Fig. 6B). It is likely because the ablation of ACT1, a universal adaptor for all IL-17 family cytokine receptors, disrupts not only IL-17 (IL-17A), but also IL-17F downstream signaling that is potentially protective in the ligature-induced inflammation. Additionally, homeostatic IL-17(A) activity itself appears to be necessary to prevent plaque-induced alveolar bone loss, because more bone loss was observed in Il17−/− mice than in WT control mice upon P. gingivalis oral infection (44). Based on our in vivo data, we concluded that the off-balance between IL-17 and IL-10 in Il10-null mice fosters an excessive IL-17 and IL-17–mediated downstream inflammatory milieu that instigates the progression of periodontitis. Interestingly, the intervention that solely blocks IL-17 activity appears to be ineffective in ameliorating colitis in mice, further suggesting the complex role of IL-17 in inflammatory diseases (9, 45).
In the ligature-induced periodontitis murine model, the progressive alveolar bone loss is closely associated with elevated innate immune cell infiltrates in the gingival tissue (25). In Il10 KO mice, the significantly higher expression of Ccr2 (Fig. 6D), a chemokine receptor for monocyte recruitment, in the ligature model suggested that macrophages play a central role in alveolar bone resorption associated with an excessive IL-17 signaling (46). In this study, we directly quantified the gingival macrophage plasticity, as reflected by the ratio of M1/M2 macrophage polarization, in situ in animals with or without IL-10. The imbalance of M1/M2 ratio is a hallmark mechanism of certain inflammatory diseases (47, 48). Higher M1/M2 ratios in the isolated gingival cells from the ligature model indicated a more M1-skewing macrophage phenotype in Il10−/− mice. In addition, IL-10 has been described as a key cytokine in M2-like macrophages polarization (49). In this report, we did not observe a higher percentage of neutrophil recruitment in the ligature model in Il10−/− mice than the WT mice. Previous studies demonstrated that IL-17 is a critical neutrophil recruitment upstream signal and indispensable for the neutrophil-engaged protection against oral Candida albicans infection (50, 51). These data suggest that, in a nonspecific pathogen-induced periodontitis model, the excessive IL-17 signal in the absence of IL-10 specifically promotes a proinflammatory M1 macrophage response that contributes to an exaggerated bone loss. The reversal of extensive bone loss by locally blocking IL-17 cytokine further confirms the essential role of excessive IL-17 in alveolar bone resorption in the ligature-induced periodontitis model.
Both bone-loss models clearly demonstrated IL-17–mediated inflammatory network was elevated in Il10−/− mouse gingiva. This elevated inflammatory network was also associated with a more bone-loss phenotype in Il10−/− w inflammation in the ligature model when IL-10 signaling is abolished.
Our ex vivo data suggest that IL-17 can stimulate innate immune cells to produce chemokines, including CXCL1 and CXCL5, whereas IL-10 dampens this IL-17–mediated inflammatory response. The data also demonstrate that gingival epithelial cells are among the major target cell populations of IL-17. Previous reports have shown that IL-10 can increase ICAM expression in bronchial epithelial cells (52). IL-10 also downregulates IFN-γ–induced MHC class II in intestinal epithelial cells (53). The reported activity of IL-10 on nonimmune cells prompted us to investigate the modulatory role of IL-10 in the gingival epithelial cells in vivo. However, the modulatory effect of IL-10 on gingival epithelia was not supported by the IL-17 downstream inflammatory effector analysis on captured murine gingival epithelial cells from Il10−/− mice upon P. gingivalis oral challenge. Together, these in vivo data from the P. gingivalis–induced bone-loss model in Il10−/− mice and ex vivo data support the conclusion that, although IL-17 elicits inflammatory response in gingival epithelial cells, the dampening effect of IL-10 on IL-17 is not mediated through gingival epithelial cells, but through other cell types such as innate immune cells.
One limitation of this report is that we did not assess the IL-10–modified IL-17–mediated response in osteoclast differentiation. IL-17 possesses a potent osteoclastogenic activity by promoting RANKL expression in synovial fibroblasts and osteoblasts (54). IL-10 can directly inhibit osteoclastogenesis process by suppressing RANKL-induced NFATc1 expression (55). Whether or how IL-10 inhibits IL-17–induced osteoclastogenic effect requires further investigation.
In conclusion, our clinical data suggest an IL-17–dominated and IL-10–modified inflammatory network is characteristically associated with a periodontitis trait. By using murine periodontitis models, we further demonstrated that the protective role of IL-10 in periodontitis is, at least in part, mediated through thwarting excessive IL-17 inflammatory network, which supports our clinical findings.
Supplementary Material
Acknowledgments
We thank Dr. Ryan Balfour Sartor at UNC-CH School of Medicine and Dr. Ulrich Sienbenlist at National Institute of Allergy and Infectious Diseases for generosity in providing Il10−/− and Act1−/− mice, respectively, for this study. We thank the Histology Research Core Facility in the Department of Cell Biology and Physiology at the UNC-CH for histological services.
This work was supported by National Institutes of Health Grants 1K99DE02786 (to S.Z.), R01DE021418 (to S.O.), R01DE023836 (to S.O.), and R01DE026728 (to Y.L.L.).
Abbreviations used in this article:
- ABC
alveolar bone crest
- ARIC
Atherosclerosis Risk in Communities
- CEJ
cementoenamel junction
- CMC
carboxymethylcellulose
- EALGE3i
extent of clinical attachment loss ≥3 mm at the interproximal site
- EPDGE4
extent of probing depth ≥4-mm site; GCF, gingival crevicular fluid
- Il10−/−
Il10-deficient
- iNOS
inducible NO synthase
- KD
knockdown
- KO
knockout
- m1
mouse first molar
- m2
mouse second molar
- PCA
principal component analysis
- pHGE
primary human gingival epithelial cell
- PIT
periodontal inflammatory trait
- shRNA
short hairpin RNA
- UNC-CH
University of North Carolina at Chapel Hill
- WT
wild-type
Footnotes
L. S.: involvement in key in vivo and ex vivo experiments and manuscript preparation; M. G.: ligature animal model and bone loss analysis; L.W.: pathogen-induced animal model; Y.J.: research design, experiment troubleshooting, and manuscript revision; E.Z.: statistical support; K.M.: cell isolation from tissues and flow cytometry; J.Z.: cell isolation from tissues and flow cytometry; J.T.M.: research design and manuscript revision; N.Y.: sample preparation and clinical participant recruitment; K.M.: major statistical analysis; Y.L.L.: research design and manuscript revision; S.O.: research design, clinical data analysis, and manuscript preparation; S.Z.: principal investigator, overall study design and oversight, and manuscript preparation.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Preshaw PM, Alba AL, Herrera D, Jepsen S, Konstantinidis A, Makrilakis K, and Taylor R. 2012. Periodontitis and diabetes: a two-way relationship. Diabetologia 55: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eke PI, Dye BA, Wei L, Thornton-Evans GO, and Genco RJ, CDC Periodontal Disease Surveillance workgroup: James Beck (University of North Carolina, Chapel Hill, USA), Gordon Douglass (Past President, American Academy of Periodontology), Roy Page (University of Washin. 2012. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res 91: 914–920. [DOI] [PubMed] [Google Scholar]
- 3.Offenbacher S, Divaris K, Barros SP, Moss KL, Marchesan JT, Morelli T, Zhang S, Kim S, Sun L, Beck JD, et al. 2016. Genome-wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum. Mol. Genet 25: 2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teles R, Sakellari D, Teles F, Konstantinidis A, Kent R, Socransky S, and Haffajee A. 2010. Relationships among gingival crevicular fluid biomarkers, clinical parameters of periodontal disease, and the subgingival microbiota. J. Periodontal 81: 89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong Y, Slade GD, Beck JD, and Offenbacher S. 2007. Gingival crevicular fluid interleukin-1beta, prostaglandin E2 and periodontal status in a community population. J. Clin. Periodontol 34: 285–293. [DOI] [PubMed] [Google Scholar]
- 6.Groeger SE, and Meyle J. 2015. Epithelial barrier and oral bacterial infection. Periodontol. 2000 69: 46–67. [DOI] [PubMed] [Google Scholar]
- 7.Malakouti M, Brown GE, Wang E, Koo J, and Levin EC. 2015. The role of IL-17 in psoriasis. J. Dermatolog. Treat 26: 41–44. [DOI] [PubMed] [Google Scholar]
- 8.Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, and Fujiyama Y. 2003. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52: 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, Blumenschein WM, Judo M, Ayanoglu G, McClanahan TK, et al. 2015. Interleukin-23-Independent IL-17 production regulates intestinal epithelial permeability. [Published erratum appears in 2015 Immunity 43: 1022.] Immunity 43: 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittal SK, and Roche PA. 2015. Suppression of antigen presentation by IL-10. Curr. Opin. Immunol 34: 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Yu X, Lin J, Hu Y, Zhao Q, Kawai T, Taubman MA, and Han X. 2017. B10 cells alleviate periodontal bone loss in experimental periodontitis. Infect. Immun 85: e00335–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu P, Hu Y, Liu Z, Kawai T, Taubman MA, Li W, and Han X. 2016. Local induction of B cell interleukin-10 competency alleviates inflammation and bone loss in ligature-induced experimental periodontitis in mice. Infect. Immun 85: e00645–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki H, Okamatsu Y, Kawai T, Kent R, Taubman M, and Stashenko P. 2004. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J. Periodontal Res 39: 432–41. [DOI] [PubMed] [Google Scholar]
- 14.Veldhoen M 2017. Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol 18: 612–621. [DOI] [PubMed] [Google Scholar]
- 15.Elter JR, Champagne CM, Offenbacher S, and Beck JD. 2004. Relationship of periodontal disease and tooth loss to prevalence of coronary heart disease. J. Periodontol 75: 782–790. [DOI] [PubMed] [Google Scholar]
- 16.Champagne CM, Buchanan W, Reddy MS, Preisser JS, Beck JD, and Offenbacher S. 2003. Potential for gingival crevice fluid measures as predictors of risk for periodontal diseases. Periodontol. 2000 31: 167–180. [DOI] [PubMed] [Google Scholar]
- 17.Zhang S, Barros SP, Moretti AJ, Yu N, Zhou J, Preisser JS, Niculescu MD, and Offenbacher S. 2013. Epigenetic regulation of TNFA expression in periodontal disease. J. Periodontol 84: 1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armitage GC 1999. Development of a classification system for periodontal diseases and conditions. Ann. Periodontol 4: 1–6. [DOI] [PubMed] [Google Scholar]
- 19.Fordham JB, Naqvi AR, and Nares S. 2015. Regulation of miR-24, miR-30b, and miR-142-3p during macrophage and dendritic cell differentiation potentiates innate immunity. J. Leukoc. Biol 98: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fordham JB, Guilfoyle K, Naqvi AR, and Nares S. 2016. MiR-142-3p is a RANKL-dependent inducer of cell death in osteoclasts. Sci. Rep 6: 24980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Offenbacher S, Barros S, Mendoza L, Mauriello S, Preisser J, Moss K, de Jager M, and Aspiras M. 2010. Changes in gingival crevicular fluid inflammatory mediator levels during the induction and resolution of experimental gingivitis in humans. J. Clin. Periodontol 37: 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livak KJ, and Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 23.Baker PJ Dixon M, and Roopenian DC. 2000. Genetic control of susceptibility to Porphyromonas gingivalis-induced alveolar bone loss in mice. Infect. Immun 68: 5864–5868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasaki H, Suzuki N, Kent R Jr., Kawashima N, Takeda J, and Stashenko P. 2008. T cell response mediated by myeloid cell-derived IL-12 is responsible for Porphyromonas gingivalis-induced periodontitis in IL-10-deficient mice. J. Immunol 180: 6193–6198. [DOI] [PubMed] [Google Scholar]
- 25.Marchesan J, Girnary MS, Jing L, Miao MZ, Zhang S, Sun L, Morelli T, Schoenfisch MH, Inohara N, Offenbacher S, and Jiao Y. 2018. An experimental murine model to study periodontitis. Nat. Protoc 13: 2247–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, Chen GY, Nunez G, Giannobile WV, Raes J, and Inohara N. 2013. In-duction of bone loss by pathobiont-mediated Nod1 signaling in the oral cavity. Cell Host Microbe 13: 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dutzan N, Abusleme L, Konkel JE, and Moutsopoulos NM. 2016. Isola-tion, characterization and functional examination of the gingival immune cell network. J. Vis. Exp DOI: 10.3791/53736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garlanda C, Dinarello CA, and Mantovani A. 2013. The interleukin-1 family: back to the future. Immunity 39: 1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones CE, and Chan K. 2002. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-I, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol 26: 748–753. [DOI] [PubMed] [Google Scholar]
- 30.Ouyang W, Kolls JK, and Zheng Y. 2008. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28: 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajishengallis G, Darveau RP, and Curtis MA. 2012. The keystonepathogen hypothesis. Nat. Rev. Microbiol 10: 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saraiva M, and O’Garra A. 2010. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol 10: 170–181. [DOI] [PubMed] [Google Scholar]
- 33.Jenkins JK, Malyak M, and Arend WP. 1994. The effects of interleukin-10 on interleukin-1 receptor antagonist and interleukin-1 beta production in human monocytes and neutrophils. Lymphokine Cytokine Res. 13: 47–54. [PubMed] [Google Scholar]
- 34.Wang L, Wang J, Jin Y, Gao H, and Lin X. 2014. Oral administration of all-trans retinoic acid suppresses experimental periodontitis by modulating the Th17/Treg imbalance. J. Periodontol. 85: 740–750. [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Shao S, Jiao Z, Guo M, Xu H, and Wang S. 2012. The Th17/Treg imbalance and cytokine environment in peripheral blood of patients with rheumatoid arthritis. Rheumatol. Int 32: 887–893. [DOI] [PubMed] [Google Scholar]
- 36.Erbel C, Akhavanpoor M, Okuyucu D, Wangler S, Dietz A, Zhao L, Stellos K, Little KM, Lasitschka F, Doesch A, et al. 2014. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol 193: 4344–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, and Russell CB. 2013. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J. Invest. Dermatol 133: 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li D, Guo B, Wu H, Tan L, Chang C, and Lu Q. 2015. Interleukin-17 in systemic lupus erythematosus: a comprehensive review. Autoimmunity 48: 353–361. [DOI] [PubMed] [Google Scholar]
- 39.Bakır B, Yetkin Ay Z, Búyúkbayram HI, Kumbul Doğuç D, Bayram D, Candan IA, and Uskun E. 2016. Effect of curcumin on systemic T helper 17 cell response; gingival expressions of interleukin-17 and retinoic acid receptorrelated orphan receptor γt; and alveolar bone loss in experimental periodontitis. J. Periodontol 87: e183–e191. [DOI] [PubMed] [Google Scholar]
- 40.Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, et al. 2018. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 10: eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mocellin S, Panelli MC, Wang E, Nagorsen D, and Marincola FM. 2003. The dual role of IL-10. Trends Immunol. 24: 36–43. [DOI] [PubMed] [Google Scholar]
- 42.Berlato C, Cassatella MA, Kinjyo I, Gatto L, Yoshimura A, and Bazzoni F. 2002. Involvement of suppressor of cytokine signaling-3 as a mediator of the inhibitory effects of IL-10 on lipopolysaccharide-induced macrophage activetion. J. Immunol 168: 6404–6411. [DOI] [PubMed] [Google Scholar]
- 43.Moretti S, Bartolommei L, Galosi C, Renga G, Oikonomou V, Zamparini F, Ricci G, Borghi M, Puccetti M, Piobbico D, et al. 2015. Fine-tuning of Th17 cytokines in periodontal disease by IL-10. J. Dent. Res 94: 1267–1275. [DOI] [PubMed] [Google Scholar]
- 44.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, Evans RT, and Gaffen SL. 2007. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood 109: 3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, Stevens E, Bigler J, Davis JA, Rottman JB, et al. 2015. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 43: 739–750. [DOI] [PubMed] [Google Scholar]
- 46.Deshmane SL, Kremlev S, Amini S, and Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res 29: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wynn TA, Chawla A, and Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lam RS ’, O’Brien-Simpson NM, Lenzo JC, Holden JA, Brammar GC, Walsh KA, McNaughtan JE, Rowler DK, Van Rooijen N, and Reynolds EC. 2014. Macrophage depletion abates Porphyromonas gingivalis-induced alveolar bone resorption in mice. J. Immunol 193: 2349–2362. [DOI] [PubMed] [Google Scholar]
- 49.Lopes RL, Borges TJ, Zanin RF, and Bonorino C. 2016. IL-10 is required for polarization of macrophages to M2-like phenotype by mycobacterial DnaK (heat shock protein 70). Cytokine 85: 123–129. [DOI] [PubMed] [Google Scholar]
- 50.Huppler AR, Conti HR, Hernandez-Santos N, Darville T, Biswas PS, and Gaffen SL. 2014. Role of neutrophils in IL-17–dependent immunity to mucosal candidiasis. [Published erratum appears in 2015 J. Immunol. 194: 1382.] J. Immunol 192: 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conti HR, and Gaffen SL. 2015. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J. Immunol 195: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bianco A, Sethi SK, Allen JT, Knight RA, and Spiteri MA. 1998. Th2 cytokines exert a dominant influence on epithelial cell expression of the major group human rhinovirus receptor, ICAM-1. Eur. Respir. J 12: 619–626. [DOI] [PubMed] [Google Scholar]
- 53.Thelemann C, Eren RO, Coutaz M, Brasseit J, Bouzourene H, Rosa M, Duval A, Lavanchy C, Mack V, Mueller C, et al. 2014. Interferon-g induces expression of MHC class II on intestinal epithelial cells and protects mice from colitis. PLoS One 9: e86844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, et al. 1999. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Invest 103: 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans KE, and Fox SW. 2007. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.