

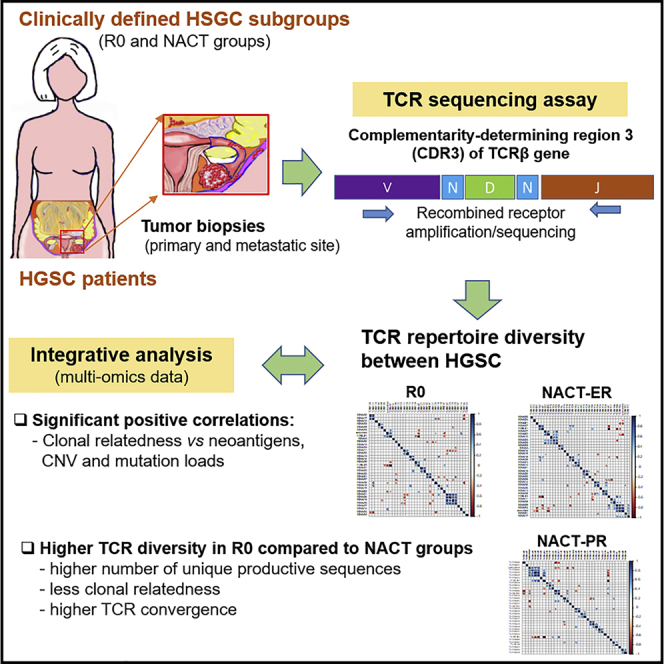
Summary
In patients with high-grade serous ovarian cancer (HGSC), it is unclear which genomic features are related to complete gross resection (R0), which is typically associated with better clinical outcomes, or response to neoadjuvant chemotherapy (NACT). In this study, we evaluated T cell receptor (TCR) repertoire diversity in primary and metastatic tumor samples (n = 90) from clinically well-annotated patients with HGSC who achieved R0 or received NACT with excellent or poor response based on a laparoscopic triage algorithm. TCR sequencing followed by an integrative analysis with comprehensive multi-omics data identified higher TCR diversity (e.g., higher number of unique productive sequences and less clonal relatedness) in the R0 than NACT groups. We found enrichment of specific TCRβ genes usage, distinct mutual exclusiveness and co-occurrence pattern of TCRβ genes among the groups. We also found significantly positive correlations between clonal relatedness and neoantigens, copy number variations, and mutation load in the groups.
Subject areas: Biological Sciences, Immunology, Cancer
Graphical abstract
Highlights
-
•
Higher TCR repertoire diversity in the R0 versus neoadjuvant chemotherapy groups
-
•
Enrichment of specific TCRβ genes usage was noted among HGSC subgroups
-
•
Distinct mutual exclusiveness of TCRβ genes was noted among HGSC subgroups
-
•
Positive correlations between clonal relatedness and copy number variations
Biological Sciences; Immunology; Cancer
Introduction
High-grade serous ovarian cancer (HGSC) is the most common and aggressive type of epithelial ovarian cancer, with a high rate of overall mortality and tumor heterogeneity and variable clinical outcomes (Jayson et al., 2014). The Cancer Genome Atlas and other analyses have identified multiple molecular abnormalities including TP53 mutations, somatic or germline BRCA mutations, and CCNE1 amplifications, in HGSC tumor samples from patients who have undergone upfront debulking surgery (Cancer Genome Atlas Research Network, 2011; Patch et al., 2015; Walsh et al., 2011). However, samples from patients who have been treated in a consistent manner needed to understand the molecular and cellular heterogeneity and drug resistance of HGSC.
A previous study used a laparoscopic triage approach (Fleming et al., 2018; Nick et al., 2015) and found much higher rates of complete gross resection (R0) in patients who were triaged to primary surgery; neoadjuvant chemotherapy (NACT) was offered as an alternative when primary debulking surgery was not feasible. However, it is unknown whether the difference in clinical outcomes resulted from aggressive surgical efforts or whether there are underlying biological differences between disease that is fully resectable and disease that is not. In addition, the genomic features that best discriminated R0 versus NACT (responders and non-responders) remain unclear, and no practical methods exist for identifying such predictive biomarkers in clinical settings.
Recently, we evaluated clinically well-annotated primary tumor and metastasis samples that had been obtained prior to definitive surgery or chemotherapy and discovered significant distinct molecular abnormalities (e.g., greater NF1 loss and less chromothripsis-like patterns) and cellular changes (e.g., increased numbers of neoantigens and infiltrated T-cells) in patients who underwent R0 surgery compared with in those who were triaged to NACT (Lee et al., 2020). However, further characterization and understanding is needed with regard to T cell repertoires, including the diversity and clinical relevance of T cells.
In the current study, we characterized T cell repertoire by detecting a rearranged complementarity-determining region 3 (CDR3) of the T cell receptor β (TCRβ) gene (Xu and Davis, 2000) on a high-throughput TCR sequencing analysis of tissue samples from clinically well-defined groups of patients with HGSC. We then performed integrative analyses, with immune assessments, high-depth whole-genome sequencing from the matched samples among the groups to interrogate the association of T cell-based immune response and clinical outcomes.
Results
Patient group and sequencing data
The study design and patient cohort was reported previously (Lee et al., 2020). In brief, a total of 30 patients in three groups was included: 10 patients who had no visible residual disease after primary surgery (R0), 10 who had an excellent response to NACT (NACT-ER), and 10 who had a poor response to NACT (NACT-PR). For each patient, one primary and two metastatic tumor samples were included. High-throughput TCR profiling via ImmunoSEQ was performed on the 90 samples (Table S1).
Similarities within and among patient groups by TCR sequencing
We performed a high-throughput TCR sequencing analysis with a mean total of 14,192 reads per sample; a mean of 7,494 unique TCRβ rearrangements from each sample was identified for the entire cohort. To compare the T cell repertoires of all samples within patient groups, we used the Bhattacharyya coefficient (Bhattacharyya, 1943) to assess the frequency-weighted similarities between each of the two samples. The Bhattacharyya coefficient ranges from 0 to 1, where 0 indicates that there is no overlap at all and 1 indicates that the TCR sequence frequencis are identical. As shown in Figure 1A, similarities of 0.11–0.79 were observed between different tumor samples within the same patient. In contrast, almost no similarities were observed among patients. The mean Bhattacharyya coefficient between different tumors from the same patients was 0.43 in the R0 group, 0.52 in the NACT-ER group, and 0.47 in the NACT-PR group; no significant differences were observed among the three groups (Figure 1B).
Figure 1.

Intrapatient and interpatient similarity by patient group
(A) Similarity matrix of TCR sequences between each of the two samples. The Bhattacharyya coefficient (0–1) was used to calculate the weighted similarity score; samples from the same patients are shown next to each other. R0, no residual disease; NACT-ER, excellent response to NACT; NACT-PR, poor response to NACT.
(B) Comparison of the similarity scores between different samples within the same patients across three groups.
Diversity evaluation of T cell repertoires
The diversity of T cell repertoires was evaluated using several metrics, including richness, clonality, evenness, and relatedness. Richness was estimated using the number of unique productive sequences (UPSs) in which the TCRβ nucleotide sequences can be translated to amino acid sequences without an early stop codon. The mean numbers of UPSs were 10,157 in the R0 group, 3,829 in the NACT-ER group, and 4,043 in the NACT-PR group. When comparing groups, we found no significant differences in the primary tumors (Figures 2A and S1A). However, the metastatic tumors in the R0 group had a significantly higher number of UPSs than did those in both the NACT-ER (p = 0.0043) and NACT-PR (p = 0.011) groups (Figures 2B and S2A). Interestingly, when we compared the primary and metastatic tumors within each group, the richness of the T cell repertoires was significantly higher in metastatic tumors than in primary tumors (p = 0.0083) in the R0 group, while no differences were found in the NACT group (Figure S3A). We also calculated the frequencies of the most abundant productive TCR sequences in primary tumors (Figures S1B and S1C) and metastatic (Figures S2B and S2C) tumors among the groups. The mean frequencies of the top productive sequences (TPSs) were 4.98% in the R0 group, 5.70% in the NACT-ER group, and 5.56% in the NACT-PR group. In addition, no significant differences were found between any of the two groups or between primary and metastatic tumors within groups (Figure S3B).
Figure 2.

Diversity evaluation of T cell repertoires by patient group
(A and B) Comparison of T cell repertoire richness in primary tumors (A) and metastatic tumors (B) across three groups. TCR richness was evaluated by the number of unique productive sequences, which were defined as sequences that are in frame and do not have an early stop codon.
(C and D) Comparison of clonality across three groups in primary tumors (C) and metastatic tumors (D). The clonality score is derived from the Shannon entropy, which range from 0 to 1, where 0 indicates that all sequences have the same frequency and 1 indicates that the repertoire is dominated by a single sequence.
(E and F) Comparison of clonal evenness across three groups in primary tumors (E) and metastatic tumors (F). The clonal evenness was evaluated by the Gini coefficient, which range from 0 to 1, where 0 indicates that all clones have identical frequencies, and 1 indicates that they are completely unequal.
(G and H), Comparison of clonal relatedness among three groups in primary tumors (G) and metastatic tumors (H). Clonal relatedness was defined as the proportion of sequences that are related to the most frequent sequence by a defined distance threshold. The value ranges from 0 to 1, where 0 indicates that no sequences are related and 1 indicates that all sequences are related.
Next, we evaluated the clonality score, which reflects the uncertainty of clonal selection in the clonal expansion process, namely whether a subset of clones is preferred in the clonal expansion. This was derived from the Shannon entropy and measured on a scale from 0 to 1, where 0 indicates that all TCR sequences have the same frequency and 1 indicates that the repertoire is dominated by a single clone. The clonality scores in the R0 group ranged from 0.02 to 0.25 (mean, 0.09) (Figures 2C and 2D). In the NACT group, the mean clonality score was 0.11 in the NACT-ER group (range, 0.04 to 0.21) and 0.11 in the NACT-PR group (range, 0.01 to 0.33). No significant differences were found between any of the groups (Figures S1D and S2D). We also observed no differences between primary and metastatic tumors (Figure S3C).
We used the Gini coefficient to measure the evenness metric, a measure of the distribution of the relative abundances of each unique TCR amino acid sequences, which captured the inequality in clonotype size across the population. Similar to the clonality score, the Gini coefficient ranges from 0 to 1, where 0 indicates that all clones have identical frequencies, and 1 indicates that they are completely unequal (i.e. sample oligoclonality). The Gini coefficient in the R0 group ranged from 0.13 to 0.65, and the average score was 0.41 (Figures 2E and 2F). The mean clonality score was 0.44 in the NACT-ER group (range, 0.22 to 0.65) and 0.43 in the NACT-PR group (range, 0.14 to 0.73). No statistically significant differences were found between the groups (Figures S1E and S2E). In addition, no differences were identified when comparing the primary and metastatic tumor samples in all three groups (Figure S3D).
Because of the limitations of the clonality and evenness scores (i.e., they do not account for sequence similarity), we further evaluated clonal relatedness by considering the nucleotide similarity between TCR sequences within each sample; clonal relatedness is defined as the fraction of unique nucleotide sequences that are related to the most frequent sequence by an edit distance threshold (Lombardo et al., 2017). The value ranges from 0 to 1 where 0 indicates that no sequences are related, and 1 indicates that all sequences are related to the most frequent sequence. Interestingly, although we did not observe any significant differences in the relatedness scores in the primary tumors between the groups (Figures 2G and S1F), the R0 group had significantly lower relatedness score in metastatic tumors than did the NACT group (p = 0.0025) (Figure S2F). As shown in Figure 2H, the mean relatedness score in metastatic tumors was 0.002 in the R0 group, 0.004 in the NACT-ER group (p = 0.0067), and 0.003 in the NACT-PR group (p = 0.013). We compared the primary and metastatic tumors within each group and found that the metastatic tumors in the R0 group showed a non-statistically significant trend toward a lower relatedness score than did the primary tumors. We found no difference between primary and metastatic tumors in the NACT groups (Figure S3E).
To further cross-validate our findings, we filtered out all the TCRs with <2 reads and repeated the analysis. The results after filtering are consistent with what we have presented. As shown in Figure S4, in the metastatic tumors, the number of UPSs is significantly higher in the R0 group compared to the NACT-ER (p = 0.0043) and NACT-PR (p = 0.0032) groups (Figure S4B), but no significant differences were found in the primary tumors (Figure S4A). Furthermore, in the metastatic tumors, the TCR clones are significantly less related in the R0 group than NACT-ER (p = 0.013) and NACT-PR (p = 0.013) groups (Figure S4D), but no differences were found in the primary tumors (Figure S4C). The conclusions after filtering remained the same for the other metrics including clonality and Gini coefficient, in which we found no differences.
Evaluation of TCR convergence
TCR convergence, which reflects TCRs that are different at the nucleotide level but identical at the amino acid level, was calculated as the aggregate frequency of clones (unique TCRβ nucleotide sequences) that share a variable gene and CDR3 amino acid with at least one other identified clone. The convergence scores ranged from 0 to 0.22 in the R0 group, with a median value of 0.12; 0.04 to 0.27 in the NACT-ER group, with a median value of 0.10; and 0.03 to 0.45 in the NACT-PR group, with a median value of 0.10. As shown in Figures S5A and S5B, no statistically significant differences were found among the R0, NACT-ER, and NACT-PR groups (Figures S5C and S5D). In addition, no differences were found when comparing primary and metastatic samples in all three groups (Figure S5E).
CDR3 length distribution
The variable CDR3 lengths of healthy individuals were conformed to normal Gaussian distributions (Kou et al., 2000; Lai et al., 2019); the degree to which the frequency distribution of CDR3 lengths deviated from normality was reported as an indication of clonal expansion and thus of reduced diversity (Long et al., 2006; Matsumoto et al., 2006). Therefore, we evaluated the distribution of CDR3 length in all three groups. The mean CDR3 length was 43.43nt (14.48aa) in the R0 group, 43.38nt (14.46aa) in the NACT-ER group, and 43.69nt (14.56aa) in the NACT-PR group (Figure 3). All of the three groups demonstrated significant deviation from a normal distribution for CDR3 length. We used the Shapiro-Wilk test and found that the W value was 0.961 in the R0 group, 0.966 in the NACT-ER group, and 0.958 in the NACT-PR group, suggesting that CDR3 length in the NACT-PR group deviated more from the normal distribution.
Figure 3.

CDR3 Length (nt) distribution and normality by patient group
(A) Distribution of CDR3 length in the R0, NACT-ER, and NACT-PR groups. The mean CDR3 length was 43.43nt (14.48aa) in the R0 group, 43.38nt (14.46aa) in the NACT-ER group and 43.69nt (14.56aa) in the NACT-PR group.
(B) Distribution of CDR3 length in the R0 group and its deviation from a normal distribution (p < 0.001).
(C) Distribution of CDR3 length in the NACT-ER group and its deviation from a normal distribution (p < 0.001).
(D) Distribution of CDR3 length in the NACT-PR group and its deviation from a normal distribution (p < 0.001).
We also performed a multidimensional analysis. First, we evaluated the distribution and correlation across different metrics, including the UPSs, clonality, Gini coefficient, frequency of TPS, clonal relatedness, average CDR3 length, and TCR convergence, as shown in Figure S6. Positive correlations were found between TCR clonality, Gini coefficient, TPS and TCR convergence. We further performed linear discriminant analysis (LDA) is to find the linear combination of all the metrics that would give the greatest separation between the groups (R0, NACT-ER, and NACT-PR groups). The results of LDA are shown in Figure S7. Based on the group-wise comparison, the LD1 values in all the three groups are significantly different, while the LD2 can separate NACT-PR from both R0 and NACT-ER groups.
TCR sequences dynamics
We further investigated the TCR amino acid sequences shared by multiple samples and a total number of 58,985 TCR amino acid sequences were found to be recurrent in at least two samples. The vast majority of the TCRs were shared by less than three samples (92.3%) and the most frequently found TCR sequences are shared by 27 of 90 samples. To show the TCR dynamics, we further generated a heatmap showing the TCR sequences that appear in more than 15% of the samples (Figure S8A). We also tracked the frequencies of six most common clones which were present in more than 20 samples in Figure S8B. For some of the clones, we observed high frequency in primary and metastatic tumor samples in the same patients, suggesting the TCRs are shared by both primary and metastatic sites. While for some TCR clones (e.g. CASSLQGNTEAFF), we observed less variation across samples. Overall, no patterns were observed to be associated with patient groups.
Comparison of usage of variable (V), diversity (D), and joining (J) region gene segments
To further measure the properties of TCR diversity in the group, we calculated the V, D and J gene usage patterns of each sample. The heatmap of the frequencies revealed that no specific patterns were observed. The usage of three V gene families, TCRBV14 (p = 0.0073), TCRBV19 (p = 0.02), and TCRBV21 (p = 0.036) (Figure S9 and Table S2), was significantly higher in the R0 group than in the NACT groups (Figure 4A). When comparing the NACT-ER and NACT-PR groups (Figure 4B), we found that the usage of two gene families was significantly higher in the NACT-ER group, including TCRBV06 (p = 0.037) and TCRBV29 (p = 0.022). In contrast, the usage of three V gene families was significantly higher in NACT-PR, including TCRBV01 (p = 0.048), TCRBV09 (p = 0.017), and TCRBV12 (p = 0.0048). In the D gene usage comparison (Figure S10 and Table S2), the NACT group showed significantly higher usage of TCRBD01-01 than did the R0 group (p = 0.036); no significant difference was found between the NACT-ER and NACT-PR groups. The usage of two J genes was significantly higher in the R0 group, including TCRBJ02-01 (p = 0.013) and TCRBJ02-02 (p = 0.011), than in the NACT groups (Figures 4C, 4D, and S11 and Table S2). The usage of TCRBJ01-01 was significantly higher in the NACT-ER group (p = 0.0115), and the usage of TCRBJ02-05 was significantly higher in NACT-PR group (p = 0.004).
Figure 4.

Comparison of the usage of V, D, and J genes among groups
(A and B) Comparison of V-gene family usage between the R0 and NACT groups (A), and between the NACT-ER and NACT-PR group (B).
(C and D) Comparison of J gene usage between the R0 and the NACT groups (C) and the NACT-ER and the NACT-PR groups (D).
(E–F)(E and G) Mutual exclusivity and co-occurrence of the usage of V, D, and J gene families in the R0 group (E), NACT-ER group (F), and NACT-PR group (G). Correlations between any of the two gene families were calculated; the significant results are shown in the figure (p < 0.01).
Next, we assessed the mutual exclusivity and co-occurrence of the usage of the V, D, and J gene families. As shown in Figure 4, specific patterns were found in corresponding groups. For example, in the R0 group (Figure 4E), significant positive correlations (p < 0.01) were found between TCRBV02 and TCRBD01, TCRBD02 and TCRBJ02, and TCRBV05 and TCRBJ01. In the NACT-ER group (Figure 4F), significant positive correlations were found between TCRBV23/25/27 and TCRBD02, TCRBV06/07/18 and TCRBJ01, and TCRBV04/09 and TCRBJ02. In the NACT-PR group (Figure 4G), significant positive correlations were found between TCRBV04/07 and TCRBD01, TCRBV21 and TCRBJ01, and TCRBV03/25/28 and TCRBJ02. Of interest, TCRBJ01 and TCRBJ02 were negatively correlated in all three groups, suggesting mutual exclusivity. The principal component analysis of V gene usage and V, D, and J gene usage revealed that all three groups largely overlapped (Figure S12).
Integrative analyses
In our previous study of the same cohort of patients, a higher level of strong-binding neoantigens was found in the R0 group than in the NACT group (Lee et al., 2020). In addition, positive correlations were found between the level of strong-binding neoantigens and mutation load and copy number variation load. Therefore, we determined the correlation between TCR clonal diversity and the neoantigen level. As shown in Figure S13, no correlations were observed between the clonality (Shannon entropy) and the number of strong-binding neoantigens in any of the three groups. Similarly, no correlation was found between clonal evenness (Gini coefficient) and the neoantigen level. However, positive correlation was found between the TCR clonal relatedness and the neoantigen level (R = 0.31, p = 0.007) (Figure 5A). TCR clonal relatedness also showed a trend toward a positive correlation with CNV load (R = 0.43, p < 0.001) and mutation load (R = 0.27, p = 0.019) (Figures 5B and 5C).
Figure 5.

Integrative analyses of TCR sequencing, WGS and immune profiling
(A–C) (A and C) Correlation between TCR clonal relatedness and neoantigen level (A), mutation load (B) and CNV load (C), as identified on whole-genome sequencing (N = 75).
(D–F) (D and F) Correlation between TCR clonal relatedness and percentage of CD8+ cells (D), CD4+ cells (E), and CD20 + cells (F) in all areas on the basis of immune profiling data (N = 53).
We also explored the correlation between TCR diversity and BRCA status, including BRCA germline mutations, BRCA somatic mutations, and any BRCA mutations (either germline or somatic). As shown in Figure S14, TCR richness estimated by the number of UPSs had no differences in BRCA status. TCR clonality was significantly higher in BRCA mutant groups for both germline (p = 0.045) and somatic (p = 0.003) mutations. TCR evenness quantified by the Gini coefficient was significantly higher in the BRCA germline mutant group than BRCA wild-type tumors (p = 0.035), and the trend is the same for BRCA somatic mutant group, but without statistical significance (p = 0.06). Lastly, TCR clonal relatedness was significantly higher in the BRCA somatic mutant group compared with the wild-type group (p = 0.025).
We further evaluated the correlation between TCR clonal diversity and the percentage of different immune cells estimated from immune profiling in our previous study (Lee et al., 2020). For the T cells in all areas, including tumor and non-tumor areas, we observed significant negative correlations between TCR clonal relatedness (Figures 5D–5F, S15A, and S15B) and the percentage of CD8+ cells (R = −0.57, p < 0.001), CD4+ cells (R = −0.5, p < 0.001), and CD20 + cells (R = −0.52, p < 0.001), whereas we observed no significant correlations between the percentages of immune cells and TCR clonality in all areas (Figure S16). For T cells from the tumor area only (Figures S17 and S18), negative correlations were found between TCR clonal relatedness and the percentages of CD8+ cells (R = −0.46, p < 0.001) and CD20 + cells (R = −0.42, p = 0.002). No significant correlations were found between TCR clonal relatedness and the other two immune cell types in the tumor area, FOXP3+-positive cells and macrophages. In addition, no significant correlations were found between TCR clonality and the percentages of any of the immune cells in tumor area.
Discussion
To our knowledge, this is the first report of the characteristics of TCR repertoires of highly clinically defined HGSC samples from primary and metastatic sites by high-throughput TCR deep sequencing; we also performed an integrated analysis of multi-omics data. Our findings provide a comprehensive understanding of TCR repertoire diversity in patients with HGSC. Importantly, we identified higher TCR β-chain CDR3 diversity (i.e., a higher number of UPSs, less clonal relatedness, and higher TCR convergence) in the R0 groups than in the NACT groups. We also found enrichment of specific V, D, and J genes usage in each group and significant unique mutual exclusivity and co-occurrence of V, D, and J genes in all three groups. In addition, we also found significant positive correlations between clonal relatedness and neoantigens, copy number variations, and mutation load by integrative analyses.
In a recent comprehensive multi-omics analysis, we identified distinct molecular differences and immune cell repertoire variations including tumor-associated neoantigens and infiltrated T cells (Lee et al., 2020) among patients with HGSC who received care under a consistently applied surgical algorithm (Fleming et al., 2018; Lee et al., 2020; Nick et al., 2015). Tumor-associated antigens have been considered for immunotherapeutic strategies, and tumor mutations and immunotherapy-targeting neoantigens were highly correlated in various solid tumors (Schumacher and Schreiber, 2015; Snyder et al., 2014). In this study, we used bulk TCR sequencing to extend our characterization of deep TCR repertoires of highly annotated HGSC groups; this technique has the advantage of having high sensitivity for identifying T cell diversity.
The characteristics of T cells in the antigenic peptide-major histocompatibility complex are primarily determined by the CDR3 region of the TCR β-chain; interrogation of the sequences of the CDR3 region measures the diversity of TCR CDR3 repertoires (Davis, 1990; Davis and Bjorkman, 1988). Interestingly, the increase in TCR diversity was associated with better clinical outcomes and immune response in patients with cancer (Cha et al., 2014; Hopkins et al., 2018; Hsu et al., 2016; Postow et al., 2015; Robert et al., 2014) and provided a measurement of tumor immunogenicity (Drake et al., 2014). Consistently, we also observed that the TCR repertoire of the R0 group is more diverse than is that of the NACT group by the measurement of multiple metrics; specifically, a higher number of UPSs and lower clonal similarity, clonal relatedness, clonality, and evenness in the R0 group than in the NACT group. In addition to TCR repertoire diversity and clonality, while we observed a trend toward a higher level of TCR convergence with no statistical significance in the R0 than in the NACT groups, TCR convergence has been estimated to be a predictive biomarker for response to checkpoint inhibitors in patients with cancer (Looney et al., 2019).
TCR junctional diversity arises from random combinatorial joins of the V, J, and D gene segments, which generate large numbers of each completed chain that defines antigen-binding specificity (Nikolich-Zugich et al., 2004). The diversity of direct measurements of TCR diversity, such as V, D, and J region genes, have been considered potential prognostic biomarkers and predictors of chemoimmunotherapy response in patients with cancer (Farah et al., 2020; Han et al., 2015; Powles et al., 2018; Reuben et al., 2020; Vardi et al., 2016). Notably, we identified significantly higher usage of the TCRBV, TCRBD, and TCRBJ genes among the groups; none of these genes have been studied in the context of predicting surgical outcome and chemotherapy response in HGSC. Further, we found significant positive co-occurrence and mutual exclusiveness of three unique gene pairs among TCRB gene segments in the R0 group, indicating important characteristic spectra of TCRB gene variation patterns between the groups.
We previously found that the R0 group had significantly higher levels of neoantigens than did the NACT groups, which are associated with tumor immune cell infiltration (Lee et al., 2020). Thus, similar to our previous observations, we found no differences in TCR CDR3 diversity between primary and metastatic disease sites in the groups, indicating that the biological processes likely arise at an early stage (Jimenez-Sanchez et al., 2017; Reiter et al., 2018). However, with integrated analysis, we identified a significant positive correlation between TCR clonal relatedness and neoantigens, CNV, and mutation load, as determined by WGS analysis (Lee et al., 2020). However, we were not able to determine the correlation between TCR clonal relatedness and immune cell populations, which may be due to the sample size.
In this study, we focused on CDR3 for TCR repertoire analyses because of the diverse V, D, and J genes and the TCR peptide interaction. Alternatively, detection of CDR1 and CDR2 could help elucidate the entire TCR structure and varieties that result from their important function in coordinating with major histocompatibility complex molecules and their influences on the sensitivity and affinity of TCR binding (Birnbaum et al., 2014; Garcia and Adams, 2005). Furthermore, the tumor heterogeneity of HGSC at the molecular and cellular levels is still not fully understood (Bast et al., 2009). Moreover, the tumor-bulk sequencing approach with no spatial information for the TCR repertoire, has limitations when addressing questions of tumor complexity and diversity in HGSC. Therefore, extended approaches such as spatial heterogeneity of the TCR repertoire and single cell analyses (Chen et al., 2016; De Simone et al., 2018; Emerson et al., 2013; Gerlinger et al., 2013; Reuben et al., 2017) could be a new avenue for understanding the deeper intra-tumor heterogeneity of TCR landscape in HGSC.
Overall, our findings in this study demonstrated distinct TCR CDR3 repertoires among three HGSC subgroups. By combing the multi-omics data from DNA, RNA, and protein in the same patients with HGSC we previously reported, the findings in this study could provide a deeper understanding of tumor heterogeneity and identify actionable molecular and cellular mechanisms for therapeutic strategies.
Limitations of the study
The tumor-bulk TCR sequencing approach without spatial information for the TCR repertoires may not allow for full understanding of tumor complexity and diversity of HGSC.
Resource availability
Lead contact
Anil K. Sood, M.D., Department of Gynecologic Oncology and Reproductive Medicine, The University of Texas MD Anderson Cancer Center, Unit 1362, 1515 Holcombe Boulevard, Houston, TX 77030, Email: asood@mdanderson.org.
Materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by corresponding authors, Sanghoon Lee (slee29@mdanderson.org) and Anil K. Sood (asood@mdanderson.org).
Data and code availability
Sequence data from the TCR sequencing in this study has been deposited in the ImmuneACCESS project repository of Adaptive Biotechnology database (https://doi.org/10.21417/LS2021iS, https://clients.adaptivebiotech.com/pub/sanghoon-2021-iscience).
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank A.M. Sutton (Department of Scientific Publications, The University of Texas MD Anderson Cancer Center, Houston, TX) for editorial assistance. We thank J. Celestino, R.A. Hajek, and M.B. Morgan (MD Anderson Cancer Center, Houston, TX) for their support on the preparation of TCR sequencing. We also acknowledge the MD Anderson Biospecimen Extraction Resource for the extraction of genomic DNA and the MD Anderson Cancer Genomics Laboratory for TCR sequencing. This work was supported in part by the MD Anderson Ovarian Cancer Moon Shot; National Institutes of Health (NIH) grants P50CA217685, R35CA209904 and P30CA016672 (the Clinical Trials Office and Cancer Genomics Laboratory); and the MD Anderson Center for Translational and Public Health Genomics (used the Biospecimen Extraction Resource). S.N.W. is supported by the Andrew Sabin Family Fellowship and the GOG Foundation Scholar Investigator Award. A.K.S. is supported by the American Cancer Society Research Professor Award and the Frank McGraw Memorial Chair in Cancer Research.
Author contributions
A.K.S. supervised the study. S.L., P.A.F., and A.K.S. conceived and designed the study. S.L. took the lead in writing the manuscript. S.L., L.Z., and A.K.S. wrote the manuscript. S.L., L.Z., P.A.F., and A.K.S. analyzed, managed and interpreted the sequencing data. L.Z., L.D.L., and J.Z. analyzed and managed the TCR sequencing data. S.L., L.Z., J.Z., P.A.F., and A.K.S. interpreted the TCR sequencing data. S.L., S.N.W., A.A.J., N.D.F., and A.K.S. contributed to the acquisition of clinical samples and data. All authors read and approved the final manuscript.
Declaration of interests
S.N.W. has clinical research grants from AstraZeneca, ArQule, Bayer, Clovis Oncology, Cotinga Pharmaceuticals, NCCN, Novartis, Roche/Genentech, and Tesaro, and consults with AstraZeneca, Circulogene, Clovis Oncology, Merck, Novartis, Pfizer, Roche/Genentech, Takeda, and Tesaro. A.A.J. consults with Roche/Genentech, Aravive, and Almac Group; has research funding from AstraZeneca, Pfizer, Bristol-Myers Squibb, Immatics, and Iovance Biotherapeutics; has honoraria from Gerson Lehrman Group; has travel support from AstraZeneca and MedImmune. N.D.F. consults with Tesaro. A.K.S. consults with Merck and Kiyatec, has research funding from M-Trap, and is a shareholder of Bio-Path Holdings. All other authors declare no competing interests.
Published: February 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102053.
Contributor Information
Sanghoon Lee, Email: slee29@mdanderson.org.
Anil K. Sood, Email: asood@mdanderson.org.
Supplemental information
References
- Bast R.C., Jr., Hennessy B., Mills G.B. The biology of ovarian cancer: new opportunities for translation. Nat. Rev. Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A. On a measure of divergence between two statistical populations defined by their probability distributions. Bull. Calcutta Math. Soc. 1943;35:99–109. [Google Scholar]
- Birnbaum M.E., Mendoza J.L., Sethi D.K., Dong S., Glanville J., Dobbins J., Ozkan E., Davis M.M., Wucherpfennig K.W., Garcia K.C. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. 2014;157:1073–1087. doi: 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha E., Klinger M., Hou Y., Cummings C., Ribas A., Faham M., Fong L. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci. Transl Med. 2014;6:238ra270. doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Zhang C., Pan Y., Xu R., Xu C., Chen Z., Lu Z., Ke Y. T cell receptor beta-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J. Pathol. 2016;239:450–458. doi: 10.1002/path.4742. [DOI] [PubMed] [Google Scholar]
- Davis M.M. T cell receptor gene diversity and selection. Annu. Rev. Biochem. 1990;59:475–496. doi: 10.1146/annurev.bi.59.070190.002355. [DOI] [PubMed] [Google Scholar]
- Davis M.M., Bjorkman P.J. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- De Simone M., Rossetti G., Pagani M. Single cell T cell receptor sequencing: techniques and future challenges. Front. Immunol. 2018;9:1638. doi: 10.3389/fimmu.2018.01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake C.G., Lipson E.J., Brahmer J.R. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson R.O., Sherwood A.M., Rieder M.J., Guenthoer J., Williamson D.W., Carlson C.S., Drescher C.W., Tewari M., Bielas J.H., Robins H.S. High-throughput sequencing of T-cell receptors reveals a homogeneous repertoire of tumour-infiltrating lymphocytes in ovarian cancer. J. Pathol. 2013;231:433–440. doi: 10.1002/path.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah M., Reuben A., Spassova I., Yang R.K., Kubat L., Nagarajan P., Ning J., Li W., Aung P.P., Curry J.L. T cell repertoire in combination with T cell density predicts clinical outcomes in patients with merkel cell carcinoma. J. Invest. Dermatol. 2020;140:2146–2156.e4. doi: 10.1016/j.jid.2020.02.031. [DOI] [PubMed] [Google Scholar]
- Fleming N.D., Nick A.M., Coleman R.L., Westin S.N., Ramirez P.T., Soliman P.T., Fellman B., Meyer L.A., Schmeler K.M., Lu K.H. Laparoscopic surgical algorithm to triage the timing of tumor reductive surgery in advanced ovarian cancer. Obstet. Gynecol. 2018;132:545–554. doi: 10.1097/AOG.0000000000002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia K.C., Adams E.J. How the T cell receptor sees antigen--a structural view. Cell. 2005;122:333–336. doi: 10.1016/j.cell.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Gerlinger M., Quezada S.A., Peggs K.S., Furness A.J., Fisher R., Marafioti T., Shende V.H., McGranahan N., Rowan A.J., Hazell S. Ultra-deep T cell receptor sequencing reveals the complexity and intratumour heterogeneity of T cell clones in renal cell carcinomas. J. Pathol. 2013;231:424–432. doi: 10.1002/path.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Liu X., Wang Y., Wu X., Guan Y., Li H., Chen X., Zhou B., Yuan Q., Ou Y. Identification of characteristic TRB V usage in HBV-associated HCC by using differential expression profiling analysis. Oncoimmunology. 2015;4:e1021537. doi: 10.1080/2162402X.2015.1021537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A.C., Yarchoan M., Durham J.N., Yusko E.C., Rytlewski J.A., Robins H.S., Laheru D.A., Le D.T., Lutz E.R., Jaffee E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight. 2018;3:e122092. doi: 10.1172/jci.insight.122092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M., Sedighim S., Wang T., Antonios J.P., Everson R.G., Tucker A.M., Du L., Emerson R., Yusko E., Sanders C. TCR sequencing can identify and track glioma-infiltrating T cells after DC vaccination. Cancer Immunol. Res. 2016;4:412–418. doi: 10.1158/2326-6066.CIR-15-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson G.C., Kohn E.C., Kitchener H.C., Ledermann J.A. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sanchez A., Memon D., Pourpe S., Veeraraghavan H., Li Y., Vargas H.A., Gill M.B., Park K.J., Zivanovic O., Konner J. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170:927–938 e920. doi: 10.1016/j.cell.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou Z.C., Puhr J.S., Rojas M., McCormack W.T., Goodenow M.M., Sleasman J.W. T-Cell receptor Vbeta repertoire CDR3 length diversity differs within CD45RA and CD45RO T-cell subsets in healthy and human immunodeficiency virus-infected children. Clin. Diagn. Lab Immunol. 2000;7:953–959. doi: 10.1128/cdli.7.6.953-959.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L., Zhou X., Chen H., Luo Y., Sui W., Zhang J., Tang D., Yan Q., Dai Y. Composition and diversity analysis of the B-cell receptor immunoglobulin heavy chain complementarity-determining region 3 repertoire in patients with acute rejection after kidney transplantation using high-throughput sequencing. Exp. Ther. Med. 2019;17:2206–2220. doi: 10.3892/etm.2019.7183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Zhao L., Rojas C., Bateman N.W., Yao H., Lara O.D., Celestino J., Morgan M.B., Nguyen T.V., Conrads K.A. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 2020;31:107502. doi: 10.1016/j.celrep.2020.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo K.A., Coffey D.G., Morales A.J., Carlson C.S., Towlerton A.M.H., Gerdts S.E., Nkrumah F.K., Neequaye J., Biggar R.J., Orem J. High-throughput sequencing of the B-cell receptor in African Burkitt lymphoma reveals clues to pathogenesis. Blood Adv. 2017;1:535–544. doi: 10.1182/bloodadvances.2016000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.A., Khalili J., Ashe J., Berenson R., Ferrand C., Bonyhadi M. Standardized analysis for the quantification of Vbeta CDR3 T-cell receptor diversity. J. Immunol. Methods. 2006;317:100–113. doi: 10.1016/j.jim.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Looney T.J., Topacio-Hall D., Lowman G., Conroy J., Morrison C., Oh D., Fong L., Zhang L. TCR convergence in individuals treated with immune checkpoint inhibition for cancer. Front. Immunol. 2019;10:2985. doi: 10.3389/fimmu.2019.02985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto Y., Matsuo H., Sakuma H., Park I.K., Tsukada Y., Kohyama K., Kondo T., Kotorii S., Shibuya N. CDR3 spectratyping analysis of the TCR repertoire in myasthenia gravis. J. Immunol. 2006;176:5100–5107. doi: 10.4049/jimmunol.176.8.5100. [DOI] [PubMed] [Google Scholar]
- Nick A.M., Coleman R.L., Ramirez P.T., Sood A.K. A framework for a personalized surgical approach to ovarian cancer. Nat. Rev. Clin. Oncol. 2015;12:239–245. doi: 10.1038/nrclinonc.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich-Zugich J., Slifka M.K., Messaoudi I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 2004;4:123–132. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- Patch A.M., Christie E.L., Etemadmoghadam D., Garsed D.W., George J., Fereday S., Nones K., Cowin P., Alsop K., Bailey P.J. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–494. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- Postow M.A., Manuel M., Wong P., Yuan J., Dong Z., Liu C., Perez S., Tanneau I., Noel M., Courtier A. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer. 2015;3:23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles R.L., Redmond D., Sotiriou C., Loi S., Fumagalli D., Nuciforo P., Harbeck N., de Azambuja E., Sarp S., Di Cosimo S. Association of T-cell receptor repertoire use with response to combined trastuzumab-Lapatinib treatment of HER2-positive breast cancer: secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol. 2018;4:e181564. doi: 10.1001/jamaoncol.2018.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J.G., Makohon-Moore A.P., Gerold J.M., Heyde A., Attiyeh M.A., Kohutek Z.A., Tokheim C.J., Brown A., DeBlasio R.M., Niyazov J. Minimal functional driver gene heterogeneity among untreated metastases. Science. 2018;361:1033–1037. doi: 10.1126/science.aat7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A., Gittelman R., Gao J., Zhang J., Yusko E.C., Wu C.J., Emerson R., Zhang J., Tipton C., Li J. TCR repertoire intratumor heterogeneity in localized lung adenocarcinomas: an association with predicted neoantigen heterogeneity and postsurgical recurrence. Cancer Discov. 2017;7:1088–1097. doi: 10.1158/2159-8290.CD-17-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben A., Zhang J., Chiou S.H., Gittelman R.M., Li J., Lee W.C., Fujimoto J., Behrens C., Liu X., Wang F. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nat. Commun. 2020;11:603. doi: 10.1038/s41467-019-14273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert L., Tsoi J., Wang X., Emerson R., Homet B., Chodon T., Mok S., Huang R.R., Cochran A.J., Comin-Anduix B. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014;20:2424–2432. doi: 10.1158/1078-0432.CCR-13-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Snyder A., Makarov V., Merghoub T., Yuan J., Zaretsky J.M., Desrichard A., Walsh L.A., Postow M.A., Wong P., Ho T.S. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi A., Agathangelidis A., Stalika E., Karypidou M., Siorenta A., Anagnostopoulos A., Rosenquist R., Hadzidimitriou A., Ghia P., Sutton L.A. Antigen selection shapes the T-cell repertoire in chronic lymphocytic leukemia. Clin. Cancer Res. 2016;22:167–174. doi: 10.1158/1078-0432.CCR-14-3017. [DOI] [PubMed] [Google Scholar]
- Walsh T., Casadei S., Lee M.K., Pennil C.C., Nord A.S., Thornton A.M., Roeb W., Agnew K.J., Stray S.M., Wickramanayake A. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. U S A. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.L., Davis M.M. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi: 10.1016/s1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data from the TCR sequencing in this study has been deposited in the ImmuneACCESS project repository of Adaptive Biotechnology database (https://doi.org/10.21417/LS2021iS, https://clients.adaptivebiotech.com/pub/sanghoon-2021-iscience).