

Abstract
Chimeric antigen receptor (CAR) αβ T cell adoptive immunotherapy has shown great promise for improving cancer treatment. However, there are several hurdles to overcome for the wide clinical application of CAR-αβ T cells therapy, including side effects and a limited T cells source from cancer patients. Therefore, we sought to identify an alternative T cell subset that could avoid these limitations and improve the effectiveness of CAR-T immunotherapy. γδ T cells are a minor subset of T cells, which share the characteristic of innate immune cells and adaptive immune cells. Vγ9Vδ2 T cells are a predominant γδ T subset in the circulating peripheral blood. In this study, we investigated the antigen-specific antitumor activity of CAR-Vγ9Vδ2 T cells targeting MUC1-Tn antigen. Vγ9Vδ2 T cells were expanded from peripheral blood mononuclear cells of healthy volunteers with zoledronic acid and interleukin-2. CAR-Vγ9Vδ2 T cells were generated by transfection of lentivirus encoding MUC1-Tn CAR. Cytotoxicity assays with various cancer cell lines revealed that CAR-Vγ9Vδ2 T cells could effectively lyse tumor cells in an antigen-specific manner, with similar or stronger effects than CAR-αβ T cells. However, CAR-Vγ9Vδ2 T cells had shorter persistence, which could be improved with the addition of IL-2 to maintain the function of CAR-Vγ9Vδ2 T cells with consecutive stimulation of tumor cells. Using a xenograft mouse model, we further showed that CAR-Vγ9Vδ2 T cells more effectively suppressed tumor growth in vivo than Vγ9Vδ2 T cells. Therefore, MUC1-Tn CAR-modified Vγ9Vδ2 T cells may represent a novel, promising ready-to-use product for cancer allogeneic immunotherapy.
Keywords: Vγ9Vδ2 T cells, chimeric antigen receptor, MUC1-Tn, cancer immunotherapy
Introduction
Chimeric antigen receptor (CAR) T cell adoptive immunotherapy has shown great achievements in the treatment of hematological malignancies [1,2]. To date, αβ T cells are the most commonly applied effector cells in adoptive immunotherapy. However, the wide application of CAR-αβ T cell therapy is hindered by many unfavorable factors, including the potential to induce cytokine release storm syndrome and neurotoxicity [3]. Moreover, as an individualized immunotherapy, patient self-derived T cells are required to prepare CAR-αβ T cells. This can be a major limitation given that the immune system of most cancer patients is often damaged due to previous rounds of therapy or by the disease itself, leading to a lack of sufficient T cells or dysfunctional T cells, which could increase the risk of failure in preparing self-derived CAR T cells or decrease the clinical efficacy [4]. Development of off-the-shelf allogeneic CAR-T products may be an ideal solution to overcome these problems.
γδ T cells are a minor subset of T cells accounting for 1-5% of total circulating T cells but are the predominant lymphocytes in the epithelial tissues [5]. γδ T cells share the characteristics of innate immune cells and adaptive immune cells, and play important roles in resisting the infection of bacteria, viruses, and the invasion and growth of tumors in the host [6]. Moreover, unlike the major histocompatibility class (MHC)-dependent antigen recognition mechanism of αβ T cells, γδ T cells can recognize danger-associated molecular signals in an MHC-independent manner [7], thereby making them suitable for allogeneic CAR-T immunotherapy. Arruda et al. [8] reported that γδ T cells play an important role in both leukemia and infection control in hematopoietic stem cell transplantation and had no association with graft versus host disease development, suggesting that γδ T cells may be an ideal candidate for allogeneic immunotherapy. Previous studies demonstrated that γδ T cells play an important role in cancer therapy, and that intratumoral γδ T cells represent the most favorable prognostic indicator across diverse cancers [9,10].
γδ T cells can be divided into two main subsets based on their δ chain usage: Vδ2+ cells prefer to co-express with Vγ9 chain, while Vδ2- cells can pair with an array of Vγ chains [11]. Vγ9Vδ2 T subtype cells are predominant in circulating peripheral blood [12], and were demonstrated to be primed for innate cytotoxicity against multiple tumors [6,9]. Vγ9Vδ2 T cells can also act as antigen-presenting cells (APCs) after activation, which may play a critical role in enhancing the immune response [13,14]. In recent years, increasing numbers of studies on CAR-modified Vγ9Vδ2 T cells have emerged. Although Vγ9Vδ2 T cells exert an innate anti-tumor effect, CAR modification can endow Vγ9Vδ2 T cells with specific anti-tumor effects and extend their scope of tumor treatment. Rischer et al. [15] reported that CAR-Vγ9Vδ2 T cells targeting GD2 showed enhanced antigen-specific tumor reactivity, and similar results were achieved with Vγ9Vδ2 T cells expressing a CD19-targeted CAR. Therefore, CAR-modified Vγ9Vδ2 T cells may represent a novel promising immunotherapeutic approach for cancer therapy.
Mucin 1 (MUC1) with the Tn epitope as a cancer marker is an antigen that is highly expressed in a form with abnormal O-glycosylation on the surface of a variety of cancer cells, including breast cancer and gastric cancer, but is not expressed or is expressed at low levels in normal tissues [16-18]. Therefore, MUC1-Tn antigen can be used as an ideal target for solid tumor therapy. Posey et al. [17] reported that 5E5-CAR T cells targeting MUC1-Tn antigen displayed effective anti-tumor activity on T cell leukemia and pancreatic cancer in vitro and in vivo.
Toward this end, in this study, we developed a method for preparation of clinical-grade CAR-Vγ9Vδ2 T cells. Vγ9Vδ2 T cells were selectively expanded from total peripheral blood mononuclear cells (PBMCs) of healthy donors by zoledronic acid (ZOL) with interleukin (IL)-2 to a clinically significant number [12]. ZOL-expanded Vγ9Vδ2 T cells for adoptive transfer have been previously reported in preclinical and clinical trials with demonstrated safety [19]. Second-generation MUC1-Tn-specific CAR (MUC1-Tn-CD28-CD3ζ)-modified Vγ9Vδ2 T cells were generated via lentiviral transfection, and their specific anti-tumor activity was investigated in vitro and in vivo. These results can therefore provide a novel and promising allogeneic strategy for cancer immunotherapy.
Material and methods
Cloning
The MUC1-Tn-specific single-chain variable fragment (scFv) was derived from the MUC1-Tn specific antibody PG926, which is provided by PersonGen BioTherapeutics (Suzhou) Co., Ltd. The CAR structure consists of a PG926 scFv, Fc hinge, CD28 transmembrane, intracellular domain, and CD3 zeta domain. The DNA sequence of the COSMC gene was acquired by reverse transcription of mRNA from human T cells, followed by polymerase chain reaction (PCR) amplification. The CAR cassette and the COSMC sequence were both subcloned into XbaI- and NotI-digested pCDH-CMV lentiviral vectors to prepare pCDH-CMV-MUC1-Tn-CAR and pCDH-CMV-COSMC lentiviral vectors, respectively.
Lentivirus preparation
pCDH-CMV-MUC1-Tn-CAR and pCDH-CMV-COSMC lentivirus were prepared as previously described [20].
Vγ9Vδ2 T and T cell activation, transduction, and expansion
PBMCs from healthy volunteers were isolated using Ficoll-Hypaque gradient centrifugation. Vγ9Vδ2 T cells were then activated by treatment with ZOL (1.75 mM; Aosaikang Pharmaceutical, Jiangsu, China) and IL-2 (200 U/mL; Novoprotein, Shanghai, China). αβ T cells were activated by TransAct (20 µL; Miltenyi Biotec, Auburn, CA, USA) in the presence of IL-7 (155 U/mL; Novoprotein) and IL-15 (190 U/mL; Novoprotein). After 48-h stimulation, both T cell subsets were transduced with CAR lentivirus at a multiplicity of infection of 20, respectively.
Cell lines
HGC-27, SUN-1, KATO III, T47D, MDA-MB-468, MDA-MB-231, Jurkat T, and 293 T cells were purchased from the Cell Bank of Shanghai Institute of Biochemistry & Cell Biology (Shanghai, China). HGC-27, SUN-1, KATO III, and Jurkat T cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum. T47D, MDA-MB-468, MDA-MB-231, and 293 T cells were cultured in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Flow cytometry
To detect the expression of MUC1-Tn on HGC-27, SUN-1, KATO III, and Jurkat T cells, the cells were stained with PG926 antibody and mouse IgG (BD) isotype antibody at 37°C for 30 min, washed three times with phosphate-buffered saline (PBS), and then stained with anti-human IgG Fc antibody (Jackson ImmunoResearch, West Grove, PA, USA) at 37°C for 30 min. To confirm the phenotype of γδ T cells, the expanded γδ T cells were stained with mouse anti-human CD3 antibody (BD, USA) and mouse anti-human δ2 antibody (BD) antibody at 37°C for 30 min, washed three times with 500 µL PBS, and then suspended in 500 µL PBS. To detect CAR expression on T cells, αβ T, CAR-αβ T, and CAR-Vγ9Vδ2 T cells were stained with anti-human IgG Fc antibody (Jackson ImmunoResearch) at 37°C for 30 min, washed three times with PBS, and then suspended in 500 µL PBS. All assays were analyzed using a flow cytometer (ACEA Biosciences, San Diego, CA, USA).
Tumor cells elimination assay
We established a green fluorescent protein (GFP)-expressing Jurkat T cell line by transfection of lentivirus encoding GFP, and stable GFP expression was confirmed by flow cytometry. To compare the tumor elimination ability of CAR-Vγ9Vδ2 T cells and CAR-αβ T cells, 2 × 105 GFP-Jurkat T cells were seeded in a 48-well plate. The GFP-Jurkat T cells were co-cultured with αβ T cells, CAR-αβ T cells, Vγ9Vδ2 T cells, or CAR-Vγ9Vδ2 T cells at an effector:target (E:T) ratio of 1:1 (n = 3). The cells were collected every 48 h for the detection of residual tumor cells by flow cytometry (ACEA Biosciences), GFP positive cells represent tumor cells. The effector cells were stained with CD3 antibody (BD) for cell number counting by adding Counting Beads (Invitrogen, Paisley, UK) and analyzed by flow cytometry (ACEA Biosciences). Subsequently, the effector cells and GFP-Jurkat T cells were re-seeded at an E:T ratio of 1:1. The same culture system was applied in each restimulation experiment.
Cytotoxicity assay
To compare the cytotoxicity of CAR-Vγ9Vδ2 T cells and CAR-αβ T cells, we applied the 5-(and-6)-carboxyfluorescein diacetate succinimidyl ester (CFSE)/7-aminoactinomycin D (7-AAD) flow cytometry method and xCELLigence real-time cell analyzer (RTCA; ACEA Biosciences) [21,22]. In the CFSE/7-AAD flow cytometry assay, the target cells (HGC-27, SUN-1, KATO III, Jurkat T, and COSMC-Jurkat cells) were stained with CFSE (Invitrogen) at 37°C for 15 min and then washed three times with PBS. The density of the CFSE-stained cells was adjusted to 2 × 105 cells/mL and the cells were then seeded into 24-well culture plates. The effector cells were added according to various E:T ratios. Twenty-four hours later, the cells were collected, resuspended in an identical volume of PBS, and then 7-AAD (BD) was added. The percentage of specific lysis (CFSE-positive and 7-AAD-positive) was calculated using flow cytometry.
In the xCELLigence RTCA (ACEA Biosciences) assay, 10,000 target cells were seeded into E-plate 16 (ACEA Biosciences), and the cell index was recorded every 5 min. After 24 h, effector cells and control media were added according to various E:T ratios, and the cell index was recorded every 15 min on the system.
Cytokine detection
For cytokine detection, the co-culture supernatants from the cytotoxicity assay were collected and analyzed for the levels of interferon gamma (IFN-γ), Granzyme B, and tumor necrosis factor alpha (TNF-α) using a cytometric bead array (CBA) system. The Human Granzyme B CBA Flex Set D7 Kit, Human IFN-γ CBA Flex Set E7 Kit, Human IL-2 CBA Flex Set A4, and Human TNF CBA Flex Set D9 were obtained from BD.
Animal experiment
Six to seven-week-old female NOD-Prkdcs-cidIl2rgtm1/Bcgen (B-NSG) mice (Biocytogen, Haimen, China) were subcutaneously inoculated with 1 × 106 HGC-27 cells on the right dorsal side. Tumor growth was assessed twice a week by measuring two perpendicular diameters with calipers. Tumor volume (V) was calculated using the following equation: V = π/6 × d2 × D (where D is the longitudinal diameter and d is the latitudinal diameter). When tumors reached a mean volume of 100 mm3, the mice were randomly assigned to the following three groups (with four mice per group): (i) intratumoral injection of PBS (control), (ii) intratumoral injection of 1 × 107 Vγ9Vδ2 T cells, (iii) and intratumoral injection of 1 × 107 CAR-Vγ9Vδ2 T cells. All mice in each group received 20,000 U of IL-2 every two days throughout the treatment period. All in vivo mouse experiments were conducted in accordance with the experimental animal management regulations of Soochow University.
Statistical analysis
GraphPad Prism5 software (GraphPad Software Inc., La Jolla, CA, USA) was used for statistical analyses. Student’s t-test was used to compare values between two groups. P < 0.05 was considered to indicate a statistically significant difference.
Results
Generation of Vγ9Vδ2 T cells
Vγ9Vδ2 T cells were selectively expanded by treatment with ZOL and IL-2 to reach a clinically significant number. After activation and expansion, flow cytometry using mouse anti-human CD3 antibody and mouse anti-human δ2 antibody showed that the purity of Vγ9Vδ2 T cells was greater than 90% (Figure 1).
Figure 1.
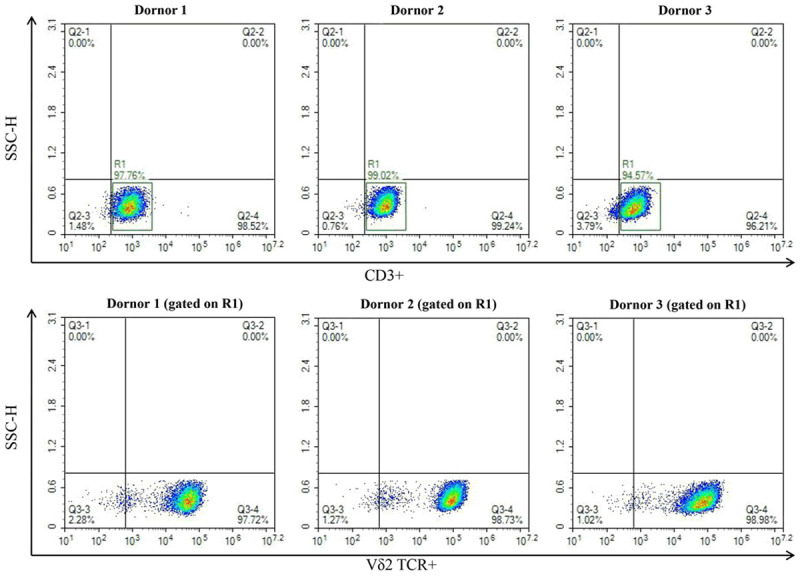
Generation of Vγ9Vδ2 T cells. Phenotype detection of Vγ9Vδ2 T cells expanded from PBMCs of three healthy volunteers by flow cytometry.
Generation of CAR-αβ T and CAR-Vγ9Vδ2 T cells
To target MUC1-Tn antigen, we constructed second-generation CAR using PG926 scFv. The schematic of CAR is shown in Figure 2A. The CAR-αβ T and CAR-Vγ9Vδ2 T cells were then generated by lentivirus transfection. Flow cytometry using mouse anti-human Fc antibody showed positive rates of CAR-αβ T cells and CAR-Vγ9Vδ2 T cells of 39.43% and 42.60%, respectively (Figure 2B), demonstrating successful establishment of a preparation method for CAR-Vγ9Vδ2 T cells.
Figure 2.
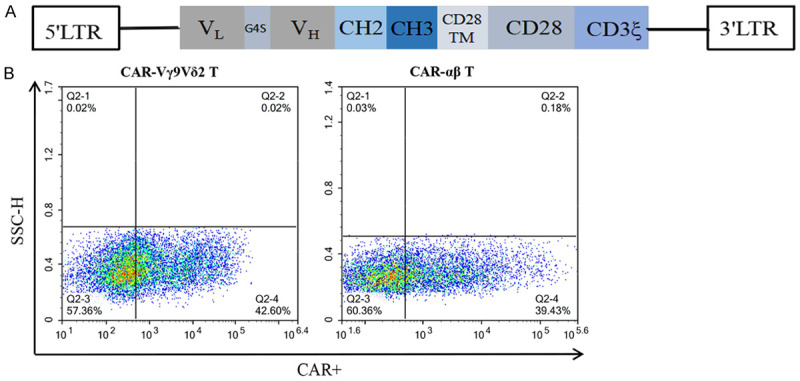
Generation of CAR-αβ T and CAR-Vγ9Vδ2 T cells. A. Lentiviral vector construct with leader sequence, anti-MUC1-Tn scFv, Fc hinge, CD28 transmembrane domain, CD28 intracellular domain, and CD3ζ. B. MUC1-Tn CAR expression on αβ T cells and Vγ9Vδ2 T cells detected by flow cytometry using mouse anti-human Fc antibody.
CAR-Vγ9Vδ2 T cells lyse tumor cells in a CAR-dependent antigen-specific manner
Several studies have revealed that COSMC gene mutations or epigenetic silencing is the main contributor to MUC1-Tn antigen formation. The COSMC gene of Jurkat T cells has a one-nucleotide deletion that results in a frameshift mutation and early truncation of the T synthase chaperone protein [17], leading to the expression of Tn antigen. We confirmed the expression of MUC1-Tn antigen in Jurkat T cells using PG926 antibody (Figure 3A). The COSMC function of Jurkat T cells was restored by transduction of a lentivirus encoding COSMC, and this cell line was designated COSMC-Jurkat T cells. Subsequently, loss of MUC1-Tn antigen expression on COSMC-Jurkat T cells was confirmed by flow cytometry using PG926 antibody (Figure 3A). The cytotoxicity assays showed that CAR Vγ9Vδ2 T cells exhibited specific cytotoxicity on Jurkat T cells and not on COSMC-Jurkat T cells (Figure 3B). The TNF-α and IFN-γ secretion levels of CAR Vγ9Vδ2 T cells were significantly higher than that of Vγ9Vδ2T cells after co-incubation with Jurkat T cells. Besides, although the secretion of TNF-α and IFN-γ of CAR Vγ9Vδ2 T cells were higher than that of Vγ9Vδ2 T cells after co-culture with COSMC-Jurkat T cells, the difference was not as large as that observed with co-culture of Jurkat T cells, and this is may due to the activation of Vγ9Vδ2 T cells by lentivirus transfection (Figure 3C). The secretion level of granzyme B of CAR Vγ9Vδ2 T cells was significantly elevated compared with that of Vγ9Vδ2T cells co-cultured with Jurkat T cells and CAR Vγ9Vδ2 T cells co-cultured with COSMC-Jurkat T cells at low E:T ratio, but there was no significant elevation of granzyme B at high E:T ratio between Jurkat T and COSMC-Jurkat T co-cultured effector cells. This suggested that secretion of granzyme B by Vγ9Vδ2 T cells may reach saturation at a high E:T ratio; indeed, the Vγ9Vδ2T and CAR Vγ9Vδ2 T cells co-cultured with COSMC-Jurkat T cells also displayed strong and similar granzyme B secretion (Figure 3C). These results demonstrated that CAR Vγ9Vδ2 T cells possess high antigen-specific cytotoxicity against tumor cells.
Figure 3.

Tumor cell lysis of CAR-Vγ9Vδ2 T cells in a CAR-dependent antigen-specific manner. A. MUC1-Tn antigen expression level on Jurkat T cells and COSMC-Jurkat T cells assessed by flow cytometry, demonstrating loss of MUC1-Tn expression on COSMC-Jurkat T cells. B. Antigen-specific cytotoxicity of CAR-Vγ9Vδ2 T cells against Jurkat T cells and COSMC-Jurkat T cells at different E:T ratios assayed by flow cytometry. C. Detection of TNF-α, granzyme B, and IFN-γ concentrations in the supernatants from the cytotoxicity assay of CAR-Vγ9Vδ2 T cells against Jurkat T cells and COSMC-Jurkat T cells. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
CAR-Vγ9Vδ2 T cells exhibit similar or stronger cytotoxicity than CAR-αβ T cells on breast cancer cells
To investigate whether the CAR modification enhanced the cytotoxicity of Vγ9Vδ2 T cells, we assayed the cytotoxicity of CAR-Vγ9Vδ2 T and CAR-αβ T cells against breast cancer cell lines with different levels of MUC1-Tn antigen expression (Figure 4A). Both CAR-Vγ9Vδ2 T and CAR-αβ T cells exhibited significantly stronger cytotoxicity against tumor cells than Vγ9Vδ2 T and αβ T cells, respectively. Moreover, CAR-Vγ9Vδ2 T cells displayed cytotoxicity that was similar to or greater than that of CAR-αβ T cells (Figure 4B). The CBA assay further revealed that CAR-Vγ9Vδ2 T cells secreted greater levels of IFN-γ, granzyme B, and TNF-α than Vγ9Vδ2 T cells when co-cultured with breast cancer cells (Figure 4C).
Figure 4.
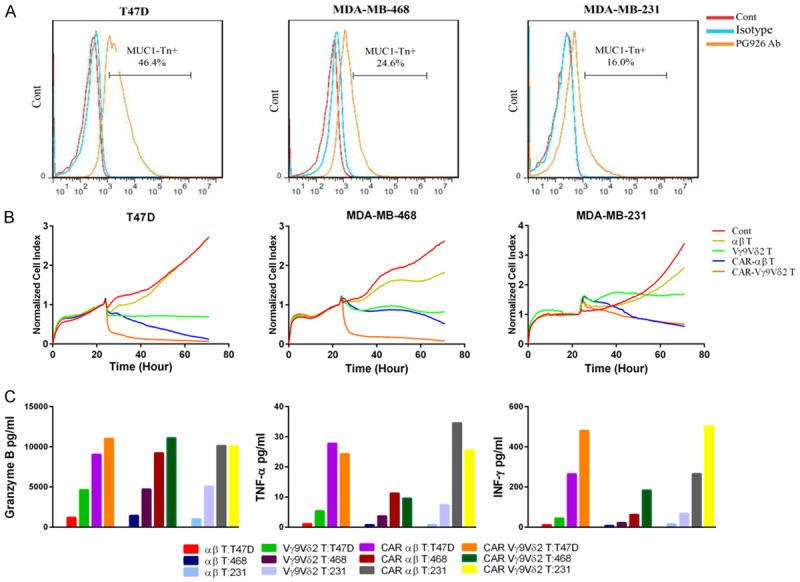
CAR-Vγ9Vδ2 T cells exhibit similar or stronger cytotoxicity than CAR-αβ T cells on breast cancer cells. A. MUC1-Tn antigen expression levels on T47D cells, MDA-MB-231 cells, and MDA-MB-468 cells. B. Cytotoxicity of CAR-Vγ9Vδ2 T cells and CAR T cells against T47D cells, MDA-MB-231 cells, and MDA-MB-468 cells detected by the RTCA assay. C. Detection of TNF-α, granzyme B, and IFN-γ concentrations in the supernatants from the cytotoxicity assays of CAR-Vγ9Vδ2 T cells and CAR-αβ T cells against T47D cells, MDA-MB-231 cells, and MDA-MB-468 cells.
CAR-Vγ9Vδ2 T cells exhibit stronger cytotoxicity than CAR-αβ T cells on gastric cancer cells
The cytotoxicity of CAR-Vγ9Vδ2 T and CAR-αβ T cells against gastric cancer cell lines were also evaluated. MUC1-Tn antigen expression in the gastric cancer cell lines HGC-27, KATO III, and SUN-1 is shown in Figure 5A. CAR-Vγ9Vδ2 T cells more effectively lysed all three gastric cancer cell lines compared to the CAR-αβ T cells (Figure 5B). These results were similar with the cytotoxicity results of CAR-Vγ9Vδ2 T and CAR-αβ T cells against breast cancer cell lines.
Figure 5.
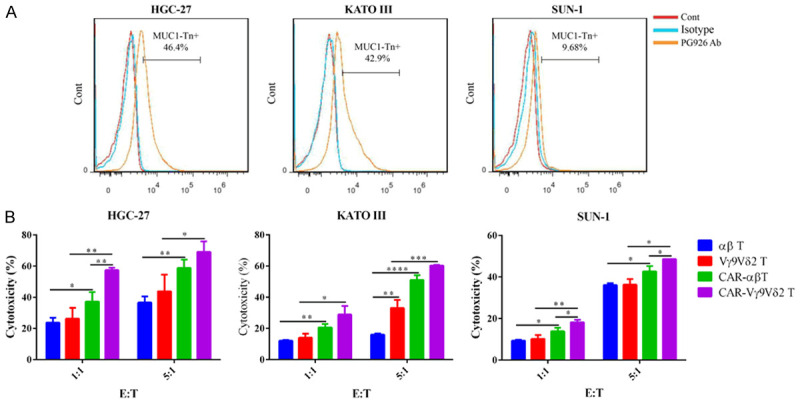
CAR-Vγ9Vδ2 T cells exhibit stronger cytotoxicity than CAR-αβ T cells on gastric cancer cells. A. MUC1-Tn antigen expression levels on HGC-27 cells, SUN-1 cells, and KATO III cells. B. Cytotoxicity of CAR-Vγ9Vδ2 T and CAR-αβ T cells against HGC-27 cells, SUN-1 cells, and KATO III cells detected by flow cytometry. Data are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
CAR-Vγ9Vδ2 T cells have deficiencies in persistence
The persistence of T cell function has a significant impact on their antitumor effect. The deficiencies in persistence may attenuate their anti-tumor effects and limit their clinical application. Therefore, we compared the ability of CAR-Vγ9Vδ2 T cells and CAR-αβ T cells to consecutively eliminate tumor cells. The stable GFP expression in lentivirus-transfected Jurkat T cells was confirmed by flow cytometry (Figure 6A). After the fourth stimulation (E:T of 1), CAR-Vγ9Vδ2 T cells could no longer eliminate the tumor cells, with ~91% residual tumor cells detected, whereas the CAR-αβ T cells could still effectively eliminate the tumor cells at this point (Figure 6B and 6C). These results indicated that CAR-Vγ9Vδ2 T cells have similar or stronger cytotoxicity than CAR-αβ T cells, but their functional persistence is weaker.
Figure 6.

CAR-Vγ9Vδ2 T cells have deficiencies in persistence. A. GFP expression level on Jurkat T cells assessed by flow cytometry. B, C. Consecutive tumor cells eliminated by CAR-Vγ9Vδ2 T cells assessed by multiple Jurkat T cells stimulation cycles; CAR-αβ T cells served as a control for comparison. Data are presented as mean ± SD (n = 3) of a representative experiment. ****P < 0.0001.
IL-2 is important for maintaining the persistence of CAR-Vγ9Vδ2 T cells
Based on the above results, we next sought to improve the persistence of Vγ9Vδ2 T cells. IL-2 is widely used in the expansion of Vγ9Vδ2 T cells in vitro; thus, we explored the effect of IL-2 on the persistence of Vγ9Vδ2 T cells. As expected, both Vγ9Vδ2 T and CAR-Vγ9Vδ2 T cells displayed enhanced cytotoxicity and persistence in the presence of IL-2, based on more effectively eliminating Jurkat T cells than their counterparts without IL-2 addition (Figures 6A and 7B). These results demonstrated that IL-2 plays an important role in maintaining the function of Vγ9Vδ2 T cells and improving their persistence, providing the basis for the experimental design of the in vivo tumor inhibition assay in the xenograft mouse model.
Figure 7.

IL-2 is important for maintaining the persistence of CAR-Vγ9Vδ2 T cells. A, B. Effect of IL-2 on consecutive elimination of tumor cells by Vγ9Vδ2 T cells assessed by multiple Jurkat T cells stimulation cycles. Data are presented as mean ± SD (n = 3). ***P < 0.001.
CAR-Vγ9Vδ2 T cells exhibit effective anti-tumor activity in vivo
A schematic representation of the in vivo experimental design is shown in Figure 8A. Both Vγ9Vδ2 T and CAR-Vγ9Vδ2 T cells could effectively suppress tumor growth, while CAR-Vγ9Vδ2 T cells exhibited a stronger anti-tumor effect (Figure 8B and 8C). The weight of mice in each group was stable throughout the experimental period, indicating that CAR-Vγ9Vδ2 T cell treatment did not cause evident toxicity in the mice (Figure 8D). These results indicated that the innate anti-tumor ability of Vγ9Vδ2 T cells was retained during stimulation and expansion in vitro, and CAR-Vγ9Vδ2 T cells retained their innate antitumor properties when acquiring antigen-specific cytotoxicity.
Figure 8.
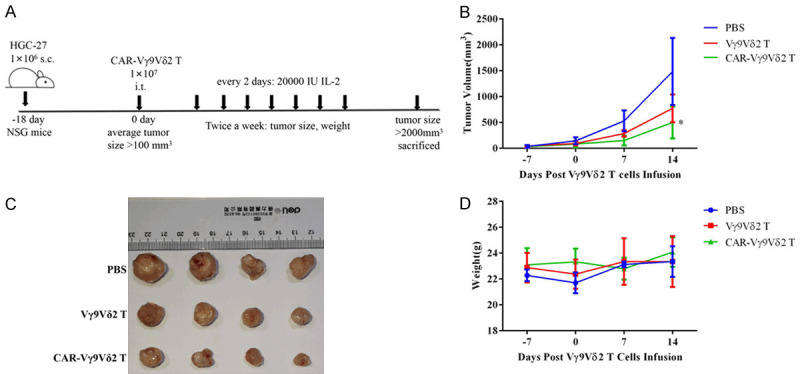
CAR-Vγ9Vδ2 T cells exhibit effective anti-tumor activity in vivo. A. Treatment scheme of the xenograft HGC-27 model. B. Tumor volume of the mice in different groups assessed twice a week. C. Photographs of the tumors of mice in different groups. D. Body weight of mice measured twice a week. Data are presented as mean ± SD (n = 4). *P < 0.05.
Discussion
In recent years, CAR-αβ T cell immunotherapy has shown impressive progress in immunotherapy against hematological malignancies [1,2], but similar success has not yet been achieved for the treatment of solid tumors [23]. To date, αβ T cells are the most commonly used effector cells for CAR-T immunotherapy, but their clinical application remains hindered due to unfavorable side effects and limitations of the cell source. To address these problems, we focused on the minor T cell subset γδ T cells as an alternative source for T cell therapy. γδ T cells can recognize antigens in an MHC-independent manner [7], thereby making them suitable for allogeneic CAR-T immunotherapy. Therefore, the application of γδ T cells may overcome the hurdles faced by CAR-αβ T cells.
Following confirmation of successful CAR expression on Vγ9Vδ2 T cells, we first investigated the antigen-specific cytotoxicity on tumor cells in vitro. Several studies have revealed that COSMC gene mutations or epigenetic silencing is the main reason for MUC1-Tn antigen formation. The COSMC gene of Jurkat T cells has a one-nucleotide deletion leading to the expression of Tn antigen [17]. In this study, the COSMC function of Jurkat T cells was restored by transfection of lentivirus encoding COSMC, and the loss of MUC1-Tn antigen expression was confirmed by flow cytometry. Jurkat T cells and COSMC-Jurkat T cells were then used as target cells to verify the specific cytotoxicity of CAR-Vγ9Vδ2 T cells. The results showed that CAR-Vγ9Vδ2 T cells could specifically lyse antigen-positive Jurkat T cells, with no specific lysis detected on antigen-negative COSMC-Jurkat T cells. These results demonstrated that MUC1-Tn CAR modification could endow Vγ9Vδ2 T cells with antigen-specific cytotoxicity.
We further compared the cytotoxicity and tumor cell inhibitory ability of CAR-Vγ9Vδ2 T cells and CAR-αβ T cells, demonstrating stronger or similar cytotoxicity of CAR-Vγ9Vδ2 T cells against a variety of cancer cell lines. However, CAR-αβ T cells displayed stronger ability to consecutively eliminate tumor cells. These results suggest that although CAR-Vγ9Vδ2T cells displayed cytotoxicity that was similar to or greater than that of CAR-αβ T cells, the shorter persistence could limit their clinical application.
To overcome this issue, we demonstrated that culture in the presence of IL-2 could significantly improve the persistence and simultaneously enhance the cytotoxicity of CAR Vγ9Vδ2 T cells, providing an important basis for the in vivo experiments. Mice were received 20,000 U IL-2 every two days in vivo antitumor experiments. CAR-Vγ9Vδ2 T cells could more effectively suppress tumor growth in vivo compared to the PBS group and Vγ9Vδ2 T cells group, although Vγ9Vδ2 T cells also showed a certain degree of anti-tumor activity. These results indicated that CAR modification endowed Vγ9Vδ2 T cells with specific anti-tumor activity. In addition, the Vγ9Vδ2 T cells showed a certain degree of cytotoxicity, indicating that the innate anti-tumor ability of Vγ9Vδ2 T cells was not lost during their stimulation and expansion in vitro. Therefore, CAR-Vγ9Vδ2 T cells could retain their innate anti-tumor properties when acquiring antigen-specific cytotoxicity.
Collectively, we successfully established a clinical-grade CAR-Vγ9Vδ2 T cells production method, providing a solid foundation for clinical application. Both the in vitro and in vivo anti-tumor experiments demonstrated that MUC1-Tn CAR modification endowed Vγ9Vδ2 T cells with antigen-specific cytotoxicity. Deficiencies in persistence of Vγ9Vδ2 T cells were improved by the addition of IL-2; however, further study is needed to explore better ways to solve this problem. Polito et al. [4] expanded γδ T cells by artificial APC with IL-2 and IL-15 to obtain a polyclonal cellular product, which containing a long-living γδ T population with anti-tumor activity against a broad range of cancers. Therefore, harnessing polyclonal γδ T cells to establish CAR-T products may be an alternative option to overcome the persistence defects of homogeneous Vγ9Vδ2 T cells.
In conclusion, Vγ9Vδ2 T cells have endogenous anti-tumor activity and can recognize antigens in an MHC-independent manner, resulting in a negligible allogeneic transplantation reaction. The present results show that MUC1-Tn-specific CAR-modified Vγ9Vδ2 T cells displayed significantly enhanced antigen-specific antitumor potency both in vitro and in vivo. Therefore, CAR-modified Vγ9Vδ2 T cells can be developed as a novel off-the-shelf allogeneic immunotherapy product to overcome the current limitations of cancer immunotherapy.
Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFC1303403), Natural Science Foundation of China (Grant Nos. 81872431 and 31471283), Priority Academic Program Development of Jiangsu Higher Education Institutions, the Collaborative Innovation Major Project (Grant No. XYXT-2015304), Six Talent Peaks Project in Jiangsu Province (No. SWYY-CXTD 010), and Natural Science Foundation of the Jiangsu Higher Education Institutions of China (General Program, Grant No. 19KJ 320003).
Disclosure of conflict of interest
None.
References
- 1.Vairy S, Garcia JL, Teira P, Bittencourt H. CTL019 (tisagenlecleucel): CAR-T therapy for relapsed and refractory B-cell acute lymphoblastic leukemia. Drug Des Dev Ther. 2018;12:3885. doi: 10.2147/DDDT.S138765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polito VA, Cristantielli R, Weber G, Bufalo FD, Belardinilli T, Arnone CM, Petretto A, Antonucci L, Giorda E, Tumino N, Pitisci A, Angelis BD, Quintarelli C, Locatelli F, Caruana I. Universal ready-to-use immunotherapeutic approach for the treatment of cancer: expanded and activated polyclonal γδ memory T cells. Front Immuno. 2019;10:2717. doi: 10.3389/fimmu.2019.02717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13:88. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, Gustafsson K, Anderson J. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther. 2017;25:1234–1247. doi: 10.1016/j.ymthe.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayday AC. γδ T cells and the lymphoid stress-surveillance response. Immunity. 2019;31:184–196. doi: 10.1016/j.immuni.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Arruda LCM, Gaballa A, Uhlin M. Impact of γδ T cells on clinical outcome of hematopoietic stem cell transplantation: systematic review and meta-analysis. Blood Adv. 2019;3:3436–3448. doi: 10.1182/bloodadvances.2019000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng WG, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, Alizadeh AA. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2005;21:938. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Front Immuno. 2014;5:636. doi: 10.3389/fimmu.2014.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bottino C, Tambussi G, Ferrini S, Ciccone E, Varese P, Mingari MC, Moretta L, Moretta A. Two subsets of human T lymphocytes expressing gamma/delta antigen receptor are identifiable by monoclonal antibodies directed to two distinct molecular forms of the receptor. J Exp Med. 1988;168:491–505. doi: 10.1084/jem.168.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo M, Sakuta K, Noguchi A, Ariyoshi N, Sato K, Sato S, Sato K, Hosoi A, Nakajima J, Yoshida Y, Shiraishi K, Nakagawa K, Kakimi K. Zoledronate facilitates large-scale ex vivo expansion of functional γδ T cells from cancer patients for use in adoptive immunotherapy. Cytotherapy. 2008;10:842–856. doi: 10.1080/14653240802419328. [DOI] [PubMed] [Google Scholar]
- 13.Gomes AQ, Martins DS, Silva-Santos B. Targeting γδ T lymphocytes for cancer immunotherapy: from novel mechanistic insight to clinical application. Cancer Res. 2010;70:10024–10027. doi: 10.1158/0008-5472.CAN-10-3236. [DOI] [PubMed] [Google Scholar]
- 14.Brandes M, Willimann K, Bioley G, Levy N, Eberl M, Luo M, Tampe R, Levy F, Romero P, Moser B. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc Natl Acad Sci U S A. 2009;106:2307–2312. doi: 10.1073/pnas.0810059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rischer M, Pscherer S, Duwe S, Vormoor J, Jurgens H, Rossig C. Human gamma delta T cells as mediators of chimaeric-receptor redirected antitumour immunity. Br J Haematol. 2004;126:583–592. doi: 10.1111/j.1365-2141.2004.05077.x. [DOI] [PubMed] [Google Scholar]
- 16.Springer GF. T and Tn, general carcinoma autoantigens. Science. 1984;224:1198–1206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- 17.Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mande U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM, Cogdill AP, Chen TJ, Song DC, Schollel J, Kranz DM, Feldman MD, Young R, Keith B, Schreiber H, Clausen H, Johnson LA, June CH. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44:1444–1454. doi: 10.1016/j.immuni.2016.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beatson R, Maurstad G, Picco G, Arulappu A, Coleman J, Wandell HH, Clausen H, Mandel U, Taylor-Papadimitriou J, Sletmoen M, Burchell JM. The breast cancer-associated glycoforms of MUC1, MUC1-Tn and sialyl-Tn, are expressed in COSMC wild-type cells and bind the C-type lectin MGL. PLoS One. 2015;10:e0125994. doi: 10.1371/journal.pone.0125994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilhelm M, Smetak M, Schaefer-Eckart K, Kimmel B, Birkmann J, Einsele H, Kunzmann V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J Transl Med. 2014;12:45. doi: 10.1186/1479-5876-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao XY, Wu H, Du KQ, Zhu YC, Meng HM, Gong ZS, Zong YH, Huang L, Lu M, Tang JR, Li YF, Zhai XC, Wang XL, Ye SS, Chen D, Yuan L, Qin L, Yang L. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci. 2016;59:386–397. doi: 10.1007/s11427-016-5024-7. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, You F, Jiang L, Li JL, Zhu XJ, Bao YY, Sun X, Tang XW, Meng HM, An GL, Zhang BZ, Yang L. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-ζ or CD64-BB-ζ receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget. 2017;8:37128. doi: 10.18632/oncotarget.16201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kho D, MacDonald C, Johnson R, Unsworth CP, O’Carroll SG, Mez E, Angel CE, Graham ES. Application of xCELLigence RTCA biosensor technology for revealing the profile and window of drug responsiveness in real time. Biosensors (Basel) 2015;5:199–222. doi: 10.3390/bios5020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, Liu H, Wu MF, Gee AP, Mei ZY, Rooney CM, Heslop HE, Brenner MK. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]