

Abstract
The Wnt/β-catenin pathway, which is associated with disease progression, is activated in many cancers. Tankyrase (TNKS) has received attention as a target molecule for Wnt/β-catenin pathway inhibition. We identified K-476, a novel TNKS inhibitor, a dual pocket binder that binds to both the nicotinamide and ADP-ribose pockets. In a human colon cancer cell line, K-476 specifically and potently inhibited TNKS and led to stabilization of the Axin protein, resulting in Wnt/β-catenin pathway suppression. Aberrant Wnt/β-catenin pathway activation was recently reported as a possible mechanism of ineffectiveness in immune checkpoint inhibitor (ICI) treatment. Because the Wnt/β-catenin pathway activation causes dendritic cell inactivation and suppresses chemokine production, resulting in a paucity of CD8+ T cells in tumor tissue, which is an important effector of ICIs. Thus, TNKS inhibitors may enhance the efficacy of ICIs. To examine whether K-476 enhances the antitumor effect of anti-PD-L1 antibodies, K-476 was administered orally with an anti-PD-L1 antibody to melanoma-bearing C57BL/6J mice. Although K-476 was ineffective as a monotherapy, it significantly enhanced the antitumor effect in combination with anti-PD-L1 antibody. In mice, intra-tumor infiltration of CD8+ T cells was increased by combination treatment. K-476 upregulated the chemokine expression (e.g., Ccl3 and Ccl4), which attracted CD8+ T cells. This was considered to contribute to the increased CD8+ T cells in the tumor microenvironment. Furthermore, while the potential gastrointestinal toxicity of TNKS inhibitors has been reported, it was not observed at effective doses. Thus, K-476 could be an attractive therapeutic option to enhance the efficacy of ICIs.
Keywords: Wnt/β-catenin, tankyrase inhibitor, immunotherapy, immune checkpoint inhibitor, PD-L1
Introduction
The Wnt/β-catenin pathway is known to regulate cell proliferation, stem cell renewal and tissue differentiation [1]. When Wnt binds to the Frizzled receptor, key transcription co-factor β-catenin forms a complex with transcription factor T-cell factor (TCF) and transcribes downstream genes. Wnt/β-catenin pathway activation is regulated by degradation of β-catenin by the β-catenin destruction complex consisting of adenomatous polyposis coli (APC), Axin, glycogen synthase kinase 3 (GSK3β) and casein kinase (CK1) α [2]. In cancer patients, due to mutation or the overexpression of Wnt/β-catenin pathway related genes [3,4], this pathway is often activated and considered to contribute to malignant transformation, tumor progression, and resistance to conventional cancer therapy [5,6]. Consequently, it is hypothesized that targeting of the Wnt/β-catenin pathway may be utilized to directly inhibit tumor cell proliferation or cancel resistance to conventional therapy.
In addition to the direct role in cancer cells in malignancies, the role of Wnt/β-catenin pathway in immuno-oncology has recently gained much attention. The advent of immune checkpoint inhibitors (ICIs), such as anti-PD-1/PD-L1 and anti-CTLA-4 antibodies, represents a paradigm shift in cancer treatment. However primary and acquired resistance to ICIs has been a clinical issue. The immune-desert tumor microenvironment, characterized by the absence of infiltrating lymphocytes including CD8+ T cells, cytotoxic effector cells for immunotherapy, is considered to be one of the main causes of resistance to ICIs [7]. Aberrant activation of the Wnt/β-catenin signaling pathway is considered to at least partially cause the formation of the immune-desert environment. This abnormal activation suppresses chemokine production from dendritic cells and inhibits the migration of CD8+ T cells into the tumor microenvironment [8].
Many approaches have been used to inhibit the Wnt/β-catenin pathway [9,10]. One important discovery was that tankyrase (TNKS) can be a target molecule for Wnt/β-catenin pathway regulation [11,12]. TNKS1 and TNKS2 belong to the poly (ADP-ribose) polymerase (PARP) family and catalyze the formation of poly-ADP-ribose chain from β-nicotinamide adenine dinucleotide (β-NAD+). In the Wnt/β-catenin pathway, Axin is poly (ADP-ribosyl) ated (PARsylated) by TNKS leading to ubiquitination by RNF146 for degradation. Thus, the application of a TNKS inhibitor causes the accumulation of Axin in the cell and promotes β-catenin degradation [13], which leads to selective Wnt/β-catenin pathway inhibition.
Thus, TNKS inhibitors may be useful in cancer immunotherapy for attracting CD8+ T cells to tumors and overcoming resistance to ICIs. Indeed, it was recently reported that G007-LK, a TNKS inhibitor, exerts antitumor effects in combination with anti-PD-1 antibody [14]. We therefore synthesized K-476, a highly potent and selective TNKS inhibitor, and evaluated its pharmacological activity to enhance the antitumor effects of ICIs. Since gastrointestinal toxicity is a major concern in the development of tankyrase inhibitors, we also carefully examined the toxicological effect of K-476 in this study.
Materials and methods
Cells and culture conditions
The DLD-1 human colon cancer cell line was obtained from the Japanese Collection of Research Bioresources, Osaka, Japan in 1998. The COLO 320DM human colon cancer cell line was obtained from ATCC in 2008. The culture methods for DLD-1 and COLO320DM cells were described previously [15]. Both cell lines were authenticated by a short tandem repeat assay at the Japanese Collection of Research Bioresources. The luciferase expressing mouse melanoma cell line, B16.F10.Luc, was kindly provided by Dr. T. Murakami (Saitama Medical University, Saitama, Japan). B16.F10.Luc.NY-ESO-1 (B16-derived melanoma) cells were generated at Takara Bio Inc., (Shiga, Japan) by the introduction of the NY-ESO-1 gene with a retrovirus vector (6321D-SYN4576). B16-derived melanoma cells were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). All cell lines were grown at 37°C under 5% CO2 and confirmed to be Mycoplasma-free.
Test articles
The step-by-step synthesis details and characterization data of K-476 are described in Supplementary Methods and Data. Anti-mouse CD274 (B7-H1, PD-L1) antibody and Rat IgG2b, kappa isotype control antibody were purchased from BioLegend (San Diego, CA, USA).
Animals
Female C57BL/6J mice (age: 6-7 weeks) were purchased from Charles River Japan, Inc. (Kanagawa, Japan). All animal studies were performed in accordance with company policy on the care and use of laboratory animals (Kyowa Kirin Co., Ltd., Shizuoka, Japan). K-476 was suspended in 0.5 w/v% methylcellulose 400 (Wako, Osaka, Japan). Anti-mouse PD-L1 antibody (clone B199373) and the isotype control (clone 10F9G2) were diluted in Dulbecco’s phosphate buffered saline (DPBS, Life Technologies (Carlsbad, CA, USA)) at a concentration of 1 mg/mL. K-476 and anti-PD-L1 antibody were administrated orally and intraperitoneally, respectively, according to the schedules described in Figure 3. B16-derived melanoma cells were suspended in DPBS and injected subcutaneously into C57BL/6J mice. The tumor size and body weight were measured every three or four days. The tumor volume was calculated as follows (DL, long diameter, DS, short diameter);
Figure 3.
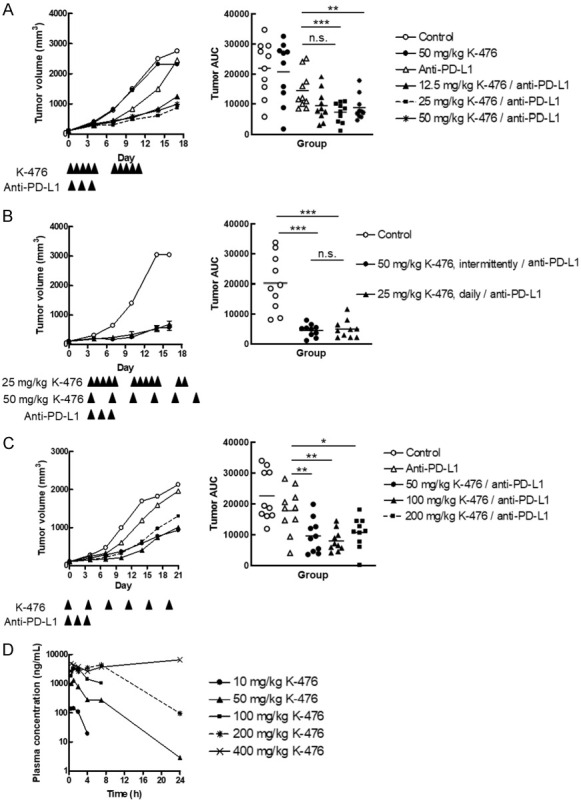
K-476 exerts an antitumor effect in combination with anti-PD-L1 antibody. B16-derived melanoma bearing mice were used for the antitumor study and pharmacokinetics evaluation. Anti-PD-L1 antibody was intraperitoneally administered once daily on days 0, 2 and 4. A-C. The left panels show the mean tumor volume (mm3) (n = 10 mice/group). The right panels show the area under tumor growth curve (tumor AUC) of each mouse and the mean (n = 10). A. The antitumor effect of the continuous administration of K-476 with or without anti-PD-L1 antibody. K-476 was orally administered once daily on days 0-4 and 7-11. B. The antitumor effect of K-476 (25 and 50 mg/kg) with anti-PD-L1 antibody. K-476 (25 mg/kg) was administered once daily on days 0-4, 7-11 and 14-15. K-476 (50 mg/kg) was administered on days 0, 4, 7, 11 and 14. C. The antitumor effect of K-476 (50, 100, 200 and 400 mg/kg, intermittent) with anti-PD-L1 antibody. K-476 was orally administered at doses of 50, 100 and 200 mg/kg on days 0, 4, 8, 12, 16 and 20 with an anti-PD-L1 antibody. D. The plasma concentration-time profiles of K-476 in mice after a single oral administration of K-476 at doses of 10, 50, 100, 200 and 400 mg/kg.
Tumor volume (mm3) = DL × DS × DS × ½
The area under the tumor growth curve (tumor AUC) was calculated for each tumor growth curve.
Reporter assay
The construction method of reporter cell lines DLD-1/TCF-luciferase (Luc) and DLD-1/mutated TCF (mtTCF)-Luc and the reporter assay method were described previously [15].
Real time RT-PCR
The gene expression analyses using human cell lines were performed as follows. COLO 320DM cells were seeded at 50,000 cells/well in 24-well plates. The next day, cells were treated with compounds. After the indicated time points, total RNA was extracted using an RNeasy plus kit (QIAGEN) and reverse-transcribed using VILO reagent (Invitrogen). Real time PCR was performed with TaqMan Universal PCR Master Mix (Applied Biosystems). Transcript levels were determined using an Applied Biosystems 7500 fast real-time PCR system (Applied Biosystems). Each mRNA level was normalized to the level of GAPDH mRNA. The primers used for real-time PCR were as follows: GAPDH (TaqMan Gene Expression Assays, Assay ID: Hs02758991_g1), FGF20 (TaqMan Gene Expression Assays, Assay ID: Hs00173929_m1), AXIN2 (TaqMan Gene Expression Assays, Assay ID: Hs00610344_m1), CD44 (TaqMan Gene Expression Assays, Assay ID: Hs01075861_m1), MYC (TaqMan Gene Expression Assays, Assay ID: Hs Hs00905030_m1), and BAMBI (TaqMan Gene Expression Assays, Assay ID: Hs03044164_m1).
The gene expression analyses using mouse tumor tissues were performed as follows. The tumor tissue of a mouse to which the compounds had been administered was disrupted with a Tissue Lyser II (Qiagen). Total RNA was extracted using a Maxwell 16LEVSimply RNA Tissue Kit (Promega) and reverse-transcribed using VILO reagent. Real time PCR was performed in the same manner as described above. Each mRNA level was normalized to the level of Hrpt mRNA. The primers used for real-time PCR were as follows: Hprt (TaqMan Gene Expression Assays, Assay ID: Mm03024075_m1), Ccl3 (TaqMan Gene Expression Assays, Assay ID: Mm99999057_m1), Ccl4 (TaqMan Gene Expression Assays, Assay ID: Mm00443111_m1), Cxcl1 (TaqMan Gene Expression Assays, Assay ID: Mm04207460_m1), and Cxcl2 (TaqMan Gene Expression Assays, Assay ID: Mm00436450_m1).
Western blotting
The method of Western blotting was described previously [15]. The tumor tissue of a mouse to which the compounds had been administered was disrupted with a Tissue Lyser II. The following antibodies were used: anti-Axin1 antibody (Cell Signaling Technology), anti-Axin2 antibody (Cell Signaling Technology), anti-unphospho-β-catenin antibody (Millipore, Billerica, MA), anti-β-actin antibody (Sigma-Aldrich), anti-rabbit IgG, HRP-Linked F (ab’)2 fragment (Donkey) (GE Healthcare) and anti-mouse IgG, HRP-Linked F (ab’)2 fragment (Sheep) (GE Healthcare).
TNKS and PARP family enzyme activity assay
The activity of each enzyme was measured using a PARP1, PARP2, PARP3, PARP6, PARP7 and PARP11 Chemiluminescent Assay Kit, and a TNKS1 and TNKS2 Histone Ribosylation Assay Kit (Antibody Detection) at BPS bioscience, San Diego, CA. The assay was performed in duplicate in accordance with the manufacturer’s instructions.
Crystallography
The catalytic domain of TNKS2, amino acids 946-1162, was expressed in E. coli and purified by a previously described method [16]. After purification, TNKS2 was stored at -80°C in buffer containing 30 mmol/L HEPES pH 7.5, 300 mmol/L NaCl, 10 v/v% glycerol, 0.5 mmol/L TCEP. Apo TNKS2 crystals were obtained using the sitting drop vapor diffusion method by mixing the protein solution with an equal volume of reservoir solution containing 30 w/v% PEG 3350, 0.2 mol/L lithium sulfate, 0.1 mol/L Tris pH 8.5 at 4°C. Complex crystals were obtained by soaking apo crystals for 10 days in soaking buffer containing 0.1 mmol/L K-476 (1 v/v% DMSO), 30 w/v% PEG 3350, 0.2 mol/L lithium sulfate, 0.1 mol/L Tris pH 8.5. The complex crystals were flash frozen in liquid nitrogen before the diffraction experiments.
The X-ray diffraction data for the complex crystal was collected at the BL17A beamline of the Photon Factory in the High Energy Accelerator Research Organization, Tsukuba, Japan. The data were processed using the XDS program [17]. The structures were solved by molecular replacement using the MOLREP program [18] from the CCP4 software suite [19]. The REFMAC5 program [20] was used to optimize the bond length and bond angle between the atoms and to calculate electronic density, such as 2Fo-Fc map and Fo-Fc map. Drawing of electronic density, modification of the structure and refinement were performed using the Coot program [21,22]. Drawing of the structure was performed using CCP4mg program [23]. The structure parameters, such as R factor [24], R free [25] were refined until it was statistically accepted and the final structure was determined. The apo-TNK2 structure registered as PDB ID: 3KR7 at Protein Data Bank (PDB) was used as a search model. The TNKS2 structure in complex with K-476 has been deposited in the Protein Data Bank with access codes 7CE4.
Cell growth inhibition assays
Cells were seeded in 96-well microplates (Nunc). The next day, cells were treated with compounds. After 144 hours, XTT reagent (Roche Diagnostics) was added to the cells. After incubation at 37°C, formation of formazan dye from tetrazolium salt XTT was measured by SpectraMax 340PC (Molecular Devices).
Immunological analyses
Tumors were isolated from mice and dissociated by Tumor Dissociation Kit, mouse and gentleMACS Dissociators according to the manufacturer’s instructions (Miltenyi Biotec). The cell suspensions were hemolyzed with BD Pharm Lyse (BD Biosciences) and filtered through 70-μm mesh. Prepared tumor infiltrating lymphocytes (TILs) were analyzed by flow cytometry.
Flow cytometry
The following antibodies were used for flow cytometry: Brilliant Violet 510™ anti-mouse CD45 antibody (Clone: 30-F11), Alexa Fluor® 700 anti-mouse CD3 antibody (Clone: 17A2), and Brilliant Violet 785™ anti-mouse CD8a antibody (Clone: 53-6.7) were purchased from BioLegend (CA, USA). Flow cytometry was carried out using a BD LSRFortessa X-20 with the FACSDiva software program (BD Biosciences).
Hematoxylin and Eosin (H&E) staining
The small intestine (jejunum and ileum) was removed from all animals. Small intestinal tissue was immediately fixed in 10% neutral buffered formalin. Formalin-fixed tissues were embedded in paraffin and sectioned at 3-5 µm in thickness. Sections were stained with hematoxylin and eosin (H&E).
Pharmacokinetic analysis
Blood samples were collected at 0.5, 1, 2, 4, 7 and 24 hours after oral administration of K-476 at doses of 10, 50, 100, 200, and 400 mg/kg (n = 2). Plasma was separated and stored frozen until use in the analysis. The plasma concentration of K-476 was determined by liquid chromatography tandem mass spectrometry (LC/MS/MS). Plasma samples were protein precipitated with internal standard-containing acetonitrile, and the extract was subjected to a liquid LC/MS/MS analysis. The analyte was separated on a C18 reverse-phase column. LC/MS/MS detection was performed by electrospray ionization in positive ion mode. The detected mass-to-charge ratios were 553/280 (Q1/Q3) for K-476. The lower limit of quantification (LLOQ) was 10 nmol/L. Measured concentrations below the LLOQ were expressed as below the LLOQ (BLQ). When the K-476 concentration was BLQ in each dosing group, the mean values were calculated regarding BLQ as 0 ng/mL.
The maximum plasma concentration (Cmax) and time to reach Cmax (tmax) were obtained directly from the observed values. The elimination half-life (t1/2) was determined using the last 2-4 time points. The area under the concentration-time curve from time zero to the last data point (t) (AUC0-t) was calculated using the trapezoidal method. The AUC after the last time point was estimated by extrapolation with the t1/2. The sum of AUC0-t and AUC after the last data point was regarded as AUC0-∞. Pharmacokinetic parameters were rounded to three significant figures.
Statistical analysis
In Figure 3A and 3C, the statistical analysis was performed by Dunnett’s test. In Figures 3B and 4C, the statistical analysis was performed by Tukey’s test. In Figure 4D, statistical significance was evaluated by Pearson’s correlation. In Figure 4E, statistical significance was determined by Welch’s t-test. All statistical tests were performed using the JMP 14 software program (SAS Institute Inc.). Asterisks indicate statistical significance as follows: *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant.
Figure 4.
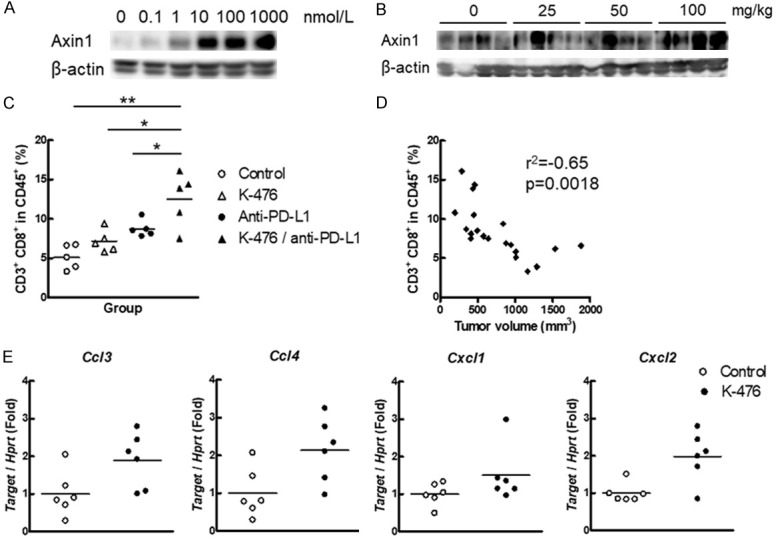
K-476 stabilizes Axin 1 and enhances the chemokine expression to increase CD8+ T cells in the TME. B16-derived melanoma bearing mice were used for B-E. A. K-476 stabilized Axin 1 in K-476-treated B16-derived melanoma cells. K-476 was added to the cells (0, 0.1, 1, 10, 100 and 1000 nmol/L). After 24 hours, the cells were collected for Western blotting. B. K-476 stabilized Axin 1 in tumors of K-476-treated mice. Tumors were collected from the mice 24 hours after the administration of K-476 (0, 25, 50, 100 mg/kg) for Western blotting. C. CD8+ T cells were increased in the tumor of mice treated with K-476 in combination with anti-PD-L1 antibody. Tumor cells were collected from K-476 (50 mg/kg)- and/or anti-PD-L1 antibody-treated mice. K-476 and anti-PD-L1 antibody was administered once daily on days 0, 3 and 6. The percentage of CD8+ cells in CD45+ CD3+ cells on day 7 were measured by flow cytometry. Each plot represents an individual value and each bar represents the mean (n = 5). D. The percentage of CD8+ T cells and tumor volume were inversely correlated in the mice used in Figure 3C. E. The expression of Ccl3, Ccl4, Cxcl1 and Cxcl2 was evaluated by real time PCR. The expression of target genes was normalized by Hprt. Tumor cells were collected from mice 24 hours after the administration of K-476 (100 mg/kg). Each plot represents an individual value and each bar represents the mean (n = 6).
Results
Novel TNKS 1/2 inhibitor K-476 binds to the ADP-ribose and the nicotinamide binding pockets of TNKS
We have previously reported a selective TNKS inhibitor K-756 that inhibits the Wnt/β-catenin pathway by stabilizing the Axin protein and promoting active β-catenin degradation [15]. X-ray crystallography demonstrated that K-756 binds to the ADP-ribose binding pocket [15], while a known TNKS inhibitor XAV939 binds to the nicotinamide binding pocket [16]. Based on this information, K-476 was designed and synthesized to bind to both the ADP-ribose and nicotinamide binding pockets, which was expected to increase the inhibitory activity against TNKS. 3-Cyano-4-fluorophenyl moiety was added to quinazoline-2-one of K-756 to bind the nicotinamide binding pocket (Figure 1A). To prove our hypothesis, we performed X-ray crystallography of TNKS2 in complex with K-476. The 3-cyano-4-fluorophenyl moiety of K-476 bound to the nicotinamide binding pocket while the quinazoline-2-one and 6,7-dimethoxyquinazoline groups bound to the ADP-ribose binding pocket Figure 1B). A hydrogen bond was formed between the oxygen on the carbonyl group of quinazoline-2-one and the main chain NH of Tyr1060. His1048 stacked with the 6,7-dimethoxyquinazoline group and the Phe1035 side chain contacted with one methoxy moiety. These interactions were equivalent to the interactions observed in a co-crystal structure of K-756 with TNKS1 [15]. Indeed, the superposition of the K-476/TNKS2 and K-756/TNKS1 complexes overlapped well, despite the introduction of 3-cyano-4-fluorophenyl moiety to K-476 (Figure 1C). As for the nicotinamide binding pocket, the cyano group of K-476 forms hydrogen bonds with the main chain NH of Gly1032 and with the side chain hydroxy oxygen atom of Ser1068. The carbonyl group of K-476 also forms hydrogen bond with the main chain NH of Tyr1060. This indicates that the 3-cyano-4-fluorophenyl moiety of K-476 functions as a chemical substituent of nicotinamide and overlapped well with the nicotinamide binding pocket of XAV939 (Figure 1D).
Figure 1.
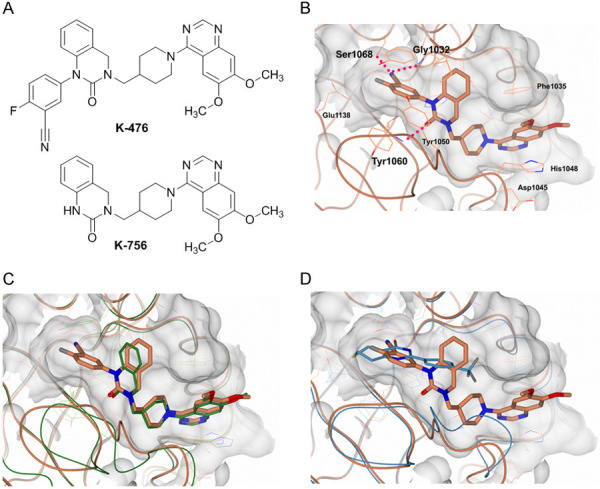
The chemical structure of K-476 and co-crystal data of K-476 binding to the dual pockets of TNKS2. A. The chemical structure of K-476 and K-756. B. The ligand binding site of the TNKS2 catalytic domain (CD) in complex with K-476. The dashed red lines indicate hydrogen bonds. C. A superposition drawing of the TNKS2/K-476 (coral) and TNKS1/K-756 complex (green). D. A superposition drawing of the TNKS2/K-476 (coral) and TNKS2/XAV939 complex (blue) (PDB ID: 3KR8).
As the ADP-ribose binding pocket only exists in TNKS among the PARP family enzymes, we concluded that K-756’s high selectivity against PARP family enzymes is due to its binding to the ADP-ribose pocket [15]. To study whether K-476 maintained its selectivity, the inhibitory activity against PARP family enzymes was tested. As a result, K-476 completely inhibited TNKS1 and TNKS2 at 1 μmol/L, while it did not inhibit other PARP family enzymes (PARP1, 2, 3, 6, 7, 8, 10, 11 and 12) by > 40%, even at 10 μmol/L (Table 1).
Table 1.
Inhibitory activity against PARP family enzymes
| PARPs | Enzyme inhibitory activity (%) | ||
|---|---|---|---|
|
| |||
| K-476 | XAV939 | ||
|
|
|
||
| 1 μmol/L | 10 μmol/L | 10 μmol/L (or 1 μmol/L*) | |
| PARP1 | 0 | 0 | 84* |
| PARP2 | 0 | 0 | 99* |
| PARP3 | 0 | 0 | 3 |
| TNKS1 | 100 | 100 | 100* |
| TNKS2 | 100 | 100 | 100* |
| PARP6 | 0 | 0 | 0 |
| PARP7 | 2 | 18 | 17 |
| PARP8 | 2 | 0 | 0 |
| PARP10 | 8 | 38 | 10 |
| PARP11 | 1 | 0 | 37 |
| PARP12 | 0 | 3 | 61 |
K-476 is a highly potent Wnt/β-catenin pathway inhibitor that stabilizes Axin proteins and inhibits downstream genes
To evaluate the Wnt/β-catenin pathway-specific inhibitory activity of K-476, luciferase assays using the DLD-1/TCF-Luc reporter cell line and the DLD-1/mtTCF-Luc counterpart cell line [15] were performed. K-476 inhibited Wnt/β-catenin pathway-dependent luciferase activity with an IC50 of 0.3 nmol/L in DLD-1/TCF-Luc cells but did not inhibit Wnt/β-catenin pathway-independent luciferase activity, even at 100 nmol/L in DLD-1/mtTCF-Luc cells (Figure 2A). To our surprise, the potency of K-476 increased more than 360 times in comparison to the IC50 of luciferase activity of K-756, which was 110 nmol/L [15].
Figure 2.

K-476 inhibits the Wnt/β-catenin pathway by stabilizing Axin proteins and decreasing active β-catenin. A. K-476 inhibited the reporter activity in DLD-1/TCF-Luc cells but not in DLD-1/mtTCF-Luc cells. DLD-1/TCF-Luc cells or DLD-1/mtTCF-Luc cells were treated with K-476. After 18 hours, their luciferase activity was measured. Each of the values represents the mean of triplicate. B. K-476 inhibited the cell growth of COLO 320DM cells. K-476 was added to the cells in 96-well plates. After 144 hours, the anti-proliferative activity was measured by an XTT assay. Each point represents the mean ± S.D. of triplicate. C. K-476 inhibited the Wnt/β-catenin downstream genes. K-476 was added to COLO 320DM cells in 24-well plates. After 24 hours, mRNA was collected for RT-PCR. D. K-476 stabilized Axin 1 and 2 and decreased active β-catenin in COLO 320DM cells. K-476 was added to COLO 320DM cells in 10-cm dishes. After 24 hours, cells were collected for Western blotting.
In our previous report, β-catenin siRNA and K-756 inhibited the cell growth of COLO 320DM cells [15]. Thus, we used this cell line as a Wnt/β-catenin-dependent cell line. K-476 showed antiproliferative activity in COLO 320DM cells at a GI50 of 1.5 nmol/L (Figure 2B). K-476 inhibited the Wnt/β-catenin downstream genes (Figure 2C) and stabilized Axin1 and Axin2 and decreased active β-catenin level (Figure 2D) at 1 nmol/L, which was consistent with the growth inhibitory activity. Hence, these results suggested that K-476 inhibited the cell growth of COLO 320DM cells via inhibition of the Wnt/β-catenin pathway.
K-476 exerts an antitumor effect in combination with anti-PD-L1 antibody
We next evaluated the in vivo antitumor effect of K-476 in cancer immunotherapy using the B16.F10.Luc.NY-ESO-1 (B16-derived melanoma) bearing mouse model. K-476 was orally administered with or without anti-PD-L1 antibody according to the schedules described in Figure 3A. Although K-476 (50 mg/kg) alone did not exert an antitumor effect, K-476 significantly enhanced the antitumor effect in combination with anti-PD-L1 antibody with a continuous administration schedule at doses of 25 and 50 mg/kg (Figure 3A). In the groups that received a combination of K-476 and anti-PD-L1 antibody, the antitumor effect was almost comparable in the groups that received 25 and 50 mg/kg of K-476.
TNKS inhibitors, in general, have the potential to induce intestinal toxicity [11,12]. Avoiding continuous inhibition is one possible approach to reduce such intestinal toxicity (see discussion). Thus, we compared the antitumor effect of the same total dose K-476 administered with continuous and intermittent administration schedules; the antitumor effect of the continuous administration of 25 mg/kg of K-476 and the intermittent administration of 50 mg/kg of K-476 (both combined with anti-PD-L1 antibody) was compared. Both treatments exerted a comparable antitumor effect (Figure 3B). Because intermittent administration can avoid sustained exposure to TNKS inhibitors, we decided to administer K-476 intermittently. Then, to examine the maximum effective dose of K-476 in an intermittent administration schedule, K-476 was administered at doses of 50, 100 or 200 mg/kg with anti-PD-L1 antibody to B16-derived melanoma bearing mice. In the combination groups, the significant antitumor effect of K-476 combined with anti-PD-L1 antibody was similar among the three examined doses (50, 100 and 200 mg/kg) (Figure 3C). This result suggested that 50 mg/kg of K-476 was a sufficient dose to exert the maximum antitumor effect in combination with anti-PD-L1 antibody.
Next, the plasma concentration of K-476 was evaluated after a single administration of K-476 in B16-derived melanoma bearing mice. At doses of > 50 mg/kg, the plasma concentration of K-476 was > 10 nmol/L (5.52 ng/mL), which was considered to be a sufficient concentration to inhibit TNKS (see Figure 3D; Table 2) for at least 24 hours. However, at 50 mg/kg, the concentration would be < 10 nmol/L after 24 hours, meaning that TNKS was not inhibited during the interval period between the administration of 50 mg/kg of the drug in an intermittent schedule. Interestingly, even in this situation, 50 mg/kg was sufficient to show a maximum antitumor effect. This suggested that continuous exposure over 24 hours was not needed to exert a sufficient antitumor effect.
Table 2.
The pharmacokinetic parameters of K-476 after oral administration to mice
| Dose of K-476 (mg/kg) | tmax (h) | Cmax (ng/mL) | t1/2 (h) | AUC0-24 a (ng•h/mL) | AUC0-∞ (ng•h/mL) |
|---|---|---|---|---|---|
| 10 | 0.75 | 147 | 0.79 | 356 | 376 |
| 50 | 0.75 | 1360 | 1.98 | 6060 | 6080 |
| 100 | 2.00 | 3350 | 3.04 | 13500 | 19900 |
| 200 | 5.50 | 5340 | 3.16 | 61200 | 61600 |
| 400 | 24.00 | 6560 | n.c. | 111000 | n.c. |
AUC0-4 for 10 mg/kg. AUC0-8 for 200 mg/kg.
n.c., not calculated.
K-476 stabilizes Axin 1 protein in tumor
To confirm the Wnt/β-catenin pathway-specific inhibitory activity of K-476 in the B16-derived melanoma mouse model, the B16-derived melanoma cell line was treated with K-476 for 24 hours in vitro. Axin 1 stabilization in the B16-derived melanoma cell line began to be observed from 1 nmol/L and became evident with K-476 concentrations of ≥10 nmol/L (Figure 4A). To investigate Axin 1 stabilization by K-476 in vivo, tumors were collected from K-476-treated B16-derived melanoma bearing mice. Axin 1 stabilization was clearly observed in the K-476 (100 mg/kg)-treated group. Stabilization was also observed in some animals of the K-476 (25 mg/kg)- or K-476 (50 mg/kg)-treated groups (Figure 4B). These results indicated that K476 exerted an inhibitory effect against TNKS in the B16-derived melanoma bearing mouse model.
K-476 upregulates the production of Ccl3 and Ccl4 in the tumor microenvironment (TME) and increases CD8+ T cells in tumors
CD8+ T cells are one of the most important immune cells for the antitumor effect in the TME [26,27]. Thus, we investigated whether the administration of K-476 with anti-PD-L1 antibody increased the percentage of CD8+ T cells in the TME. As shown in Figure 4C, K-476 combined with anti-PD-L1 antibody significantly increased the percentage of CD8+ T cells in comparison to anti-PD-L1 antibody. In addition, the percentage of CD8+ T cells and tumor volume were inversely correlated (Figure 4D), suggesting that the antitumor effect observed in this study was related to the CD8+ T-cell function.
To investigate the mechanism of the CD8+ T-cell increase in the TME after the administration of K-476, we evaluated the chemokine expression in tumors. Ccl3, Ccl4 and Cxcl1 are known to attract CD8+ T cells [28,29]. The expression levels of Ccl3, Ccl4, Cxcl1 and Cxcl2 are also known to be upregulated by Wnt/β-catenin pathway inhibition [8]. Therefore, we evaluated the expression of these four genes in tumors after the administration of K-476. The significant upregulation of Ccl3 and Ccl4 was confirmed in tumors after the administration of K-476. Although not statistically significant, Cxcl1 tended to be increased by K-476 (P = 0.1714) (Figure 4E). These results suggested that CD8+ T cells were attracted by Ccl3, Ccl4 and Cxcl1, leading to enhancement of the antitumor effect. The expression of Cxcl2 was also significantly increased by K-476 (Figure 4E). Since dendritic cells express Cxcl1 and Cxcl2 [30], the administration of K-476 may have attracted not only CD8+ T cells but also dendritic cells into tumors. The increased dendritic cells may have contributed to the increased number of CD8+ T cells in the TME.
The intermittent administration of K-476 was not associated with intestinal toxicity at doses higher than the effective dose
We finally examined the tolerable dose of K-476 with or without anti-PD-L1 antibody. K-476 was administered intermittently to C57BL/6J mice at doses of 0, 50, 100, 200 or 400 mg/kg. Pathological analyses by H&E staining showed no intestinal injury with the administration of K-476 at doses of up to 200 mg/kg with or without anti-PD-L1 antibody (Figure 5). Some observations, such as villi and crypt atrophy, desquamation of the mucosal epithelium, and the infiltration of inflammatory cells were found in the jejunum, ileum and cecum in the K-476 (400 mg/kg)-treated group (Figure 5; Table 3). These results suggested that K-476 did not show small intestinal toxicity at the effective dose.
Figure 5.
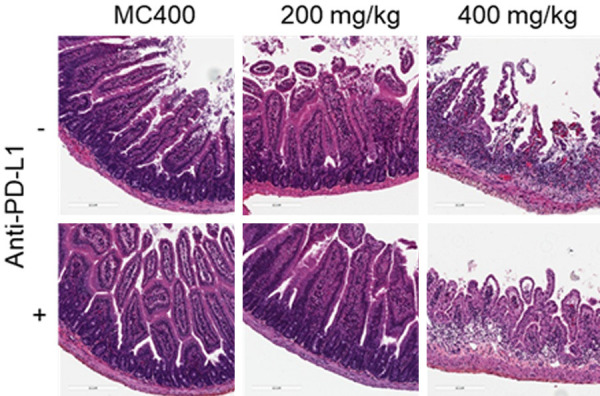
K-476 was administered on day 0, 4, 7, 10 and 14. Anti-PD-L1 antibody was administered on day 0, 2 and 4. Animals were euthanized on day 17 (three days after K-476 final administration). The intermittent administration of K-476 at an effective dose did not disrupt the tissue morphology of the small intestine. The histopathology of the non-treated small intestine, K-476-treated small intestine, PD-L1-treated small intestine and PD-L1 and K-476-treated small intestine.
Table 3.
Pathological findings in the mouse small intestine
| Dose of K-476 (mg/kg) | 0 | 50 | 100 | 200 | 400 | ||
|---|---|---|---|---|---|---|---|
| K-476 | Number of evaluated animals | 5 | 1 | 1 | 5 | 5 | |
| Jejunum | Mucosal epithelium desquamation | 0 | 0 | 0 | 0 | 2 | |
| Mononuclear cell infiltration | 0 | 0 | 0 | 0 | 2 | ||
| Hyperplasia/Regeneration | 0 | 0 | 0 | 0 | 2 | ||
| Atrophy | 0 | 0 | 0 | 0 | 4 | ||
| Ileum | Mucosal epithelium abrasion | 0 | 0 | 0 | 0 | 3 | |
| Mononuclear cell infiltration | 0 | 0 | 0 | 0 | 4 | ||
| Atrophy | 0 | 0 | 0 | 0 | 4 | ||
| K-476/anti-PD-L1 | Number of evaluated animals | 5 | 0 | 2 | 5 | 5 | |
| Jejunum | Mucosal epithelium desquamation | 0 | 0 | 0 | 0 | 1 | |
| Mononuclear cell infiltration | 0 | 0 | 0 | 0 | 1 | ||
| Hyperplasia/Regeneration | 0 | 0 | 0 | 0 | 2 | ||
| Edema | 0 | 0 | 0 | 0 | 1 | ||
| Atrophy | 0 | 0 | 0 | 0 | 1 | ||
| Ileum | Mucosal epithelium abrasion | 0 | 0 | 0 | 0 | 2 | |
| Mononuclear cell infiltration | 0 | 0 | 0 | 0 | 2 | ||
| Hyperplasia/Regeneration | 0 | 0 | 0 | 0 | 1 | ||
| Atrophy | 0 | 0 | 0 | 0 | 2 | ||
Describes the number of animals with pathological findings among the evaluated animals.
Discussion
Since the discovery of K-756 [15], we have aimed to increase its potency while maintaining the selectivity against other PARP family enzymes. The first reported TNKS inhibitor, XAV939, was a nicotinamide pocket binder that worked as a chemical substituent of the nicotinamide moiety of βNAD+ [13]. Although XAV939 was the first TNKS inhibitor reported, its selectivity was not high enough due to its binding to the nicotinamide binding pocket, which is conserved among other PARP families. We previously reported a second type of TNKS inhibitor, K-756, which bound to the ADP-ribose binding pocket [15]. K-756 showed high selectivity for TNKS because its binding site was specific to TNKS. After the discovery of K-756, we focused on generating a dual-pocket binding compound that would enhance binding to TNKS. We conserved the structure of K-756 to maintain selectivity and added 3-cyano-4-fluorophenyl moiety, which would bind to the nicotinamide binding pocket. As a result, we discovered dual-pocket binding K-476, which was demonstrated to be a highly potent and selective TNKS inhibitor, and to show higher Wnt/β-catenin pathway inhibitory activity in comparison to K-756. K-756 interacts with TNKS mainly by the stacking of benzene ring of dimethoxy quinazoline with His1048 and Phe1035, and the binding of the benzene ring of quinazoline-2-one to the hydrophobic area of TNKS. The introduction of 3-cyano-4-fluorophenyl moiety in K-476 resulted in the acquisition of additional interactions with Ser1068 and Gly1032 of the nicotinamide pocket. These interactions are almost equivalent to those of the pyrimidin-4-one moiety scaffold of XAV939, which partly explains the high potency of K-476. Actually, the TNKS inhibitory activity of K-476 is 360 times more potent than the original lead compound, K-756. We consider it noteworthy that the 3-cyano-4-fluorophenyl group of K-476 bound to the nicotinamide pocket of TNKS without causing any conformational or structural rearrangements within the K-756-based moiety or the residues in the ADP ribose binding pocket.
We also showed that the intermittent administration of K-476 enhances the antitumor effect of anti-PD-L1 antibody in the B16-derived melanoma bearing mouse model. The data on the plasma concentration of K-476 indicated that K-476 enhanced the effect of anti-PD-L1 antibody, even during the period without K-476 exposure. One of the reasons why K-476 is effective without sustained exposure is that it improves host immunity. We revealed the upregulation of Ccl3, Ccl4, Cxcl1 and Cxcl2 in the tumor following the administration of K-476. Ccl3, Ccl4 and Cxcl1 are chemokines that are known to induce CD8+ T cells [28,29]. The administration of K-476 increased the proportion of CD8+ T cells in TIL in this mouse model. Our results suggested that the increased expression levels of Ccl3, Ccl4 and Cxcl1 induced by K-476 improved the immune environment of the tumor and contributed to the enhancement of the antitumor effect of anti-PD-L1 antibody. The administration of K-476 also upregulated Cxcl2, suggesting that dendritic cells were also increased in tumor by K-476 [30].
The clinical efficacy of ICIs is significantly higher when more infiltrating lymphocytes, especially CD8+ T cells, are present in the tumor [31-33]. K-476 was shown to act on Wnt/β-catenin signal activated cells, and induce CD8+ T cells into tumors to enhance the efficacy of ICIs in our preclinical model. Since the activation of Wnt/β-catenin signaling is cancer-specific [34,35], K-476 is expected to enhance the efficacy of ICIs without the excessive activation of systemic host immunity.
We indicated that K-476 (50 mg/kg) was sufficient to exert the maximum antitumor effect and no toxicological effects were observed at doses of up to 200 mg/kg in combination with anti-PD-L1 antibody in mice. Because TNKS inhibitors cause intestinal toxicity [11,12], it has been considered difficult to separate efficacy from toxicity. However, in the present study, K-476 was effective without intestinal toxicity, as it exerted its efficacy at low doses in combination with anti-PD-L1 antibody. In addition, human intestinal cells are continuously renewed every 3-4 days [36]. Even intestinal toxicity is caused by TNKS inhibition, the toxicity is expected to be manageable since the antitumor effect of K-476 is maintained with an intermittent administration schedule. In this study, we suggest that K-476 has the potential to be a safe and attractive therapeutic agent that enhances the efficacy of ICIs.
Acknowledgements
We thank Dr. T. Murakami (Saitama Medical University, Saitama, Japan) for providing the B16.F10.Luc cell line, S. Nagata and R. Ebihara (Kyowa Kirin Co., Ltd., Shizuoka, Japan) for supporting the animal studies.
Disclosure of conflict of interest
Haruka Kinosada, Ryoko Okada-Iwasaki, Kana Kunieda, Minami Suzuki-Imaizumi, Yuichi Takahashi, Hikaru Miyagi, Michihiko Suzuki, Keiichi Motosawa, Miwa Watanabe, Motoya Mie, Toshihiko Ishii, Hiroshi Ishida, Jun-ichi Saito and Ryuichiro Nakai are employees of Kyowa Kirin Co., Ltd.
Supporting Information
References
- 1.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 2.Verheyen EM, Gottardi CJ. Regulation of Wnt/β-catenin signaling by protein kinases. Dev Dyn. 2010;239:34–44. doi: 10.1002/dvdy.22019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 4.Hamada F. Wnt signaling and cancer. Kaibogaku Zasshi. 2009;84:111–112. [PubMed] [Google Scholar]
- 5.Matsuda Y, Schlange T, Oakeley EJ, Boulay A, Hynes NE. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 2009;11:1–16. doi: 10.1186/bcr2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin-Orozco E, Sanchez-Fernandez A, Ortiz-Parra I, Ayala-San Nicolas M. WNT signaling in tumors: the way to evade drugs and immunity. Front Immunol. 2019;10:1–21. doi: 10.3389/fimmu.2019.02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, Zitvogel L. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 9.Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, Fischer M, Chaudhari A, Ji M, Kapoun AM, Lam A, Lazetic S, Ma S, Mitra S, Park IK, Pickell K, Sato A, Satyal S, Stroud M, Tran H, Yen WC, Lewicki J, Hoey T. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109:11717–22. doi: 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, Kasibhatla S, Schuller AG, Li AG, Cheng D, Li J, Tompkins C, Pferdekamper A, Steffy A, Cheng J, Kowal C, Phung V, Guo G, Wang Y, Graham MP, Flynn S, Brenner JC, Li C, Villarroel MC, Schultz PG, Wu X, McNamara P, Sellers WR, Petruzzelli L, Boral AL, Seidel HM, McLaughlin ME, Che J, Carey TE, Vanasse G, Harris JL. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110:20224–9. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norum JH, Skarpen E, Brech A, Kuiper R, Waaler J, Krauss S, Sørlie T. The tankyrase inhibitor G007-LK inhibits small intestine LGR5+ stem cell proliferation without altering tissue morphology. Biol Res. 2018;51:1–13. doi: 10.1186/s40659-017-0151-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menon M, Elliott R, Bowers L, Balan N, Rafiq R, Costa-Cabral S, Munkonge F, Trinidade I, Porter R, Campbell AD, Johnson ER, Esdar C, Buchstaller HP, Leuthner B, Rohdich F, Schneider R, Sansom O, Wienke D, Ashworth A, Lord CJ. A novel tankyrase inhibitor, MSC2504877, enhances the effects of clinical CDK4/6 inhibitors. Sci Rep. 2019;9:1–16. doi: 10.1038/s41598-018-36447-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 14.Waaler J, Mygland L, Tveita A, Strand MF, Solberg NT, Olsen PA, Aizenshtadt A, Fauskanger M, Lund K, Brinch SA, Lycke M, Dybing E, Nygaard V, Bøe SL, Heintz KM, Hovig E, Hammarström C, Corthay A, Krauss S. Tankyrase inhibition sensitizes melanoma to PD-1 immune checkpoint blockade in syngeneic mouse models. Commun Biol. 2020;3:1–13. doi: 10.1038/s42003-020-0916-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada-Iwasaki R, Takahashi Y, Watanabe Y, Ishida H, Saito J, Nakai R, Asai A. The Discovery and characterization of K-756, a Novel Wnt/b-Catenin pathway inhibitor targeting tankyrase. Mol Cancer Ther. 2016;15:1525–34. doi: 10.1158/1535-7163.MCT-15-0938. [DOI] [PubMed] [Google Scholar]
- 16.Karlberg T, Markova N, Johansson I, Hammarström M, Schütz P, Weigelt J, Schüler H. Structural basis for the interaction between tankyrase-2 and a potent Wnt-signaling inhibitor. J Med Chem. 2010;53:5352–5355. doi: 10.1021/jm100249w. [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 18.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–5. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brünger AT, Kuriyan J, Karplus M. Crystallographic R factor refinement by molecular dynamics. Science. 1987;235:458–460. doi: 10.1126/science.235.4787.458. [DOI] [PubMed] [Google Scholar]
- 25.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–5. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 26.Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. 2019;17:1–13. doi: 10.1016/j.csbj.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 28.Patterson SJ, Pesenacker AM, Wang AY, Gillies J, Mojibian M, Morishita K, Tan R, Kieffer TJ, Verchere CB, Panagiotopoulos C, Levings MK. T regulatory cell chemokine production mediates pathogenic T cell attraction and suppression. J Clin Invest. 2016;126:1039–1051. doi: 10.1172/JCI83987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engeman T, Gorbachev AV, Kish DD, Fairchild RL. The intensity of neutrophil infiltration controls the number of antigen-primed CD8 T cells recruited into cutaneous antigen challenge sites. J Leukoc Biol. 2004;76:941–9. doi: 10.1189/jlb.0304193. [DOI] [PubMed] [Google Scholar]
- 30.Svedova J, Tsurutani N, Liu W, Khanna KM, Vella AT. TNF and CD28 signaling play unique but complementary roles in the systemic recruitment of innate immune cells after staphylococcus aureus enterotoxin A inhalation. J Immunol. 2016;196:4510–21. doi: 10.4049/jimmunol.1600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e551. doi: 10.1016/S1470-2045(16)30406-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2015;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenzel PJ, Schindeldecker M, Tagscherer KE, Foersch S, Herpel E, Hohenfellner M, Hatiboglu G, Alt J, Thomas C, Haferkamp A, Roth W, Macher-Goeppinger S. Prognostic and predictive value of tumor-infiltrating leukocytes and of immune checkpoint molecules PD1 and PDL1 in clear cell renal cell carcinoma. Transl Oncol. 2020;13:336–345. doi: 10.1016/j.tranon.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schatoff EM, Leach BI, Dow LE. Wnt signaling and colorectal cancer emma. Physiol Behav. 2016;176:139–148. [Google Scholar]
- 35.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 36.Darwich AS, Aslam U, Ashcroft DM, Rostami-Hodjegan A. Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab Dispos. 2014;42:2016–2022. doi: 10.1124/dmd.114.058404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.