

Abstract
DEAD (Asp-Glu-Ala-Asp) box protein 5 (DDX5), a prototypical member of the DEAD/H-box protein family, has been involved in several human malignancies. However, the expression and biological role of DDX5 in esophageal cancer (EC) remain largely unknown. In this study, we examined the role of DDX5 in regulating EC cell proliferation and tumorigenesis and explored its possible molecular mechanism. We found that DDX5 was overexpressed in human EC cell lines. In addition, knockdown of DDX5 significantly inhibited the proliferation of EC cells in vitro and the growth of EC xenografts in vivo. Knockdown of DDX5 also suppressed the migration/invasion and epithelial-to-mesenchymal transition (EMT) phenotype in EC cells. Furthermore, we observed that knockdown of DDX5 inhibited the expression of β-catenin, c-Myc, and cyclin D1 in EC cells. In conclusion, our findings provide the first evidence that siRNA-DDX5 inhibited the proliferation and invasion of EC cells through suppressing the Wnt/β-catenin signaling pathway. Therefore, DDX5 may be a novel potential therapeutic target for the prevention and treatment of EC.
Key words: DEAD (Asp-Glu-Ala-Asp) box protein 5 (DDX5), Esophageal cancer (EC), Proliferation, Invasion
INTRODUCTION
Esophageal cancer (EC) is a major cause of cancer death in the world. It often leads to dysphagia, pain, and other symptoms. It is more common in developing countries, especially China1. Despite improvements in therapy, including radical surgery, chemotherapy, and radiotherapy, EC frequently shows fast local invasion or regional lymph node metastasis at diagnosis, and the 5-year survival rate of EC patients is less than 30%2–5. Thus, there is an urgent need to further understand the molecular mechanisms underlying EC tumorigenesis for the treatment of EC.
DEAD (Asp-Glu-Ala-Asp) box protein 5 (DDX5) is a prototypical member of the DEAD/H-box protein family, which is characterized by a region of nine conserved amino acid motifs including DEAD (Asp-Glu-Ala-Asp)6. Several studies have shown that DDX5 is expressed in all dividing cells of different vertebrates and participates in nearly all aspects of RNA metabolism, including RNA splicing, translation, ribosome biogenesis, and miRNA processing7–9. DDX5 also plays an important role as a transcriptional coactivator of transcription factors that are themselves highly regulated10. More recently, DDX5 was found to be involved in tumor progression by promoting cell proliferation and epithelial-to-mesenchymal transition (EMT)11. For example, Wang et al. confirmed that DDX5 was significantly overexpressed in non-small cell lung cancer (NSCLC) tissues, and overexpression of DDX5 promoted proliferation of NSCLC cells in vitro and growth of NSCLC xenografts in vivo12. However, the expression and biological role of DDX5 in EC remain largely unknown. In this study, we examined the role of DDX5 in regulating EC cell proliferation and tumorigenesis and explored its possible molecular mechanism.
MATERIALS AND METHODS
Cell Culture
Four human EC cell lines (EC9706, EC109, TE13, and ECA109) and human esophageal epithelial cells (HEEC) were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Gibco, Rockville, MD, USA) and 1% (v/v) penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, USA) under standard conditions at 37°C in a 5% CO2 humidified atmosphere.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from the cells using the TRIzol reagent kit (Invitrogen, Carlsbad, CA, USA). Total RNA (1 μg) was then reverse transcribed with the RevertAid™ First-Strand cDNA Synthesis Kit. PCR amplification was carried out by ABI PRISM 7900 thermocycler using SYBR Premix Taq (Applied Biosystems, Foster City, CA, USA). The following primer pairs were used for the PCR amplification: DDX5, 5′-CACATCAATCATCAGCCATTCCT-3′ (forward) and 5′-CAAACAAATAGGCCCATCGC-3′ (reverse); GADPH, 5′-ATCCCATCACCATCTTCCAG-3′ (forward) and 5′-CCATCACGCCACAGTTTCC-3′ (reverse). The PCR procedure was as follows: 94°C for 5 min; 94°C for 20 s, 55°C for 30 s, and 72°C for 20 s; 2 s for plate reading for 35 cycles; melting curve, 65°C–95°C. GAPDH was used as a control for normalizing the gene expression. The data obtained were analyzed by the 2−ΔΔCt method.
Western Blot
EC cells were harvested and washed twice with PBS and then lysed in RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) with protease and phosphatase inhibitors on ice for 10 min. The protein concentration was determined using a Bradford protein assay (Takara Biotechnology, Dalian, P.R. China). Equal amounts of protein sample were separated by 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Boston, MA, USA). Proteins on PVDF membranes were blocked with a 5% skim milk solution and then incubated with various primary antibodies [DDX5, E-cadherin, N-cadherin, vimentin, β-catenin, c-Myc, and cyclin D1 or GAPDH from Santa Cruz Biotechnology (Santa Cruz, CA, USA)] at 4°C overnight. Subsequently, membranes were washed extensively with TBST and incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. The membranes were visualized using enhanced chemiluminescence (ECL) reagent. Developed films were digitized by scanning, and the optical densities were analyzed using the ImageJ software.
Short Hairpin RNA and Cell Transfection
DDX5 short hairpin RNA (shRNA-DDX5) and control shRNA (scramble) were purchased from Shanghai Sangon Co., Ltd. (Shanghai, P.R. China). For in vitro transfection, EC cells were seeded in each well of 24-well microplates, grown for 24 h to reach 50% confluence, and transfected with shRNA-DDX5 or scramble using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Cell Proliferation Assay
The cell counting kit-8 (CCK-8) assay was performed to determine cell proliferation. In brief, EC cells transfected with shRNA-DDX5 or scramble were seeded into 96-well plates (1 × 104 cells/well) and cultured at 24-h intervals for 4 days, and then incubated with WST-8 dye at 37°C. Optical density was determined at a wavelength of 490 nm using microplate spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA).
Cell Migration and Invasion Assays
Cell migration was measured using the Transwell assay. In brief, EC cells transfected with shRNA-DDX5 or scramble suspended in DMEM were added to the upper chamber. The lower chamber was filled with 600 μl of DMEM containing 10% FBS. After incubation at 37°C, nonmigrating cells on the upper side of the filter were wiped off. The cells that migrated through the membrane were fixed in absolute methanol for 10 min, stained with 0.1% crystal violet for 30 min, and quantified by counting five independent visual fields under a microscope (Olympus, Tokyo, Japan). The cell invasion assay was performed by the same procedure, except that the membrane was coated with Matrigel to form a matrix barrier.
In Vivo Xenograft Tumor Assay
Five-week-old BALB/c female nude mice were purchased from the Experimental Animal Centre of The Second Affiliated Hospital of Medical School, Xi’an Jiaotong University (P.R. China). The mice were housed under specific pathogenic-free conditions and were allowed to adjust to local conditions for 1 week before injection. All experimental procedures were approved by the Institutional Animal Care and Use Committee at The Second Affiliated Hospital of Medical School, Xi’an Jiaotong University (P.R. China). For the in vivo study, EC9706 cells at 1 × 106 cells/0.1 ml transfected with shRNA-DDX5 or scramble were resuspended in PBS (0.1 ml) and injected subcutaneously into the flank of nude mice. The size of the tumors was measured once every 5 days and calculated according to the formula [tumor volume = (L × W 2) × 0.5] using digital calipers. Approximately 26 days following implantation, mice were euthanized by subcutaneous injection with sodium pentobarbital (40 mg/kg), and the tumors were weighed.
Statistical Analysis
The results are expressed as mean ± SD. A one-way ANOVA followed by a Student’s t-test was used to determine if the results were statistically significant. A value of p < 0.05 was considered to be statistically significant.
RESULTS
DDX5 Is Highly Expressed in Human EC Cell Lines
We first examined the mRNA expression of DDX5 in four EC cell lines and a normal human esophageal epithelial cell line (HEEC) by qRT-PCR assay. The results demonstrated that the mRNA expression levels of DDX5 were higher in EC cell lines compared to HEEC (Fig. 1a). Similarly, Western blot revealed that DDX5 protein levels in the EC cell lines were higher than that in the HEEC cells (Fig. 1b).
Figure 1.
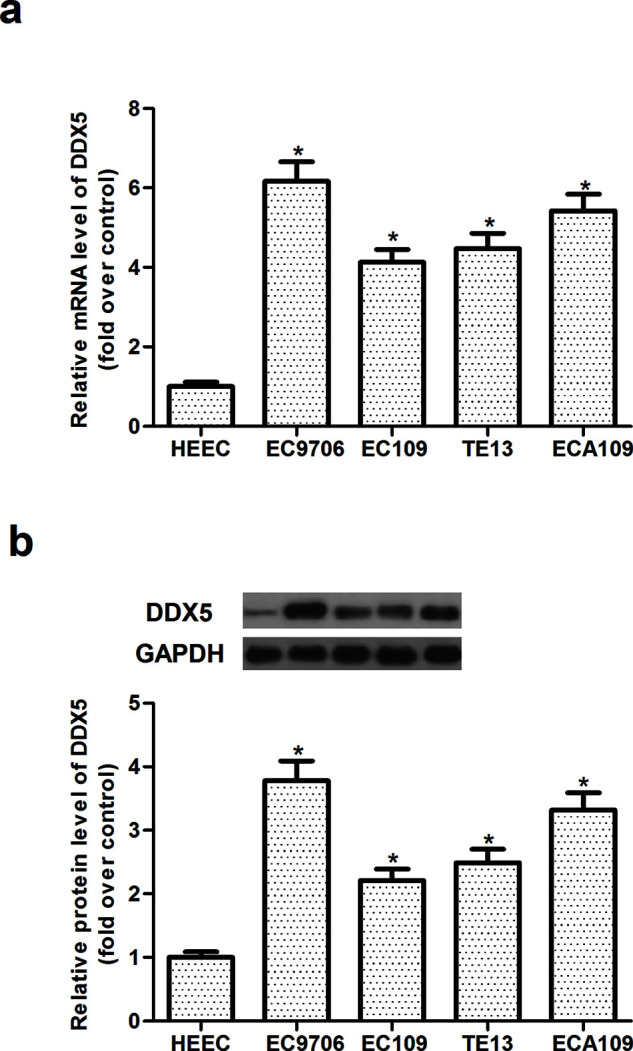
DDX5 is highly expressed in human EC cell lines. (a) The mRNA expression levels of DDX5 were determined by qRT-PCR in four human EC cell lines (EC9706, EC109, TE13, and ECA109) and human esophageal epithelial cells (HEEC). (b) The protein expression levels of DDX5 were determined by Western blot in four human EC cell lines (EC9706, EC109, TE13, and ECA109) and human esophageal epithelial cells (HEEC). The values shown represent the mean ± SD. *p < 0.05 versus HEEC group.
Knockdown of DDX5 Inhibits EC Cell Proliferation
To investigate the critical role of DDX5 in regulating EC, we transfected shRNA-DDX5 or scramble into EC9706 and ECA109 cells, respectively, finding that the shRNA-DDX5 significantly reduced the expression of DDX5 in EC9706 and ECA109 cells (Fig. 2a and b).
Figure 2.

Knockdown of DDX5 inhibits EC cell proliferation. EC9706 and ECA109 cells were transfected with shRNA-DDX5 or scramble for 48 h, respectively. The corresponding transfection efficiency was detected by Western blot in EC9706 (a) and ECA109 cells (b). Cell proliferation was examined by the CCK-8 assay at the indicated time in EC9706 (c) and ECA109 cells (d). The values shown represent the mean ± SD. *p < 0.05 versus scramble group.
In addition, the results of the CCK-8 assay showed that knockdown of DDX5 dramatically inhibited proliferation in EC9706 and ECA109 cells (Fig. 2c and d).
Knockdown of DDX5 Inhibits EC Cell Migration and Invasion
We then performed Transwell assays and a Matrigel invasion assay to assess the effects of DDX5 on cell migration and invasion. The Transwell migration assay indicated that, compared to control parental cells, knockdown of DDX5 markedly decreased the cell migration rate in both EC9706 and ECA109 cells (Fig. 3a and b). Moreover, the invasion assay showed that the invasiveness of cells was significantly lower in EC9706 and ECA109 cells (Fig. 3c and d) transfected with siRNA-DDX5 or scramble than that of control parental cells.
Figure 3.

Knockdown of DDX5 inhibits EC cell migration and invasion. EC9706 and ECA109 cells were transfected with shRNA-DDX5 or scramble for 48 h, respectively. The migratory potential of EC9706 (a) and ECA109 cells (b) was detected by Transwell assay. The invasive ability of EC9706 (c) and ECA109 cells (d) was detected by the Transwell assay with Matrigel. The values shown represent the mean ± SD. *p < 0.05 versus scramble group.
Knockdown of DDX5 Inhibits the Epithelial-to-Mesenchymal (EMT) Phenotype in EC Cells
Given that shRNA-DDX5 inhibits EC cell metastasis, we wonder whether DDX5 plays an important role in regulating the EMT phenotype in EC cells. To test this hypothesis, we evaluated the expression of EMT-related markers using Western blot. Knockdown of DDX5 in EC9706 cells led to an increase in epithelial cell markers, such as E-cadherin, and a decrease in mesenchymal cell markers, such as N-cadherin and vimentin (Fig. 4a). Similar results were observed in ECA109 cells (Fig. 4b).
Figure 4.

Knockdown of DDX5 inhibits the epithelial-to-mesenchymal transition (EMT) phenotype in EC cells. EC9706 and ECA109 cells were transfected with shRNA-DDX5 or scramble for 48 h, respectively. (a) The protein expression levels of E-cadherin, N-cadherin, and vimentin were determined by Western blot in EC9706 cells. (b) The protein expression levels of E-cadherin, N-cadherin, and vimentin were determined by Western blot in ECA109 cells. The values shown represent the mean ± SD. *p < 0.05 versus scramble group.
Knockdown of DDX5 Inhibits EC Tumor Growth
To further confirm the effects of DDX5 on tumor growth in vivo, EC9706 cells infected with shRNA-DDX5 or scramble were implanted subcutaneously into the flank of nude mice. We found that knockdown of DDX5 significantly inhibited the weight of the EC tumor (Fig. 5a), as well as reduced the volume of EC tumors (Fig. 5b).
Figure 5.
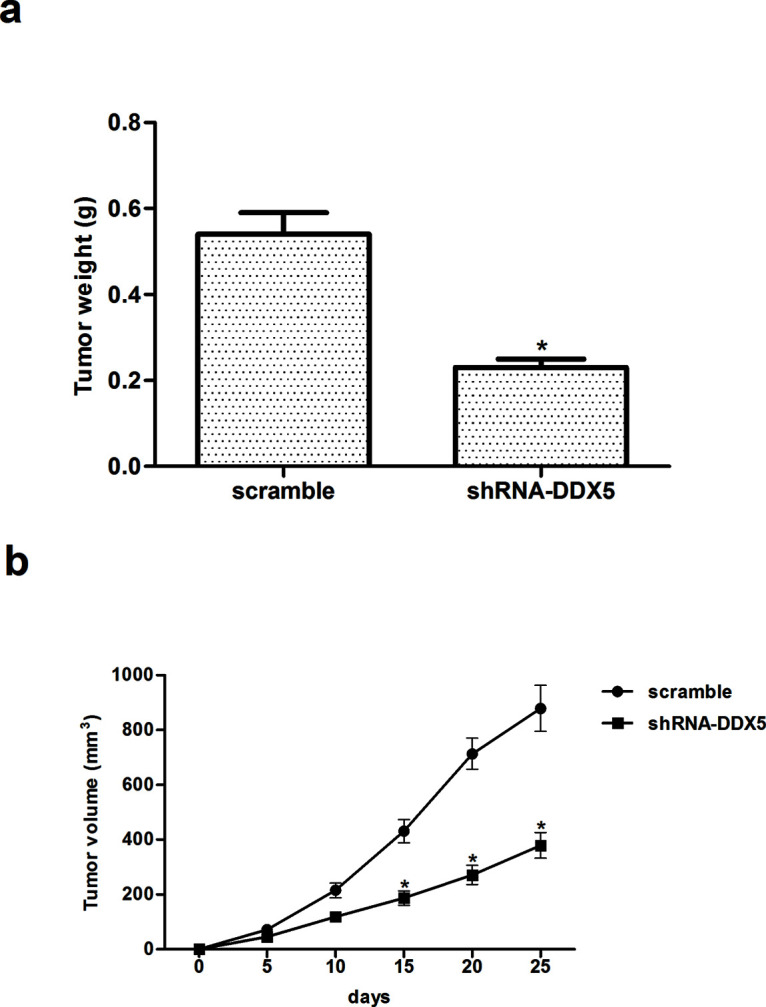
Knockdown of DDX5 inhibits EC tumor growth. EC9706 cells infected with shRNA-DDX5 or scramble were implanted subcutaneously into the flank of nude mice. (a) Tumor weights of the two groups. (b) Growth curves of tumor size. The values shown represent the mean ± SD. *p < 0.05 versus scramble group.
Knockdown of DDX5 Inhibits the Activation of Wnt/β-Catenin Pathway in EC Cells
To further elucidate the molecular mechanism by which DDX5 contributes to these malignant features, we examined the effect of DDX5 on the expression of β-catenin, cyclin D1, and c-Myc involved in the Wnt/β-catenin signaling pathway. Western blot analysis revealed that knockdown of DDX5 dramatically decreased the expression of β-catenin, cyclin D1, and c-Myc in EC9706 cells, compared with the scramble group (Fig. 6).
Figure 6.
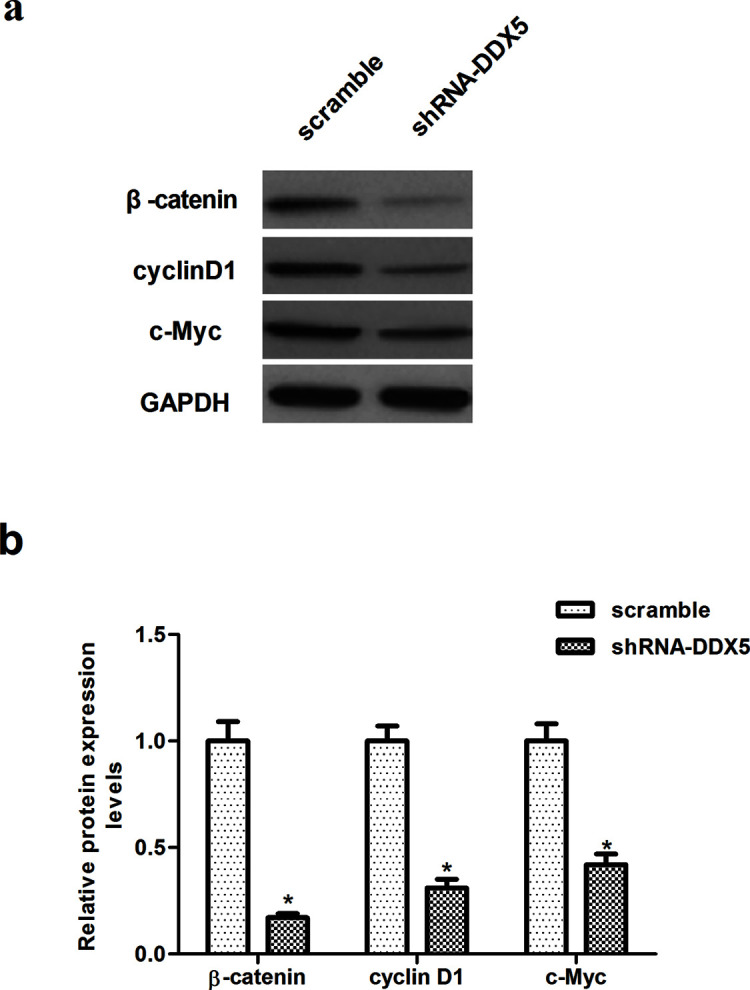
Knockdown of DDX5 inhibits the activation of the Wnt/β-catenin pathway in EC cells. EC9706 cells were transfected with shRNA-DDX5 or scramble for 48 h. (a) The protein expression levels of β-catenin, cyclin D1, and c-Myc were determined by Western blot. (b) Quantitative analysis of protein expression levels of β-catenin, cyclin D1, and c-Myc using the Image-Pro Plus 6.0 software and normalized to GAPDH. The values shown represent the mean ± SD. *p < 0.05 versus scramble group.
DISCUSSION
In this study, we found that DDX5 was overexpressed in human EC cell lines. In addition, knockdown of DDX5 significantly inhibited the proliferation of EC cells in vitro and the growth of EC xenografts in vivo. Knockdown of DDX5 also suppressed the migration/invasion and EMT phenotype in EC cells. Furthermore, we observed that knockdown of DDX5 inhibited the expression of β-catenin, c-Myc, and cyclin D1 in EC cells.
Emerging evidence supports the finding that DDX5 functions as an oncogene in several tumors13–16. DDX5 was found to be highly expressed in prostate cancer compared with matched normal tissues13. Another study showed that the expression of DDX5 was significantly increased in colon cancer, and knockdown of DDX5 in colon cancer cells inhibits their proliferation and diminishes their ability to form tumors in vivo17. In accordance with these reports, in the present study we demonstrate that DDX5 was overexpressed in human EC cell lines, and knockdown of DDX5 significantly inhibited the proliferation of EC cells in vitro and EC tumor growth in vivo. Collectively, these results further support the notion that DDX5 functions as an oncogene in the development and progression of EC.
EMT plays an important role in EC metastasis18,19. It is characterized by the loss of epithelial differentiation and acquisition of mesenchymal-like cellular competence of tumor cells. Previous studies have demonstrated that an increased level of EMT is associated with the progression and metastasis of tumors20,21. In the present study, we found that knockdown of DDX5 significantly inhibited the migration and invasion of EC cells. In addition, we observed that knockdown of DDX5 led to an increase in E-cadherin and a decrease in N-cadherin and vimentin. These data suggest that DDX5 might be an important contributor to EMT progression in EC cells, thus facilitating the migration and invasion of EC cells.
The Wnt/β-catenin signaling pathway regulates multiple fundamental cellular processes including cell proliferation, migration, and tumorigenesis22–25. β-Catenin is a multifunctional protein that mediates cell–extracellular matrix adhesion and promotes tumor proliferation and metastasis26,27. Reduced β-catenin expression was observed in patients with EC28,29. Recently, cyclin D1 and c-Myc have been identified as target genes of β-catenin30,31. Therefore, it is reasonable to assume that inhibition of the Wnt/β-catenin signaling pathway may be a good way to prevent the development and progression of EC. It was reported that Wnt2 could promote EC cell growth by activating the Wnt/β-catenin signaling pathway and, subsequently, upregulated cyclin D1 and c-Myc expression32. Furthermore, studies have shown that DDX5 directly interacted with β-catenin, promoted its nuclear translocation, and coactivated the expression of cyclin D1 and c-Myc in NSCLC cells12. DDX5 was also involved in breast cancer progression by maintaining a positive feedback loop with the β-catenin/transcription factor 4 signaling pathway33. In the present study, we found that knockdown of DDX5 significantly inhibited the expression of the Wnt/β-catenin pathway components (β-catenin, c-Myc, and cyclin D1) in EC cells. These results suggest that siRNA-DDX5 suppresses the proliferation and invasion of EC cells mediated through the Wnt/β-catenin signaling pathway.
In conclusion, our findings provide the first evidence that DDX5 may play an important role in EC growth and metastasis. Therefore, DDX5 may be a novel potential therapeutic target for the prevention and treatment of EC.
ACKNOWLEDGMENT
The authors declare no conflicts of interest.
Footnotes
The authors declare no conflicts of interest.
REFERENCES
- 1. Chen J, Kwong D, Cao T, Hu Q, Zhang L, Ming X, Fu L, Guan X. Esophageal squamous cell carcinoma (ESCC): Advance in genomics and molecular genetics. Dis Esophagus 2015;28:84–9. [DOI] [PubMed] [Google Scholar]
- 2. Kimura M, Ishiguro H, Tanaka T, Takeyama H. Advanced esophageal cancer with tracheobronchial fistula successfully treated by esophageal bypass surgery. Int J Surg Case Rep. 2015;9:115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lagergren J. Oesophageal cancer in 2014: Advances in curatively intended treatment. Nature Rev Gastroenterol Hepatol. 2015;12:74–5. [DOI] [PubMed] [Google Scholar]
- 4. Lin J, Kligerman S, Goel R, Sajedi P, Suntharalingam M, Chuong MD. State-of-the-art molecular imaging in esophageal cancer management: Implications for diagnosis, prognosis, and treatment. J Gastrointest Oncol. 2015;6:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- 6. Linder P, Jankowsky E. From unwinding to clamping—The DEAD box RNA helicase family. Nat Rev Mol Cell Biol. 2011;12:505–16. [DOI] [PubMed] [Google Scholar]
- 7. Fuller-Pace FV. DExD/H box RNA helicases: Multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006;34:4206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jalal C, Uhlmann-Schiffler H, Stahl H. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res. 2007;35:3590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caretti G, Lei EP, Sartorelli V. The DEAD-box p68/p72 proteins and the noncoding RNA steroid receptor activator SRA: Eclectic regulators of disparate biological functions. Cell Cycle 2007;6:1172–6. [DOI] [PubMed] [Google Scholar]
- 10. Fuller-Pace FV, Ali S. The DEAD box RNA helicases p68 (Ddx5) and p72 (Ddx17): Novel transcriptional co-regulators. Biochem Soc Trans. 2008;36:609–12. [DOI] [PubMed] [Google Scholar]
- 11. Wang H, Gao X, Yang JJ, Liu Z-R. Interaction between p68 RNA helicase and Ca2+-calmodulin promotes cell migration and metastasis. Nat Commun. 2013;4:1354–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Z, Luo Z, Zhou L, Li X, Jiang T, Fu E. DDX5 promotes proliferation and tumorigenesis of non-small-cell lung cancer cells by activating β-catenin signaling pathway. Cancer Sci. 2015;106:1303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang D, Huang J, Hu Z. RNA helicase DDX5 regulates microRNA expression and contributes to cytoskeletal reorganization in basal breast cancer cells. Mol Cell Proteomics 2012;11:M111.011932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mazurek A, Luo W, Krasnitz A, Hicks J, Powers RS, Stillman B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2012;2:812–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Janknecht R. Multi-talented DEAD-box proteins and potential tumor promoters: p68 RNA helicase (DDX5) and its paralog, p72 RNA helicase (DDX17). Am J Transl Res. 2010;2:223–4. [PMC free article] [PubMed] [Google Scholar]
- 17. Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res. 2007;67:7572–8. [DOI] [PubMed] [Google Scholar]
- 18. Li W, Jiang G, Zhou J, Wang H, Gong Z, Zhang Z, Min K, Zhu H, Tan Y. Down-regulation of miR-140 induces EMT and promotes invasion by targeting Slug in esophageal cancer. Cell Physiol Biochem. 2014;34:1466–76. [DOI] [PubMed] [Google Scholar]
- 19. Rees JR, Onwuegbusi BA, Save VE, Alderson D, Fitzgerald RC. In vivo and in vitro evidence for transforming growth factor-beta1-mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res. 2006;66:9583–90. [DOI] [PubMed] [Google Scholar]
- 20. Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metast Rev. 2009;28:15–33. [DOI] [PubMed] [Google Scholar]
- 22. Yao H, Ashihara E, Maekawa T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin Ther Targets 2011;15:873–87. [DOI] [PubMed] [Google Scholar]
- 23. Li J, Ying J, Fan Y, Wu L, Ying Y, Chan AT, Srivastava G, Tao Q. WNT5A antagonizes WNT/β-catenin signaling and is frequently silenced by promoter CpG methylation in esophageal squamous cell carcinoma. Cancer Biol Ther. 2010;10:617–24. [DOI] [PubMed] [Google Scholar]
- 24. Zhou C, Liu S, Zhou X, Xue L, Quan L, Lu N, Zhang G, Bai J, Wang Y, Liu Z. Overexpression of human pituitary tumor transforming gene (hPTTG), is regulated by beta-catenin/TCF pathway in human esophageal squamous cell carcinoma. Int J Cancer 2005;113:891–8. [DOI] [PubMed] [Google Scholar]
- 25. Ge XS, Ma HJ, Zheng XH, Ruan HL, Liao XY, Xue WQ, Chen YB, Zhang Y, Jia WH. HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 2013;104:1675–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9:15–21. [DOI] [PubMed] [Google Scholar]
- 27. Kudo Y, Kitajima S, Ogawa I, Hiraoka M, Sargolzaei S, Keikhaee MR, Sato S, Miyauchi M, Takata T. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous beta-catenin. Clin Cancer Res. 2004;10:5455–63. [DOI] [PubMed] [Google Scholar]
- 28. Zhao X-J, Li H, Chen H, Liu Y-X, Zhang L-H, Liu S-X, Feng Q-L. Expression of e-cadherin and beta-catenin in human esophageal squamous cell carcinoma: Relationships with prognosis. World J Gastroenterol. 2003;9:225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voutilainen KA, Anttila MA, Sillanpää SM, Ropponen KM, Saarikoski SV, Juhola MT, Kosma V-M. Prognostic significance of E-cadherin-catenin complex in epithelial ovarian cancer. J Clin Pathol. 2006;59:460–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 1999;398:422–6. [DOI] [PubMed] [Google Scholar]
- 31. He T-C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998;281:1509–12. [DOI] [PubMed] [Google Scholar]
- 32. Fu L, Zhang C, Zhang L-Y, Dong S-S, Lu L-H, Chen J, Dai Y, Li Y, Kong KL, Kwong DL. Wnt2 secreted by tumour fibroblasts promotes tumour progression in oesophageal cancer by activation of the Wnt/β-catenin signalling pathway. Gut 2011;60:1635–43. [DOI] [PubMed] [Google Scholar]
- 33. Guturi KKN, Sarkar M, Bhowmik A, Das N, Ghosh MK. DEAD-box protein p68 is regulated by β-catenin/transcription factor 4 to maintain a positive feedback loop in control of breast cancer progression. Breast Cancer Res. 2014;16:496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]