

Abstract
Ovarian cancer is the most lethal disease among gynecological malignancies. More effective therapy is required to counter high recurrence rates and chemotherapy resistance. We investigated the efficacy and molecular mechanisms of three combined treatments (TCTs)—a novel histone deacetylase (HDAC) inhibitor OBP-801/YM753, 5-fluorouracil (5-FU), and paclitaxel (PTX)—in human ovarian cancer SKOV-3 and OVCAR-3 cells. The inhibition of cell growth was stronger with TCTs than with each single agent and with two combined treatments. The TCTs significantly induce G2 phase arrest in both cell lines. We then analyzed the molecular mechanisms and found that the TCTs increased the phosphorylation of p38 (Thr180/Tyr182), decreased the expression of CDC25C, and increased the phosphorylation of CDC2 (Tyr15), an inactive form of CDC2. To examine the responsibilities of the p38 pathway for G2 phase arrest induced by the TCTs, we employed the p38 inhibitor SB203580. SB203580 inhibited G2 phase arrest, suppression of CDC25C, and phosphorylation of CDC2 (Tyr15) induced by the TCTs. These results suggest that the TCTs can induce G2 phase arrest through activation of the p38 signaling pathway. We therefore believe that this combination is promising as a novel therapeutic strategy against ovarian cancer.
Key words: Histone deacetylase (HDAC) inhibitor, 5-Fluorouracil (5-FU), Paclitaxel (PTX), G2 phase arrest, p38, Ovarian cancer
INTRODUCTION
Ovarian cancer is the leading cause of death among gynecological cancers1. Early stage ovarian cancer often shows no symptoms, and the majority of patients are diagnosed at an advanced stage. The treatment strategy is surgical therapy for reduction of the tumor and chemotherapy including platinum-based drug and paclitaxel (PTX)2. In ovarian cancer, the response rate of first-line chemotherapy is approximately 70%. However, in the majority of cases, relapse occurs within 5 years, and additional chemotherapy is needed3. In stage III and IV patients, the median duration of progression-free survival after first-line chemotherapy is 18 months4. Since relapsed cancer is usually resistant to platinum-based drugs and PTX, these patients are treated with other agents: 5-fluorouracil (5-FU), topotecan, etoposide, gemcitabine, or doxorubicin. However, the response rate of second-line chemotherapy is only 10%–20%5. Therefore, there is an urgent need for a novel treatment strategy for the management of ovarian cancer.
Histone deacetylase (HDAC) inhibitor is promising for the treatment of various cancers because of its ability to induce cell cycle arrest and apoptosis6,7. The FDA has already approved several HDAC inhibitors (vorinostat, romidepsin, and belinostat) for the treatment of cutaneous T-cell lymphoma or peripheral T-cell lymphoma8,9. In ovarian cancer cells, HDAC inhibitor has also been reported to induce cell cycle arrest and apoptosis10,11. A novel HDAC inhibitor, OBP-801/YM753, which was originally identified as spiruchostatin A (an enhancer of plasminogen activator inhibitor-1 gene expression12), was also identified as a novel HDAC inhibitor using p21 promoter reporter screening13. OBP-801/YM753 showed the most potent HDAC-inhibitory activity in our study; it was about 50 times more effective than vorinostat13. This novel HDAC inhibitor, OBP-801/YM753, is under clinical trial in the US.
As mentioned above, PTX is used in first-line chemotherapy in ovarian cancer treatment, and 5-FU is used in second-line chemotherapy. However, a previous report revealed that 5-FU inhibited PTX-induced M phase arrest and apoptosis in breast cancer cells14. Therefore, the combination of PTX and 5-FU would not show a synergistic effect as a chemotherapy treatment.
On the other hand, HDAC inhibitor augmented the effectiveness of various traditional cytotoxic drugs including 5-FU and PTX15. For example, a previous report showed that HDAC inhibitor enhanced the sensitivity of 5-FU16. On the other hand, it was reported that the combination treatment of HDAC inhibitor and PTX synergistically inhibited cell growth in ovarian cancer cells17.
On the basis of these previous reports, we speculated that HDAC inhibitor could augment the effects of 5-FU and PTX despite the offsetting effect between 5-FU and PTX. We therefore investigated the efficacy of three combined treatments (TCTs)—OBP-801/YM753, PTX, and 5-FU—against ovarian cancer cells with elucidation of the molecular mechanisms of these treatments.
MATERIALS AND METHODS
Cell Culture
Human ovarian cancer SKOV-3 and OVCAR-3 cells were purchased as cell lines of NCI-60 from the NCI Developmental Therapeutics Program (Bethesda, MD, USA). These cell lines were expanded and placed in stock within a month of receipt. For the experiments, cells were used fewer than 3 months after resuscitation. SKOV-3 and OVCAR-3 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 4 mM glutamine, 50 U/ml penicillin, and 100 μg/ml streptomycin. All cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Reagents
OBP-801/YM753 was provided by Oncolys Biopharma (Tokyo, Japan). 5-FU and PTX were purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). SB203580 was purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell Viability Assay
The number of viable cells was determined using a cell counting kit-8 assay (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. After incubation for 72 h with the indicated concentrations of various reagents, the kit reagent WST-8 was added to the medium, and the cells were incubated for a further 4 h. The absorbance of samples (450 nm) was determined using a multiwell spectrophotometer (Viento; DS Pharma Biomedical Co., Ltd., Osaka, Japan).
Analysis of Cell Cycle
For cell cycle analysis, cells were incubated with OBP-801/YM753 and/or PTX and/or 5-FU at the indicated concentrations for 24 h and then harvested. After washing with PBS, the cells were permeabilized with 0.1% Triton X-100, and the nuclei were stained with propidium iodide (Sigma-Aldrich, St. Louis, MO, USA). The DNA content was measured using FACSCalibur™ (Becton-Dickinson, Franklin Lakes, NJ, USA), and the data were analyzed with ModFit LT™ (Verity Software House, Topsham, ME, USA) and the Cell Quest software package (Becton-Dickinson).
Western Blot Analysis
Cells were lysed with a lysis buffer (50 mM Tris-HCl, 1% SDS, 2 μg/ml leupeptin, 2 μg/ml aprotinin, 0.1% 2-mercaptoethanol, and 1 mM phenylmethylsulfonyl fluoride). The protein extract was loaded onto a polyacrylamide gel, subjected to electrophoresis, and transferred to a PVDF membrane (EMD Millipore, Billerica, MA, USA). The blots were blocked in 5% skim milk/TBS-T for 1 h at room temperature and incubated with the appropriate primary antibodies. Primary antibodies were obtained for the following proteins: phospho-CDC2 (Tyr15), CDC2, cyclin B1, phospho-histone H3 (Ser10), histone H3, phospho-histone H2A.X (Ser139), histone H2A.X, CDC25C, phospho-CHK1 (Ser345), CHK1, phospho-CHK2 (Thr68), CHK2, phospho-p38 (Thr180/Tyr182), p38 (Cell Signaling Technology), GAPDH (HyTest Ltd., Turku, Finland), and β-actin (Sigma-Aldrich). An appropriate secondary antibody, anti-rabbit IgG, HRP-linked antibody, or anti-mouse IgG, HRP-linked antibody (GE Healthcare, Piscataway, NJ, USA), was incubated for 1 h at room temperature. The signals were detected with Chemi-Lumi One (Nacalai Tesque, Kyoto, Japan) or Western Chemiluminescent HRP Substrate (EMD Millipore). The band intensity was assessed by ImageJ software18.
Statistical Analysis
Significance was assessed by Student’s t-test. Differences were considered significant with a value of p < 0.05.
RESULTS
The TCTs (OBP-801/YM753, 5-FU, and PTX) Significantly Inhibit Cell Growth With G2/M Phase Arrest in Ovarian Cancer Cells
We first investigated the effect of OBP-801/YM753, 5-FU, or PTX, individually, on the cell growth of human ovarian cancer, SKOV-3 and OVCAR-3 cells, using a cell counting kit-8 assay. OBP-801/YM753, 5-FU, or PTX inhibited the cell growth of SKOV-3 (Fig. 1A–C) cells in a dose-dependent manner. Moreover, the TCTs (OBP-801/YM753, 5-FU, and PTX) showed a stronger inhibitory effect on growth than each single agent and two combined treatments (Fig. 1D). Similar results were found in another human ovarian cancer, OVCAR-3 cells (Fig. 1E–H).
Figure 1.

Three combined treatments (TCTs)—OBP-801/YM753, 5-FU, and PTX—significantly inhibit the growth of human ovarian cancer SKOV-3 and OVCAR-3 cells. Human ovarian cancer SKOV-3 cells were treated with DMSO (as control) and OBP-801/YM753 (A), 5-FU (B), or PTX (C) at the indicated concentrations. Human ovarian cancer OVCAR-3 cells were treated with DMSO (as control) and OBP-801/YM753 (E), 5-FU (F), or PTX (G) at the indicated concentrations. SKOV-3 (D) and OVCAR-3 (H) cells were treated with DMSO or OBP-801/YM753 (3 nM) and/or 5-FU (100 nM) and/or PTX (2 nM). After incubation for 72 h, viable cells were counted using a cell counting kit-8 assay. Data represent means ± SD of three determinations. *p < 0.01 compared with the TCTs versus DMSO-treated controls.
The TCTs Induce G2 Phase Arrest With the Induction of the Phosphorylation of CDC2 and Suppression of the Expression of CDC25C
Since it is known that 5-FU usually induces S phase arrest, PTX induces M phase arrest, and HDAC inhibitors induce both G1 and/or G2 phase arrest, we investigated the effect of the TCTs on the cell cycle progression of SKOV-3 cells using flow cytometry. As a result, TCTs more significantly induced G2/M phase arrest than each single agent and two combined treatments (Fig. 2A).
Figure 2.

The TCTs induce G2 phase arrest. (A) SKOV-3 cells were treated with DMSO or OBP-801/YM753 (3 nM) and/or 5-FU (100 nM) and/or PTX (2 nM) for 24 h and were analyzed by flow cytometry. Data represent means ± SD of three determinations. *p < 0.01 compared with TCTs versus DMSO-treated controls (indicated for the G2/M phase only). (B) Western blotting for p-CDC2 (Tyr15), CDC2, cyclin B1, p21, CDC25C, p-histone H3 (Ser10), and histone H3 was performed with the lysate of cells treated for 24 h with DMSO or OBP-801/YM753 (3 nM) and/or PTX (2 nM) and/or 5-FU (100 nM) in SKOV-3 cells. GAPDH and β-actin were used as loading controls.
To investigate whether the cell cycle arrest induced by the TCTs was G2 or M phase arrest, we evaluated the phosphorylation of histone H3 (Ser10), a well-known M phase marker, the phosphorylation of CDC2 (Tyr15), and the expression of p21, cyclin B1, and CDC25C (Fig. 2B). After 24 h, the TCTs did not increase the phosphorylation of histone H3. On the other hand, the TCTs increased the phosphorylation of CDC2 and decreased the expression of CDC25C. The expression of cyclin B1 was unchanged. The expression of p21 was increased in all treatments, including each single agent, but there were no accumulative effects in the two or TCTs. These results suggest that the TCTs induced G2 phase arrest, but not M phase arrest, with the induction of the phosphorylation of CDC2 and the suppression of CDC25C expression.
The TCTs Increase the Phosphorylation of p38, but Neither the Phosphorylation of CHK1 nor CHK2, Following DNA Damage
We also analyzed the phosphorylation of histone H2A.X, a well-known DNA damage marker. The TCTs increased the phosphorylated form of histone H2A.X (Fig. 3A and B). This result suggests that the TCTs induced DNA damage.
Figure 3.

The TCTs induce the phosphorylation of p38, but neither the phosphorylation of CHK1 nor CHK2, following DNA damage. (A) Western blotting for p-histone H2A.X (Ser139), histone H2A.X, p-CHK1 (Ser345), CHK1, p-CHK2 (Thr68), CHK2, p-p38 (Thr180/Tyr182), and p38 was performed with the lysate of cells treated for 24 h with DMSO or OBP-801/YM753 (3 nM) and/or 5-FU (100 nM) and/or PTX (2 nM) in SKOV-3 cells. GAPDH was used as a loading control. The phosphorylation ratio of histone H2A.X (B), CHK1 (C), CHK2 (D), and p38 (E) was determined as a ratio of each phosphorylated protein compared to each total protein and expressed as fold induction relative to control. Band intensity was quantified with ImageJ.
To investigate the relationship between the G2 phase arrest and the DNA damage caused by the TCTs, we analyzed the effects on the phosphorylation of CHK1, CHK2, and p38, all of which are known to be activated by DNA damage and subsequently suppress the expression of CDC25C. The TCTs remarkably increased the phosphorylation of p38, but neither the phosphorylation of CHK1 nor CHK2 (Fig. 3A and C–E). These results suggest that the TCTs activate the p38 pathway, but not the CHK1 or CHK2 pathways, following DNA damage.
The p38 Inhibitor Suppresses the G2 Phase Arrest Induced by the TCTs
To evaluate the contribution of the activation of p38 pathway in the G2 phase arrest induced by the TCTs, we employed a p38 selective inhibitor, SB203580. SB203580 significantly suppressed the G2 phase arrest caused by the TCTs (Fig. 4A). Moreover, SB203580 suppressed the phosphorylation of CDC2 caused by the TCTs (Fig. 4B). In another human ovarian cancer, OVCAR-3 cells, the TCTs induced G2 phase arrest (Fig. 4C) and the phosphorylation of both p38 and CDC2 (Fig. 4D). Furthermore, SB203580 significantly suppressed the G2 phase arrest and the phosphorylation of CDC2 caused by the TCTs (Fig. 4C and D). These results suggest that the TCTs can induce G2 phase arrest through the activation of the p38 signaling pathway in human ovarian cancer cells.
Figure 4.

p38 inhibitor suppresses G2 phase arrest and the phosphorylation of CDC2 caused by the TCTs. SKOV-3 (A) and OVCAR-3 (C) cells were treated with or without the TCTs for 24 h and/or pretreatment with SB203580 (20 μM) for 2 h. The population, in terms of the phases of the cell cycle, was analyzed by flow cytometry. Data represent means ± SD of three determinations. *p < 0.01 compared with the TCTs versus the TCTs with SB203580 (indicated for the G2/M phase only). SB, SB203580; combo, TCTs (OBP-801/YM753, PTX, and 5-FU). Western blotting for CDC25C, p-CDC2 (Tyr15), CDC2, p-p38 (Thr180/Tyr182), and p38 was performed with the lysate of cells treated with or without the TCTs for 24 h and/or pretreatment with SB203580 (20 μM) for 2 h in SKOV-3 (B) and OVCAR-3 (D) cells. GAPDH was used as a loading control.
DISCUSSION
In this study, we demonstrated that the TCTs—a novel HDAC inhibitor OBP-801/YM753, 5-FU, and PTX—significantly induced G2 phase arrest in human ovarian cancer cells. It is known that CDC2 and CDC25C regulate the transition from G2 to M phase, called the “G2/M checkpoint”19. The cell cycle transition from G2 to M phase is regulated by the dephosphorylation of CDC2 by CDC25C19,20. In our data, the TCTs increased the phosphorylation of CDC2 with the suppression of CDC25C but did not increase the phosphorylation of histone H3, a well-known M phase marker. These results suggest that the TCTs induced G2 phase arrest at the G2/M checkpoint.
Since the G2/M checkpoint is known to arrest the cell cycle at the G2 phase in order to repair damaged DNA, we speculated that the TCTs may cause DNA damage. As a result, the TCTs induced the phosphorylated form of histone H2A.X, a well-known DNA damage marker. Since HDAC inhibitors are known to increase DNA damage by the downregulation of DNA repair proteins such as Rad51, Ku-70, Ku-86, and DNA-PKcs21,22, our results suggest that OBP801/YM753, an HDAC inhibitor, of the TCTs may prolong DNA damage, resulting in activation of the G2/M checkpoint.
In response to DNA damage, phosphorylation and the inactivation of CDC25C by CHK1/2 play a major role in G2 phase arrest23–25. However, following DNA damage, p38 also phosphorylates and inactivates CDC25C, inducing G2 phase arrest26,27. Therefore, we examined whether CHK1/2 or the p38 pathway is responsible for this G2 phase arrest followed by DNA damage. Our data indicate that the TCTs increased the phosphorylation of p38, but not the phosphorylation of CHK1/2. Moreover, SB203580, a p38 selective inhibitor, suppressed both the phosphorylation of CDC2 and G2 phase arrest caused by the TCTs. These data suggest that G2 phase arrest caused by the TCTs depends on the p38 pathway, not the CHK1/2 pathway, following DNA damage. Although previous studies showed that 5-FU or PTX inactivated CDC25C28–30, we could detect neither DNA damage nor the inactivation of CDC25C by each single treatment at the concentrations we used. Therefore, it is of interest to elucidate further the mechanisms of the specific activation of the p38 pathway following DNA damage by the TCTs.
On the basis of our results, we considered that the TCTs caused DNA damage followed by the activation of p38 and the inactivation of CDC2, consequently leading to cell growth inhibition with G2 phase arrest in ovarian cancer cells (Fig. 5). Recently, it was reported that the inefficient combination of oxaliplatin and cetuximab against colorectal cancer in clinical trials was due to the inactivation of p38 by cetuximab31. This report and our results suggest the importance of the activation of the p38 pathway in the efficacy of chemotherapy.
Figure 5.
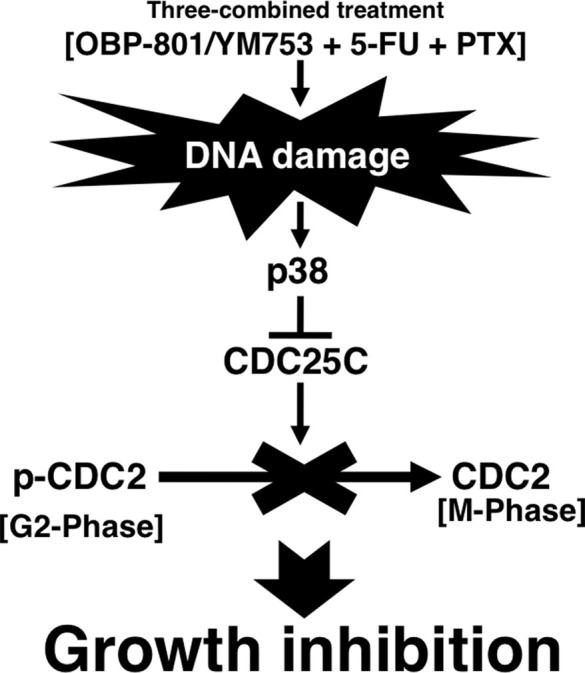
The scheme of mechanisms of inducing G2 phase arrest through the p38 pathway by the TCTs.
In conclusion, this is the first report to demonstrate that treatment with three combined agents—a novel HDAC inhibitor OBP-801/YM753, 5-FU, and PTX—induces G2 phase arrest through the p38 pathway in human ovarian cancer cells. We believe that this treatment is a promising therapy for ovarian cancer.
ACKNOWLEDGMENT
The authors would like to acknowledge the funding support from the research fund of Kyoto Prefectural University of Medicine.
REFERENCES
- 1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. [DOI] [PubMed] [Google Scholar]
- 2. Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J Clin Oncol. 2002;20:1248–59. [DOI] [PubMed] [Google Scholar]
- 3. Markman M, Markman J, Webster K, Zanotti K, Kulp B, Peterson G, Belinson J. Duration of response to second-line, platinum-based chemotherapy for ovarian cancer: Implications for patient management and clinical trial design. J Clin Oncol. 2004;22:3120–5. [DOI] [PubMed] [Google Scholar]
- 4. McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334:1–6. [DOI] [PubMed] [Google Scholar]
- 5. Markman M. New, Expand, and modified use of approved antineoplastic agents in ovarian cancer. Oncologist 2007;12:186–90. [DOI] [PubMed] [Google Scholar]
- 6. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006;6:38–51. [DOI] [PubMed] [Google Scholar]
- 7. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. [DOI] [PubMed] [Google Scholar]
- 8. Porcu P, Wong HK. We should have a dream: Unlocking the workings of the genome in cutaneous T-cell lymphomas. Clin Lymphoma Myeloma 2009;9:409–11. [DOI] [PubMed] [Google Scholar]
- 9. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007;12:1247–52. [DOI] [PubMed] [Google Scholar]
- 10. Strait KA, Warnick CT, Ford CD, Dabbas B, Hammond EH, IIstrup SJ. Histone deacetylase inhibitors induce G2-checkpoint arrest and apoptosis in cisplatinum-resistant ovarian cancer cells with overexpression of Bcl-2-related protein Bad. Mol Cancer Ther. 2005;4:603–11. [DOI] [PubMed] [Google Scholar]
- 11. Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, Rice VM. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: Implications for therapy. Cancer Biol Ther. 2007;6:795–801. [DOI] [PubMed] [Google Scholar]
- 12. Masuoka Y, Nagai A, Shin-ya K, Furihata K, Nagai K, Suzuki K, Hayakawa Y, Seto H. Spiruchostatins A and B, novel gene expression enhancing substances produced by Pseudomonas sp. Tetrahedron Lett. 2001;42:41–4. [Google Scholar]
- 13. Shindoh N, Mori M, Terada Y, Oda K, Amino N, Kita A, Taniguchi M, Sohda KY, Nagai K, Sowa Y, Masuoka Y, Orita M, Sasamata M, Matsushime H, Furuichi K, Sakai T. YM753, a novel histone deacetylase inhibitor, exhibits antitumor activity with selective, sustained accumulation of acetylated histones in tumors in WiDr xenograft model. Int J Oncol. 2008;32:545–55. [PubMed] [Google Scholar]
- 14. Johnson KR, Wang L, Miller MC III, Willingham MC, Fan W. 5-Fluorouracil interferes with paclitaxel cytotoxicity against human solid tumor cells. Clin Cancer Res. 1997;3:1739–45. [PubMed] [Google Scholar]
- 15. Na YS, Kim SM, Jung KA, Yang SJ, Hong YS, Ryu MH, Ro S, Cho DH, Kim JC, Jin DH, Lee JS, Kim TW. Effects of the HDAC inhibitor CG2 in combination with irrinotecan, 5-fluorouracil, or oxaliplatin on HCT116 colon cancer cells and xenografts. Oncol Rep. 2010;24:1509–15. [DOI] [PubMed] [Google Scholar]
- 16. Lee JH, Park JH, Jung Y, Kim JH, Jong HS, Kim TY, Bang YJ. Histone deacetylase inhibitor enhances 5-fluorouracil cytotoxicity by down-regulating thymidylate synthase in human cancer cells. Mol Cancer Ther. 2006;5:3085–95. [DOI] [PubMed] [Google Scholar]
- 17. Zuco V, De Cesare M, Cincinelli R, Nannei R, Pisano C, Zaffaroni N, Zunino F. Synergistic antitumor effects of novel HDAC inhibitors and paclitaxel in vitro and in vivo. PLOS One 2011;6:e29085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 Years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer 2009;9:153–66. [DOI] [PubMed] [Google Scholar]
- 20. Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–66. [DOI] [PubMed] [Google Scholar]
- 21. Mueller S, Yang X, Sottero TL, Gragg A, Prasad G, Polley MY, Weiss WA, Matthay KK, Davidoff AM, DuBois SG, Haas-Kogan DA. Cooperation of the HDAC inhibitor vorinostat and radiation in metastatic neuroblastoma: Efficacy and underlying mechanisms. Cancer Lett. 2011;306:223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shoji M, Ninomiya I, Makino I, Kinoshita J, Nakamura K, Oyama K, Nakagawara H, Fujita H, Tajima H, Takamura H, Kitagawa H, Fushida S, Harada S, Fujimura T, Ohta T. Valproic acid, a histone deacetylase inhibitor, enhances radiosensitivity in esophageal squamous cell carcinoma. Int J Oncol. 2012;40:2140–6. [DOI] [PubMed] [Google Scholar]
- 23. Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: Linkage of DNA damage to Cdk regulation through Cdc25. Science 1997;277:1497–501. [DOI] [PubMed] [Google Scholar]
- 24. Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997;277:1501–5. [DOI] [PubMed] [Google Scholar]
- 25. Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998;282:1893–7. [DOI] [PubMed] [Google Scholar]
- 26. Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001;411:102–7. [DOI] [PubMed] [Google Scholar]
- 27. Hirose Y, Katayama M, Stokoe D, Haas-Kogen DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol. 2003;23:8306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yoshikawa R, Kusunoki M, Yanagi H, Noda M, Furuyama JI, Yamamura T, Hashimoto-Tamaoki T. Dual antitumor effects of 5-fluorouracil on the cell cycle in colorectal carcinoma cells: A novel target mechanism concept for pharmacokinetic modulating chemotherapy. Cancer Res. 2001;61:1029–37. [PubMed] [Google Scholar]
- 29. Matuo R, Sousa FG, Escargueil AE, Grivicich I, Garcia-Santos D, Chies JA, Saffi J, Larsen AK, Henriques JA. 5-Fluorouracil and its active metalolite FdUMP cause DNA damage in human SW620 colon adenocarcinoma cell line. J Appl Toxicol. 2009;29:308–16. [DOI] [PubMed] [Google Scholar]
- 30. Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation factor 4E-binding protein 1 through a Cdk-dependent mechanism. Oncogene 2005;24:4851–60. [DOI] [PubMed] [Google Scholar]
- 31. Santoro V, Jia R, Thompson H, Nijhuis A, Jeffery R, Kiakos K, Silver AR, Hartley JA, Hochhauser D. Role of reactive oxygen species in the abrogation of oxaliplatin activity by cetuximab in colorectal cancer. J Natl Cancer Inst. 2015;108:djv394. [DOI] [PMC free article] [PubMed] [Google Scholar]