

Abstract
There is an urgent need to improve the clinical management of hepatocellular carcinoma (HCC), one of the most common causes of global cancer-related deaths. Zingibar officinale is a medicinal herb used throughout history for both culinary and medicinal purposes. It has antioxidant, anticarcinogenic, and free radical scavenging properties. Previously, we proved that the crude flavonoid extract of Z. officinale (CFEZO) inhibited growth and induced apoptosis in several cancer cell lines. However, the effect of the CFEZO on an HCC cell line has not yet been evaluated. In this study, we explored the anticancer activity of CFEZO against an HCC cell line, HepG2. CFEZO significantly inhibited proliferation and induced apoptosis in HepG2 cells. Typical apoptotic morphological and biochemical changes, including cell shrinkage and detachment, nuclear condensation and fragmentation, DNA degradation, and comet tail formation, were observed after treatments with CFEZO. The apoptogenic activity of CFEZO involved induction of ROS, depletion of GSH, disruption of the mitochondrial membrane potential, activation of caspase 3/9, and an increase in the Bax/Bcl-2 ratio. CFEZO treatments induced upregulation of p53 and p21 expression and downregulation of cyclin D1 and cyclin-dependent kinase-4 expression, which were accompanied by G2/M phase arrest. These findings suggest that CFEZO provides a useful foundation for studying and developing novel chemotherapeutic agents for the treatment of HCC.
Key words: Hepatocellular carcinoma (HCC), Apoptosis, Reactive oxygen species (ROS), Glutathione (GSH), Mitochondrial potential
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the five most common cancers worldwide and the third leading cancer phenotype resulting in mortality among all cancers1. Despite its particularly high prevalence in Asian countries owing to endemic hepatitis B virus infections, its incidence is also rising in Western countries as a result of increasing hepatitis C virus infections2. Currently, therapeutic strategies for HCC include multimodal treatments, doxorubicin, cisplatin, and 5-fluorouracil (5-FU) as the primary choices for treating HCC cases. Nonetheless, the response rate and overall 5-year survival remain poor3. Furthermore, clinical applications of chemotherapy are limited due to their severe side effects4. Recent targeted cancer therapy agents, such as sorafenib, exhibited an improved clinical outcome in advanced liver cancer cases5; nonetheless, the overall mortality rate of this disease still exceeds 90% worldwide1. Therefore, the search for novel therapeutic options is imperative.
An overwhelming body of evidence accumulated in the last decades has indicated that a common denominator in all cancer phenotypes, including HCC, is the evasion of apoptotic cell death6. Therefore, apoptosis induction is considered to be a practical approach for the successful eradication of cancer cells, and searching for bioactive agents with the ability to induce apoptosis in cancer cells is the ultimate aim of chemoprevention and/or chemotherapy. Apoptosis is an ordered and orchestrated cell suicide program that plays a crucial role in development and tissue homeostasis7. It can be triggered through the extrinsic (death receptor) pathway, which is initiated from outside the cell through proapoptotic receptors on the cell surface, or the intrinsic (mitochondrial) pathway, which is initiated from within the cell7. In the intrinsic pathway, the mitochondria play a central role in the integration and circulation of death signals (such as oxidative stress, drug, and DNA damage) initiated inside the cells in regulating cell death pathways8. The perturbation of mitochondria appears to be the trigger for the initiation of apoptotic cascades, where the loss of mitochondrial membrane potential facilitates the release of apoptotic factors (such as cytochrome c and AIF) from the mitochondrial intermembrane space into the cytoplasm9. The release of these factors constitutes the “point of no return”; it induces the propagation of the apoptotic cascades and execution of cell death8. However, the process of apoptotic factor release is tightly controlled by members of the Bcl-2 family10. The fine-tuning of the balance between the pro- and antiapoptotic members within this family in a cell leads to programmed cell death or survival7. Upon receiving apoptotic signals, proapoptotic Bcl-2 family proteins, such as Bax and Bak, are activated, resulting in outer mitochondrial membrane permeabilization and, consequently, apoptotic factor release. On the other hand, antiapoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL, prevent this permeabilization. The released mitochondrial factors trigger the activation of further downstream effectors, caspase family members, which are cysteine proteases stored in most cells as procaspases and the actual executioners of apoptotic process11. Among the caspase family, procaspases 8 and 9 are the major actors; they are activated through the extrinsic and intrinsic pathways, respectively7. Activated cascades by both caspases mediate activation of procaspase 3. The latter is responsible for the cleavage of key cellular proteins, such as cytoskeletal proteins, DNA repair proteins, and inhibitory subunits of endonucleases, which lead to the cardinal morphological changes observed in cells committing apoptosis12.
An exponential increase in the number of studies demonstrates that excessive reactive oxygen species (ROS) can directly activate the mitochondrial permeability transition and result in mitochondrial membrane potential loss, leading to apoptotic cell death8. ROS is produced by all aerobic cells to regulate cellular processes. Under physiological conditions, low levels of ROS play a role as an intracellular messenger in regulating many molecular events, including cell proliferation and apoptosis13. However, generation of a large amount or sustained levels of ROS can cause serious damage to lipids, proteins, and DNA, cumulatively known as oxidative stress. This stress provokes cellular events leading to growth arrest, senescence, and, eventually, apoptotic and necrotic cell death14. Most cancer cells, even at rest, have comparatively higher levels of intracellular ROS than normal cells owing to their higher metabolic rate. These high levels of ROS play an essential role in maintaining cancerous phenotypes due to their stimulating effects on cell growth and proliferation15. Accordingly, this high level of ROS causes high oxidative stress in cancer cells, rendering them more vulnerable to ROS-induced insults and apoptosis15. These circumstances might provide an opportunity to exploit the cell-killing potential of ROS by using exogenous ROS-stressing agents to increase the intracellular ROS to a toxic level, or the threshold that triggers cancer cell death. Consistent with this notion, promoting ROS generation in the mitochondria was found to effectively kill cancer cells and to be a practical approach for developing antitumor agents16. For example, human leukemia cells with intrinsic oxidative stress have been found to be highly sensitive to ROS stress induced by 2-methoxyestradiol and arsenic trioxide17. Likewise, the anticancer potentials of taxol and doxorubicin have been attributed to their ROS-promoting activities.
Cumulative studies have shown that, under normal physiological conditions, cells possess an adequate antioxidant defense system to neutralize ROS. Within this system, reduced glutathione (GSH) acts as a major antioxidant, protecting cells from the damaging effects of ROS and from apoptotic cell death18. GSH exists in all cells and is the only available scavenger to hydrogen peroxide in mitochondria, and its depletion leads to severe mitochondrial damage19. It is present in a reduced form (GSH) and an oxidized form (GSSG), and the GSH/GSSG ratio serves as an indicator of changes in intracellular reduction and oxidation reactions19. Increased GSH levels have been found to play a crucial role in cancer progression and resistance20. Conversely, depletion of cellular GSH levels in tumor cells through chemotherapeutic intervention decreases the drug resistance and increases the therapeutic response21,22.
An important aim of cancer research is to find therapeutic compounds having a high specificity for cancerous cells/tumor and fewer side effects than the currently used cytostatic/cytotoxic agents. In this regard, a number of biologically active phytochemical-derived medicinal herbs are gaining momentum. Among these medicinal herbs, Zingiber officinale (ginger) has drawn great attention. It is used in traditional medicine to treat various ailments including atherosclerosis, rheumatoid arthritis, high cholesterol, ulcers, depression, and impotence23. Although the herb belongs to the Zingiberaceae family grown in Southeast Asia, it has been introduced to many parts of the globe24. Most of the notable anticancer properties of the herb were ascribed to phenolic ingredients (gingerol, shogaol, paradol, and those derivatives), which have an antioxidant potential23. In addition to these phenolics, the herb also contains a huge amount of flavonoid compound25,26. However, no report has yet addressed the anticancer activity of these flavonoids.
Flavonoids are a group of bioactive compounds extensively found in foodstuffs of plant origin. They possess a remarkable spectrum of pharmacological properties, such as antimicrobial, antiviral, anti-inflammatory, antiallergic, analgesic, antioxidant, and hepatoprotective activities. The use of flavonoids for both cancer prevention and treatment has increased dramatically over the past decade27,28. Numerous epidemiological studies have validated the inverse relation between the consumption of flavonoids and the risk of cancer29. Flavonoids have been found to inhibit carcinogenesis at multiple levels. For instance, they inhibit proliferation and induce cell cycle arrest and apoptosis in cancer cells. They also inhibit activation of procarcinogens, metastasis, and angiogenesis; stimulate the immune response against cancer cells; and sensitize cells to anticancer drugs29. The observed anticancer properties of flavonoids were rendered to their frank apoptogenic potentialities through modulating different key targets involved in both apoptotic pathways27. Although the molecular mechanisms by which flavonoids induce apoptosis have not been fully clarified, several mechanisms were reported, including increase in ROS, depletion of GSH, modulation of signaling pathways, and expression levels of Bcl-2 family proteins, activation of caspases, and DNA fragmentation27.
MATERIALS AND METHODS
Herbal Material and Extract Preparation
For the herb used in this study, the mature and healthy rhizomes of Z. officinale were purchased from the local market of Jeddah, KSA. They were properly washed, thinly grated, and used to prepare the extract. The powder (500 g) was subjected to maceration in 75% ethanol for 7 days, filtered, and dried under a vacuum. The extract was prepared as previously described30,31. Briefly, the concentrated extract was defatted using pet ether. The aqueous portion was separated, collected, and then fractionated with N-butanol-saturated water. The N-butanol portion was separated and collected. The aqueous portion was then discarded, and the N-butanol portion was fractionated with 1% KOH. The 1% KOH portion was then separated, collected, and dried. The N-butanol portion was discarded. The KOH portion was then fractionated with dilute HCl and N-butanol-saturated water, and the N-butanol portion was separated and collected. The dilute HCl portion was discarded and the N-butanol portion (crude flavonoids fraction) was then separated, collected, dried, and dissolved in DMSO. Our initial experiments indicated that all tested cell lines in this study could tolerate as much as 0.4% to 0.6% DMSO without showing any toxicity. So to avoid a toxic effect from DMSO and to assure that cell toxicity was only due to crude flavonoid extract of Z. officinale (CFEZO) constituents, the final concentrations of DMSO in all experiments were kept as low as 0.2%. To achieve this goal, the crude dried CFEZO extract was aliquoted into stocks of increasing weights (5, 7.5, 10.0, and 12.5 g); each stock was then dissolved in 100 μl of 20% DMSO (diluted in virgin media). Including only 1 μl of any of these stocks into cells growing in 100 μl of tissue culture media safeguards that the final concentrations of CFEZO stocks are 50, 75, 100, and 125 μg/ml, and the final concentration of DMSO is limited to 0.2%. In all control experiments, cells growing in 100 μl of media were treated with 1 μl of 20% DMSO.
Cell Culture and Treatments
All cell lines in this study, including human liver cancer cells (HepG2), breast cancer cells (MCF-7), cervical cancer cells (HeLa), and nonneoplastic human epithelial cells (OEC) were obtained from King Fahed Center for Medical Research, King Abdulaziz University, KSA. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin antibiotics in tissue culture flasks under a humidifying atmosphere containing 5% CO2 and 95% air at 37°C. The cells were subcultured at 3- to 4-day intervals.
The effect of CFEZO on the viability of HepG2 cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT; Cayman Chemicals, Ann Arbor, MI, USA) assay following the manufacturer’s instructions. Early log phase cells were trypsinized and regrown in 96-well cell culture plates at a concentration of 104 cells/ml in 100 μl of complete culture medium. Twenty-four hours later, the medium was removed and replaced with fresh medium with or without CFEZO. MTT was then added to each well and incubated for 4 h, after which the plates were centrifuged at 1,800 rpm for 5 min at 4°C. After careful removal of the medium, 0.1 ml of PBS was added to each well, and the plates were shaken. The absorbance of converted dye was measured at a wavelength of 570 nm, and the increased absorbance was directly proportional to cell viability. The effects of CFEZO on growth inhibition were assessed as percent cell viability, where DMSO water-treated cells were taken as 100% viable. For these studies, all experiments were repeated three or more times.
Clonogenic Assay
This assay measures the ability of tumor cells to grow and form foci in a manner unrestricted by growth contact inhibition as is characteristically found in normal, untransformed cells. Approximately 500 cells were seeded into six-well plates in triplicate and allowed to adhere overnight. Thereafter, cell culture medium was changed, and cells were treated with increasing doses of CFEZO. The cells were allowed to incubate at 37°C in the incubator undisturbed for 15 days. During this period, each individual surviving cell would proliferate and form colonies. On day 15, the colonies were washed with cold PBS, fixed with cold 70% ethanol, and stained with 0.25% Giemsa stain.
Apoptotic Assay
The nuclear morphological changes associated with apoptosis were analyzed using Hoechst 33342 or acridine orange/ethidium bromide (AO/EtBr) staining as previously described32. HepG2 cells were suspended at a final concentration of 150 × 103 cells/ml in 24-well plates and allowed to adhere to the bottom of the wells for 24 h before CFEZO treatment. Cells were then exposed to the indicated concentrations of the CFEZO for 24 h before being washed with PBS and stained with Hoechst 33342 or AO/EtBr for 15 min at 37°C. Cells were subsequently washed with PBS and viewed under an inverted fluorescence microscope (Leica, Germany).
DNA Fragmentation Assay
DNA gel electrophoresis was used to determine the presence of internucleosomal DNA cleavage. Briefly, the HepG2 cells (106 cells/100-mm dish) treated with various concentrations of CFEZO for the indicated periods were collected, washed in PBS, and resuspended in lysis buffer (0.5% Triton X-100 in 10 mM EDTA, and 10 mM Tris-HCl, pH 8.0) on ice for 30 min. Genomic DNA was then extracted as previously detailed30,31. The DNA was then resolved by electrophoresis on a 2% agarose gel. After electrophoresis at 80–100 V, the gel was stained with ethidium bromide, and DNA was visualized by a UV transilluminator (Bio-Rad, Hercules, CA, USA).
Comet Assay (Single-Cell Gel Electrophoresis)
CFEZO-induced DNA damage was determined using a comet assay. Cells were treated with the indicated concentrations of CFEZO for 24 h in complete medium, harvested, resuspended in ice-cold PBS, and processed under a dimmed light, as described previously30,31. Prepared comet slides were viewed, and nuclei images were visualized and captured at 100× magnification with an Axioplan 2 fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with a CCD camera (Optronics, Goleta, CA, USA).
Determination of ROS Production
The production of ROS was monitored by fluorescence microscopy using DCFH-DA (Cayman Chemicals), following the manufacturer’s instructions. This dye is a stable compound that readily diffuses into cells and is hydrolyzed by intracellular esterase to yield DCFH, which is trapped within cells. Hydrogen peroxide or low-molecular-weight hydroperoxides produced by cells oxidize DCFH to the highly fluorescent compound 2′,7′-dichlorofluorescein (DCF). Thus, the intensity of the fluorescence is proportional to the amount of peroxide produced by the cells. For this assay, cells (104) in 96-well plates were loaded with 10 μM DCFH-DA for 30 min at 37°C in the dark, washed twice with PBS, detached with trypsin, and washed in PBS. After centrifugation, the cell pellet was suspended in 200 μl of PBS. The cells were observed under a fluorescence microscope (Leica). At the same time, fluorescence intensity of the cell was measured using a microplate reader (BioTek Synergy) with excitation at 485/20 nm and emission at 528/20 nm.
Analysis of the Mitochondrial Membrane Potential (Δψm)
Mitochondrial membrane potential (Δψm) was measured using a cytofluorimetric, lipophilic cationic dye, 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide, JC-1 (Cayman Chemicals), following the manufacturer’s instructions. Briefly, 1 × 104 cells per well were seeded onto a 96-well plate. Cells were treated with CFEZO at the indicated concentrations for 24 h. JC-1 dye contained in the JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman Chemicals) was added into live culture for 30 min. Cells were washed with wash buffer as described by the manufacturer’s instructions. Stained cells were visualized and acquired using a microplate reader (BioTek Synergy) with excitation at 535 nm and emission at 595 nm.
Detection of Activity of Caspases
Caspase 3/7, caspase 9, and caspase 8 activities were determined using the Caspase-Glo® 3/7, Caspase-Glo® 9, and Caspase-Glo® 8 Assay kits, respectively (Promega, Madison, WI, USA). The HepG2 cells were seeded in 100 μl of medium in 96-well plates (104 cells/well) and treated with the indicated concentrations of CFEZO for 24 h. The activities of caspase 3/7 were then measured according to the manufacturer’s instructions. Briefly, 100 μl of the assay reagents was added to each well, and the contents of the wells were mixed using a plate shaker at 300–500 rpm for 3 h. The luminescence of each sample was measured using a plate-reading luminometer (BioTek Synergy).
Cell Cycle Analysis
Briefly, HepG2 cells (300 × 103) were plated in culture flasks (25 cm2), allowed to attach overnight, and exposed to the indicated concentrations of HepG2 for 24 h. The cells were then trypsinized, harvested, washed with cold PBS, and centrifuged; the pellets were then fixed in 2 ml of ice-cold 70% ethanol overnight at 4°C. Next, the cell suspensions were centrifuged, and pellets were resuspended overnight at 4°C in 800 μl of PBS and RNase A at a final concentration of 10 μg/ml. The cells were incubated with propidium iodide (20 μg/ml final concentration) for 2 h at 4°C in the dark and finally subjected to flow cytometry analysis using NovoCyte™ Flow Cytometer (ACEA Biosciences, San Diego, CA, USA).
Total RNA Extraction and Quantitative Reverse Transcription PCR
Cells were seeded (20 × 104/well) onto six-well plates and treated with the indicated concentrations of CSENS for 24 h. After this period, floating and adherent cells were collected, washed with PBS, and pelleted by centrifugation (700 × g, 5 min). For RNA extraction and reverse transcriptase PCR, gene-specific primers were used as previously described32.
Western Blot Analysis
For Western blot analysis, polyacrylamide gel electrophoresis and immunoblotting were performed as previously described32.
Statistical Analysis
All experiments were repeated from three to five times independently and in triplicate. The results are presented as mean ± standard deviation (SD). The statistical significance of the results was determined by Student’s t-test using the GraphPad Prism 6 program (La Jolla, CA, USA), and a value of p < 0.05 was considered statistically significant. In all experiments, statistical significance is indicated as p < 0.05, p < 0.01, p < 0.001, and p < 0.0001 compared to that of the control.
RESULTS
CFEZO Inhibits HepG2 Cell Proliferation and Colony Growth
MTT assays were performed to investigate the effects of CFEZO on the proliferation of HepG2 cells. Cells were treated with increasing concentrations (50, 75, 100, and 125 μg/ml) of CFEZO for 24, 48, or 72 h. CFEZO showed a dose- and time-dependent inhibitory effect on growth of HepG2 cells (Fig. 1A). This caused 50% growth inhibition (IC50) at around 115, 65, and 40 μg/ml for 24, 48, and 72 h, respectively. To confirm these results, we repeated the above experiments using human breast and cervical cancer cell lines MCF-7 and HeLa cells, respectively. We observed that CFEZO recapitulated its potential for growth inhibition in the contexts of both cell lines (Fig. 1A). The effect of CFEZO on the viability of nonmalignant human esophageal epithelial cells (OEC) was then investigated to determine whether CFEZO-dependent growth inhibition is selective toward cancer cells. The pairwise comparison between the effect of CFEZO treatments on cancer cells and on OEC cells indicates that sensitivity of OEC cells to the cytotoxic effects of CFEZO is marginal (Fig. 1A).
Figure 1.

CFEZO inhibits HepG2 cell proliferation and colony growth. (A) The displayed cell lines were seeded at a density of 104/well in 96-well plates and treated with the indicated concentrations of CFEZO for 24 (dotted line), 48 (solid line), and 72 h (dashed line). The inhibition of cell proliferation was assessed by the MTT as detailed in the Materials and Methods section. The experiments were repeated five times in triplicates, and cell viabilities at each dose of the extract were expressed in terms of percentage of control and reported as the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to that of control. (B) HepG2 cells were seeded onto a six-well plate at 1,000 cells/well and treated with the indicated concentrations of CFEZO and assayed as has been detailed in the Materials and Methods section.
Next, the antiproliferative potential of CFEZO on HepG2 cells was further determined and verified using colony formation assay (clonogenicity). Volumes of 50, 75, 100, and 125 μg/ml of CFEZO reduced colony formation and size in HepG2 (Fig. 1B). Taken together, these findings indicate that CFEZO has an apparent potential to inhibit the growth of HepG2 cells.
CFEZO Induces Morphological Features of Apoptosis in HepG2 Cells
To test whether the decrease in cell viability observed after treatment with CFEZO is due to apoptosis, the treated cells were investigated directly under a microscope. The images in Figure 2A show that cells treated with CFEZO exhibited typical apoptotic signs including cell shrinkage, loss of contact with neighboring cells, rounding up, substrate detachment, membrane blebbing, nuclear condensation, and formation of apoptotic bodies. All these changes indicate the occurrence of apoptotic events. On the other hand, the control cells showed a high confluency of monolayer cells.
Figure 2.

CFEZO treatments induced morphologic features of apoptosis in HepG2 cells. The cells were treated with the indicated concentrations of CFEZO for 24 h, after which cells were examined for emergence of apoptotic hallmarks. (A) Light microscopy photomicrographs showing morphological changes in HepG2 cells after incubation with CFEZO. Cellular shrinkage, detachment, and irregularity of cell shape are notable [magnification: 20× (I), 40× (II)]. (B) Cells stained with 1 μg/ml AO/EtBr for 15 min at 37°C and visualized by fluorescence microscope [magnification: 20× (I), 60× (II)]. (C) Cells were stained with vital nuclear stain (Hoechst 33342) for 15 min at 37°C and visualized by a fluorescence microscope (magnification: 20×).
To confirm these findings, cells were stained with AO/EtBr fluorescence staining after exposure to CFEZO. In this assay, cells stained green are viable cells, fragmented green nuclei represent early apoptotic cells, and yellow/green dots of condensed nuclei represent late apoptosis. Our results indicated that the nuclei of untreated control cells were found to be intact, round in shape, and stained green (Fig. 2B). On the other hand, cells treated with 50 and 100 μg/ml of CFEZO exhibited early apoptotic signs where some cells were stained green, and some displayed orange nuclei. At the highest dose of CSENS (125 μg/ml), an increase in the number of cells with red-colored nuclei (necrotic/late apoptotic) was predominate. Thus, the morphological analysis of AO/EtBr-stained HepG2 cells indicated significant morphological changes.
Finally, cells were treated with Hoechst 33342. CFEZO-treated cells exhibited typical apoptotic nuclear morphology including nuclear shrinkage, DNA condensation, and fragmentation (Fig. 2C). On the other hand, control cells appeared normal. Collectively, these results indicate that CFEZO treatment induced apoptotic cell death in HepG2 cells.
CFEZO Induces Accumulation of ROS and Depletion of GSH in HepG2
ROS generation has been reported to be associated with phytochemical-induced apoptosis in many cancer cells33 so we further investigated whether ROS production is involved in CFEZO-induced apoptosis. The production of ROS was monitored by fluorescence microscopy using DCFH-DA. This dye is a stable nonpolar compound that readily diffuses into cells and is hydrolyzed by intracellular esterase to yield DCFH, which is trapped within cells. Hydrogen peroxide or low-molecular-weight peroxides produced by cells oxidize DCFH to the highly fluorescent compound DCF. Thus, the intensity of fluorescence is proportional to the amount of peroxide produced by the cells. Treatment with CFEZO induced an increase in DCFH-DA fluorescence in a time-dependent manner (Fig. 3A). Compared with control cells, HepG2 cells treated with CFEZO at a volume of 75, 100, and 125 μg/ml for 24 h markedly increased ROS generation 2-, 3.3-, and 4.6-fold, respectively. These data support our hypothesis that CFEZO treatment leads to an increase in ROS production, which may represent a critical step in CFEZO-induced apoptosis in HepG2 cells.
Figure 3.
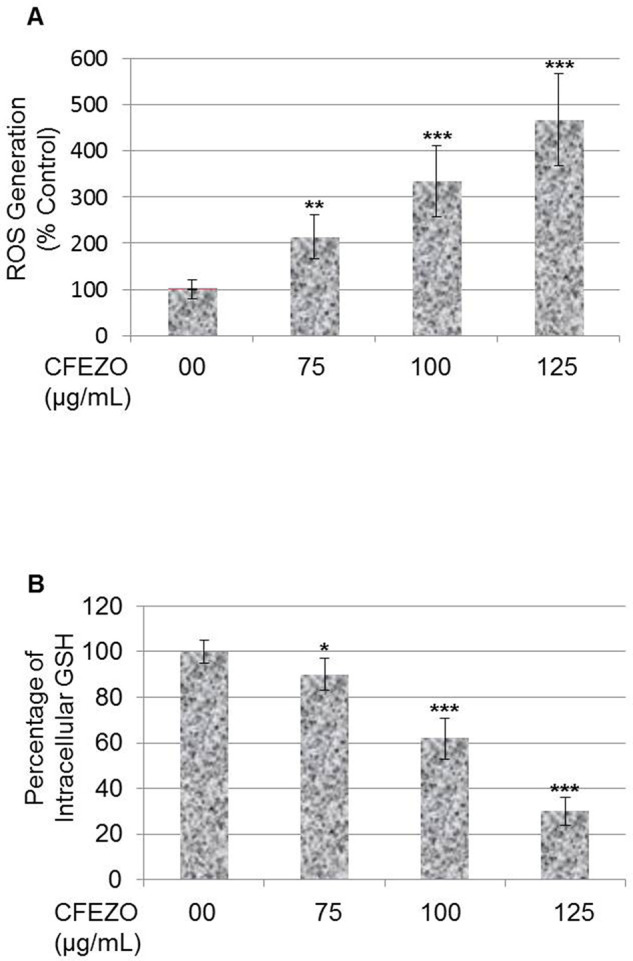
CFEZO induces accumulation of ROS and depletion of GSH in HepG2. (A) Cells were treated with the indicated concentrations of CFEZO for 24 h and then stained with DCFH-DA, and the DCF fluorescence intensity was measured by a fluorescence spectrophotometer. Data are representative results from three independent experiments. (B) Cells were treated with the indicated concentrations of CFEZO for 24 h and then assayed for GSH levels as detailed in the Materials and Methods section. The results are presented as the mean ± SD of three independent experiments. Statistical significance in ROS/GSH levels of *p < 0.05, **p < 0.01, and ***p < 0.001 compared to that of control.
To confirm the above results, we studied the possibility that CFEZO could deplete intracellular GSH. When cells are exposed to increased levels of oxidative stress, GSSG will accumulate, and the ratio of GSH to GSSG will decrease. CFEZO treatment remarkably depleted the intracellular GSH level in a dose-dependent manner (Fig. 3B). The GSH levels were significantly decreased by 10%, 40%, and 70% with 75, 100, and 125 μg/ml of CFEZO. Collectively, these results show that CFEZO can induce oxidative stress and deplete GSH levels in HepG2 cells.
CFEZO Induces Loss of Mitochondrial Membrane Potential in HepG2 Cells
Disruption of mitochondrial membrane potential (Δψm) is one of the earliest intracellular events to occur following the induction of apoptosis. The vital mitochondrial dye JC-1 is a useful tool for investigating mitochondrial function. This fluorescent dye accumulates rapidly and selectively within the mitochondria, depending on the membrane potential, and it undergoes a reversible change in fluorescence emission from red to green as Δψm decreases. Cells with high membrane potential promote the formation of dye aggregates (red fluorescence), and cells with low membrane potential contain monomeric JC-1 (green fluorescence). To investigate the loss of Δψm during the early phase of apoptosis induced by CFEZO, cells were stained with JC-1 and monitored with a fluorescent microscope. Untreated cells gave off a bright red fluorescence in control cells, indicating that JC-1 was accumulated in untreated control cells, and there was high membrane potential present (Fig. 4A). When cells were treated with CFEZO, the mitochondrial membrane potential (Δψm) began to decrease, and fluorescence shifted from red to green, indicating disruption of mitochondrial function. The ratio of red fluorescence intensity to green fluorescence intensity was used to quantitatively analyze the disruption of Δψm (Fig. 4B).
Figure 4.

CFEZO induces loss of mitochondrial membrane potential in HepG2 cells. (A) The HepG2 cells were treated with the indicated concentrations of CFEZO for 24 h. Then cells were stained with a mitochondria-specific dye, JC-1, and its fluorescence was monitored using fluorescence microscopy. (B) Mitochondrial membrane potential state in treated cells measured by spectrofluorometry. The results are presented as the mean ± SD of three independent experiments. Statistical significance in mitochondrial potential of ***p < 0.001 compared to that of control.
CFEZO Induces Activities of Caspases 3 and 9 in HepG2 Cells
Apoptosis is known to be generally associated with the sequential activation of caspases. To identify whether the caspases were involved in the mechanism of apoptosis, herein the activities of caspases 3/7, 9, and 8 were investigated in CFEZO-treated cells using Caspase-Glo® assays. After treatment with CFEZO (75, 100, and 125 μg/ml) for 24 h, there were obvious dose-dependent increases in activities of caspases 3 and 9 (Fig. 5). At a concentration of 125 μg/ml, the activities of caspases 3 and 9 increased 9- and 5.2-fold, respectively. On the other hand, CFEZO insignificantly altered the activity of caspase 8. These results indicate that CFEZO induced apoptosis in HepG2 cells via the intrinsic (mitochondrial) pathway.
Figure 5.

CFEZO induces activities of caspases 3 and 9 in HepG2 cells. HepG2 cells were seeded on a 96-well luminometer plate (104 cells/well) and treated with indicated concentrations of CFEZO for 24 h. Then activities of caspases 3/7, 9, and 8 were evaluated as detailed in the Materials and Methods section. Values present the mean of triplicates ± SD, and each experiment was performed three times. Statistical significance in caspase activities of *p < 0.05, **p < 0.01, and ***p < 0.001 compared to that of control.
CFEZO Induces DNA Damage and Oligonucleosomal Degradation
A distinct biochemical hallmark of apoptosis is the fragmentation of genomic DNA34. To elucidate whether CFEZO decreased cell survival by induction of DNA fragmentation, genomic DNA was isolated from HepG2 cells following exposure to 50, 75, 100, and 125 μg/ml of CFEZO, and DNA integrity was monitored by agarose gel electrophoresis of DNA. Intact genomic DNA was found in control cells (Fig. 6A). DNA fragmentation became apparent at 50 μg/ml of CFEZO treatment for 24 h, and these DNA fragmentation responses were dose dependent. When cells were treated with 50 μg/ml of CFEZO, DNA ladders were just visible as early as 6 h in HepG2 cells after treatment, and gradually increasing DNA fragmentation was observed from 6 to 24 h (Fig. 6B). The efficacious induction of apoptosis was observed at 50 μg/ml for 24 h, suggesting that cell death induced by CFEZO treatment was mainly caused by apoptosis.
Figure 6.

CFEZO induces DNA damage and oligonucleosomal degradation. HepG2 cells were treated with the indicated concentration of CFEZO for 24 h (A) or with 50 μg/ml for the shown time intervals (B) before being harvested, assayed, and electrophoresed through agarose gels and showed that CFEZO treatments induced oligonucleosomal degradation of the genomic DNA. Lane M indicates the DNA marker ladder. (C) Comet assay showing DNA damage in HepG2 cells treated with CFEZO. Magnification: 10× (top), 100× (bottom). Notice that the treated cells (bottom) show a clear appearance of a fan-like comet formation, which is a typical characteristic of apoptotic phenomenon.
To further confirm that CFEZO treatment induces DNA degradation, we carried out a comet assay. This assay is a sensitive method for monitoring single-strand DNA breaks at the single-cell level and is used as a biomarker of apoptosis. Untreated HepG2 control cells had no detectable comet tails or had shorter comet tails, whereas cells treated with CFEZO exhibited significant comet tail formations in a dose-dependent manner (Fig. 6C). This result suggests that CFEZO treatment markedly induced DNA damage in HepG2 cells.
CFEZO Modulates Expression of Apoptosis- and Cell Cycle-Regulating Proteins
To further clarify the involvement of mitochondria-mediated apoptosis induced by CFEZO in HepG2 cells, qRT-PCR analysis was used to measure the mRNA expression of Bcl-2 and Bax. A significant increase in the mRNA expression level of Bax was observed in 75, 100, and 125 μg/ml CFEZO-treated HepG2 cells when compared to control HepG2 cells. On the other hand, a significant decrease in the mRNA expression level of Bcl-2 was observed when compared to HepG2 control cells (Fig. 7A).
Figure 7.

CFEZO modulates the expression of the apoptosis- and cell cycle-regulating proteins. The HepG2 cells were incubated with the indicated concentrations of the CFEZO for 24 h, and then total RNA and protein lysates were prepared and subjected to qRT-PCR and Western analyses. (A) CFEZO treatment upregulates the expression of the Bax mRNA (A) and protein (B) and downregulates the expression of the Bcl-2 mRNA (A) and protein (B). The histograms show Bax/Bcl-2 ratios; the relative amount of Bax/Bcl-2 mRNAs (A) or proteins (B) was normalized to the amount of GAPDH (A) or β-actin (B). (C) CFEZO treatments modulate expression levels of the displayed cell cycle-regulating proteins. The experiments were repeated several times, and typical results from independent experiments are shown. The histograms represent fold change (increase/decrease) in each protein normalized to corresponding control treatment (arbitrarily set 1) in each individual experiment. Statistical significance in mRNA (A) and protein (B, C) levels of *p < 0.05, **p < 0.01, and ***p < 0.001 compared to that of control. (D) CFEZO treatment induced G2/M cell cycle arrest in HepG2 cells. Cells were treated with the indicated concentrations of CFEZO for 24 h, harvested, stained with annexin V and propidium iodide dye, and analyzed by flow cytometry for cell cycle distribution. Representative cell cycle profiles (top) and quantitation of percentages of the cells (bottom) in each phase of the cell cycle are shown. The results are presented as the mean ± SD of three independent experiments. Statistical significance in cell number of *p < 0.05 and **p < 0.01 compared to that of control.
To confirm these findings, the expression level of the Bax and Bcl-2 proteins was measured by Western blot analysis. CFEZO treatment resulted in the downregulation of Bcl-2 and the upregulation of Bax in a dose-dependent manner, leading to an increase in the Bax/Bcl-2 ratio (Fig. 7B). These results indicate that CFEZO induced apoptosis through increasing the Bax/Bcl-2 ratio.
Deregulation of cell cycle progression is a hallmark of cancer6. In response to DNA damage, the p53 protein is activated and induces cell cycle arrest through its downstream effector p21. Meanwhile, cyclin D1 functions as a regulator of CDK4/6 and is required for cell cycle G1/S transition35. To search for the indication of mechanisms involved in the cell cycle, we decided to investigate whether CFEZO could affect the expression of these proteins. Cells were incubated with the increasing concentrations of CFEZO for 24 h. Total protein lysates were then prepared and subjected to Western analysis. Treatment with CFEZO markedly increased the level of p53 and p21 proteins, which was accompanied by a decrease in the expression of cyclin D1 and CDK4 (Fig. 7C). Thus, these findings suggest that CFEZO may inhibit cell cycle progression.
To analyze whether CFEZO treatments mediate alterations in cell cycle distribution, we analyzed the percentage of HepG2 cells in the different stages of cell cycle. Exposure of HepG2 cells to CFEZO resulted in an accumulation of cells in the G2/M phase, which was associated with a reduction of the G1/S cell population with a modest decrease in the number of cells in the S phase (Fig. 7D). These results suggest that CFEZO could inhibit cell cycle progression, which might be part of the CFEZO-mediated anticancer effect on HepG2 cells.
DISCUSSION
Despite the efforts of innumerable researchers worldwide to ameliorate the dismal outcomes of HCC, an effective systemic therapy for this disease is still lacking. There is growing body of scientific evidence indicating that flavonoids play a beneficial role in the prevention of many cancer phenotypes including leukemia, melanoma, colon, breast, lung, and prostate27. In addition, we previously reported that crude flavonoid extract used in this study exerted an antiproliferative potential against glioblastoma and colon cancer cell lines30,31. In this study, we demonstrated that CFEZO inhibited the proliferation of HCC cells and induced apoptosis. The MTT assay proved that the cytotoxic effect of CFEZO against HepG2 cells exhibited dose and time dependency, with an IC50 equal to 115, 65, and 40 μg/ml for 24, 48, and 72 h, respectively. Coherent to its antiproliferative and cytotoxic effects, CFEZO efficiently ablated the potentiality of HepG2 cells to form colonies as confirmed by the anchorage-dependent colony formation assay. Since this assay measures the ability of tumor cells to grow and form foci in a manner unrestricted by growth contact inhibition, as is characteristically found in untransformed cells, this assay is further evidence demonstrating the anticancer potential of CFEZO. Furthermore, CFEZO suppressed growth of human breast and cervical carcinoma cell lines, MCF-7 and HeLa, respectively, indicating that the cytotoxic effect of CFEZO extends to a wide range of cancer cell lines.
Apoptosis plays a central role in the etiology, pathogenesis, and therapy of a variety of human malignancies, including HCC, where evasion of apoptotic cell death is one of the initial changes in a cell that leads to malignant transformation6. As a result, cumulative studies indicate that current chemotherapeutic agents, including γ-irradiation, immunotherapy, and suicide gene therapy, mostly exert their anticancer potentials with the induction of apoptosis36. Consistent with these studies, CFEZO inhibited growth of HepG2 cells through induction of apoptotic events, which was confirmed by characteristic morphological changes, DNA degradation, and ladder formation, as well as increase in caspase activity. The CFEZO-treated cells exhibited typical morphological features of cells committing apoptosis; they appeared to be losing viability, detached, rounded, balloon-like in shape, and shrunken with an irregularity in cellular shape. Conversely, untreated HepG2 cells assumed typical epithelial morphology when attaching to the substrate. The morphological changes of the CFEZO-treated cells were also perceived through fluorescence microscopy after staining cells with double fluorescent stains AO/EtBr. Staining of apoptotic cells with AO/EtBr is considered to be the correct method for distinguishing between viable and nonviable cells, based on membrane integrity37. In this assay, AO, but not EtBr, can cross the plasma membrane of vital cells and stain the nuclei green. When the cell is viable and has an intact cell membrane, the AO enters the cell and intercalates into the DNA, giving the cell a green appearance. Conversely, when the cell is nonviable and losing membrane integrity (late apoptotic/necrotic), EtBr also intercalates into the DNA, making the cell appear orange, since EtBr overwhelms AO staining. Our results (depicted in Fig. 2C) indicate that control cells displayed bright green nuclei. On the other hand, cells treated with the lowest doses of CFEZO (50 and 75 μg/ml) exhibited orange nuclei, indicating the emergence of early apoptotic signs. At the highest dose of CFEZO (125 μg/ml), an increase in the number of cells with red-colored nuclei predominated, indicating late apoptotic/necrotic death. Thus, the morphological analysis of AO/EtBr-stained HepG2 cells indicated the incidence of apoptotic events.
There is considerable evidence demonstrating that most anticancer agents either directly induce DNA damage or indirectly induce secondary stress-responsive (e.g., ROS production) signaling pathways to trigger apoptosis by activation of the mitochondrial apoptotic pathway36. Based on this notion, we investigated the effect of CFEZO on the mitochondria and DNA. To this end, we evaluated the levels of intracellular ROS and GSH after CFEZO treatments. Both ROS and GSH are tightly balanced inside the cell. When this balance is disrupted by excessive ROS production and/or GSH depletion, oxidative stress may occur. This stress might initiate the early stages of apoptosis14. In fact, many chemotherapeutics such as ionizing radiation, etoposide, arsenates, and flavonoids rely on their ability to stimulate ROS production, which alters cellular redox balance leading to oxidative stress and subsequent induction of apoptosis in cancer cells33,38. The findings herein display that, after CFEZO treatment, HepG2 cell death was accompanied by a consistent and monotonic increase in the ROS level, indicating that CFEZO has a potential to increase levels of intracellular ROS to a critical threshold, leading to oxidative stress and eventual apoptotic events. CFEZO treatment has also dose-dependently depleted levels of GSH. This deserves attention because intracellular GSH content has a decisive impact on anticancer drug-induced apoptosis, where increased levels of cellular GSH promote the survival of tumor cells and impede apoptotic cell death induced by chemotherapeutics39,40. Accordingly, one approach to minimize drug resistance and maximize therapeutic response is through the depletion of GSH levels in tumor cells21. Therefore, depletion of GSH by CFEZO could be exploited to increase the response of HCC cells to current therapeutics. This is an important finding since HCC cells express high intracellular levels of GSH, which facilitate the growth of HCC cells41 and confer resistance to current therapies42. In view of the fact that cells can undergo apoptosis in response to GSH depletion, CFEZO treatment might trigger events leading to an accumulation of intracellular free radicals and initiation of apoptotic cascades, which may contribute, at least in part, to the reduction of HepG2 cell viability.
It is generally accepted that the molecular mechanism underlying apoptosis induction by excessive production of ROS and/or depletion of GSH is the oxidative damage to the mitochondrial membrane14. Disruption of the mitochondrial membrane leads to enhancement of proapoptotic Bax over Bcl-2 proteins, which enhances the permeability of the mitochondrial membrane and, consequently, the release of potentially apoptogenic factors. These factors cascade events, culminating at the activation of caspases 9 and 8 and, ultimately, activation of caspase 37. The latter triggers downstream proteases and nucleases that execute degradation of cell contents and the appearance of apoptotic features12. In this study, the results of JC-1 staining of treated cells showed the change from red to green fluorescence after CFEZO treatment, indicating that CFEZO has a potential to disrupt mitochondrial membrane integrity. Consistent with mitochondrial disruption, CFEZO treatment enhanced the activities of caspases 3 and 9, indicating that CFEZO engages in the intrinsic (mitochondrial) pathway. On the other hand, CFEZO treatment did not alter the activity of caspase 8, suggesting the disengagement of the extrinsic pathway in the apoptosis caused by CFEZO. Other seminal findings in the current study involve the upregulation of mRNA and protein expression levels of Bax and the downregulation of Bcl-2 expression by CFEZO. Since fine-tuning of the balance between the Bax and Bcl-2 factors within apoptotic pathways in a cell leads to apoptosis or survival, CFEZO treatments tipped the balance in favor of the apoptotic trend. Increase in the Bax/Bcl-2 ratio by CFEZO also deserves attention since Bax has been found to be downregulated in HCC43. Conversely, overexpression of Bcl-2 is frequently observed in several cancer phenotypes and is often associated with an unfavorable outcome44,45. Impairment of the Bcl-2 gene expression is a hallmark of cancer and can promote resistance to different drug-induced activations of the mitochondria apoptotic pathway46. As a result, the downregulation of Bcl-2 expression has been found to enhance apoptotic response to anticancer drugs47. Therefore, modulation of Bcl-2 and Bax expressions by CFEZO would offer a practical approach for sensitizing HCC cells to chemotherapeutic-induced HCC apoptosis. Collectively, these findings indicate that CFEZO increased the ROS level, depleted the GSH level, reduced the mitochondrial membrane potential, and increased the Bax/Bcl-2 ratio, which provoked apoptotic cascades in HepG2 cells via the mitochondria pathway.
Fragmentation of DNA at the internucleosomal linker regions is an irreversible event in apoptosis and has been observed in cells undergoing apoptosis induced by a variety of agents34. It usually takes place at the end of apoptosis or at the apoptosis/necrosis stage and, due to its characteristic patterns revealed by agarose gel electrophoresis, is widely used as a distinctive marker in the apoptosis process34. In this study, we observed a ladder-like appearance of DNA in the HepG2 cells treated with CFEZO, and the effect was dose and time dependent. Another assay demonstrates that CFEZO-induced DNA damage in HepG2 cells stems from a comet assay. It has been found that the comet assay is more sensitive than the DNA ladder assay in detecting DNA damage and distinguishes apoptosis from necrosis, which makes it a reliable assay for the detection of apoptotic cell death48. The results of the comet assay show that treatment of HepG2 cells with CFEZO resulted in the formation of a comet tail and that its movement increased in a dose-dependent manner. In addition, at the highest dose, the comet showed a typical apoptotic head (stained clusters of apoptotic bodies), and the comet tail showed a clear appearance of a fan-like comet shape, which are typical characteristics of apoptotic phenomenon detected by this assay49. These findings add further proof that CFEZO treatment mediates DNA damage in HepG2 cells. Multiple lines of evidence indicate that, when damaged DNA cannot be repaired, cells are likely to proceed to apoptosis50. Therefore, the damage to genomic DNA in HepG2 cells caused by the CFEZO treatment mostly triggered events leading to the initiation of an apoptotic cascade, which may contribute, at least in part, to the diminution of cell viability in HepG2 cells.
One of the seminal findings in this study is the increasing expression level of the tumor-suppressor protein p53 by CFEZO, indicating a pivotal role of p53 in the CFEZO-triggering apoptosis. Many chemopreventive agents are known to exert their anticancer effects through the induction of apoptosis via p53-dependent mechanisms51. p53 is the guardian of the genome; in response to cellular stress leading to DNA damage, the p53 pathway is activated to maintain the integrity of the genome52. When DNA damage cannot be repaired, p53 provokes signaling cascades, leading to apoptosis and the elimination of mutated or DNA-damaged cells52. If p53 signaling does not respond to DNA damage, cells with DNA damage can escape apoptosis and turn into cancer cells53. However, 50% of human cancers overall carry inactive p53 protein due to a mutation in the p53 gene (TP53). Furthermore, many human cancers retain a wild-type p53, but impairment in p53 signaling or misregulation (e.g., overexpression of p53− regulators) inactivates its function, which facilitates cancer progression by protecting cancer cells from p53-dependent responses54. Related to this notion, inactivating mutations or impaired signaling of p53 is most frequently found in HCC55 and was significantly associated with poor patient outcomes56. Therefore, the restoration of p53 function in tumors could be an attractive approach for treating this cancer. In this regard, the results showing CFEZO-upregulated expression of p53 could be a practical approach to amend the misregulated activity of p53 in HCC. It is widely accepted that p53 exerts its apoptotic function through transcription-dependent and -independent activities57. In its transcription-independent apoptotic activity, p53 physically interacts with the antiapoptotic Bcl-2 family of proteins, Bcl-XL and Bcl-2, inhibiting their antiapoptotic function58. In its transcription-dependent apoptotic activity, p53 activates the expression of a myriad of genes encoding proapoptotic proteins (such as Bax, Puma, Noxa, Fas, and others) or cell cycle arrest proteins, mainly p21, in addition to other factors57. p21 is considered to be one of the most important and potent effectors induced by p53 in response to DNA damage52, where its induction potently inhibits proliferation in mammalian cells. Consistent with this, overexpression of p21 inhibits HCC growth59, and the expression level of p21 serves as an independent and good survival factor for HCC, where patients with a positive expression of p21 in HCC had a longer disease-specific survival after resection60. At the molecular level, p21 inhibits cell cycle progression by association with cyclin/CDK complexes, inhibiting their functional role35. It can inhibit the activity of all cyclin/CDK complexes, indicating that it is a universal cyclin/CDK inhibitor61. Importantly, it is a potent inhibitor of cyclin D1/CDK4 complexes, which are well known to be overexpressed in many human cancers, including HCC62. In relation to this, an inhibition of cyclin D1 levels has been shown to induce apoptosis in HCC59,63. These studies prompted us to study the expression profiles of cyclin D1, CDK4, and p21 in the present study. We found that CFEZO downregulated the expression levels of cyclin D1 and CDK4 and, conversely, upregulated the expression of p21, suggesting that CFEZO targets cell cycle progression. To determine the antimitogenic effect of CFEZO, we analyzed the percentage of HepG2 cells in the different stages of cell cycle following CFEZO treatment. We found that CFEZO blocked cells in the G2/M phase, indicating that cell cycle arrest might be part of the CFEZO-mediated inhibition of HepG2 cellular proliferation. The G2/M checkpoint prevents the entry of DNA-damaged cells from entering mitosis and allows repairing of the DNA that was damaged in late S or G2 phases. It is also a potential target for anticancer drugs in chemotherapy. However, the mechanism by which CFEZO-induced downregulation of cyclin D1 and CDK4 expression contributes to the cell cycle G2/M arrest in HepG2 cells is unknown at present. This is because the G2/M phase transition is driven by cyclin B–Cdc235. Nonetheless, accumulating evidence indicates that both cyclin D1 and CDK4 are highly expressed in the G2 phase, and their high expression is essential for continuing cell cycle progression through the next G1 phase64. Consistent with these, mounting evidence indicates that there is an association between the downregulation of cyclin D1 and G2/M arrest. For example, a derivative of 6-mercaptopurine, 6-[(1-naphthylmethyl) sulfanyl]-9H-purine, has been found to induce the G2/M phase via downregulation of cyclin D1 and CDK4 in HepG2 cells65. Similarly, natural polyketides, called annonaceous acetogenins, have been found to induce G2/M arrest in human HCC BEL-7402/5-FU and HepG2/ADM cell lines through downregulation of cyclin D166. More recently, 24-acetylisodahurinol-3-O-β-D-xylopyranoside, a cycloartane triterpenoid isolated from Cimicifuga foetida, significantly induced the G2/M phase arrest in breast cancer cells, SW527, through the downregulation of cyclin D167. Maple polyphenols, ginnalins A–C, also induced the G2/M arrest in MCF-7 breast cancer cells via downregulation of the cyclin D1 protein level68. Likewise, CDK4 plays an important role outside of the G1/S phase transition, where its activity is necessary for normal cell cycle progression through the G2 phase into mitosis and the fidelity of mitosis69. Consistent with this, 5-hydroxy-6,7,8,40-tetramethoxyflavone, a hydroxylated polymethoxyflavone mainly found in citrus plants, has been found to induce G2/M phase arrest through downregulation of CDK4 levels70. Therefore, the results herein showing CFEZO induced G2/M arrest through downregulation of cyclin D1 and CDK4, along with increasing expression of p21, agree with the previous studies. However, we cannot rule out the fact that other cell cycle-related proteins, probably responsible for CFEZO-induced G2/M phase arrest, may also be involved. Further investigation is needed in order to study the effect of CFEZO on G2/M regulatory proteins.
In conclusion, the present study demonstrated that CFEZO could inhibit proliferation of the HCC cell line, HepG2, and the effect was in a time- and dose-dependent manner. It induced apoptosis of HepG2 cells through a mitochondria-mediated apoptosis pathway involving production of ROS, depletion of GSH, and an increase in the ratio of Bax/Bcl-2. Other molecular mechanisms of CFEZO entailed in the activation of caspases 9 and 3 are upregulation of p53 and p21 proteins and downregulation of cyclin D1 and CDK4, leading to blocking at the G2/M stage. Therefore, results from this study provide critically important experimental facts to suggest that CFEZO may be a potential therapeutic agent for treating HCC.
ACKNOWLEDGMENTS
This project was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, Saudi Arabia, under Grant No. 1433/130/26. The authors thank DSR for the technical and financial support.
Footnotes
The authors declare no conflicts of interest.
REFERENCES
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. [DOI] [PubMed] [Google Scholar]
- 2. El-Serag HB, Davila JA, Petersen NJ, Mc-Glynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: An update. Ann Intern Med. 2003;139:817–23. [DOI] [PubMed] [Google Scholar]
- 3. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072–9. [DOI] [PubMed] [Google Scholar]
- 4. Peck-Radosavljevic M. Drug therapy for advanced-stage liver cancer. Liver Cancer 2014;3:125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jelic S, Sotiropoulos GC. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v59–v64. [DOI] [PubMed] [Google Scholar]
- 6. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 7. Wong RS. Apoptosis in cancer: From pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. [DOI] [PubMed] [Google Scholar]
- 9. Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis 2007;12:815–33. [DOI] [PubMed] [Google Scholar]
- 10. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004;305:626–9. [DOI] [PubMed] [Google Scholar]
- 11. Reed JC. Mechanisms of apoptosis. Am J Pathol. 2000;157:1415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cruchten SV, Den Broeck WV. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol. 2002;31:214–23. [DOI] [PubMed] [Google Scholar]
- 13. Fruehauf JP, Meyskens FL Jr. Reactive oxygen species: A breath of life or death? Clin Cancer Res. 2007;13:789–94. [DOI] [PubMed] [Google Scholar]
- 14. Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis 2007;12:913–22. [DOI] [PubMed] [Google Scholar]
- 15. Glasauer A, Chandel NS. Targeting antioxidants for cancer therapy. Biochem Pharmacol. 2014;92:90–101. [DOI] [PubMed] [Google Scholar]
- 16. Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. [DOI] [PubMed] [Google Scholar]
- 17. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003;101:4098–104. [DOI] [PubMed] [Google Scholar]
- 18. Franco R, Cidlowski JA. Apoptosis and glutathione: Beyond an antioxidant. Cell Death Differ. 2009;16:1303–14. [DOI] [PubMed] [Google Scholar]
- 19. Lushchak VI. Glutathione homeostasis and functions: Potential targets for medical interventions. J Amino Acids 2012;2012:736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, Marinari UM, Domenicotti C. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2012;2013:972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Backos DS, Franklin CC, Reigan P. The role of glutathione in brain tumor drug resistance. Biochem Pharmacol. 2012;83:1005–12. [DOI] [PubMed] [Google Scholar]
- 22. Liu FL, Hsu JL, Lee YJ, Dong YS, Kung FL, Chen CS, Guh JH. Calanquinone A induces anti-glioblastoma activity through glutathione-involved DNA damage and AMPK activation. Eur J Pharmacol. 2014;730:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baliga MS, Haniadka R, Pereira MM, D’Souza JJ, Pallaty PL, Bhat HP, Popuri S. Update on the chemopreventive effects of ginger and its phytochemicals. Crit Rev Food Sci Nutr. 2011;51:499–523. [DOI] [PubMed] [Google Scholar]
- 24. Park EJ, Pizzuto JM. Botanicals in cancer chemoprevention. Cancer Metastasis Rev. 2002;21:231–55. [DOI] [PubMed] [Google Scholar]
- 25. Ghasemzadeh A, Jaafar HZ, Rahmat A. Identification and concentration of some flavonoid components in Malaysian young ginger (Zingiber officinale Roscoe) varieties by a high performance liquid chromatography method. Molecules 2010;15:6231–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Han JS, Lee S, Kim HY, Lee CH. MS-based metabolite profiling of aboveground and root components of Zingiber mioga and officinale. Molecules 2015;20:16170–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chahar MK, Sharma N, Dobhal MP, Joshi YC. Flavonoids: A versatile source of anticancer drugs. Pharmacogn Rev. 2011;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kozłowska A, Szostak-Węgierek D. Flavonoids: Food sources and health benefits. Rocz Panstw Zakl Hig. 2014;65:79–85. [PubMed] [Google Scholar]
- 29. Kale A, Gawande S, Kotwal S. Cancer phytotherapeutics: Role for flavonoids at the cellular level. Phytother Res. 2008;22:567–77. [DOI] [PubMed] [Google Scholar]
- 30. Elkady AI, Hussein RA, Abu-Zinadah AO. Effects of crude extracts from medicinal herbs Rhazya stricta and Zingiber officinale on growth and proliferation of human brain cancer cell line in vitro. Biomed Res Int. 2014;2014:260210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Elkady AI, Hussein RA, Abu-Zinadah AO. Differential control of growth, apoptotic activity and gene expression in human colon cancer cells by extracts derived from medicinal herbs, Rhazya stricta and Zingiber officinale and their combination. World J Gastroenterol. 2014;20:15275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elkady AI, Hussein RA, El-Assouli SM. Mechanism of action of Nigella sativa on human colon cancer cells: The suppression of AP-1 and NF-κB transcription factors and the induction of cytoprotective genes. Asian Pac J Cancer Prev. 2015;16:7943–57. [DOI] [PubMed] [Google Scholar]
- 33. Antosiewicz J, Ziolkowski W, Kar S, Powolny AA, Singh SV. Role of reactive oxygen intermediates in cellular responses to dietary cancer chemopreventive agents. Planta Med. 2008;74:1570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–8. [DOI] [PubMed] [Google Scholar]
- 35. Vermeulen K, Van Bockstaele DR, Berneman ZN. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 2003;36:131–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khan N, Afaq F, Mukhtar H. Apoptosis by dietary factors: The suicide solution for delaying cancer growth. Carcinogenesis 2007;28:233–9. [DOI] [PubMed] [Google Scholar]
- 37. Archana M, Bastian, Yogesh TL, Kumaraswamy KL. Various methods available for detection of apoptotic cells—A review. Indian J Cancer 2013;50:274–83. [DOI] [PubMed] [Google Scholar]
- 38. Oh SH, Lim SC. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetyl cysteine mediated catalase upregulation. Toxicol Appl Pharmacol. 2006;212:212–23. [DOI] [PubMed] [Google Scholar]
- 39. Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22:343–52. [DOI] [PubMed] [Google Scholar]
- 40. Park WH, Kim SH. MAPK inhibitors augment gallic acid-induced A549 lung cancer cell death through the enhancement of glutathione depletion. Oncol Rep. 2013;30:513–9. [DOI] [PubMed] [Google Scholar]
- 41. Huang ZZ, Chen C, Zeng Z, Yang H, Oh J, Chen L, Lu SC. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001;15:19–21. [DOI] [PubMed] [Google Scholar]
- 42. Ye CG, Yeung JH, Huang GL, Cui P, Wang J, Zou Y, Zhang XN, He ZW, Cho CH. Increased glutathione and mitogen-activated protein kinase phosphorylation are involved in the induction of doxorubicin resistance in hepatocellular carcinoma cells. Hepatol Res. 2013;43:289–99. [DOI] [PubMed] [Google Scholar]
- 43. Lachenmayer A, Alsinet C, Chang CY, Llovet JM. Molecular approaches to treatment of hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walensky L. BCL-2 in the crosshairs: Tipping the balance of life and death. Cell Death Differ. 2006;13:1339–50. [DOI] [PubMed] [Google Scholar]
- 45. Sasi N, Hwang M, Jaboin J, Csiki I, Lu B. Regulated cell death pathways: New twists in modulation of BCL2 family function. Mol Cancer Ther. 2009;8:1421–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adams J, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 2007;26:1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Losert D, Pratscher B, Soutschek J, Geick A, Vornlocher HP, Muller M, Wacheck V. Bcl-2 downregulation sensitizes nonsmall cell lung cancer cells to cisplatin, but not to docetaxel. Anticancer Drugs 2007;18:755–61. [DOI] [PubMed] [Google Scholar]
- 48. Yasuhara S, Zhu Y, Matsui T, Tipirneni N, Yasuhara Y, Kaneki M, Rosenzweig A, Martyn JA. Comparison of comet assay, electron microscopy, and flow cytometry for detection of apoptosis. J Histochem Cytochem. 2003;51:873–85. [DOI] [PubMed] [Google Scholar]
- 49. Chakraborty S, Kundu T, Dey S, Bhattacharya KR, Siddiqi M, Roy M. Tea-induced apoptosis in human leukemia K562 cells as assessed by comet formation. Asian Pacific J Cancer Prev. 2006;7:201–7. [PubMed] [Google Scholar]
- 50. Zhou BB, Elledge SJ. The DNA damage response: Putting checkpoints in perspective. Nature 2000;408:433–9. [DOI] [PubMed] [Google Scholar]
- 51. Amin AR, Karpowicz PA, Carey TE, Arbiser J, Nahta R, Chen ZG, Dong JT, Kucuk O, Khan GN, Huang GS, Mi S, Lee HY, Reichrath J, Honoki K, Georgakilas AG, Amedei A, Amin A, Helferich B, Boosani CS, Ciriolo MR, Chen S, Mohammed SI, Azmi AS, Keith WN, Bhakta D, Halicka D, Niccolai E, Fujii H, Aquilano K, Ashraf SS, Nowsheen S, Yang X, Bilsland A, Shin DM. Evasion of anti-growth signaling: A key step in tumorigenesis and potential target for treatment and prophylaxis by natural compounds. Semin Cancer Biol. 2015;35(Suppl):S55–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Selivanova G. Wild type p53 reactivation: From lab bench to clinic. FEBS Lett. 2014;588:2628–38. [DOI] [PubMed] [Google Scholar]
- 53. Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell 2009;137:413–31. [DOI] [PubMed] [Google Scholar]
- 54. Sabapathy K. The contrived mutant p53 oncogene—Beyond loss of functions. Front Oncol. 2015;5:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Meng X, Franklin DA, Dong J, Zhang Y. MDM2–p53 pathway in hepatocellular carcinoma. Cancer Res. 2014;74:7161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kirstein MM, Vogel A. The pathogenesis of hepatocellular carcinoma. Dig Dis. 2014;32:545–53. [DOI] [PubMed] [Google Scholar]
- 57. Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene 2008;27:6507–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chi SW. Structural insights into the transcription-independent apoptotic pathway of p53. BMB Rep. 2014;47:167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cai X, Hu X, Cai B, Wang Q, Li Y, Tan X, Hu H, Chen X, Huang J, Cheng J, Jing X. Metformin suppresses hepatocellular carcinoma cell growth through induction of cell cycle G1/G0 phase arrest and p21CIP and p27KIP expression and downregulation of cyclin D1 in vitro and in vivo. Oncol Rep. 2013;30:2449–57. [DOI] [PubMed] [Google Scholar]
- 60. Kao JT, Chuah SK, Huang CC, Chen CL, Wang CC, Hung CH, Chen CH, Wang JH, Lu SN, Lee CM, Changchien CS, Hu TH. P21/WAF1 is an independent survival prognostic factor for patients with hepatocellular carcinoma after resection. Liver Int. 2007;27:772–81. [DOI] [PubMed] [Google Scholar]
- 61. Abukhdeir AM, Park BH. p21 and p27: Roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masaki T, Shiratori Y, Rengifo W, Igarashi K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H, Watanabe S, Omata M, Kuriyama S. Comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology 2003;37:534–43. [DOI] [PubMed] [Google Scholar]
- 63. Relja B, Meder F, Wilhelm K, Henrich D, Marzi I, Lehnert M. Simvastatin inhibits cell growth and induces apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int J Mol Med. 2010;26:735–41. [DOI] [PubMed] [Google Scholar]
- 64. Yang K, Hitomi M, Stacey DW. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006;1:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang XG, Bao YL, Huang YX, Sun LG, Zhang YW, Yu CL, Wu Y, Li YX. 6-[(1-naphthylmethyl)sulfanyl]-9H-purine induces G2/M phase arrest and apoptosis in human hepatocellular carcinoma HepG2 cells. Eur J Pharmacol. 2012;695:27–33. [DOI] [PubMed] [Google Scholar]
- 66. Qian JQ, Sun P, Pan ZY, Fang ZZ. Annonaceous acetogenins reverses drug resistance of human hepatocellular carcinoma BEL-7402/5-FU and HepG2/ADM cell lines. Int J Clin Exp Pathol. 2015;8:11934–44. [PMC free article] [PubMed] [Google Scholar]
- 67. Kong Y, Li F, Nian Y, Zhou Z, Yang R, Qiu MH, Chen C. KHF16 is a leading structure from Cimicifuga foetida that suppresses breast cancer partially by inhibiting the NF-kB signaling pathway. Theranostics 2016;6:875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. González-Sarrías A, Ma H, Edmonds ME, Seeram NP. Maple polyphenols, ginnalins A–C, induce S- and G2/M-cell cycle arrest in colon and breast cancer cells mediated by decreasing cyclins A and D1 levels. Food Chem. 2013;136:636–42. [DOI] [PubMed] [Google Scholar]
- 69. Burgess A, Wigan M, Giles N, Depinto W, Gillespie P, Stevens F, Gabrielli B. Inhibition of S/G2 phase CDK4 reduces mitotic fidelity. J Biol Chem. 2006;281:9987–95. [DOI] [PubMed] [Google Scholar]
- 70. Qiu P, Dong P, Guan H, Li S, Ho CT, Pan MH, McClements DJ, Xiao H. Inhibitory effects of 5-hydroxy polymethoxyflavones on colon cancer cells. Mol Nutr Food Res. 2010;Suppl 2:S244–52. [DOI] [PubMed] [Google Scholar]