

Abstract
Objectives:
To draw attention towards fructose-1,6-bisphosphatase (FBPase) deficiency as an important cause of hypoglycemia and lactic acidosis and to implement preventive strategies.
Methods:
This observational, cross-sectional study was conducted on 7 Saudi patients with genetically confirmed FBPase deficiency from 2008 to 2018 at Prince Sultan Military Medical City, Riyadh, Saudi Arabia.
Results:
Participants ranged in age from 1-10 years, and all presented with recurrent hypoglycemia. All but one had associated severe metabolic acidosis, and 3 patients (42.9%) presented with hypoglycemia and severe acidosis since birth. The mean duration from presentation to diagnosis was 39.4 months, as other diagnoses, like glycogen storage diseases and mitochondrial diseases needed to be ruled out. Development was normal apart from speech delay in one patient with a novel variant of the FBP1 gene. All patients have homozygous variants in the FBP1 gene.
Conclusion:
Fructose-1,6-bisphosphatase is an important cause of hypoglycemia and acidosis; therefore, it is important to offer early molecular diagnostics in any child presenting with these symptoms. Molecular diagnostics should always be undertaken to confirm the diagnosis and for further preventive strategies.
Keywords: Fructose-1,6-bisphosphatase; FBPase; FBP1 gene; fructose; hypoglycemia
Deficiency of hepatic fructose-1,6-bisphosphatase (FBPase) is an autosomal recessive disorder with impaired gluconeogenesis leading to hypoglycemia and lactic acidosis. It was first described in 1970.1
Fructose-1,6-bisphosphatase1 gene is composed of 8 exons located on chromosome 9q22.2-q22.3.2 Fructose-1,6-bisphosphatase enzyme is crucial for the conversion of FBP to fructose 6-phosphate and inorganic phosphate.3 Patients could develop seizures and coma.3 The metabolic decompensation usually occurs with fasting, infections, and stress. Fructose-1,6-bisphosphatase deficiency is also associated with impaired purine catabolism and this is the cause of hyperuricemia in these patients.4 Other derangements with metabolic decompensations include increased liver enzymes and increased urinary ketones.
With early diagnosis and strict management, the long-term prognosis for FBPase deficiency is excellent which highlights the importance of early molecular confirmation.5,6
The aim of this study was to draw the attention of pediatricians towards FBPase deficiency as an important cause of recurrent hypoglycemia and lactic acidosis and to emphasize the importance of molecular diagnostics to confirm the diagnosis.
Methods
This study was observational, cross-sectional, according to principles of Helsinki Declaration, carried out in Prince Sultan Military Medical City, Riyadh, Saudi Arabia, on October 2018 by reviewing medical records of 7 Saudi patients with genetically confirmed FBPase deficiency from 2008-2018.
All patients presenting with hypoglycemia or acidosis with confirmed molecular diagnosis of FBPase deficiency were recruited in the study. A total of 7 patients with genetically confirmed diagnosis were included. Those without molecular confirmation were excluded. Data were collected from medical records, including scheduled outpatient clinics and inpatient admission records. Ethical approval from our Institutional Review Board was obtained before starting this study.
Statistical analysis
The data were analyzed using Statistical Package for Social Science, version 20 (IBM Corp, Armonk, NY, USA). Qualitative data were presented in numbers and percentages. PubMed search was used to find prior related researches.
Results
Case 1
A female patient was a product of first-degree cousin marriage, presenting on the 1st day of life with respiratory distress, hypoglycemia, and acidosis. Sepsis was excluded. Systemic examination was unremarkable apart from laryngomalacia. She had normal development. Investigations revealed blood glucose of 0.8 mmol/l (3.3-8), lactate 12.6 mmol/l (0.5-2.2), PH 7.18 (7.35-7.45), carbonate (HCO3) 9.9 mmol/l (22-26), and high uric acid level 728 umol/l (110-390). Ammonia, creatine phosphokinase (CK), lipid profile, hepatic profile, Tandem mass spectrometry (MS), and urine organic acids were normal. At first the suspected diagnosis was glycogen storage disease (GSD) and mitochondrial disease (MD), which were excluded by molecular studies. Then the diagnosis was confirmed by whole exome sequence (WES) at the age of 41 months, homozygous for FBP1 gene: c.114-115 ins CTGCAC (p.L38delinsLCT).7
Case 2
The male patient is the brother of case 1 and he had the same presentation as his sister with normal systemic examination. His blood glucose 2.2 mmol/l, PH 7.23, HCO3 11 mmol/l, and lactate 14.4 mmol/l. Ammonia, CK, lipid profile, and Tandem MS were normal. Hepatic profile showed slightly increased gamma-glutamyl transferase level 88 U/L(7-32 U/L) other liver enzymes were normal. Urine organic acid showed highly elevated lactic acid, pyruvic acid, moderately elevated 3-hydroxybutyric acid, and 2-hydroxybutyric acid. Mitochondrial disease was excluded. The final diagnosis was confirmed at the age of 18 months by WES revealing same variant as his sister.
Case 3
The female patient was an outcome of consanguineous marriage, she presented on the first day of life with hypoglycemia and severe metabolic acidosis. System examination showed just palpable liver. Her development was normal. Her blood glucose 2.2 mmol/l, PH 7.09, sodium bicarbonate 5.4 mmol/l, lactate 13.4mmol/l, CK 377 U/L (50-170 U/L). Urate, lipid profile, liver enzymes, ammonia, and Tandem MS were all normal. Urine organic acids revealed increased lactic acid, 3 hydroxybutyric acid and 2 hydroxybutyric acid. Genetic study for GSD comprehensive panel by massively parallel sequencing at the age of 34 months revealed homozygous for c. 114-119dup (p. C39-T40dup) in the FBP1 gene, exon 1. Her elder sister aged 15 years also had recurrent attacks of hypoglycemia and acidosis starting at birth. The same variant was proved by targeted gene sequence.
Case 4
The female patient was also an outcome of consanguineous marriage, referred to our hospital from Al Madina Almunnawarah, Saudi Arabia with recurrent attacks of hypoglycemia and lactic acidosis starting at one year. Her development was normal. Systemic examination was unremarkable apart from palpable liver. Her blood glucose was 1.3 mmol/l, lactate 7.8 mmol/l, and urate 689 umol/l (143-339 umol/l). Lipid profile, hepatic profile, Tandem MS, and urine organic acids were normal. Whole exome sequence at 33 months confirmed a homozygous variant in the FBP1 gene c. 841G›T (p.Glu281*) leading to premature stop codon.7 The period between presentation until diagnosis was 21 months.
Case 5
The patient was an outcome of consanguineous marriage, presented with recurrent attacks of hypoglycemia and lactic acidosis since the age of one year. His systemic examination was unremarkable with normal development. His glucose 1mmol/l, lactate 10.8 mmol/l. Urate, hepatic profile, lipid profile, and Tandem MS were normal. Urine organic acids once showed raised lactic acid, 2 hydroxybutyric and 3 hydroxybutyric acid. Fructose-1,6-bisphosphatase1 comprehensive gene sequencing at the age of 9 years and 5 months revealed homozygous mutation c. 114-119dup (p.C39-T40dup), exon 1.
Case 6
The male patient was referred from Jeddah, Saudi Arabia as a case of recurrent hypoglycemia possibly GSD for further work up. He was an outcome of consanguineous parents. Hypoglycemic episodes started at the age of 5 months. His systemic examination showed palpable liver. He had delayed speech and otherwise normal development. His CK, lactate, hepatic profile, lipid profile, Tandem MS, and urine organic acid were normal. Diagnosis was confirmed at the age of 3 years and 7 months when GSD panel was carried out (BCM-Mitome NGS) and the patient proved to be homozygous for c. 334-2 A›T in the FBP1 gene, novel variant.
Case 7
The male patient was referred from Hafr Albatin, Saudi Arabia for recurrent attacks of hypoglycemia and acidosis since the age of 2 years. His parents are first cousins and had one sibling who died at the age of one and a half year with the same symptoms. His glucose 2.3 mmol/l, lactate 5.2 mmol/l. Hepatic profile, lipid profile, Tandem MS, and ammonia were normal. Urine for organic acids showed high lactic acid, 2-hydroxybutyric acid and 3-hydroxybutyric acid. Fructose-1,6-bisphosphatase1 gene sequencing revealed homozygous variant (c.959dupG) which is predicted to result in translational frame shift and premature protein termination (p.Ser321Lefs*13). This variant has been reported in both the homozygous and compound heterozygous state in several FBPase deficiency patients.8-10 This variant has alternately been described in the literature using designations such as G857GGGG981 and InsG 960/961.
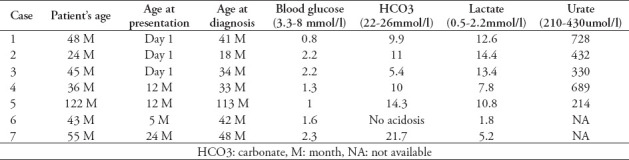
Table 1 summarizes the presentation and laboratory results of the patients with the normal reference values.
Table 1.
Summary of clinical and laboratory findings in the patients.
Discussion
Although several reports in the literature about FBPase deficiency, including reports from Kingdom of Saudi Arabia,1-4,11 this diagnosis still not entertained by the pediatricians, which makes a delay in the management of patients. In this study although number of patients is limited, the delineation of genomic variants has helped in reaching the diagnosis and in future preventive strategies including pre-implantation genetic diagnosis for the families and carrier screening for other family members.
This study was positive familial history for the condition in 5 of our patients (71.4%), which could be explained by the high rate of consanguinity among Saudi population.
Three of the patients (42.9%) presented at birth with respiratory distress, severe acidosis, and hypoglycemia. In a study of 4 patients with FBPase deficiency carried out by Niu Li et al,3 2 patients (50%) presented on day 1 and 3 with symptoms.
The mean duration from the patients’ presentation until diagnosis was 39.4 months. The earliest age at diagnosis was 1.5 years, whereas the latest age of diagnosis was 9 years and 5 months (Table 1). Diagnosis was made late due to the limited molecular testing facilities available in our institute 8 years back and because MD or GSD were first suspected in other institutions. Once negative, they were referred to us, with FBPase deficiency having remained unsuspected as a possible cause in the other institutions.
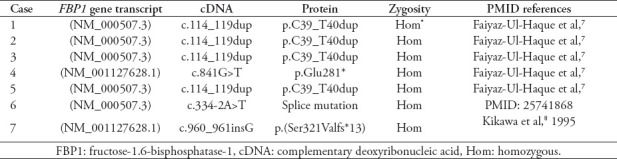
In all patients, the diagnosis was confirmed by molecular testing, as shown in Table 2. Homozygous mutation in FBP1 gene with duplication of 2 amino acids (c.114-115 ins CTGCAC) was documented in 4 patients, namely, case 1, 2, 3, and 5. This mutation was reported before by Faiyaz-Ul-Haque et al.7 One of our patients had a novel mutation of FBP1 gene (c. 334-2 A›T). The mutation site was in intron 3; this variant change the invariant splice site and is thus categorized as likely pathogenic.12 This novel variant needs further molecular and functional studies.
Table 2.
The genotype of the 7 patients with fructose-1,6-bisphosphatase deficiency.
All patients were managed on fructose, sucrose free diet with added cornstarch and frequent feeding, with glucose monitoring at home with excellent prognosis. Families offered PGD in order to prevent disease recurrence. Pinto et al,11 studied the international practices in the dietary management of patients with FBPase deficiency and concluded that in emergencies all agreed upon restriction of sucrose and fructose, but the use of cornstarch varied widely especially in older patients where it may not be necessary.
In conclusion, fructose-1,6-bisphosphatase deficiency is an overlooked cause of hypoglycemia and acidosis in our community, since GSD and MD were initially suspected, which led to a delay in the diagnosis averaging approximately 3 years in duration in this study. It would be important to consider this diagnosis early for any child with recurrent hypoglycemia and acidosis, and to increase the awareness of general pediatricians towards this disease as it is easy to treat with good prognosis. It is important to offer early molecular diagnostics particularly for implementing preventive strategy like PGD.
Furthermore, the wide variation of the clinical presentation needs to be elaborated with further research, including the search for any correlations between genotype and phenotype with further functional studies specially for the novel variants.
Acknowledgment
The authors gratefully acknowledge the patients and their families for their patience until a final diagnosis was reached, their laboratory staff and dieticians for their endless support, and the American manuscript editors for their help with English editing.
Footnotes
References
- 1.Baker L, Winegrad AI. Fasting hypoglycemia and metabolic acidosis associated with deficiency of hepatic fructose-1,6-diphosphatase activity. Lancet. 1970;2:13–16. doi: 10.1016/s0140-6736(70)92474-8. [DOI] [PubMed] [Google Scholar]
- 2.El-Maghrabi MR, Lange AJ, Jiang W, Yamagata K, Stoffel M, Takeda J, et al. Human fructose-1,6-bisphosphatase gene (FBP1):exon-intron organization, localization to chromosome bands 9q22.2q22.3, and mutation screening in subjects with fructose-1,6-bisphosphatase deficiency. Genomics. 1995;27:520–525. doi: 10.1006/geno.1995.1085. [DOI] [PubMed] [Google Scholar]
- 3.Niu Li, Guoying Chang, Yufei Xu, Yu Ding, Guoqiang Li, Tingting Yu, et al. Clinical and Molecular Characterization of Patients with Fructose 1,6Bisphosphatase Deficiency. Int J Mol Sci. 2017;18:857. doi: 10.3390/ijms18040857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christel Tran. Inborn errors of fructose metabolism. What can we learn from them? Nutrients. 2017;9:356. doi: 10.3390/nu9040356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kılıç M, Kasapkara ÇS, Yılmaz DY, Özgül RK. Exon 2 deletion represents a common mutation in Turkish patients with fructose-1,6-bisphosphatase deficiency. Metab Brain Dis. 2019;34:1487–1491. doi: 10.1007/s11011-019-00455-8. [DOI] [PubMed] [Google Scholar]
- 6.Ijaz S, Zahoor MY, Imran M, Ramzan K, Bhinder MA, Shakeel H, et al. Genetic analysis of fructose-1,6-bisphosphatase (FBPase) deficiency in nine consanguineous Pakistani families. J Pediatr Endocrinol Metab. 2017;30:1203–1210. doi: 10.1515/jpem-2017-0188. [DOI] [PubMed] [Google Scholar]
- 7.Faiyaz-ul-Haque M, Al-Owain MA, Al-Dayel FA, Al-Hassnan ZN, Al-Zaidan H, Rahbeeni ZA, et al. Novel FBP1 gene mutations in Arab patients with fructose-1,6-bisphosphatase deficiency. Eur J Pediatr. 2009;168:1467–1471. doi: 10.1007/s00431-009-0953-9. [DOI] [PubMed] [Google Scholar]
- 8.Kikawa Y, Inuzuka M, Jin BY, Kaji S, Yamamoto Y, Shigematsu Y, et al. Identification of a genetic mutation in a family with fructose-1,6-bisphosphatase deficiency. Biochem Biophys Res Commun. 1995;210:797–804. doi: 10.1006/bbrc.1995.1729. [DOI] [PubMed] [Google Scholar]
- 9.Kikawa Y, Inuzuka M, Jin BY, Kaji S, Koga J, Yamamoto Y, et al. Identification of genetic mutations in Japanese patients with fructose-1,6-bisphosphatase deficiency. Am J Hum Genet. 1997;61:852–861. doi: 10.1086/514875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santer R, du Moulin M, Shahinyan T, Vater I, Maier E, Muntau AC, et al. A summary of molecular genetic findings in fructose-1,6-bisphosphatase deficiency with a focus on a common long-range deletion and the role of MLPA analysis. Orphanet J Rare Dis. 2016;11:44. doi: 10.1186/s13023-016-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto A, Alfadhel M, Akroyd R, Atik Altınok Y, Bernabei SM, Bernstein L, et al. International practices in the dietary management of fructose-1,6-biphosphatase deficiency. Orphanet J Rare Dis. 2018;13:21. doi: 10.1186/s13023-018-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]