

Abstract
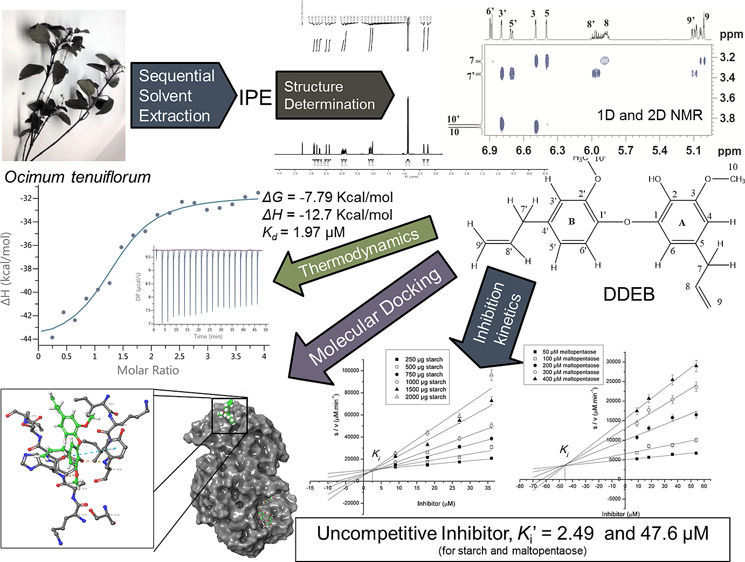
Use of human pancreatic α-amylase (HPA) inhibitors is one of the effective antidiabetic strategies to lower postprandial hyperglycemia via reduction in the dietary starch hydrolysis rate. Many natural products from plants are being studied for their HPA inhibitory activity. The present study describes isolation of dehydrodieugenol B (DDEB) from Ocimum tenuiflorum leaves using sequential solvent extraction, structure determination by one-dimensional (1D) and two-dimensional (2D) NMR analyses, and characterization as an HPA inhibitor using kinetics, binding thermodynamics, and molecular docking. DDEB uncompetitively inhibited HPA with an IC50 value of 29.6 μM for starch and apparent Ki′ of 2.49 and Ki of 47.6 μM for starch and maltopentaose as substrates, respectively. The circular dichroism (CD) study indicated structural changes in HPA on inhibitor binding. Isothermal titration calorimetry (ITC) revealed thermodynamically favorable binding (ΔG of −7.79 kcal mol–1) with a dissociation constant (Kd) of 1.97 μM and calculated association constant (Ka) of 0.507 μM. Molecular docking showed stable HPA–inhibitor binding involving H-bonds and Pi-alkyl, alkyl–alkyl, and van der Waals (vDW) interactions. The computational docking results support the noncompetitive nature of DDEB binding. The present study could be helpful for exploration of the molecule as a potential antidiabetic drug candidate to control postprandial hyperglycemia.
Introduction
Diabetes mellitus is a complex metabolic disorder caused by impairment of pancreatic β-cells resulting in either inadequate levels of insulin or the inability of cells to respond to it. It afflicts up to 425 million people globally, and India ranks second in the world with 72.9 million people being diagnosed with the major prevalent form, type 2 or non-insulin-dependent diabetes mellitus (NIDDM) associated with postprandial hyperglycemia.1
An effective therapeutic approach for management of diabetes is to decrease meal-associated hyperglycemia by decreasing the digestion of ingested carbohydrates via inhibition of carbohydrate-degrading enzymes. Pancreatic α-amylase or α-1,4-glucan-4-hydrolase (E.C.3.2.1.1), a key therapeutic target in the digestive system, catalyzes the initial step in the hydrolysis of starch to maltose, which are further degraded by α-glucosidases to glucose. Thus, retardation of starch hydrolysis achieved by reducing the action of α-amylase plays an important role in regulating the glucose spike in postprandial hyperglycemia.
Current α-glucosidase inhibitors, namely, acarbose, voglibose, and migitol, have side effects like gastrointestinal disturbances and liver and kidney dysfunction.2 Plants, such as Curcuma longa (turmeric), Momordica charantia (bitter gourd), Cinnamomum tamala, have been an exemplary source of medicines since ancient times with the Indian Ayurvedic medicinal system mentioning their use in treatment of diabetes.3 Different herbal extracts popularly used have been reported to exhibit hypoglycemic effects, are cheaper with lesser side effects than the conventional medicine, and thus offer an attractive alternative. A vast diversity of chemical entities with pharmacological relevance previously uncharacterized are present in plants and offer a good option for obtaining lead compounds.
One such perennial plant, Ocimum sp., grown in India and South East Asia has been used regularly for its medicinal properties. Alloxanized diabetic rats fed with aqueous Ocimum leaf extract displayed reduction in blood glucose levels,4 while another study5 reported a reduction in levels of fasting blood glucose, uric acid, total amino acid, total cholesterol, triglyceride, and total lipids. However, validation of the antidiabetic potential with respect to the target molecule and the mechanism of action remains to be deciphered.
Our earlier study6 showed inhibition of porcine pancreatic α-amylase (PPA) by crude 2-propanol extract of Ocimum tenuiflorum. Therefore, the present study was undertaken to isolate and characterize the bioactive component(s) and to ascertain its possible mode of action as a human pancreatic amylase (HPA) inhibitor. The bioactive molecule has been characterized using spectroscopic studies, while its mode of action has been determined using enzyme kinetics and biophysical and in silico docking studies.
Results and Discussion
Several small antidiabetic molecule inhibitors with a defined structure have been isolated from medicinal plants, but information on their targets and the mechanism of action is scarce.7 This study reports the isolation, characterization, and mode of action of a small-molecule HPA inhibitor from O. tenuiflorum.
Structure Determination of the Inhibitor
The detailed structure determination of dehydrodieugenol B (DDEB) isolated from O. tenuiflorum is given in the Supporting Information (SI) (Pages S2–S4, Figures S1–S12, Table S2). This data was compared with dehydrodieugenol B (DDEB) isolated from Nectandra leucantha reported earlier8 and found to be well in agreement. However, to date, there is no report on isolation of dehydrodieugenol B from O. tenuiflorum. The following structure (Figure 1) has been assigned to DDEB using one-dimensional (1D) and two-dimensional (2D) NMR experiments such as 1H, 13C, 13C-DEPT, 1H–1H COSY, 1H–13C HSQC, HMBC analyses.
Figure 1.

Structure of DDEB with numbering.
Human Pancreatic α-Amylase Inhibition
The sigmoidal fit of DDEB suggested a concentration-dependent inhibition with an IC50 value of 29.6 μM (9.68 μg mL–1) (Figure 2) as compared to the crude extract of 8.9 μg mL–1 acarbose, used as a positive control, that showed an IC50 value of 13.85 μM. In a previous study from our lab, bisdemethoxycurcumin (BDMC) from C. longa inhibited both porcine pancreatic amylase (PPA) and HPA with sigmoidal fits with IC50 values of 26 and 25 μM, respectively.9
Figure 2.

HPA inhibition with DDEB showing IC50 and stoichiometry (inset).
Based on the inhibition obtained at differing molar ratios of DDEB/HPA, a stoichiometry of 1:1 for inactivation was obtained, suggesting the binding of one molecule of compound with one molecule of HPA (Figure 2, inset). A similar stoichiometry has been observed for inactivation of HPA with BDMC9 and a disaccharide analogue, α-glucosyl epi-cyclophellitol,10 and of PPA with an anthocyanin, cyanidin-3-glucoside.11
Inhibition Kinetics of HPA by the Pure Compound
The effect of pure compound on the kinetics of HPA-catalyzed hydrolysis of maltopentaose and starch was studied at differing inhibitor concentrations. The double reciprocal Lineweaver–Burk plots revealed the mode of inhibition as uncompetitive for both starch and maltopentaose with a decrease in both the apparent Km and Vmax on increasing inhibitor concentration (Figure S13, Table 1).
Table 1. Kinetic Parameters of the Pure Compound on HPA with Starch and Maltopentaose as Substrates.
| substratea | DDEB (μM) | KMb | Vmax (μM min–1) | Kcat (min–1) | Kcat/KMc | Ki′ (μM) |
|---|---|---|---|---|---|---|
| starch | control | 7056.04 ± 352.80 | 0.641 ± 0.032 | 0.115 ± 0.0058 | 1.63 ± 0.08 × 10–5 | 2.49 |
| 9 | 1707.48 ± 85.38 | 0.154 ± 0.008 | 0.027 ± 0.0014 | 1.61 ± 0.08 × 10–5 | ||
| 18 | 697.56 ± 34.88 | 0.066 ± 0.033 | 0.012 ± 0.0006 | 1.69 ± 0.08 × 10–5 | ||
| 27 | 394.69 ± 19.74 | 0.038 ± 0.002 | 0.007 ± 0.00035 | 1.70 ± 0.09 × 10–5 | ||
| 36 | 256.16 ± 12.81 | 0.025 ± 0.001 | 0.004 ± 0.0002 | 1.72 ± 0.09 × 10–5 | ||
| maltopentaose | control | 132.48 ± 6.62 | 0.040 ± 0.002 | 0.0071 ± 0.0004 | 5.39 ± 0.27 × 10–5 | 47.6 |
| 9 | 97.98 ± 4.90 | 0.028 ± 0.001 | 0.0050 ± 0.0003 | 5.11 ± 0.26 × 10–5 | ||
| 18 | 72.29 ± ± 3.62 | 0.021 ± 0.001 | 0.0038 ± 0.0019 | 5.24 ± 0.26 × 10–5 | ||
| 36 | 62.71 ± 3.14 | 0.018 ± 0.0009 | 0.0032 ± 0.0002 | 5.06 ± 0.25 × 10–5 | ||
| 54 | 52.21 ± 2.61 | 0.015 ± 0.0008 | 0.0027 ± 0.0014 | 5.22 ± 0.26 × 10–5 |
Starch (1–5 mg mL–1), maltopentaose (50–400 μM), and 5.59 μM HPA were used for assays.
Unit of KM is μg for starch and μM for maltopentaose.
Unit of Kcat/KM is min–1 μg–1 for starch and min–1 μM–1 for maltopentaose.
To determine the inhibitor constant (Ki′), Bowden plots of increasing inhibitor concentrations [I] vs S/V values were plotted (Figure S13) for starch and maltopentaose, respectively. The plots of S/V against [I] provide good estimates of Ki′ as S/V does not vary greatly with V if V is distributed with uniform variance.12Ki′ values with starch and maltopentaose are found to be 2.49 and 47.60 μM, respectively. There are very few reports on compounds exhibiting uncompetitive type of inhibition. A proteinaceous inhibitor from proso millet (Panicum miliaceum) uncompetitively inhibited HPA with a Ki of 0.17 μM.13 BDMC showed uncompetitive inhibition of HPA with Ki′ values of 3 and 10.1 μM with starch and maltopentaose as substrates, respectively,9 while pinto bean peptides (PBP3, PBP6, and PBP7) also showed uncompetitive inhibition.14
However, the standard acarbose exhibited mixed mode of inhibition with Ki′ values of 2.71 and 0.866 μM for human salivary amylase (HSA) and PPA, respectively.15 Many plant-derived polyphenols and flavonoids such as myricetin, luteolin, fisetin, and quercetin are known to be effective mixed or competitive inhibitors.16 Two limonoids from Azadirachta indica, azadiradine and gedunin, showed mixed mode of HPA inhibition with maltopentaose (Ki′ = 42.2, 18.6 μM) and starch (Ki′ = 75.8, 37.4 μM) as substrates.17 Kinetic analysis of montbretin A isolated from the extract of Crocosmia crocosmiiflora demonstrated it to be a tight-binding competitive inhibitor of HPA when the 2-chloro-4-nitrophenyl a-d-maltotrioside substrate was used in the assay (Ki = 8.1 nM).18 Trans-chalcone, a flavonoid intermediate in plants, competitively inhibited α-amylase with a Ki′ of 48 μM.19
Inhibitors with an uncompetitive mode of inhibition, though rare, are warranted as such inhibition cannot be overcome by substrate concentrations and most of them have higher affinity for the enzyme at substrate saturation as they bind to the ES complex.20 Thus, the inhibitor from Ocimum being uncompetitive with an effective Ki′ suggests its potential use as an HPA inhibitor.
Circular Dichroism (CD)
CD spectroscopy was carried out to ascertain the changes in secondary and tertiary structures of HPA, if any, upon interaction of DDEB with the enzyme (Figure 3A,B).
Figure 3.

Ligand binding study. (A) Far- and (B) near-UV CD spectra of HPA with (···) and (__) without DDEB and (C) fitted isothermal titration calorimetry (ITC) curve of the ternary complex with the isotherm shown in the inset.
The far-UV CD spectra (195–250 nm) for HPA in this study were in accordance with the reported spectra for mammalian amylases.15,18 No significant change in the molar ellipticity was observed in the far-UV range (Figure 3A), suggesting that the secondary structure of the protein remains unaltered on binding of DDEB to HPA. This is similar to the earlier report of binding of BDMC, azadiradione, and gedunin to HPA.17 The CD spectrum of a protein in the near-UV spectral region of 250–350 nm is sensitive to perturbations of the tertiary structure. A major shift in the intensity and wavelength occurred on binding of DDEB to HPA (Figure 3B) at wavelengths 260–300 nm. This indicates the involvement of aromatic amino acid residues (Phe, Tyr, and Trp) in interactions with DDEB, which cause changes in the tertiary structure of HPA on binding. In an earlier study, the binding of Cys3glc to PPA resulted in an alteration in the environment of phenylalanine residues,11 while similar changes were noted for BDMC, azadiradione, and gedunin.17
It is to be noted that a reasonably good reversible inhibitor while altering the tertiary conformation of the enzyme (which could be reflected in a change in the active site conformation) should not alter its secondary structure. Alterations in the secondary structure would lead to destabilization and unfolding of the enzyme, in turn affecting the dissociation constant of the inhibitor. In fact, a number of polyphenols have been reported to alter the α-helical and β-sheet components in the protein,21 while in this study, no secondary structural change in HPA was noted on binding of the inhibitor.
Isothermal Titration Calorimetry
Since the inhibitor is uncompetitive, it would bind to the E–S complex. Hence, the titration of HPA bound to the ligand (acarbose, a noncleavable pseudo-tetrasaccharide and a known HPA inhibitor) at the active site was studied. The calorimetric titration curve of the ITC experiment is shown in Figure 3C. The binding of DDEB with the HPA–acarbose (binary) complex is exothermic as the heat change measured was negative and is shown after correction for the heat generated upon titration of DDEB in the buffer. During analysis, data (Figure 3C inset) was fitted in a single-site binding model from the Analysis software. The N (stoichiometry) of binding was found to be 1.32, which is in accordance with in vitro results. The ΔG value was −7.91, and ΔH was −12.7 kcal mol–1. The negative sign of ΔG and ΔH indicates thermodynamically favorable binding of DDEB to HPA. Based on the values of ΔH and ΔG from the ITC experiment, the ΔS (entropy) was found to be −0.016 kcal mol–1 K–1. The dissociation constant, Kd, was 1.97 μM with a calculated Ka of 0.63 μM. Similarly, studies carried out on binding of DDEB to HPA alone are given in SI and suggest similar thermodynamic values. In a previous study,17 from fluorescence quenching experiments, thermodynamic parameters for binding of azadiradione and gedunin to HPA were obtained. The free energy of binding (ΔG°) of −5.08 to −5.06 kcal mol–1, the enthalpy (ΔH) of −10.00 and −33.44 kcal mol–1, and the entropy (ΔS) of −16.75 and −95.09 kcal mol–1 K–1 were determined for azadiradione and gedunin, respectively.17 Also, the curuminoid derivative from C. longa BDMC had Kd, Gibbs free energy ΔG, enthalpy ΔH, and entropy ΔS values of 11.77 μM, −6.72, −15.77, and −30.25 kcal mol–1, respectively.7 In a study21 on the effect of young apple polyphenols on PPA inhibition, Kd values were found to be 8.740, 3.517, and 1.124 mM–1 while ΔH values were −1.74, −0.87, and −0.46 kcal mol–1 and ΔS values were 0.0014, 0.0026, and 0.0033 kcal mol–1 K–1 for tannic acid, chlorogenic acid, and caffeic acid, respectively. Another study indicated that the binding of a peptide amylase inhibitor (PAMI) to PPA is favored by enthalpic (ΔH = −10.8 kcal mol–1) and disfavored by entropic (−T ΔS = 3.7 kcal mol–1) contributions.22 Values of thermodynamic constants obtained from this study suggest that binding may be both enthalpically and entropically favorable.
Molecular Docking
SiteMap identified eight consensus surface pockets (Site 1–Site 8) on the HPA structure (Figure S16). Site 1 had a partial overlap with the known active site of HPA with residues Asp197, Glu233, and Asp300 being common, while the pockets Site 2–Site 8 were distinctly away from the active site.
The ligand preparation and optimization resulted in generation of two stable conformers with the least potential energy (Figure S17A,B). For each conformer, a total of eight docking simulations were performed (corresponding to the eight pockets identified), thus producing 16 sets of results. Analysis of the docked poses revealed that, in each set, conformer 1 showed energetically favorable binding to the receptor as compared to conformer 2. The HPA–DDEB docked complex structures with conformer 1 were analyzed further. The two top-ranking docked complexes of conformer 1 having the docking scores <−4 Kcal mol–1 were selected (Table S3). The docking and Glide score values for the Site 4 complex are more favorable than those of the Site 1 complex though the Emodel score for the Site 1 complex is marginally better than that of the Site 4 complex. Thus, the docked complex wherein DDEB is docked at Site 4 of the HPA is considered as the potential binding site.
The MM-GBSA approach facilitates computation of free energies of binding and ranking of series of ligands.23−25 This approach was used to further resolve the choice of the most probable binding site of DDEB on the HPA structure. The results showed that the docked complex with DDEB bound at Site 4 of HPA has a significantly lower (hence favorable) free energy of binding (ΔG) as compared to the Site 1 complex (Table S3). Prior studies have shown that the MM-GBSA-derived ΔG values and experimentally derived values have a good statistical correlation though the absolute values of the computationally derived ΔG are lower than the experimentally derived values such as those from ITC.23,26,27 This has been attributed to the contribution of various terms in energy expressions used to calculate ΔG values such as electrostatic components. Since the experimental data indicated uncompetitive mode of inhibition, which is also seen in computational docking studies, docking of DDEB with the cocrystal structure of HPA bound with substrate maltohexaose at the active site (PDB ID: 5TD4) was performed to generate a ternary complex. The results of this docking revealed that DDEB docked at Site 4 with favorable docking scores. Although the pose of DDEB at Site 4 in the ternary complex was slightly different from that in the binary complex (Figure 4C), the docking score for the ternary complex was more favorable, while ΔG computed with the MM-GBSA approach was comparable with that of the binary complex of HPA–DDEB (Table S3). In fact, it was observed that the Glide Emodel value (mainly used to rank the poses of ligand in the binding site) was more favorable for the ternary complex as compared to the binary complex (Glide Emodel values of −40.898 and −43.007 kcal mol–1 and ΔGMMGBSA of −59.58 and −59.84 kcal mol–1 for the binary and the ternary complex, respectively). This is in agreement with the uncompetitive mode of inhibition observed experimentally wherein the inhibitor binds to the ES complex. Analyses of HPA–DDEB interactions using Ligand Interaction Diagram utility in Glide and LIGPLOT version 4.5.3 showed that the docked complex with DDEB bound at Site 4 on HPA has several stabilizing interactions, including H-bonds and Pi–Pi and hydrophobic interactions (Table S4, Figure 4 A–D). In the ternary complex HPA–maltohexaose–DDEB, the π electron clouds of one of the aromatic rings of DDEB and that of Tyr468 have a stabilizing pi–pi interaction. There are two H-bonds present involving backbone N atoms of the amino acids Tyr468 and His476, with O4 and O3 atoms of DDEB, respectively. Apart from these, a number of hydrophobic interactions are also seen (Table S4). A comparison of the noncovalent interactions formed in binary and ternary complexes indicates a more favorable binding of DDEB in the presence of the substrate bound at the active site.
Figure 4.

Molecular docking of DDEB with HPA at Site 4. (A) and (C) Full views and close-up views of HPA–DDEB binary and HPA–maltohexaose–DDEB ternary complexes, respectively, showing H-bonds (beige dotted lines) and pi–pi interactions (cyan dotted line). In full view, DDEB is shown with CPK rendering and maltohexaose (in C) is shown as ball-and-stick rendering with carbons (green), oxygens (red), and hydrogens (white). HPA is rendered as solvent-accessible surface. The interactions of HPA and DDEB in binary (B) and ternary (D) complexes identified using LigPlot with H-bonds shown as green dotted lines and hydrophobic contacts shown as spoked arcs.
Conclusions
Many herbal extracts serve to complement current therapies, have been reported for their antidiabetic activities, and are currently being used for the treatment of diabetes. However, such medicinal plants have not gained much importance as medicines due to the lack of sustained scientific evidence. HPA is one of the important pharmacological targets for treatment of type 2 diabetes, and small-molecule inhibitors with a defined structure and mechanism of action are scarce. The mode of action of the component responsible for this and its target of action need to be validated for a typical drug discovery module. From O. tenuiflorum leaves, known to possess antidiabetic property, the isolation and identification of dehydrodieugenol B and its mechanism of HPA inhibition were studied.
The compound inhibited HPA uncompetitively with a 1:1 stoichiometry and a low Ki′. Ligand binding of DDEB with HPA indicated that the inhibitor binds at a site away from the active site of HPA. The binary complex of DDEB bound to HPA had N = 1.07, ΔG = −7.79, ΔH = −15.5 kcal mol–1, and Kd = 1.97 μM. Thermodynamics of HPA–acarbose with DDEB (ternary complex) showed N = 1.32, ΔG = −7.91, ΔH = −12.7 kcal mol–1, and Kd = 1.59 μM. Compared to the binary HPA–DDEB complex, the thermodynamic parameters of the ternary complex of HPA–acarbose–DDEB are slightly more energetically favorable, which is in accordance with uncompetitive mode of inhibition. The findings from computational studies also agree well with in vitro kinetics data.
Molecular docking and MM-GBSA studies suggest that DDEB binds at a pocket Site 4, which is distant from the active site of HPA and has favorable docking and Emodel scores as well as ΔGMMGBSA values. Two of the four aromatic amino acid residues involved in the binding site, viz., Tyr468 and His476, form pi–pi stacking and H-bonding interactions with DDEB that stabilize the complex, as indicated in the results of CD spectra of the HPA–DDEB complex.
The present study suggests that DDEB could be a promising candidate for controlling postprandial hyperglycemia and could be developed as a lead antidiabetic compound.
Experimental Section
Maltopentaose and HPA were purchased from Sigma Aldrich. Soluble starch was procured from SRL, Mumbai, India. d-Maltose and 3,5-dinitrosalicylic acid (DNSA) were obtained from HiMedia Laboratories, Mumbai, India. Solvents like methanol, 2-propanol, n-hexane, and ethyl acetate were obtained from Merck India Ltd., Mumbai. Chemicals and solvents were of AR and HPLC grades.
Collection and Authentication of Plant Material
Plant material was collected from Western Ghats in Maharashtra, India. A herbarium of the plant sample was prepared; submitted to the Botanical Survey of India (BSI), Pune, India, for authentication (voucher specimen number PDAOCT1); and identified as O. tenuiflorum.
Isolation of the Bioactive Component from O. tenuiflorum
The HPA inhibitor was purified from 2-propanol extract (IPE) of O. tenuiflorum leaves using sequential solvent extraction followed by silica gel column chromatography and semipreparative HPLC as described in the Supplementary Information (SI).
Structure Determination of the Bioactive Compound
The structure of the pure HPA inhibitor was determined by Fourier transform infrared (FTIR) spectroscopy, high-resolution mass spectrometry (HRMS), and NMR spectroscopy as given in SI.
Enzyme Inhibition Assay
The inhibition assay was performed as described in SI. One unit of enzyme activity is defined as the amount of enzyme required to release one micromole of maltose from starch or maltopentaose per minute under the assay conditions. Relative activity (%) and inhibition (%) were calculated as given in equations (1) and (2) in SI.
Determination of IC50
The IC50 value was defined as the concentration of inhibitor resulting in 50% inhibition of HPA. To determine IC50 and stoichiometry, the assay was performed with HPA (5.59 μM) incubated with varying concentrations of the inhibitor (5–50 μM) for 15 min followed by calculating percent HPA inhibited. The data was analyzed using a sigmoid fit in Origin 8.0 (Origin Lab Corporation, Northampton).
Kinetics of Enzyme Inhibition
The mode of HPA inhibition by inhibitor was determined through kinetic experiments. Starch (1–5 mg mL–1) as the physiological substrate and maltopentaose (50–400 μM) were incubated independently with 9–36 and 9–54 μM inhibitor for starch (10 min) and maltopentaose (2.5 min), respectively. The residual enzyme activity was determined by DNSA28 and Nelson–Somogyi′s method29 for starch and maltopentaose, respectively. Dixon and secondary Bowden plots12 were used to determine the inhibition constant (Ki′).
Circular Dichroism (CD) Spectroscopy
CD spectra of HPA in the presence and absence of the pure HPA inhibitor were recorded using a J-815 spectrometer (Jasco International Co. Ltd., Japan), and molar ellipticity was calculated from recorded ellipticity as detailed in SI.
Isothermal Titration Calorimetry (ITC)
Isothermal calorimetry was carried out using an isothermal titration microcalorimeter (Microcal PEAQ-ITC, Malvern Instruments Ltd., U.K.) as described in SI. Results were analyzed by Malvern PAQC Analysis software for calculations and graphical representation of the data.
Molecular Docking Studies
The crystal structure of native HPA30 (PDB ID: 1HNY) and the molecular structure of the inhibitor DDEB were used for molecular docking. Subsequently, for the generation of ternary complex (HPA–substrate–inhibitor), the HPA structure cocrystallized with maltohexaose at the active site31 (PDB ID: 5TD4) was used for docking. Standardization and validation of docking parameters, protein and ligand preparation, grid generation, docking and energy calculations, etc. were done as per SI.
Statistical Analysis
All assays were repeated as three independent sets with each set in triplicates. Results are expressed as means S.E.M. with number of observations (n). The best-fit values were achieved by applying either linear fit or nonlinear least-squares regression using software Microcal Origin 8.0. (Origin Lab Corporation, Northampton).
Acknowledgments
The authors thank the Botanical Survey of India (BSI), Pune, for authentication of the collected plant samples; the Central Instrumentation Facility, Savitribai Phule Pune University for FTIR, HRMS, CD, and NMR facilities; the Garware Research Centre for technical support, the DST-FIST program at IBB for ITC facility; and the Department of Biotechnology, Government of India, and SPPU for computational infrastructure support.
Glossary
Abbreviations
- HPA
human pancreatic α-amylase
- DDEB
dehydrodieugenol B
- CWE
cold water extract
- HWE
hot water extract
- ME
methanol extract
- IPE
2-propanol extract
- TLC
thin layer chromatography
- HPLC
high-performance liquid chromatography
- ODS
octyldecasilica
- CD
circular dichroism
- ITC
isothermal titration calorimetry
- vDW
van der Waals
- NIDDM
non-insulin-dependent diabetes mellitus
- PPA
porcine pancreatic α-amylase
- BDMC
bisdemethoxycurcumin
- HSA
human salivary amylase
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00617.
Methods and results for extraction and bioactivity-guided purification of HPA inhibitor and structure determination of the inhibitor using FTIR, HRMS, and 1D and 2D NMR experiments; methods for the enzyme inhibition assay and ITC; molecular docking; assignments and correlations of protons and carbons; docking scores and binding energies of the binary and ternary docked complexes; stabilizing interactions between HPA and the inhibitor complex at Site 4 (PDF)
Author Contributions
The isolation of bioactive compound was carried out by P.D.D., A.S.K., and S.R.C. The structure analysis was carried out by D.D.D., P.D.D., A.S.K., and S.R.C. The enzyme and protein studies were carried out by P.D.D. and A.R.K., while the computational work was carried out by P.P.K. and S.V.S. The work was conceived and planned by A.R.K. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors thank UGC UPE PHASE-II program at SPPU for financial support. P.D.D. received financial aid provided by the program.
The authors declare no competing financial interest.
Supplementary Material
References
- Cho N. H.; Shaw J. E.; Karuranga S.; Huang Y.; da Rocha Fernandes J. D.; Ohlrogge A. W.; Malanda B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. 10.1016/j.diabres.2018.02.023. [DOI] [PubMed] [Google Scholar]
- Cheng A. Y. Y.; Fantus I. G. Oral antihyperglycemic therapy for type 2 diabetes mellitus. Can. Med. Assoc. J. 2005, 172, 213–226. 10.1503/cmaj.1031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman B. V.; Naga V. K.; Rao N.; Saradhi M. P.; Rao M. V. B. Plants with antidiabetic Activities And their Medicinal Values. Int. Res. J. Pharm. 2012, 3, 11–15. [Google Scholar]
- Vats V.; Grover J. K.; Rathi S. S. Evaluation of anti-hyperglycemic and hypoglycemic effect of Trigonella foenum-graecum Linn, Ocimum sanctum Linn and Pterocarpus marsupium Linn in normal and alloxanized diabetic rats. J. Ethnopharmacol. 2002, 79, 95–100. 10.1016/S0378-8741(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Rai V.; Iyer U.; Mani U. V. Effect of Tulasi (Ocimum sanctum) leaf powder supplementation on blood sugar levels, serum lipids and tissue lipid in diabetic rats. Plant Foods Hum. Nutr. 1997, 50, 9–16. 10.1007/BF02436038. [DOI] [PubMed] [Google Scholar]
- Sudha P.; Zinjarde S. S.; Bhargava S. Y.; Kumar A. R. Potent a-amylase inhibitory activity of Indian Ayurvedic medicinal plants. BMC Complementary Altern. Med. 2011, 11, 5 10.1186/1472-6882-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy S.; Zinjarde S.; Bhargava S.; Kulkarni-Kale U.; Sawant S.; Ravikumar A. Deciphering the inactivation of human pancreatic α-amylase, an antidiabetic target, by bisdemethoxycurcumin, a small molecule inhibitor, isolated from Curcuma longa. Nat. Prod. J. 2013, 3, 15–25. 10.2174/2210315511303010005. [DOI] [Google Scholar]
- da Costa-Silva T. A.; Grecco S. S.; de Sausa F. S.; Lago J. H. G.; Martins E. G. A.; Terrazas C. A.; Varikuti S.; Owens K. L.; Beverley S. M.; Satoskar A. R.; Tempone A. G. Immunomodulatory and antileishmanial activity of Phenylpropanoid Dimers Isolated from Nectandra leucantha. J. Nat. Prod. 2015, 78, 653–657. 10.1021/np500809a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnusamy S.; Zinjarde S.; Bhargava S.; Rajamohanan P. R.; Ravikumar A. Discovering Bisdemethoxycurcumin from Curcuma longa rhizome as a potent small molecule inhibitor of human pancreatic a-amylase, a target for type-2 diabetes. Food Chem. 2012, 135, 2638–2642. 10.1016/j.foodchem.2012.06.110. [DOI] [PubMed] [Google Scholar]
- Caner S.; Zhang X.; Jiang J.; Chen H.; Nguyen N. T.; Overkleeft H.; Brayer G. D.; Withers S. G. Glucosyl epi-cyclophellitol allows mechanism based inactivation and structural analysis of human pancreatic α-amylase. FEBS Lett. 2016, 590, 1143–1151. 10.1002/1873-3468.12143. [DOI] [PubMed] [Google Scholar]
- Wiese S.; Gartner S.; Rawel H. M.; Winterhalter P.; Kullinga S. E. Protein interactions with cyanidin-3-glucoside and its influence on α-amylase activity. J. Sci. Food Agric. 2009, 89, 33–40. 10.1002/jsfa.3407. [DOI] [Google Scholar]
- Eisenthal R.; Cornish-Bowden A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem. J. 1974, 139, 715–720. 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj R. H.; Pattabiraman T. N. Purification and properties of an alpha-amylase inhibitor specific for human pancreatic amylase from proso (Panicium miliaceum) seeds. J. Biosci. 1985, 7, 257–268. 10.1007/BF02716789. [DOI] [Google Scholar]
- Ngoh Y.; Tye G. J.; Gan C. The investigation of α-amylase inhibitory activity of selected Pinto bean peptides via preclinical study using AR42J cell. J. Funct. Foods 2017, 35, 641–647. 10.1016/j.jff.2017.06.037. [DOI] [Google Scholar]
- Yoon S. H.; Robyt J. F. Study of the inhibition of four α-amylases by acarbose and its 4IV-α-maltohexaosyl and 4IV-α-maltododecaosyl analogues. Carbohydr. Res. 2003, 338, 1969–1980. 10.1016/S0008-6215(03)00293-3. [DOI] [PubMed] [Google Scholar]
- Tadera K.; Minami Y.; Takamatsu K.; Matsauka T. Inhibition of α-Glucosidase and α-amylase by flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. 10.3177/jnsv.52.149. [DOI] [PubMed] [Google Scholar]
- Ponnusamy S.; Haldar S.; Mulani F.; Zinjarde S.; Thulasiram H.; RaviKumar A. Gedunin and azadiradione: human pancreatic alpha-amylase inhibiting limonoids from Neem (Azadirachta indica) as Anti-Diabetic Agents. PLoS One 2015, 10, e0140113 10.1371/journal.pone.0140113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling C. A.; Woods K.; Zhang R.; Brastianos H. C.; Brayer G. D.; Andersen R. J.; Withers S. G. The search for novel human pancreatic α-Amylase inhibitors: high-throughput screening of terrestrial and marine natural product extracts. ChemBioChem 2008, 9, 433–438. 10.1002/cbic.200700470. [DOI] [PubMed] [Google Scholar]
- Najafian M.; Ebrahim-Habibi A.; Hezareh N.; Yaghmaei P.; Parivar K.; Larijani B. Trans-chalcone: a novel small molecule inhibitor of mammalian α-amylase. Mol. Biol. Rep. 2011, 38, 1617–1620. 10.1007/s11033-010-0271-3. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.Reversible Modes of Inhibitor Interactions with Enzymes. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; John Wiley and Sons, 2005; pp 68–69. [PubMed] [Google Scholar]
- Sun L.; Warren F. J.; Gidley M. J.; Guo Y.; Miao M. Mechanism of binding interactions between young apple polyphenols and porcine pancreatic α-amylase. Food Chem. 2019, 283, 468–474. 10.1016/j.foodchem.2019.01.087. [DOI] [PubMed] [Google Scholar]
- Dolečková-Marešová L.; Pavlík M.; Horn M.; Mareš M. De novo design of α-amylase inhibitor: a small linear mimetic of macromolecular proteinaceous ligands. Chem. Biol. 2005, 12, 1349–1357. 10.1016/j.chembiol.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Rastelli G.; Del Rio A.; Degliesposti G.; Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. 10.1002/jcc.21372. [DOI] [PubMed] [Google Scholar]
- Hou T.; Wang J.; Li Y.; Wang W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. 10.1002/jcc.21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgobba M.; Caporuscio F.; Anighoro A.; Portioli C.; Rastelli G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440. 10.1016/j.ejmech.2012.10.024. [DOI] [PubMed] [Google Scholar]
- Moraca F.; Rinaldo D.; Smith A. B. 3rd; Abrams C. F. Specific noncovalent interactions determine optimal structure of a buried ligand moiety: QM/MM and pure QM modeling of complexes of the small-molecule CD4 mimetics and HIV-1 gp120. ChemMedChem 2018, 13, 627–633. 10.1002/cmdc.201700728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Li L.; Hurley T. D.; Meroueh S. O. Molecular recognition in a diverse set of protein-ligand interactions studied with molecular dynamics simulations and end-point free energy calculations. J. Chem. Inf. Model. 2013, 53, 2659–2670. 10.1021/ci400312v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G. L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. 10.1021/ac60147a030. [DOI] [Google Scholar]
- Somogyi M. Notes on sugar determination. J. Biol. Chem. 1952, 195, 19–23. [PubMed] [Google Scholar]
- Brayer G. D.; Luo Y.; Withers S. G. The structure of human pancreatic alpha-amylase at 1.8 A resolution and comparisons with related enzymes. Protein Sci. 1995, 4, 1730–1742. 10.1002/pro.5560040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Caner S.; Kwan E.; Li C.; Brayer G. D.; Withers S. G. Evaluation of the significance of starch surface binding sites on human pancreatic α-Amylase. Biochemistry 2016, 55, 6000–6009. 10.1021/acs.biochem.6b00992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.