

Abstract

With the emergence of multi-drug-resistant strains of Mycobacterium tuberculosis, there is a pressing need for new oral drugs with novel mechanisms of action. A number of scaffolds with potent anti-tubercular in vitro activity have been identified from phenotypic screening that appear to target MmpL3. However, the scaffolds are typically lipophilic, which facilitates partitioning into hydrophobic membranes, and several contain basic amine groups. Highly lipophilic basic amines are typically cytotoxic against mammalian cell lines and have associated off-target risks, such as inhibition of human ether-à-go-go related gene (hERG) and IKr potassium current modulation. The spirocycle compound 3 was reported to target MmpL3 and displayed promising efficacy in a murine model of acute tuberculosis (TB) infection. However, this highly lipophilic monobasic amine was cytotoxic and inhibited the hERG ion channel. Herein, the related spirocycles (1–2) are described, which were identified following phenotypic screening of the Eli Lilly corporate library against M. tuberculosis. The novel N-alkylated pyrazole portion offered improved physicochemical properties, and optimization led to identification of a zwitterion series, exemplified by lead 29, with decreased HepG2 cytotoxicity as well as limited hERG ion channel inhibition. Strains with mutations in MmpL3 were resistant to 29, and under replicating conditions, 29 demonstrated bactericidal activity against M. tuberculosis. Unfortunately, compound 29 had no efficacy in an acute model of TB infection; this was most likely due to the in vivo exposure remaining above the minimal inhibitory concentration for only a limited time.
Introduction
Mycobacterium tuberculosis,1 the causative agent of tuberculosis (TB), can be fatal if not properly treated and disproportionately affects the poor in developing countries. In 2015, TB became the world’s most deadly infectious disease, killing 1.4 million people (1.2 million HIV-negative and 0.3 million HIV-positive) in 2019.2 The current 6 month treatment results in high default rates, increased transmission, and drug resistance.3−5 In order to reduce treatment length, TB treatments working through novel mechanisms are needed.6−8 However, identifying novel drugs remains a significant challenge, and high-quality leads are still urgently required.9,10
Target-directed TB drug discovery programs have historically been largely unsuccessful in delivering high-quality late-stage leads.7 To address this issue, cell-based phenotypic screening became a focus for identifying active starting points. Many of the most potent phenotypic hits target membrane proteins such as DprE1 and MmpL3 that are involved in cell wall biosynthesis.11 MmpL3 is required for the export of trehalose monomycolates (TMM) to the periplasmic space and outer membrane of M. tuberculosis. A number of structurally diverse putative MmpL3 inhibitor series have been reported.12−29 Several of these series inhibit MmpL3-mediated TMM export but may also have pleiotropic effects targeting the proton motive force.13
The scaffolds of MmpL3 inhibitors are typically lipophilic, which facilitates partitioning into hydrophobic membranes, and several contain basic amine groups. Highly lipophilic basic amines are typically cytotoxic against mammalian cell lines and have associated off-target risks, such as inhibition of human ether-à-go-go related gene (hERG) and IKr potassium current modulation. One particular spirocyclic series has been reported as a potential MmpL3 inhibitor following a phenotypic screen of the GSK library against Mycobacterium bovis.19 Although further exploration of this original hit identified compounds with excellent in vivo activity against M. tuberculosis (3), the series was discontinued because of concerns over safety related to the lipophilicity and basic nature of the scaffold.30 Of note, the original authors highlighted that further exploration may be able to design around the series liabilities while retaining the remarkable in vivo potency30
Herein, we report on a novel pyrazole spirocyclic amine series (1 and 2) with a putative MmpL3 mechanism of action that is structurally related to 3 (Table 1). The novel pyrazole portion offered improved physicochemical properties, and optimization led to identification of a zwitterionic series, with improved selectivity over both HepG2 cytotoxicity and hERG inhibition. The zwitterionic series retained potent M. tuberculosis whole cell activity, with large shifts against MmpL3 mutant strains. Unfortunately, the series representative with the best overall properties, 29, failed to show efficacy in an acute model of TB infection. As such, further work on this series was put on hold. We feel that the approach presented indicates useful insights into mechanisms for reduction of metabolism and hERG liabilities while highlighting the challenge within drug discovery of balancing these properties with potency.
Table 1. In Vitro Profile of Early Hits against a Reported MmpL3 Inhibitor.
| confirmed hit 1 | confirmed hit 2 | GSK-SPIRO 3 | early lead 4 | |
|---|---|---|---|---|
| MICa (μM) | 0.22 | 0.14 | 0.083 | 0.11 |
| MIC MmpL3 F255L mut. (μM) | 31 | 16 | 1.7 | 2.6 |
| hERGb,c IC50 (μM) | 2.9c | 1.1c | 3.1c | 3.1c |
| HepG2d (μM) | 36 | 32 | 20 | 38 |
| LLE | 4.5 | 4.5 | 3.0 | 4.3 |
| SFI | 5.2 | 5.5 | 7.1 | 5.4 |
| kin. solubility (μM) | >250 | >250 | 39 | >250 |
| MW | 379 | 373 | 475 | 373 |
| clog DpH7.4 | 2.2 | 2.5 | 4.1 | 2.5 |
| TPSA (Å2) | 30 | 30 | 31 | 30 |
| mouse Cle (mL/min/g) | >50 | >50 | 7.5 | >50 |
| human Clf (mL/min/g) | ND | ND | 1.7 | 4.6 |
MIC is the minimum concentration required to inhibit the growth of M. tuberculosis (H37Rv) in liquid culture. All MIC values are an average of at least two measurements.
hERG functional thallium flux inhibitory concentration (IC50).
hERG functional Q-patch inhibitory concentration (IC50).
HepG2 inhibitory concentration (IC50) is the concentration required to inhibit growth of HepG2 cells by 50%.
Intrinsic clearance (Cli) using CD1 mouse liver microsomes.
Intrinsic clearance (Cli) using pooled human liver microsomes. LLE is the lipophilic ligand efficiency; SFI is the solubility forecast index; TPSA is the total polar surface area. Estimations of clog DpH7.4 and TPSA were calculated using StarDrop (http://www.optibrium.com).
Results and Discussion
Structure–Activity Relationship
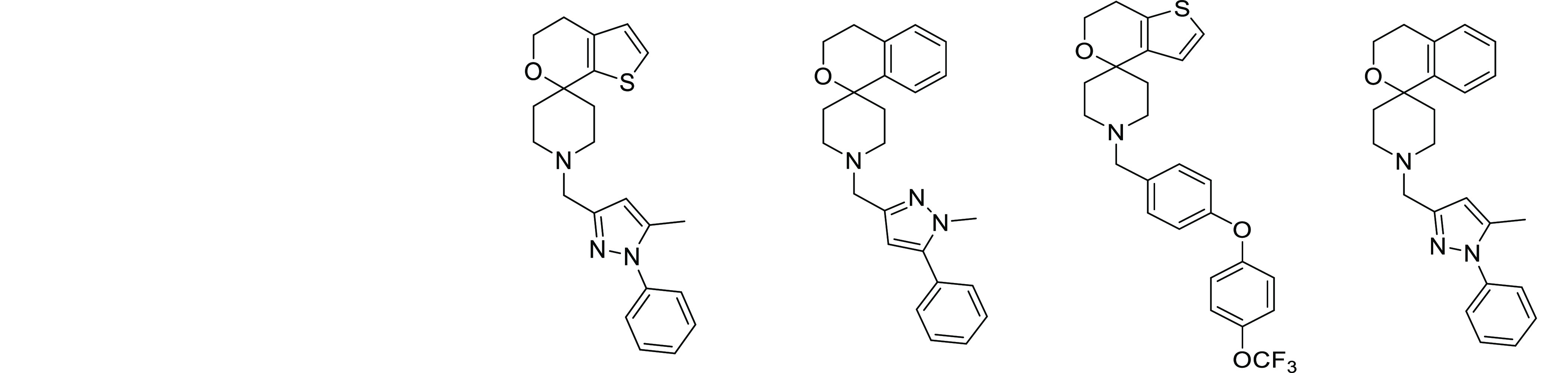
To identify novel anti-tubercular agents, an aerobic whole cell phenotypic screen was undertaken, evaluating the Eli Lilly corporate screening deck against M. tuberculosis strain H37Rv. One of the outcomes of this screen was the identification of a cluster of pyrazole-containing spirocyclic amine analogues; the spirocyclic portion of the molecule showed a clear structural resemblance to 3, a previously reported spirocycle series (Table 1).19,30 The initial hits had good anti-tubercular activity with a minimum inhibitory concentration (MIC) of 0.22 and 0.14 μM for 1 and 2, respectively (Table 1). As the original spirocycle compound was considered to be an MmpL3 inhibitor, we tested the activity of these two compounds against a strain containing a mutation in MmpL3 (F255L) and observed a large shift in the MIC confirming the likely on-target activity (Table 1).
According to the literature, 3 had not been developed further because “it suffered from a high clog P value, with the consequent potential liabilities for further development”.19,30,31 Compounds 1 and 2 offered an attractive alternative to 3 because the central aromatic ring in both was a pyrazole not a phenyl; this modification was predicted to reduce the clog DpH7.4 by around 1 log unit. The reduced lipophilicity translated into a better solubility forecast index and M. tuberculosis-derived LLE,32−35 providing optimism for improving off-target liabilities, metabolic stability, as well as solubility profile. Unfortunately, as a result of the basic nature of the piperidine group, hERG inhibition remained an issue for 1 and 2; moreover, despite the lower clog DpH7.4, both showed unexpectedly high mouse microsomal clearance.
Initial emphasis for expansion of the confirmed hits 1 and 2 was placed on understanding the scope for improving hERG channel inhibition and microsomal stability. The pyrazole of 1 and the spirocycle of 2 were combined to afford 4, which showed marginally improved selectivity over the hERG channel, although the mouse microsomal clearance remained high (Tables 1 and 2). In addition, the human microsomal metabolic stability was higher for 4 than the published molecule 3, although both values were lower than the equivalent mouse data. As a first step to attempt to reduce metabolism of 4, a mouse microsomal metabolite identification study was carried out to evaluate why the pyrazole series had worse microsomal stability than 3 despite a lower clog DpH7.4 (Table 1). This showed that 4 was metabolized rapidly to four main metabolites primarily involving hydroxylation. After a 3 min incubation in mouse microsomes, only 29% of the parent ion remained, while 61% of the detectable ions were associated with three metabolites that resulted from hydroxylation associated with regions of the molecule around the spirocycle group (Table S1; Figures S1 and S2). Because phenyl groups are known to be prone to hydroxylation, which can be prevented by fluorination,36 the phenyl substituent on the spirocycle was replaced by a series of different fluoro-substituted phenyl groups (Table 2). Unfortunately, compounds that retained good MIC activity remained very unstable in mouse microsomes, while the compounds with improved metabolic stability had significantly decreased potency (Table 2).
Table 2. In Vitro Evaluation of Spirocycle Analogues.
| R1 | MICa μM | clog DpH7.4 | hERGa IC50 μM | HepG2a IC50 μM | mouse Clia (mL/min/g) | |
|---|---|---|---|---|---|---|
| 4 | phenyl | 0.11 | 2.5 | 3.1a | 38 | >50 |
| 5 | no group | 4.7 | 1.7 | >30a | 28 | |
| 6 | 6-fluorophenyl | 0.16 | 2.7 | 3.5a | 48 | >50 |
| 7 | 7-fluorophenyl | 0.12 | 2.7 | 4.7a | 32 | >50 |
| 8 | 6,7-difluorophenyl | 0.69 | 2.8 | 1.8a | 14 | >50 |
| 9 | 6-CF3-phenyl | >20 | 3.1 | 0.8a | 7 | 9 |
| 10 | 7,8-difluorophenyl | 2.5 | 2.8 | 1.7a | 16 | >50 |
To examine the metabolic stability further, isoform-specific cytochrome P450 studies were carried out on 4, evaluating CYP3A4 and CYP2D6 as the two most abundant human CYP450 enzymes. Pyrazole 4 was more rapidly cleared by CYP2D6 bactosomes (0.34 min–1) than CYP3A4 bactosomes (0.07 min–1). As the CYP2D6-active site is known to be smaller in comparison to CYP3A4,37 it was proposed that increasing the size of substituents on either the 5-methyl pyrazole or the phenyl in 4 could potentially lead to a decrease in CYP2D6 metabolism and thereby improve the metabolic stability to be more in line with the larger compound 3.
To test the above hypothesis, the structure–activity relationship (SAR) around the methyl and phenyl substituents was expanded. Initial modifications to the 5-methyl pyrazole had little effect on MIC, hERG channel inhibition, or metabolic stability (11–14), and so further exploration of this substituent was put on hold. For the phenyl substituted compounds 15–17, there was a trend toward improved metabolic stability, in particular with the bulky OCF3 blocking group of 17 despite it having a high clog DpH7.4 (Table 3). In an attempt to reduce or maintain as low a clog DpH7.4 as possible, the SAR around a 1-pyridyl substituent was explored. For the pyridyl-substituted compounds 18–20, again there was a trend toward improved metabolic stability, in particular with bulkier and more lipophilic groups such as the CF3 in 20 (Table 3).
Table 3. In Vitro Evaluation of Spirocycle Analogues.
| R2 | R3 | MICa (μM) | clog DpH7.4 | hERGa IC50 (μM) | HepG2a IC50 (μM) | mouse Clia (mL/min/g) | |
|---|---|---|---|---|---|---|---|
| 11 | phenyl | OMe | 0.12 | 2.5 | 2.1a | 23 | >50 |
| 12 | phenyl | CyPr | 0.10 | 3.0 | 2.2a | 22 | >50 |
| 13 | phenyl | CHF2 | 0.20 | 3.1 | 3.6a | 15 | >50 |
| 14 | phenyl | CF3 | 0.09 | 3.1 | 4.7a | 30 | >50 |
| 15 | 4-(hydroxymethyl) phenyl | Me | 1.5 | 2.0 | 5.0a | >50 | 8.5 |
| 16 | 4-methylphenyl | Me | 0.08 | 2.6 | 2.5a | 11 | >50 |
| 17 | 4-(trifluoromethoxy) phenyl | Me | 0.36 | 3.4 | 1.9a | 8.6 | 7 |
| 18 | 6-methoxy pyridin-3-yl | Me | 0.37 | 2.0 | 3.5a | >50 | 44 |
| 19 | 6-(difluoromethoxy)-pyridin-3-yl | Me | 0.21 | 2.5 | 0.52a | 18 | 25 |
| 20 | 6-(trifluoromethyl) pyridin-3-yl | Me | 0.51 | 2.6 | 2.7a | 42 | 10 |
| 21 | 2-benzoic acid | Me | >20 | 0.6 | >30a | >50 | <0.5 |
| 22 | 3-benzoic acid | Me | 3.7 | 0.5 | 10.2a | >50 | 1.6 |
| 23 | 4-benzoic acid | Me | 4.8 | 0.6 | >30a | >50 | 0.5 |
| 24 | 4-benzoic acid | Et | 0.77 | 0.9 | >30a | >50 | 1.1 |
| 25 | 4-benzoic acid | iPr | 3.8 | 1.4 | >30a | >50 | 1.2 |
| 26 | 4-benzoic acid | tBu | 5.5 | 1.3 | >30a | >50 | 1.1 |
| 27 | 4-benzoic acid | CHF2 | 1.4 | 1.3 | >30a | >50 | 0.7 |
| 28 | 4-benzoic acid | CF3 | 0.65 | 1.5 | >30a | >50 | 1 |
| 29 | 4-benzoic acid | CyPr | 0.66 | 1.2 | >30a | >50 | 1.1 |
| 30 | 4-benzoic acid | CyBu | 1.8 | 1.5 | >30a | >50 | 1.6 |
| 31 | 4-benzoic acid | OMe | 1.9 | 0.6 | >30a | >50 | 1.3 |
| 32 | 4-benzoic acid | OEt | 0.53 | 0.9 | >30a | >50 | 3.8 |
| 33 | 4-benzoic acid | N-pyrrolidino- | 13 | 1.3 | >50 | 1.5 | |
| 34 | 4-benzoic acid | N-morpholino- | >20 | 1.3 | >50 | <0.5 |
As an alternative phenyl substitution, the addition of a carboxylic acid group was explored (Table 3). Such a modification adds bulk, introduces polarity, and the presence of a zwitterion has been shown previously to overcome hERG channel inhibition.38 While the ortho-acid 21 was not tolerated, the modest M. tuberculosis whole cell activity of 22 and 23 was encouraging especially because the para-acid 23 showed a dramatic improvement in the compound’s liabilities including hERG channel inhibition, mouse metabolic stability, and HepG2 cytotoxicity. Although the overall properties of 23 were an improvement on 4, this came at a significant reduction in antibacterial potency (∼40 fold). To continue optimization of 23, we reassessed modifications of the 5-methyl pyrazole as these had been tolerated previously on 4. In general, substitutions 29–34 were well tolerated, apart from cyclic amines 33 and 34, with good profiles in relation to hERG inhibition, mouse metabolic stability, and HepG2 cytotoxicity. Substituted alkyl as well as fluoroalkyl resulted in notable improvements in the whole cell potency for cyclopropyl 29, trifluoromethyl 28, ethyl 24, and ethoxy 32 (Table 3).
Biological Profiling
Compound 29 was not cytotoxic to HepG2 cells, grown in either glucose or galactose media, nor the THP-1 macrophage-like cell line (data not shown). 29 had good potency against intracellular bacteria in THP-1 cells [IC50 = 1.5 ± 1.3]. Compound 29 was also shown to retain good activity against clinical samples from the four main lineages and strains containing resistance mutations to either isoniazid or rifampicin (Table 4).
Table 4. Activity against M. tuberculosis Clinical Strains of Different Lineage and Resistancea.
| MIC90 (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| H37Rv | N0157 L1 | N0052 L2 | N0004 L3 | N0136 L4 | INH-R2 | RIF-R2 | |
| 23 | 5.1 | >50 | 7.3 | 24 | 9.6 | 8.6 | 13 |
| 29 | 0.78 | 2.8 | 2.2 | 5.2 | 2.4 | 0.37 | 0.76 |
| rifampicin | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | >50 |
| isoniazid | ND | ND | ND | ND | ND | >200 | 0.58 |
MIC is the minimum concentration required to inhibit the growth of M. tuberculosis (H37Rv) in liquid culture L1 is TB lineage 1 and so forth.
Minimum bactericidal concentration (MBC) and kill kinetics were evaluated under both aerobic (replicating) and starvation (nonreplicating) conditions. Under replicating conditions, 29 showed concentration-dependent kill of M. tuberculosis with an MBC of 3 μM (Figure 1A). At later stages, outgrowth was observed because of either compound instability over long incubation periods or the appearance of resistant mutants. Under nonreplicating conditions, 29 showed no bactericidal effect against M. tuberculosis (Figure 1B).
Figure 1.

Kill kinetics for 29 against replicating and nonreplicating M. tuberculosis. Bacterial viability in the presence of compound was determined by cfu over 28 days (A) under replicating conditions and (B) under nonreplicating conditions. The dashed lines represent the upper and lower limits of detection.
Compound 29 showed a 10-fold decrease in activity against the MmpL3F255L strain of M. tuberculosis, indicating that it retained an MmpL3-related mechanism of action. An impact on this pathway was also indicated using a hypomorph strain (P606-5C-mmpL3) that underexpresses MmpL3 when grown in the absence of anhydrotetracycline (Figure 2). Initially, it was confirmed that SQ109, a known inhibitor of MmpL3, was more active against the hypomorph strain with a four-fold improvement in MIC50, while ethambutol, a cell wall inhibitor targeting a different pathway (arabinosyl transferases), was equally effective against both the wild-type and hypomorph strains. We tested two representatives from the series (4 and 23); both showed significant shifts in potency, with the MmpL3 hypomorph being at least eight-fold more sensitive. These data support the conclusion that, as shown in the literature for 3, the series works through an MmpL3-related mechanism.
Figure 2.

Decreased MmpL3 expression results in hypersensitivity to the spirocycle series. Removal of anhydrotetracycline (atc) results in transcriptional repression of mmpL3. Growth in the presence of a negative control ethambutol (A) and a positive control SQ109 (B) as well as representative series compound, 4 (C) and 23 (D) are recorded relative to DMSO-treated samples. Data are representative of two independent experiments.
In Vivo Analysis
Based on the hit to lead SAR, representative compound 29 was selected for follow-up. A fluorinated derivative, 35, was also prepared as such modifications had been shown previously (Table 2) to not have a detrimental effect on MIC activity but could potentially improve metabolic stability and in vivo exposure. As expected, 35 had a similar MIC activity to 29 (Table 5). Although there was no significant change in metabolic stability, neither mouse nor human, the fluorination did result in an unexpected increase in activity against the hERG ion channel. The in vivo exposure of both compounds was evaluated in female C57BL/6 mice (n = 3/dose level) (Table 5). Both compounds had moderate in vivo blood clearance, moderate volume of distribution, moderate half-life, and low bioavailability. Although the exposure of 35 was greater than 29, it was not substantially improved enough to make it worthwhile to risk the increase in hERG channel inhibition. Therefore, further work focused on 29. As the initial preliminary pharmacokinetic analysis was done on the free-base form of the molecule, a hydrochloride salt form of 29 was prepared and evaluated at higher doses to determine whether the compound was suitable for an in vivo efficacy assessment. The HCl salt of 29 was dosed at 200 and 400 mg/kg in 1% carboxymethylcellulose (CMC) generating Cmax values of 22,266 and 85,278 ng/mL, respectively. Moreover, at both the 200 and 400 mg/kg doses, the exposure, both total and free (plasma Fu = 0.2), was above the reported MIC (0.66 μM/292 ng/mL) for >6 h.
Table 5. Biology and ADME/PK Profiles for Selected Best Molecules.

| 29 | 35 | |
|---|---|---|
| MICa μM | 0.66 | 0.79 |
| hERGa IC50 μM | >30a | 10a |
| HepG2a μM | >50 | >50 |
| clog DpH7.4 | 1.2 | 1.4 |
| microsomal Cla mL/min/g | 1.1 | 0.9 |
| human micro Cla mL/min/g | 0.6 | 0.5 |
| C57 mouse PK at 3 mg/kg iv and 10 mg/kg po | ||
| Cmax po (ng/mL) | 164 | 294 |
| T1/2 (h) | 2 | 2.6 |
| AUC0–24 po (ng-min/mL) | 20,728 | 36,486 |
| Clb (mL/min/kg) | 56 | 53 |
| Vdss (L/kg) | 2 | 3 |
| % F | 12 | 19 |
Based on the results from the PK studies, 29 was assessed in an acute mouse model of TB infection using Balb/c mice in a direct comparison with the previously reported compound 3.30 As expected, 3 was very active in the study promoting a >2.5 log10 reduction in colony forming units (CFUs), reducing lung burdens in infected mice near to or below the limit of detection (Figure 3). This was similar to the control drug rifampicin given at a dose of 20 mg/kg (Figure 3). In contrast, 29 showed no appreciable reduction in lung CFUs relative to the untreated control in this experiment. Plasma samples for PK from these infected animals were taken during steady state at 1 and 24 h after dosing. Although both compounds showed free plasma concentrations well above MIC at 1 h post-dosing, only 3 remained above MIC for the full 24 h period. Thus, one potential explanation for the difference in efficacy was inadequate drug exposure above MIC for 29 compared to 3.
Figure 3.

Efficacy in a mouse model of acute TB infection. BALB/c mice were infected with M. tuberculosis H37Rv via a low-dose aerosol exposure. Treatment was started 7 days post-aerosol and continued for 12 consecutive days. Drugs were administered once daily by oral gavage at 100 mg/kg (3) and 300 mg/kg (29).
Chemistry
Synthetic Routes
Synthesis of related spirocyclic amines has been previously reported.19,39 The general synthetic routes employed for the synthesis of pyrazole containing spirocyclic amines are shown below (Scheme 1). The appropriately substituted R1 aryls or heteroaryls were reacted with 1-[(4-methoxyphenyl)methyl]piperidin-4-one to form 48, which was then deprotected to form amine 49. An alternative route involved reacting the appropriately substituted R1 aryls or heteroaryls with the protected piperidin-4-one to form diols 40–43. Cyclic dehydration afforded 44–47, which were deprotected to afford amines 50–53. Substituted pyrazole aldehydes 70–77 were prepared from the esters 63–69 (Scheme S1 in Supporting Information) by reduction to the alcohol and then oxidation to the aldehydes. R2- and R3-substituted pyrazole esters 63–69 were prepared in three main ways: from R3-substituted pyrazole 60, using copper-mediated coupling of suitable R2 boronic acids, by cyclo-dehydration of ethyl 4-(R3)-2,4-dioxo-butanoates 55–59 with R2-substituted hydrazines, and by reaction of dimethyl but-2-ynedioate with R2-substituted hydrazines. In the final step, reductive amination with appropriate R2-and R3-substituted pyrazole aldehydes 70–77 afforded target compounds 1–14, 17, 18, 20, and 21. Further Chan–Lam coupling of the NH pyrazole 78 with appropriate boronic acids afforded target compounds 15, 16, and 19.
Scheme 1. General Synthetic Routes for the Synthesis of Compounds 1–21.
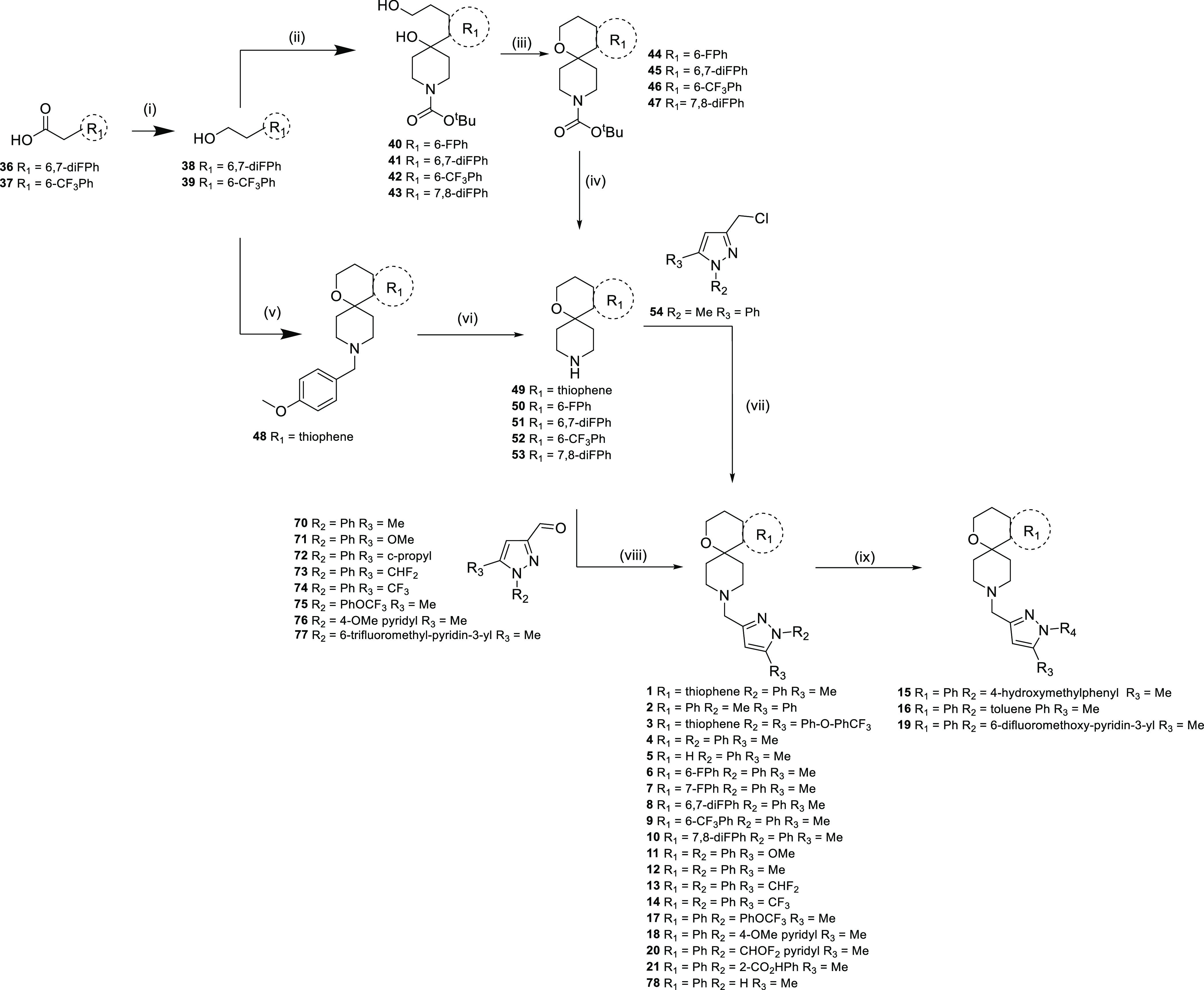
Reagents and conditions: (i) BH3.THF, THF, 100 °C, and 1 h; (ii) nBuLi, THF, −78 °C, 1 h, then tert-butyl 4-oxopiperidine-1-carboxylate −78 °C—rt, and 18 h; (iii) MeSO2Cl, Et3N, DCM, reflux, and 1.5 h; (iv) TFA, DCM, and 18 h; (v) 1-[(4-methoxyphenyl)methyl]piperidin-4-one, MeSO3H, PhMe, reflux, Dean–Stark, and 18 h; (vi) 1-chloroethyl carbonochloridate, DCM, 0 °C then MeOH, reflux, and 1 h; (vii) DIPEA, DMSO, rt, and 18 h; (viii) AcOH or EtOH, reflux, and 2 h; (ix) R2B(OH)2, Cu(OAc)2, pyridine, DCM, and 2 h; (x) Et2O, rt, 1 h, then NaOMe, MeOH, rt, 18 h, then H2SO4, MeOH, reflux, and 48 h; (xi) PPh3, DIAD, THF, MeOH, rt, and 18 h; (xii) DIBAL, DCM, −78 °C—rt, then MnO2, DCM, rt, and 48 h; (xiii) AcOH, DCM, then NaBH(OAc)3, and 18 h; (xiv) R4B(OH)2, Cu(OAc)2, pyridine, DCM, rt, and 3–18 h.
For compounds containing a carboxylic acid moiety on the phenyl ring attached to the pyrazole core, a different approach was needed. For these compounds, some could be synthesized by carrying the carboxylic acid through the synthesis protected as an oxazoline (Scheme 2) and some by a late-stage conversion from the bromide (Scheme 3). Carboxylic acid substituted phenyl hydrazines could be reacted with suitable triketo compounds to afford R3-substituted pyrazole esters 87, 88, and 90–92. Because of carrying an ester in these molecules, ester protection of the acid was not a plausible route at this stage, and so to synthetically distinguish the different functional groups, the acids were protected as oxazolines. Amide coupling with ethanolamine followed by chlorination and cyclization easily provided oxazolines 93–98, which could then be carried through the synthesis without problem. Reduction to aldehydes 99–104 as before followed by reductive amination to 105–109 and simple deprotection with 2 M HCl afforded the target compounds 22, 23, and 26–28. Compound 24 followed the majority of this route; however, the carboxylate 95 was synthesized via reaction of 4-aminobenzoic acid with ethyl 2-chloro-3-oxo-butanoate and subsequent cyclization to the pyrazole. Alternatively, when the substituted phenyl hydrazine could be reacted with R3-substituted diethoxy diketo compounds 81–83, then only one synthetic step was needed to afford compounds 25, 29, 30, and 35.
Scheme 2. General Synthetic Routes for the Synthesis of Compounds 22–30 and 35.
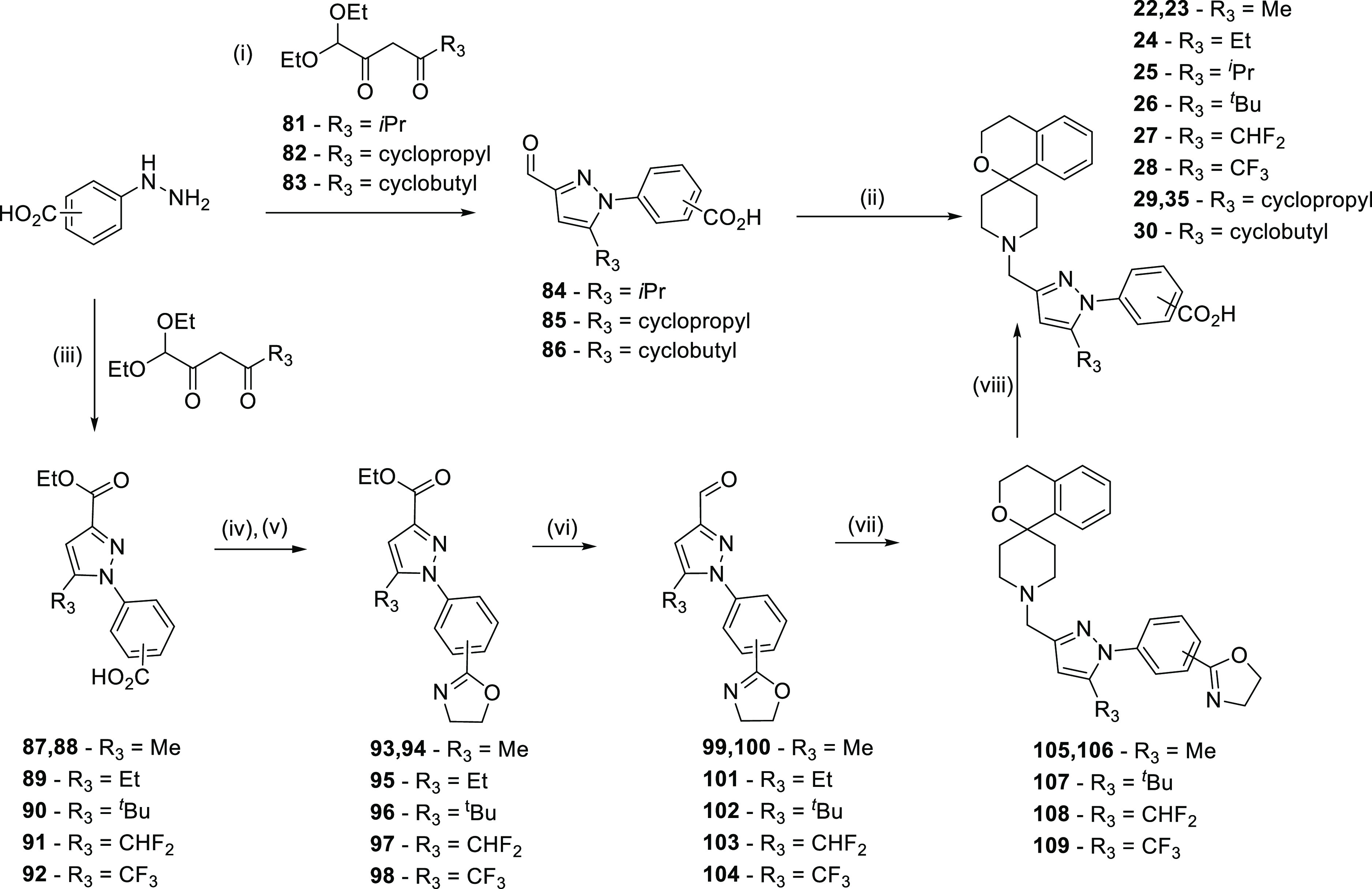
Reagents and conditions: (i) EtOH, AcOH, reflux, and 1–18 h; (ii) spiro[isochromane-1,4′-piperidine], AcOH, DCM, then NaBH(OAc)3, and 18 h; (iii) EtOH, H2O, 35 °C, and 18 h; (iv) SOCl2, pyridine, 50 °C, 2 h then ethanolamine, DCM, rt, 3 h, then SOCl2, rt, and 18 h; (v) NaH, THF, 0 °C, and 3 h; (vi) DIBAL, DCM, −78 °C—rt, then MnO2, DCM, rt, and 48 h; (vii) spiro[isochromane-1,4′-piperidine], AcOH, DCM, then NaBH(OAc)3, 18 h; and (viii) 3 M HCl, 100 °C, and 18 h.
Scheme 3. General Synthetic Routes for the Synthesis of Compounds 31–34.
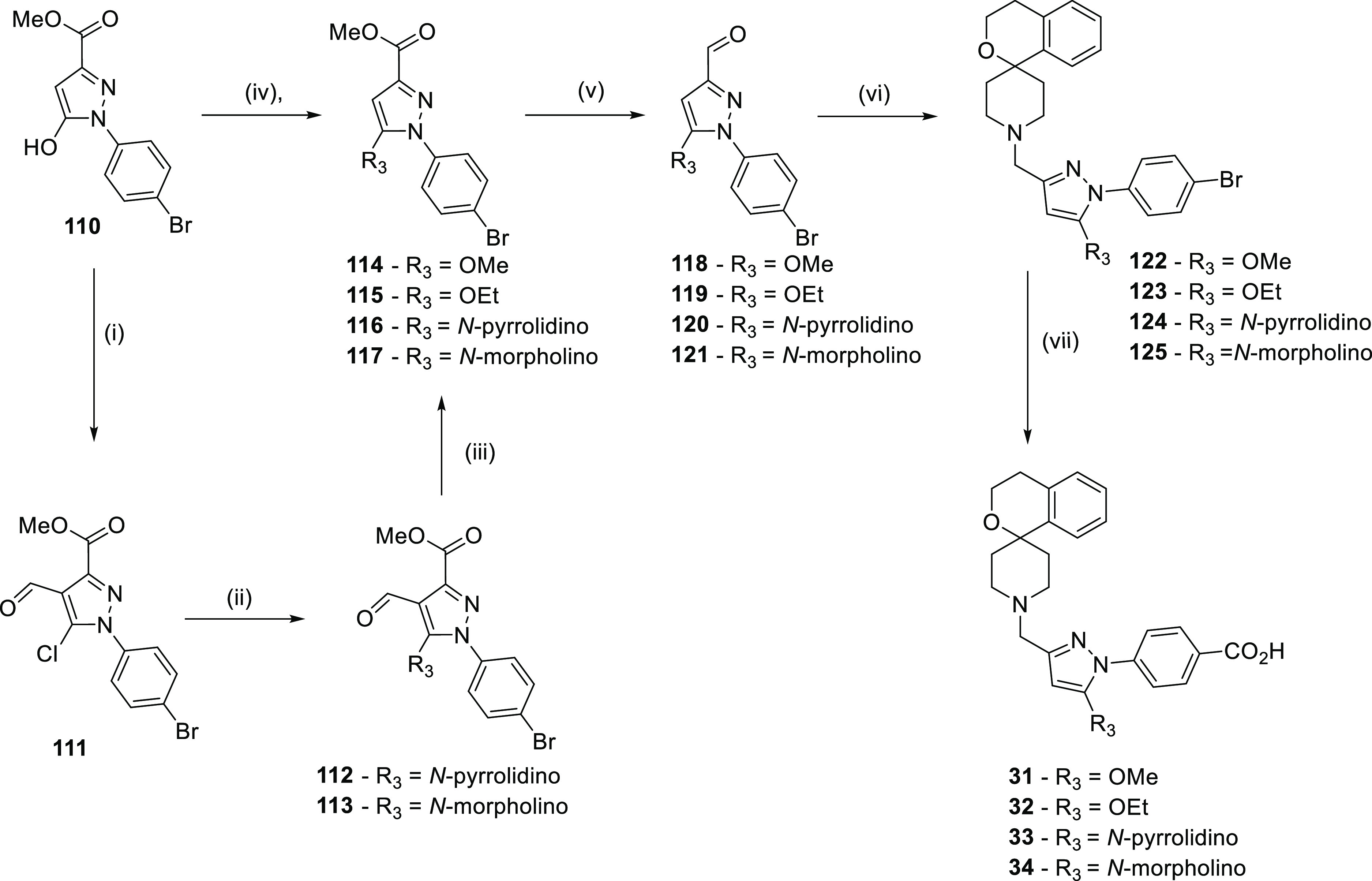
Reagents and conditions: (i) POCl3, DMF, 100 °C, and 2 h; (ii) R3H, K2CO3, DMF, microwave, 120 °C, and 1 h; (iii) pTsOH, MeOH, microwave, 120 °C, and 1 h; (iv) R3I, K2CO3, DMF, 0 °C—rt, and 2 h; (v) DIBAL, DCM, −78 °C—rt, then MnO2, DCM, rt, and 48 h; (vi) AcOH, DCM, then NaBH(OAc)3, and 18 h; and (vii) N-formylsaccharin, Pd(OAc)2, Xantphos, KF, DMF, 80 °C, and 18 h then H2O.
The carboxylic acid compounds could also be realized from the bromides. Reaction of 4-bromophenylhydrazines with dimethyl but-2-ynedioate afforded pyrazole 110. Where the desired R group was an alkoxy group, the hydroxyl moiety could be simply alkylated. Where R3 was an amine, this was introduced via a Vilsmeier reaction, followed by SNAr and removal of the aldehyde, to form compounds 114–117. As in the previous schemes, reduction to aldehydes 118–121 followed by reductive amination afforded spirocyclic amino pyrazoles 122–125. In order to convert the bromide to carboxylic acid, compounds 122–125 were carbonylated using N-formylsaccharin as a source of carbon monoxide, which is slowly released in situ as described in the literature.40 The acyl fluoride formed in the reaction as a result of the caesium fluoride base was quenched with H2O to form the final target compounds 31–34. For all the compounds discussed in this report, no alerts were found when they were processed through a PAINS filter.
Conclusions
Following on from the previous publication of a spirocycle series with potent inhibition of M. tuberculosis but limited by safety concerns,30 the optimization of a related but novel pyrazole spirocyclic amine is described. This report identified a zwitterionic series, exemplified by lead 29 that when compared to the original series, 3 had improved selectivity over HepG2, as well as reduced hERG channel inhibition. As with the original series, 29 was a putative MmpL3 inhibitor as large shifts against MmpL3 mutant strains were observed. Although 29 has a better selectivity profile than the earlier molecules, this came at the expense of reduced potency; as such, while 3 had excellent activity in an acute invivo model of TB infection, 29 was found not to be efficacious in the same model. The lack of efficacy of 29 was hypothesized to be as a result of the need for improved in vivo exposure to ensure maximum coverage above the MIC. Thus, despite the original authors highlighting the possibility of eliminating series liabilities while retaining the remarkable in vivo potency,30 given the complexity of balancing metabolic stability and hERG channel inhibition with MIC activity for this series, no further work was planned to try and improve exposure.
Experimental Section
Determination of MIC
MICs were determined against M. tuberculosis H37Rv (ATCC 25618) and mutant strains grown in Middlebrook 7H9 medium containing 10% v/v OADC (oleic acid, albumin, dextrose, and catalase) supplement (Becton Dickinson) and 0.05% w/v Tween 80 (7H9-Tw-OADC) under aerobic conditions as previously described. Bacterial growth was measured after 5 days of incubation at 37 °C.41
MmpL3 Hypomorph
Wild-type H37Rv and P606-5C (in which the native copy of mmpL3 was replaced with kanR and a tetracycline-regulated copy of mmpL3 was inserted at the att-L5 site and also contains zeoR) were each cultured in 10 mL of Middlebrook 7H9 [with kanamycin and zeocin at 25 μg/mL and anhydrotetracycline (atc) at 500 ng/mL for the mutant strain] supplemented with 0.2% (v/v) glycerol, 0.05% (v/v) tyloxapol, and ADNaCl (0.5% [w/v] BSA, 0.2% [w/v] dextrose, and 0.85% [w/v] NaCl) in a 25 cm2 tissue culture flask with a vented cap. After approximately 7 days at 37 °C and 5% CO2 in a humidified incubator, growing to mid-log to late-log phase, each of the cultures was washed with fresh 7H9 and suspended to an OD580 of 0.05 in 30 mL of 7H9 (with selection antibiotics for the mutant but without atc to deplete the levels of MmpL3) in a 75 cm2 tissue culture flask with a vented cap and was incubated for further 14 days with a passage to OD580 = 0.05 in 30 mL of 7H9 at day 7. Bacteria were then washed once in fresh medium and single-cell suspensions were prepared to a final OD580 of 0.01 in 7H9. Compounds were solubilized in DMSO and dispensed into 384-well plates using an HP D300e Digital Dispenser as 11-point, 4-fold dilution series in triplicate. DMSO at a final concentration of 1% was used as no-drug control. In all, 50 μL of single-cell suspension was pipetted to each well and cultures were incubated for 7–14 d at 37 °C in the same conditions as mentioned above. Final OD580 values were normalized to no-drug control.
M. tuberculosis Kill Kinetics
For replicating conditions, late-log phase bacteria were exposed to compounds in 5 mL medium under aerobic conditions in standing cultures over 21 days. For starvation conditions, bacteria were resuspended in phosphate-buffered saline plus 0.05% tyloxapol for 14 days before compound addition. Viable bacteria were measured by plating serial dilutions and counting cfus after 4 weeks.
Intracellular Activity
THP-1 cells were propagated in RPMI-1640 supplemented with 10% FBS, 2 mM glutagro, and 1 mM sodium pyruvate medium in a humidified atmosphere of 37 °C with 5% CO2. Cells were differentiated with 80 nM PMA treatment overnight and infected with M. tuberculosis (H37Rv LuxG13) at a multiplicity of infection of 1. Infected cells were harvested with Accutase, 5 mM EDTA, washed twice with PBS, seeded into 96-well plates at 4 × 104 cells per well, and incubated for 24 h. Compounds were added as a 10-point three-fold serial dilution (0.5% DMSO final concentration). Bacterial viability was measured after 72 h. Growth inhibition curves were fitted using the Levenberg–Marquardt algorithm. IC50 and IC90 were defined as the compound concentrations that inhibited 50 or 90% of the intracellular growth, respectively.
THP-1 Cytotoxicity
THP-1 cells were propagated, differentiated into macrophages, and harvested as described above and seeded into 96-well plates at 4 × 104 cells per well for 24 h. Compounds were added as a 10-point three-fold serial dilutions (0.5% DMSO final concentration). Viability was measured after 72 h using CellTiter-Glo. Growth inhibition curves were fitted using the Levenberg–Marquardt algorithm. IC50 was defined as the compound concentration that reduced cell viability by 50%.
HepG2 Cytotoxicity
Compound dilution curves were plated directly using a Labcyte Echo 550 acoustic dispenser (125 nL) in 384-well white clear-bottomed plates (Greiner). HepG2 cells (ECACC 85011430) were cultured in minimum essential medium (supplemented with glutamax) with 10% FCS and plated (25 μL) using a WellMate dispenser (1 × 105 per well) and incubated for 72 h. Doxorubicin was used as a positive control drug. Resazurin was then added to each well at a final concentration of 45 μM, and fluorescence was measured using PHERAstar LS (BMG Labtech) after 4 h of further incubation (excitation of 528 nm and emission of 590 nm). Raw data were normalized to controls and expressed as % growth. IC50 was defined as the compound concentration that resulted in 50% inhibition.
Intrinsic Clearance (Cli) Experiments
Test compound (0.5 μM) was incubated with female CD1 mouse liver microsomes (Xenotech LLC) or pooled human liver microsomes (Life Technologies) at a final concentration of 0.5 mg/mL 50 mM potassium phosphate buffer, pH 7.4, and the reaction started with addition of excess NADPH (8 mg/mL 50 mM potassium phosphate buffer, pH 7.4). Immediately, at time zero, then at 3, 6, 9, 15, and 30 min, an aliquot (50 μL) of the incubation mixture was removed and mixed with acetonitrile (100 μL) to stop the reaction. Internal standard was added to all samples, the samples were centrifuged to sediment precipitated protein, and the plates were then sealed prior to ultra-performance liquid chromatography–mass spectrometry (UPLC/MS/MS) analysis using a Quattro Premier XE (Waters Corporation, USA). XLfit (IDBS, UK) was used to calculate the exponential decay and consequently the rate constant (k) from the ratio of peak area of test compound to internal standard at each time point. The rate of intrinsic clearance (CLi) of each test compound was then calculated using the following calculation
where V (mL/mg protein) is the incubation volume/mg protein added and microsomal protein yield is taken as 52.5 mg protein/g liver. Verapamil (0.5 μM) was used as a positive control to confirm acceptable assay performance.
Isoform-specific metabolism studies were performed as mentioned above except that mouse liver microsomes were replaced with incubation mixtures containing EasyCYP bactosomes (50 pmol/mL, 0.5 mg/mL Cypex).
Aqueous Solubility
The aqueous solubility of the test compounds was measured using laser nephelometry. Compounds were subject to serial dilution from 10 to 0.5 mM in DMSO. An aliquot was then mixed with Milli-Q water to obtain an aqueous dilution plate with a final concentration range of 250–12 μM with a final DMSO concentration of 2.5%. Triplicate aliquots were transferred to a flat-bottomed polystyrene plate, which was immediately read on the NEPHELOstar (BMG Lab Technologies). The amount of laser scatter caused by insoluble particulates (relative nephelometry units, RNU) was plotted against compound concentration using a segmental regression fit, with the point of inflection being quoted as the compound’s aqueous solubility (μM).
Mouse Pharmacokinetics
The test compound was dosed as a bolus solution intravenously or dosed orally by gavage to female C57 black mice (n = 3/dose level). Blood samples were collected from each mouse tail vein at predetermined time points post-dose, mixed with two volumes of distilled water, and stored frozen until UPLC/MS/MS analysis. Pharmacokinetic parameters were derived from the blood concentration time curve using PK Solutions software v2.0 (Summit Research Services, USA). All regulated procedures, at the University of Dundee, on living animals were carried out under the authority of a project license issued by the Home Office under the Animals (Scientific Procedures) Act 1986, as amended in 2012 (and in compliance with EU Directive EU/2010/63).
hERG Assays
These were performed as previously described42,43 in brief: thallium flux high-throughput testing, hERG functional activity was measured in an inducible hERG T-RExTM-CHO Cell line (Thermo Fisher #K1237) using thallium influx as a surrogate indicator of potassium ion channel activity. Thallium enhances the fluorescent signal of BTC-AM dye (Thermo Fisher #B6791). Cells were loaded with the dye for 90 min in a low potassium buffer, dye removed, and compound added to the cells in a high potassium buffer in a 6 pt. dose response. After 30 min of compound incubation, channel activity was recorded upon addition of thallium buffer using a Tetra plate reader. The slope of the kinetic read was used to calculate channel activity. For QPatch testing, HEK-293 cells stably transfected with the hERG channel were tested in either QPatch (Sophion) or PatchXpress (Molecular Devices), automated planar patch-clamp systems. The cells were added to each chamber, negative pressure was applied to obtain intracellular access, and cells were exposed to either three or four ascending concentrations of drug. hERG tail current was measured as the difference in amplitude between a −50 mV pre-pulse and the end of a five-second test pulse to −50 mV, preceded by a depolarizing pulse (+20 mV). The hERG IC50 value was determined from tail current.
Murine Model of Acute TB Infection
All infections were performed at Colorado State University in a certified ABSL3 facility in accordance with the guidelines of the Colorado State University Institutional Animal Care and Use Committee. Six to eight week old female specific pathogen-free BALB/c mice were purchased from Charles River Laboratories (Wilmington, MA). The mice were infected with M. tuberculosis H37Rv via a low-dose aerosol exposure in a Glas-Col aerosol generation device (Glas-Col Inc., Terre Haute, IN).44 Each treatment group consisted of six mice. 29 was formulated in 1% CMC (Sigma). 3 was formulated in 1% MC (Sigma). Rifampin was prepared using a mortar and pestle in sterile water. The treatment was started 7 days post-aerosol and continued for 12 consecutive days. Drugs were administered by oral gavage in a 0.2 mL volume at 300, 100, and 20 mg/kg for 29, 3, and rifampicin, respectively. For endpoint analysis, mice were euthanized 3 days following the end of treatment and lungs were collected. The left lung lobe (1/3rd of the lung by weight) was homogenized for enumeration of cfu by plating dilutions of the organ homogenates on Middlebrook 7H11 medium supplemented with 10% (v/v) OADC, 0.01 mg/mL of cycloheximide, and 0.05 mg/mL of carbenicillin. The data were expressed as mean log 10 cfu ± the standard error of the mean for each group. Statistical analysis was done by one-way analysis of variance with Dunnet’s post-test to control for multiple comparisons (SigmaPlot, San Jose, CA). Values were considered significant at the 95% confidence level.
General Chemistry Methods
Chemicals and solvents were purchased from commercial vendors and used as received unless otherwise stated. Dry solvents were purchased in Sure Seal bottles stored over molecular sieves. Unless otherwise stated herein, reactions have not been optimized. Analytical thin-layer chromatography (TLC) was performed on precoated TLC plates (Kieselgel 60 F254, BDH). Developed plates were air-dried and analyzed under a UV lamp (UV 254/365 nm) and/or KMnO4 was used for visualization. Flash chromatography was performed using Combiflash Companion Rf (Teledyne ISCO) and prepacked silica gel columns purchased from Grace Davison Discovery Science or SiliCycle. Mass-directed preparative HPLC separations were performed using a Waters HPLC (2545 binary gradient pumps, 515 HPLC make-up pump, and 2767 sample manager) connected to a Waters 2998 photodiode array and a Waters 3100 mass detector. Preparative HPLC separations were performed with a Gilson HPLC (321 pumps, 819 injection module, and 215 liquid handler/injector) connected to a Gilson 155 UV/vis detector. On both instruments, HPLC chromatographic separations were conducted using Waters XBridge C18 columns, 19 mm × 100 mm, 5 μm particle size, using 0.1% ammonia in water (solvent A) and acetonitrile (solvent B) as the mobile phase. 1H NMR spectra were recorded on a Bruker ADVANCE II 500 or 400 spectrometer operating at 500 and 400 MHz (unless otherwise stated) using CDCl3, DMSO-d6, or CD3OD solutions. Chemical shifts (δ) are expressed in ppm recorded using the residual solvent as the internal reference in all cases. Signal splitting patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m), broadened (br), or a combination thereof. Coupling constants (J) are quoted to the nearest 0.1 Hertz (Hz). Low-resolution electrospray mass spectra were recorded on a Bruker Daltonics MicrOTOF mass spectrometer run in the positive mode. High-resolution mass spectroscopy (HRMS) was performed using a Bruker Daltonics MicroTof mass spectrometer. LC–MS analysis and chromatographic separation were conducted with either a Bruker Daltonics MicroTOF mass spectrometer connected to an Agilent diode array detector or a Thermo Dionex Ultimate 3000 RSLC system with a diode array detector, the column used was a Waters XBridge column (50 mm × 2.1 mm, 3.5 μm particle size), and the compounds were eluted with a gradient of 5–95% acetonitrile/water + 0.1% ammonia or with an Advion Expression Mass Spectrometer connected to a Thermo Dionex Ultimate 3000 HPLC with a diode array detector, the column used was a Waters XBridge column (50 mm × 2.1 mm, 3.5 μm particle size), or a Waters X-select column (30 mm × 2.1 mm, 2.5 μm particle size) with a gradient of 5–90% acetonitrile/water + 0.1% formic acid. All final compounds showed chemical purity of ≥95% as determined by the UV chromatogram (190–450 nm) obtained by LC–MS analysis.
General Method 1
Respective spirocyclic amines (1 equiv) and aldehydes (1 equiv) were combined in DCM (0.1–0.2 M), one drop of acetic acid was added, and the mixtures were stirred at rt for 1 h. Sodium triacetoxyborohydride (1.5–2 equiv) was added and the mixtures were stirred at rt for 18 h. The mixtures were washed with NaHCO3 (saturated solution) and the organics were concentrated in vacuo. The crude materials were purified by preparative HPLC (XBridge column, 0.1% NH4OH modifier) and the fractions were concentrated in vacuo to afford the titled compounds.
General Method 2
Respective pyrazoles (1 equiv), boronic acids (2 equiv), and copper acetate (1.5 equiv) were combined in DCM (0.1 M) and pyridine (20 equiv) and stirred at rt for 3–18 h. The solvents were removed in vacuo and the crude products were purified by either flash chromatography (0–100% EtOAc in heptane) or preparative HPLC (XBridge column, 0.1% NH4OH modifier), and the fractions were concentrated in vacuo to afford the titled compounds.
General Method 3a
Respective hydrazines (1 equiv) and 2,4-dioxo-butanoates (1 equiv) were dissolved in EtOH (0.15 M) and heated to reflux for 1–3 h. The solvent was removed in vacuo and H2O was added. The products were extracted with EtOAc and concentrated in vacuo, and the resulting crude products were purified by flash chromatography (5–50% EtOAc in heptane) to afford the titled compounds.
General Method 3b
Respective hydrazines or hydrazine hydrochlorides (1 equiv) and 2,4-dioxo-butanoates (1 equiv) were dissolved in AcOH (0.7 M), and the reaction mixtures were heated to reflux for 2 h and then allowed to cool to rt. The solvent was removed in vacuo, and the residue was partitioned between DCM and 1 M NaOH and then passed through a hydrophobic frit. The crude materials were purified by flash chromatography (0–100% EtOAc in heptane) to afford the titled compounds.
General Method 4
Oxazolines (1 equiv) were dissolved in 3 M HCl (30 equiv) and heated to 100 °C for 18 h and then allowed to cool to rt. The reaction mixtures were neutralized with NaHCO3 (saturated solution) and extracted with DCM. The organics were separated, dried through a hydrophobic frit, and concentrated in vacuo to afford the titled compounds.
General Method 5
Bromides (1 equiv), aldehydes (1.2 equiv), Pd(OAc)2 (3 mol %), Xantphos (4.5 mol %), and KF (2.5 equiv) were combined in DMF (0.12 M) in sealed tubes. The reaction mixtures were heated to 80 °C for 18 h. The reactions were quenched with H2O and stirred at rt for 10 min. DCM was added and the mixtures were filtered through Celite and washed with further DCM. The combined organics were concentrated in vacuo and purified by preparative HPLC (XBridge column, 0.1% NH4OH modifier), and the fractions were concentrated in vacuo to afford the titled compounds.
General Method 6a
Esters (1 equiv) were dissolved in DCM (0.2–0.4 M) and cooled to −78 °C under N2. DIBAL (2.5 equiv, 1 M in DCM) was added dropwise over 20 min, and the reactions were stirred for 1 h at −78 °C. The reaction mixtures were quenched carefully with H2O and MeOH (1:1, 10 mL) and allowed to warm to rt. DCM was added and the mixtures were passed through hydrophobic frits. The organics were concentrated in vacuo, and the crude products were purified by flash chromatography (0–100% EtOAc in heptane) to afford the titled compounds.
General Method 6b
Esters (1 equiv) were dissolved in DCM (0.2–0.4 M) and cooled to −78 °C under N2. DIBAL (2.5 equiv, 1 M in DCM) was added dropwise over 20 min and the reactions were stirred for 1 h at −78 °C. The reaction mixtures were quenched carefully with H2O and MeOH (1:1, 10 mL) and allowed to warm to rt. DCM was added and the mixtures were passed through hydrophobic frits. The organics were concentrated in vacuo to afford the alcohols. The residues were redissolved in DCM and MnO2 (10 equiv) was added. The reaction mixtures were stirred at rt for 18 h, then filtered through Celite cartridges, and washed well with DCM and MeOH. The solvents were removed in vacuo to afford the titled compounds.
Synthesis of 1-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)-4′,5′-dihydrospiro[piperidine-4,7′-thieno[2,3-c]pyran] (1) (Scheme 1)
5-Methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70)
Ethyl 5-methyl-1-phenyl-1H-pyrazole-3-carboxylate (2.00 g, 8.69 mmol) and DIBAL (1 M in DCM) (21.72 mL, 21.72 mmol) were combined in DCM (20 mL) according to General Method 6a. Work-up and purification afforded the title compound as a pale yellow oil (678 mg, 41%). LCMS (ESI) m/z: 189 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.50–7.42 (m, 4H), 7.42–7.37 (m, 1H), 6.22 (s, 1H), 4.72 (d, J = 5.9 Hz, 2H), 2.34 (s, 3H), 2.10 (br s, 1H).
1-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)-4′,5′-dihydrospiro[piperidine-4,7′-thieno[2,3-c]pyran] (1)
5-Methyl-1-phenyl-pyrazole-3-carbaldehyde (70) (40 mg, 0.21 mmol), spiro[4,5-dihydrothieno[2,3-c]pyran-7,4′-piperidine] (43 mg, 0.20 mmol), and sodium triacetoxyborohydride (91 mg, 0.43 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound (30 mg, 35%). HRMS (ESI) m/z: calcd for C22H26N3OS [M + H+], 380.1791; found, 380.1794. 1H NMR (500 MHz, CDCl3): δ 7.47 (d, J = 4.4 Hz, 4H), 7.39–7.35 (m, 1H), 7.15 (d, J = 5.0 Hz, 1H), 6.78 (d, J = 5.0 Hz, 1H), 6.24 (s, 1H), 3.94 (dd, J = 5.5, 5.5 Hz, 2H), 3.67 (s, 2H), 2.88 (d, J = 11.6 Hz, 2H), 2.72 (dd, J = 5.5, 5.5 Hz, 2H), 2.58–2.51 (m, 2H), 2.36 (s, 3H), 2.07–2.03 (m, 4H).
Synthesis of 1′-((1-Methyl-5-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (2) (Scheme 1)
Spiro[isochromane-1,4′-piperidine] (98 mg, 0.48 mmol), 3-(chloromethyl)-1-methyl-5-phenyl-1H-pyrazole (54) (50 mg, 0.24 mmol), and diisopropylethylamine (93 mg, 0.73 mmol) were combined in DMSO (1 mL) and stirred at rt for 18 h. Water (0.3 mL) was added, the mixture was purified by preparative HPLC (XBridge column, 0.1% NH4OH modifier), and the fractions were concentrated in vacuo to afford the title compound (40 mg, 42%). HRMS (ESI) m/z: calcd for C24H28N3O [M + H+], 374.2232; found, 374.2180. 1H NMR (500 MHz, CDCl3): δ 7.46–7.40 (5H, m), 7.22–7.12 (3H, m), 7.08 (1H, d, J = 7.3 Hz), 6.31 (1H, s), 3.91 (2H, t, J = 5.5 Hz), 3.88 (3H, s), 3.64 (2H, s), 2.90–2.87 (2H, m), 2.83 (2H, t, J = 8.1 Hz), 2.55–2.48 (2H, m), 2.14–2.06 (2H, m), 1.91 (2H, d, J = 11.8 Hz).
Synthesis of 1-(4-(4-(Trifluoromethoxy)phenoxy)benzyl)-6′,7′-dihydrospiro[piperidine-4,4′-thieno[3,2-c]pyran] (3) (Scheme 1)
1-(4-Methoxybenzyl)-6′,7′-dihydrospiro[piperidine-4,4′-thieno[3,2-c]pyran] (48)
1-[(4-Methoxyphenyl)methyl]piperidin-4-one (1.71 g, 7.80 mmol) and 2-(thiophen-2-yl)ethan-1-ol (1.00 g, 7.80 mmol) were mixed in toluene (70 mL) and methanesulfonic acid (1 mL, 15.60 mmol) was added. The reaction mixture was heated to 130 °C in Dean–Stark apparatus for 18 h. The reaction mixture was allowed to cool to rt and toluene was removed in vacuo. The residue was dissolved in MeOH and loaded onto a 20 g SCX cartridge, eluting with MeOH and 3 M NH3/MeOH. The crude material was purified further by flash chromatography (0–8% MeOH in DCM with NH3 modifier) to afford the title compound (940 mg, 35%). 1H NMR (500 MHz, CDCl3): δ 6.73 (d, J = 2.7 Hz, 3H), 6.55 (d, J = 1.7 Hz, 1H), 6.35 (dd, J = 10.8, 10.8 Hz, 2H), 6.31–6.28 (m, 1H), 4.77 (d, J = 2.4 Hz, 1H), 3.43–3.38 (m, 2H), 3.29 (d, J = 2.3 Hz, 3H), 2.98 (s, 2H), 2.30 (dd, J = 5.0, 5.0 Hz, 2H), 2.19–2.16 (m, 2H), 1.44 (dd, J = 12.1, 12.1 Hz, 2H), 1.32 (d, J = 12.8 Hz, 2H).
6′,7′-Dihydro-1,2-spiro[piperidine-4,4′-thieno[3,2-c]pyran] (49)
1′-[(4-Methoxyphenyl)methyl]spiro[6,7-dihydrothieno[3,2-c]pyran-4,4′-piperidine (48) (940 mg, 2.85 mmol) was dissolved in DCM (15 mL) under N2 and cooled to 0 °C. 1-Chloroethyl carbonochloridate (530 mg, 3.71 mmol) in DCM (3 mL) was then added dropwise, and the reaction mixture was stirred at 0 °C for 30 min and then allowed to warm up to rt. The volatiles were removed in vacuo, and the residue was dissolved in MeOH (15 mL) and heated to reflux for 1 h. After cooling to rt, the reaction mixture was passed through a 20 g SCX cartridge, eluting the amine with 3 M NH3/MeOH to afford the title compound as a pale orange gum, which solidified on standing at rt (480 mg, 76%). 1H NMR (400 MHz, CDCl3): δ 7.10 (d, J = 5.2 Hz, 1H), 6.83 (d, J = 5.2 Hz, 1H), 3.97 (dd, J = 5.4, 5.4 Hz, 2H), 3.11–3.03 (m, 2H), 2.95–2.83 (m, 4H), 1.88–1.83 (m, 4H).
1′-[[4-[4-(Trifluoromethoxy)phenoxy]phenyl]methyl]spiro[6,7-dihydrothieno[3,2-c]pyran-4,4′-piperidine] (3)
4-(Trifluoromethoxy)phenol (1.0 g, 5.61 mmol) and 4-fluorobenzaldehyde (697 mg, 5.61 mmol) were mixed in DMF (10 mL) and K2CO3 (931 mg, 6.74 mmol) was added. The reaction mixture was heated at 140 °C for 8 h and then allowed to cool to rt. H2O was added and the product was extracted with Et2O. The combined organics were washed with brine, dried (MgSO4), and concentrated in vacuo to afford 4-(4-(trifluoromethoxy)phenoxy)benzaldehyde as a yellow oil (1.40 g, 80%). 1H NMR (500 MHz, CDCl3): δ 9.98 (s, 1H), 7.90 (d, 2H), 7.30 (m, 2H), 7.11 (m, 4H). 4-[4-(Trifluoromethoxy)phenoxy]benzaldehyde (60 mg, 0.21 mmol), 6′,7′-dihydro-1,2-spiro[piperidine-4,4′-thieno[3,2-c]pyran] (49) (45 mg, 0.21 mmol), and sodium triacetoxyborohydride (90 mg, 0.43 mmol) were combined in DCM (3 mL) according to General Method 1. Work-up and purification afforded the title compound (52 mg, 51%). LCMS (ESI) m/z: 476 [M + H]+. HRMS (ESI) m/z: calcd for C25H25F3NO3S [M + H+], 476.1507; found, 476.1489. 1H NMR (500 MHz, CDCl3): δ 7.34 (d, J = 8.6 Hz, 2H), 7.19–7.15 (m, 2H), 7.07 (d, J = 5.2 Hz, 1H), 7.02–6.96 (m, 4H), 6.81 (d, J = 5.2 Hz, 1H), 3.93 (t, J = 5.4 Hz, 2H), 3.54 (s, 1H), 2.83 (t, J = 5.4 Hz, 2H), 2.75–2.70 (m, 2H), 2.40 (td, J = 12.1, 2.6 Hz, 2H), 1.97 (td, J = 13.4, 4.4 Hz, 2H), 1.89–1.83 (m, 2H).
Synthesis of 1′-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (4) (Scheme 1)
Spiro[isochromane-1,4′-piperidine] (200 mg, 0.98 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (183 mg, 0.98 mmol), and sodium triacetoxyborohydride (417 mg, 1.97 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound (244 mg, 65%). LCMS (ESI) m/z: 374 (M + H)+. HRMS (ESI) m/z: calcd for C24H28N3O [M + H+], 374.2227; found, 374.2235. 1H NMR (400 MHz, CDCl3): δ 7.47–7.43 (m, 4H), 7.38–7.32 (m, 1H), 7.23–7.11 (m, 3H), 7.10–7.06 (m, 1H), 6.22 (s, 1H), 3.90 (t, J = 5.6 Hz, 2H), 3.66 (s, 1H), 2.89–2.80 (m, 4H), 2.52 (t, J = 12.1 Hz, 2H), 2.34 (d, J = 0.7 Hz, 3H), 2.08 (td, J = 13.2, 4.5 Hz, 2H), 1.95–1.87 (m, 2H).
Synthesis of 9-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)-1-oxa-9-azaspiro[5.5]undecane (5) (Scheme 1)
1-Oxa-9-azaspiro[5.5]undecane hydrochloride (50 mg, 0.26 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (51 mg, 0.27 mmol), and sodium triacetoxyborohydride (110 mg, 0.52 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 326 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.45–7.42 (m, 3H), 7.36–7.32 (m, 1H), 6.17 (s, 1H), 3.63 (t, J = 5.2 Hz, 2H), 3.58 (s, 2H), 2.63 (m, 2H), 2.44 (m, 2H), 2.32 (d, J = 0.5 Hz, 3H), 1.89 (d, J = 13.4, 2H), 1.63–1.48 (m, 6H), 1.47–1.41 (m, 2H).
Synthesis of 6-Fluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (6) (Scheme 1)
tert-Butyl 4-(4-Fluoro-2-(2-hydroxyethyl)phenyl)-4-hydroxypiperidine-1-carboxylate (40)
2-(2-Bromo-5-fluorophenyl)ethan-1-ol (6.48 g, 29.58 mmol) was dissolved in THF (10 mL) and cooled to −78 °C. 2.5 M n-BuLi solution (23.67 mL, 59.17 mmol) was added dropwise and continuously stirred at −78 °C for 1 h. tert-Butyl 4-oxopiperidine-1-carboxylate (5.89 g, 29.58 mmol) was dissolved in THF (10 mL), and the solution was added dropwise to the reaction mixture. The mixture was continuously stirred at −78 °C for 1 h before allowing to warm slowly to rt over 18 h. The reaction was quenched with NH4Cl (saturated solution), EtOAc was added, and the organic layer was separated and dried (hydrophobic frit). The crude material was purified by flash chromatography (20–50% EtOAc in heptane) to afford the title compound. LCMS (ESI) m/z: 240 (M-CO2tBu + H)+. 1H NMR (500 MHz, CDCl3): δ 7.24 (dd, J = 8.9, 5.9 Hz, 1H), 6.89 (dd, J = 10.0, 2.8 Hz, 1H), 6.84 (ddd, J = 8.9, 7.8, 2.8 Hz, 1H), 3.94 (br s, 2H), 3.89–3.84 (m, 2H), 3.31–3.19 (br m, 4H), 1.94–1.86 (m, 2H), 1.82 (d, J = 12.7 Hz, 2H), 1.80–1.75 (m, 2H), 1.44 (s, 9H).
tert-Butyl 6-Fluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (44)
tert-Butyl 4-[4-fluoro-2-(2-hydroxyethyl)phenyl]-4-hydroxy-piperidine-1-carboxylate (40) (3.48 g, 10.25 mmol) and triethylamine (2.86 mL, 20.51 mmol) were combined in DCM (15 mL) and cooled to 0 °C. Methanesulfonyl chloride (0.79 mL, 10.25 mmol) was added dropwise and the reaction was allowed to warm to rt and then heated to reflux for 90 min. The reaction was then allowed to cool to rt and washed with H2O. The organics were separated and concentrated in vacuo. The crude material was purified by flash chromatography (10–50% EtOAc in heptane) to afford the title compound (1.96 g, 59%). 1H NMR (400 MHz, CDCl3): δ 7.03, (dd, J = 8.7, 5.5 Hz, 1H), 6.88 (td, J = 8.5, 2.7 Hz, 1H), 6.79 (dd, J = 9.3, 2.7 Hz, 1H), 3.99 (br s, 2H), 3.89 (t, J = 5.5 Hz, 2H), 3.13 (br s, 2H), 2.81 (t, J = 5.5 Hz, 2H), 1.90–1.76 (m, 4H), 1.49 (s, 9H).
6-Fluorospiro[isochromane-1,4′-piperidine] (50)
tert-Butyl 6-fluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (44) (1.96 g, 6.10 mmol) was dissolved in DCM (5 mL) and trifluoroacetic acid (4.17 mL, 54.50 mmol) was added. The reaction was stirred at rt for 18 h. The reaction mixture was diluted with MeOH and loaded onto an SCX cartridge eluting with MeOH and then 3 M NH3 in MeOH to elute the product. The solvents were removed in vacuo to afford the title compound (1.21 g, 90%). LCMS (ESI) m/z: 222 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.13 (dd, J = 8.7, 5.7 Hz, 1H), 6.88 (td, J = 8.7, 2.7 Hz, 1H), 6.78 (dd, J = 9.3, 2.7 Hz, 1H), 3.89 (t, J = 5.5 Hz, 2H), 3.11–3.02 (m, 2H), 2.93–2.86 (m, 2H), 2.81 (t, J = 5.5 Hz, 2H), 1.89–1.81 (m, 4H).
6-Fluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (6)
6-Fluorospiro[isochromane-1,4′-piperidine] (50) (40 mg, 0.18 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (34 mg, 0.18 mmol), and sodium triacetoxyborohydride (77 mg, 0.36 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 392 (M + H)+. HRMS (ESI) m/z: calcd for C24H27FN3O [M + H+], 392.2138; found, 392.2080. 1H NMR (500 MHz, CDCl3): δ 7.46–7.43 (m, 4H), 7.38–7.32 (m, 1H), 7.15 (dd, J1 = 8.7 Hz, J2 = 5.6 Hz, 1H), 6.87 (td, J1 = 8.6 Hz, J2 = 2.7 Hz, 1H), 6.77 (dd, J1 = 9.3 Hz, J2 = 2.7 Hz, 1H), 6.21 (s, 1H), 3.87 (t, J = 5.5 Hz, 2H), 3.65 (s, 2H), 2.85 (m, 2H), 2.80 (t, J = 5.5 Hz, 2H), 2.51 (t, J = 13.1 Hz, 2H), 2.33 (d, J = 0.6 Hz, 3H), 2.10–2.00 (m, 2H), 1.91–1.84 (m, 2H).
Synthesis of 7-Fluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (7) (Scheme 1)
7-Fluorospiro[isochromane-1,4′-piperidine] (40 mg, 0.18 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (34 mg, 0.18 mmol), and sodium triacetoxyborohydride (77 mg, 0.36 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 392 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.48–7.42 (m, 4H), 7.38–7.32 (m, 1H), 7.05–7.00 (m, 1H), 6.91 (dd, J1 = 10.3 Hz, J2 = 2.6 Hz, 1H), 6.83 (td, J1 = 8.4 Hz, J2 = 2.6 Hz, 1H), 6.22 (s, 1H), 3.87 (t, J = 5.5 Hz, 2H), 3.64 (s, 2H), 2.85 (m, 2H), 2.77 (t, J = 5.5 Hz, 2H), 2.55–2.46 (m, 2H), 2.43 (d, J = 0.6 Hz, 3H), 2.07–1.97 (m, 2H), 1.93–1.86 (m, 2H).
Synthesis of 6,7-Difluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (8) (Scheme 1)
2-(2-Bromo-4,5-difluoro-phenyl)ethanol (38)
2-(2-Bromo-4,5-difluoro-phenyl)acetic acid (36) (1.04 g, 4.14 mmol) was dissolved in THF (20 mL) and BH3.THF (1 M solution, 10.8 mL, 10.8 mmol) was added dropwise. The resultant solution was heated to 100 °C for 1 h. The solution was then allowed to cool to rt and H2O (15 mL) was carefully added until effervescence is ceased, followed by K2CO3 portionwise until effervescence is ceased. Et2O (20 mL) was then added and the layers were separated. The organic liquors were washed with NaHCO3 (saturated aqueous solution) and brine, dried (MgSO4), separated via a hydrophobic frit and then concentrated in vacuo to afford the crude product. Purification by flash chromatography (10–20% EtOAc in heptane) afforded the title compound. 1H NMR (400 MHz, CDCl3): δ 7.38 (dd, J1 = 11.9 Hz, J2 = 9.7 Hz, 1H), 7.14 (dd, J1 = 10.9 Hz, J2 = 8.2 Hz, 1H), 3.89–3.82 (m, 2H), 2.95 (t, J = 6.5 Hz, 2H), 1.62 (t, J = 5.4 Hz, 1H).
tert-Butyl 4-[4,5-Difluoro-2-(2-hydroxyethyl)phenyl]-4-hydroxy-piperidine-1-carboxylate (41)
2-(2-Bromo-4,5-difluoro-phenyl)ethanol (38) (954 mg, 4.02 mmol) was dissolved in THF (15 mL) and cooled to −78 °C. n-BuLi (2.5 M solution, 3.22 mL, 8.05 mmol) was added dropwise and the mixture was stirred at −78 °C for 1 h. tert-Butyl 4-oxopiperidine-1-carboxylate (0.80 g, 4.02 mmol) was dissolved in THF (10 mL), and the solution was added dropwise to the reaction mixture. The mixture was continuously stirred at −78 °C for 1 h before being allowed to warm slowly to rt over 18 h. The reaction mixture was then quenched with saturated aqueous ammonium chloride (10 mL), EtOAc (20 mL) was added, and then the organic layer was separated via a hydrophobic frit, dried (MgSO4), and concentrated in vacuo to afford the crude product. This product was purified by flash chromatography (20–50% EtOAc in heptane) to afford the title compound. LCMS (ESI) m/z: 358 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.11 (dd, J1 = 12.6 Hz, J2 = 8.5 Hz, 1H), 7.01 (dd, J1 = 11.2 Hz, J2 = 8.4 Hz, 1H), 4.16–3.89 (m, 4H), 3.48 (d, J = 5.2 Hz, 2H), 3.34–3.18 (m, 4H), 1.94–1.77 (m, 4H), 1.47 (s, 9H).
tert-Butyl 6,7-Difluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (45)
tert-Butyl 4-[4,5-difluoro-2-(2-hydroxyethyl)phenyl]-4-hydroxy-piperidine-1-carboxylate (41) (185 mg, 0.52 mmol) was dissolved in DCM (6 mL) and Et3N (154 μL, 1.11 mmol) was added. The reaction mixture was cooled to 0 °C and then methanesulfonyl chloride (43 μL, 0.55 mmol) was added dropwise. Once the addition was complete, the reaction was allowed to warm to rt for 45 min before heating to reflux for 90 min. The reaction mixture was allowed to cool to rt and washed with H2O. The volatiles were removed in vacuo to afford a pale yellow oil. This product was purified by flash chromatography (10–50% EtOAc in heptane) to afford the title compound. 1H NMR (500 MHz, CDCl3): δ 6.92–6.82 (m, 2H), 4.17–3.80 (m, 4H), 3.28–2.94 (m, 2H), 2.82–2.68 (m, 2H), 1.93–1.70 (m, 4H), 1.49 (s, 9H).
6,7-Difluorospiro[isochromane-1,4′-piperidine] (51)
tert-Butyl 6,7-difluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (45) (106 mg, 0.31 mmol) was dissolved in DCM (2 mL) and to the solution was added TFA (24 μL, 0.31 mmol) at rt for 18 h and then concentrated in vacuo. The residue was loaded onto a 1 g SCX cartridge using MeOH, eluting with 3 M NH3 in MeOH. The solvents were removed in vacuo to afford the title compound. LCMS (ESI) m/z: 240 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 6.96 (dd, J1 = 11.6 Hz, J2 = 7.9 Hz, 1H), 6.86 (dd, J1 = 10.7 Hz, J2 = 8.0 Hz, 1H), 3.86 (t, J = 5.5 Hz, 2H), 3.10–3.00 (m, 2H), 2.96–2.87 (m, 2H), 2.80–2.60 (m, 3H), 1.91–1.75 (m, 4H).
6,7-Difluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (8)
6,7-Difluorospiro[isochromane-1,4′-piperidine] (51) (27 mg, 0.11 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (21 mg, 0.11 mmol), and sodium triacetoxyborohydride (48 mg, 0.23 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 410 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.50–7.45 (m, 4H), 7.41–7.35 (m, 1H), 7.06–6.97 (m, 1H), 6.99–6.84 (m, 1H), 6.23 (s, 1H), 3.89 (t, J = 5.6 Hz, 2H), 3.66 (s, 2H), 2.87 (m, 2H), 2.77 (t, J = 5.4 Hz, 2H), 2.51 (td, J1 = 11.9 Hz, J2 = 2.8 Hz, 2H), 2.36 (d, J = 0.5 Hz, 3H), 2.06–1.95 (m, 2H), 1.94–1.86 (m, 2H).
Synthesis of 1′-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)-6-(Trifluoromethyl)spiro[isochromane-1,4′-piperidine] (9) (Scheme 1)
2-[2-Bromo-5-(trifluoromethyl)phenyl]ethanol (39)
2-[2-Bromo-5-(trifluoromethyl)phenyl]acetic acid (37) (850 mg, 3.00 mmol) was dissolved in THF (10 mL) and BH3.THF (1 M solution, 3.90 mL, 3.90 mmol) was added dropwise. The resultant solution was heated to 100 °C for 1 h. The solution was then allowed to cool to rt and H2O (15 mL) was carefully added until effervescence is ceased, followed by K2CO3 portionwise until effervescence is ceased. Et2O (20 mL) was then added and the layers were separated. The organics were washed with NaHCO3 (saturated aqueous solution) and brine, dried (MgSO4), separated via a hydrophobic frit, and then concentrated in vacuo to afford the crude product. This product was purified by flash chromatography (10–20% EtOAc in heptane) to afford the title compound. 1H NMR (500 MHz, CDCl3): δ 7.70–7.66 (m, 1H), 7.56–7.52 (m, 1H), 7.37–7.32 (m, 1H), 3.91 (t, J = 6.6 Hz, 2H), 3.08 (t, J = 6.6 Hz, 2H).
tert-Butyl 4-Hydroxy-4-[2-(2-hydroxyethyl)-4-(trifluoromethyl)phenyl]piperidine-1-carboxylate (42)
2-[2-Bromo-5-(trifluoromethyl)phenyl]ethanol (39) (780 mg, 2.90 mmol) was dissolved in THF (15 mL) and cooled to −78 °C. n-BuLi (2.5 M solution, 2.32 mL, 5.80 mmol) was added dropwise and the mixture was stirred at −78 °C for 1 h. tert-Butyl 4-oxopiperidine-1-carboxylate (578 mg, 2.90 mmol) dissolved in THF (10 mL) and the solution was added dropwise to the reaction mixture. The mixture was continuously stirred at −78 °C for 1 h before being allowed to warm slowly to rt over 18 h. The reaction mixture was then quenched with saturated aqueous ammonium chloride (10 mL). EtOAc (20 mL) was added and the organic layer was separated via a hydrophobic frit, dried (MgSO4), and concentrated in vacuo to afford the crude product. This product was purified by flash chromatography (20–50% EtOAc in heptane) to afford the title compound. 1H NMR (400 MHz, CDCl3): δ 7.46–7.38 (m, 3H), 4.31 (br s, 1H), 4.10–3.88 (m, 3H), 3.52–3.19 (m, 6H), 2.03–1.77 (m, 4H), 1.47 (s, 9H).
tert-Butyl 6-(Trifluoromethyl)spiro[isochromane-1,4′-piperidine]-1′-carboxylate (46)
tert-Butyl 4-hydroxy-4-[2-(2-hydroxyethyl)-4-(trifluoromethyl)phenyl]piperidine-1-carboxylate (42) (342 mg, 0.88 mmol) was dissolved in DCM (6 mL) and Et3N (245 μL, 1.76 mmol) was added. The reaction mixture was cooled to 0 °C and then methanesulfonyl chloride (68 μL, 0.88 mmol) was added dropwise. Once the addition was complete, the reaction was allowed to warm to rt for 45 min before heating to reflux for 90 min. The reaction mixture was allowed to cool to rt and washed with H2O. The organics were concentrated in vacuo to afford a pale yellow oil. This product was purified by flash chromatography (10–50% EtOAc in heptane) to afford the title compound. LCMS (ESI) m/z: 372 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.43 (d, J = 8.2 Hz, 1H), 7.36 (s, 1H), 7.19 (d, J = 8.2 Hz, 1H), 4.17–3.87 (m, 4H), 3.15 (br s, 2H), 2.93–2.83 (m, 2H), 1.93–1.78 (m, 4H), 1.49 (s, 9H).
6-(Trifluoromethyl)spiro[isochromane-1,4′-piperidine] (52)
tert-Butyl 6-(trifluoromethyl)spiro[isochromane-1,4′-piperidine]-1′-carboxylate (46) (169 mg, 0.46 mmol) was dissolved in DCM (2 mL) and to the solution was added TFA (0.5 mL, 6.53 mmol) at rt for 18 h and then concentrated in vacuo. The residue was loaded onto a 1 g SCX cartridge using MeOH, eluting with 3 M NH3 in MeOH. The solvents were removed in vacuo to afford the title compound. LCMS (ESI) m/z: 272 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.45–7.24 (m, 3H), 3.91–3.85 (m, 2H), 3.79 (br s, 1H), 3.12–3.02 (m, 2H), 2.99–2.90 (m, 2H), 2.89–2.79 (m, 2H), 2.00–1.78 (m, 4H).
1′-((5-Methyl-1-phenyl-1H-pyrazol-3-yl)methyl)-6-(trifluoromethyl)spiro[isochromane-1,4′-piperidine] (9)
6-(Trifluoromethyl)spiro[isochromane-1,4′-piperidine] (52) (31 mg, 0.11 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (70) (21 mg, 0.11 mmol), and sodium triacetoxyborohydride (48 mg, 0.23 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 442 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.50–7.42 (m, 5H), 7.40–7.32 (m, 3H), 6.24 (s, 1H), 3.93 (t, J = 5.5 Hz, 2H), 3.68 (s, 2H), 2.94–2.85 (m, 2H), 2.60–2.49 (m, 2H), 2.36 (d, J = 0.7 Hz, 3H), 2.16–2.04 (m, 2H), 1.96–1.88 (m, 2H).
Synthesis of 7,8-Difluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (10) (Scheme 1)
tert-Butyl 4-[4,5-Difluoro-2-(2-hydroxyethyl)phenyl]-4-hydroxy-piperidine-1-carboxylate (43)
2-(2-Bromo-4,5-difluoro-phenyl)ethanol (954 mg, 4.02 mmol) was dissolved in THF (15 mL) and cooled to −78 °C. n-BuLi (2.5 M solution, 3.22 mL, 8.05 mmol) was added dropwise and the mixture was stirred at −78 °C for 1 h. tert-Butyl 4-oxopiperidine-1-carboxylate (0.80 g, 4.02 mmol) dissolved in THF (10 mL) and the solution was added dropwise to the reaction mixture. The mixture was continuously stirred at −78 °C for 1 h before being allowed to warm slowly to rt over 18 h. The reaction mixture was then quenched with saturated aqueous ammonium chloride (10 mL). EtOAc (20 mL) was added and the organic layer was separated via a hydrophobic frit, dried (MgSO4), and concentrated in vacuo to afford the crude product. This product was purified by flash chromatography (20–50% EtOAc in heptane) to afford the title compound. LCMS (ESI) m/z: 358 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.02–6.93 (m, 1H), 6.87–6.80 (m, 1H), 4.00–3.75 (m, 4H), 3.61–3.04 (m, 6H), 2.47–2.31 (m, 2H), 1.68–1.56 (m, 2H), 1.46 (s, 9H).
tert-Butyl 7,8-Difluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (47)
tert-Butyl 4-[4,5-difluoro-2-(2-hydroxyethyl)phenyl]-4-hydroxy-piperidine-1-carboxylate (43) (198 mg, 0.55 mmol) was dissolved in DCM (6 mL) and Et3N (155 μL, 1.11 mmol) was added. The reaction mixture was cooled to 0 °C and then methanesulfonyl chloride (43 μL, 0.55 mmol) was added dropwise. Once the addition was complete, the reaction was allowed to warm to rt for 45 min before heating to reflux for 90 min. The reaction mixture was allowed to cool to room temperature (rt) and washed with water. The organic liquors were concentrated in vacuo to afford a pale yellow oil. This product was purified by flash chromatography (10–50% EtOAc in heptane) to afford the title compound. 1H NMR (500 MHz, CDCl3): δ 7.02–6.95 (m, 1H), 6.88–6.82 (m, 1H), 4.15–3.83 (m, 4H), 3.31–3.01 (m, 2H), 2.84–2.72 (m, 2H), 2.32–2.22 (m, 2H), 1.89–1.67 (m, 2H), 1.49 (s, 9H).
7,8-Difluorospiro[isochromane-1,4′-piperidine] (53)
tert-Butyl 7,8-difluorospiro[isochromane-1,4′-piperidine]-1′-carboxylate (47) (81 mg, 0.24 mmol) was dissolved in DCM (2 mL) and to the solution was added TFA (18 μL, 0.24 mmol) at rt for 18 h and then concentrated in vacuo. The residue was loaded onto a 1 g SCX cartridge using MeOH, eluting with 3 M NH3 in MeOH. The solvents were removed in vacuo to afford the title compound. LCMS (ESI) m/z: 240 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.01–6.92 (m, 1H), 6.86–6.79 (m, 1H), 3.86 (t, J = 5.5 Hz, 2H), 3.40 (br s, 1H), 3.17–3.05 (m, 2H), 3.01–2.90 (m, 2H), 2.80–2.71 (m, 2H), 2.35–2.22 (m, 2H), 1.85–1.76 (m, 2H).
7,8-Difluoro-1′-((5-methyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (10)
7,8-Difluorospiro[isochromane-1,4′-piperidine] (53) (26 mg, 0.11 mmol), 5-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde (20 mg, 0.11 mmol), and sodium triacetoxyborohydride (46 mg, 0.22 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound. LCMS (ESI) m/z: 410 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.48–7.42 (m, 4H), 7.38–7.31 (m, 1H), 7.02–6.92 (m, 1H), 6.85–6.79 (m, 1H), 6.24 (s, 1H), 3.85 (t, J = 5.5 Hz, 2H), 3.65 (s, 2H), 2.86–2.73 (m, 4H), 2.59–2.41 (m, 4H), 2.34 (d, J = 0.7 Hz, 3H), 1.85–1.77 (m, 2H).
Synthesis of 1′-((5-Methoxy-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (11) (Scheme 1)
Methyl 5-Hydroxy-1-phenyl-1H-pyrazole-3-carboxylate (62)
Phenylhydrazine (5.00 g, 46.24 mmol) in Et2O (100 mL) was added over 30 min to a mixture of dimethyl but-2-ynedioate (61) (6.57 g, 46.24 mmol) in Et2O (50 mL). After stirring for 1 h at rt, the reaction mixture was concentrated in vacuo and redissolved in MeOH (150 mL). This solution was added dropwise over 1 h to NaOMe (25 wt %) (3.99 mg, 184.94 mmol) in MeOH (150 mL) and the mixture was stirred at rt for 18 h. The solvent was removed in vacuo and 5 N HCl (100 mL) was added. The resulting precipitate was collected by filtration, washed with DCM and Et2O, and air-dried to afford 5-hydroxy-1-phenyl-1H-pyrazole-3-carboxylic acid (8.00 g, 76%). 1H NMR (400 MHz, DMSO): δ 7.74 (d, J = 7.5 Hz, 2H), 7.51 (dd, J = 7.9, 7.9 Hz, 2H), 7.36 (dd, J = 7.4, 7.4 Hz, 1H), 5.93 (s, 1H). 5-Oxo-1-phenyl-4H-pyrazole-3-carboxylic acid (8.00 g, 39.18 mmol) was mixed in MeOH (150 mL), and a few drops of concentrated H2SO4 were added. The reaction was heated to reflux for 48 h and then allowed to cool to rt. The volatiles were removed in vacuo and the crude material was purified by flash chromatography (50–100% EtOAc in heptane) to afford the title compound as a pale brown solid (4.00 g, 42%). 1H NMR (400 MHz, DMSO): δ 12.15 (m, 1H), 7.74 (d, J = 7.6 Hz, 2H), 7.51 (dd, J = 7.9, 7.9 Hz, 2H), 7.38 (dd, J = 7.4, 7.4 Hz, 1H), 5.97 (s, 1H), 3.81 (s, 3H).
Methyl 5-Methoxy-1-phenyl-1H-pyrazole-3-carboxylate (63)
Methyl 5-hydroxy-1-phenyl-1H-pyrazole-3-carboxylate (62) (500 mg, 2.29 mmol) was dissolved in THF (10 mL), and PPh3 (901 mg, 3.44 mmol) and MeOH (0.12 mL, 2.98 mmol) were added. The reaction mixture was cooled to 0 °C and DIAD (0.68 mL, 3.44 mmol) was added dropwise. The reaction mixture was allowed to warm to rt for 18 h. The reaction mixture was partitioned between H2O and DCM, passed through a hydrophobic frit, and concentrated in vacuo. The crude material was purified by flash chromatography (0–100% EtOAc in heptane) to afford the title compound as a yellow gum. DIAD side product coeluted but used in the next step with no further purification. 1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.1 Hz, 2H), 7.46 (dd, J = 7.8, 7.8 Hz, 2H), 7.36 (dd, J = 7.4, 7.4 Hz, 1H), 6.26 (s, 1H), 4.00 (s, 3H)3.96 (s, 3H).
5-Methoxy-1-phenyl-1H-pyrazole-3-carbaldehyde (71)
Methyl 5-methoxy-1-phenyl-pyrazole-3-carboxylate (63) (600 mg, 2.58 mmol) was mixed with THF (8 mL) and cooled to 0 °C. LiAlH4 (2.58 mL 2.58 mmol) was added dropwise over 10 min. After 1 h, the reaction was quenched with H2O (2 mL) and 2 M NaOH (2 mL). The resulting slurry was filtered through a Celite cartridge and concentrated in vacuo. The crude material was purified by flash chromatography (0–100% EtOAc in heptane) to afford (5-methoxy-1-phenyl-1H-pyrazol-3-yl)methanol as a pale yellow gum (180 mg, 32%). 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.6 Hz, 2H), 7.44 (dd, J = 7.9, 7.9 Hz, 2H), 7.29 (m, 1H) 5.74 (s, 1H), 4.69 (d, J = 5.3 Hz, 2H), 3.97 (s, 3H), 2.60 (s, 1H). (5-Methoxy-1-phenyl-pyrazol-3-yl)methanol (300 mg, 1.47 mmol) was dissolved in DCM (10 mL) and MnO2 (319 mg, 3.67 mmol) was added. The reaction mixture was heated to reflux for 48 h and then allowed to cool to rt. The reaction mixture was filtered through a Celite cartridge and washed with DCM. The filtrate was concentrated in vacuo and purified by flash chromatography (0–100% EtOAc in heptane) to afford the title compound (220 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 9.92 (s, 1H), 7.76 (d, J = 7.8 Hz, 2H), 7.51 (dd, J = 7.8, 7.8 Hz, 2H), 7.41 (dd, J = 7.4, 7.4 Hz, 1H), 6.22 (s, 1H), 4.02 (s, 3H).
1′-((5-Methoxy-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (11)
Spiro[isochromane-1,4′-piperidine] (40 mg, 0.20 mmol), 5-methoxy-1-phenyl-1H-pyrazole-3-carbaldehyde (71) (40 mg, 0.20 mmol), and sodium triacetoxyborohydride (83 mg, 0.39 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded 1′-((5-methoxy-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] as a colorless gum (40 mg, 50%). HRMS (ESI) m/z: calcd for C24H28N3O2 [M + H+], 390.2181; found, 390.2165. 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 7.6 Hz, 1H), 7.43 (dd, J = 8.0, 8.0 Hz, 1H), 7.27–7.09 (m, 2H), 3.97 (s, 2H), 3.92 (dd, J = 5.5, 5.5 Hz, 1H), 3.63 (s, 1H), 2.89–2.83 (m, 2H), 2.60–2.52 (m, 1H), 2.16–2.03 (m, 1H), 1.94 (d, J = 12.3 Hz, 1H).
Synthesis of 1′-((5-Cyclopropyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (12) (Scheme 1)
Methyl 5-Cyclopropyl-1-phenyl-1H-pyrazole-3-carboxylate (64)
Phenylhydrazine (634 mg, 5.88 mmol) was added to a solution of methyl 4-cyclopropyl-2,4-dioxo-butanoate (55) (1.00 g, 5.88 mmol) in EtOH (40 mL) according to General Method 3a. Work-up and purification afforded the title compound (1.16 g, 81%). LCMS (ESI) m/z: 243 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.63–7.59 (m, 2H), 7.51–7.48 (m, 2H), 7.44–7.40 (m, 1H), 6.50 (s, 1H), 3.92 (s, 3H), 1.81–1.74 (m, 1H), 1.03–0.97 (m, 2H), 0.82–0.76 (m, 2H).
5-Cyclopropyl-1-phenyl-1H-pyrazole-3-carbaldehyde (72)
Methyl 5-cyclopropyl-1-phenyl-1H-pyrazole-3-carboxylate (64) (1.16 g, 4.79 mmol) and DIBAL (1 M in DCM) (11.97 mL, 11.97 mmol) were combined in DCM (25 mL) according to General Method 6a. Work-up and purification afforded the title compound (719 mg, 67%). LCMS (ESI) m/z: 213 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 9.97 (s, 1H), 7.66–7.62 (m, 2H), 7.55–7.50 (m, 2H), 7.48–7.44 (m, 1H), 6.48 (s, 1H).
1′-((5-Cyclopropyl-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (12)
Spiro[isochromane-1,4′-piperidine] (48 mg, 0.24 mmol), 5-cyclopropyl-1-phenyl-1H-pyrazole-3-carbaldehyde (72) (50 mg, 0.24 mmol), and sodium triacetoxyborohydride (100 mg, 0.47 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound as a colorless gum (20 mg, 20%). LCMS (ESI) m/z: 400 (M + H)+. HRMS (ESI) m/z: calcd for C26H30N3O [M + H+], 400.2383; found, 400.2389. 1H NMR (500 MHz, CDCl3): δ 7.65–7.61 (m, 2H), 7.48–7.43 (m, 2H), 7.34 (tdd, J = 7.5, 1.5, 1.2 Hz, 1H), 7.21 (dd, J = 7.8, 1.6 Hz, 1H), 7.18 (td, J = 7.3, 1.6 Hz, 1H), 7.13 (td, J = 7.3, 1.6 Hz, 1H), 7.08 (d, 7.3 Hz, 1H), 5.99 (s, 1H)3.89 (t, J = 5.6 Hz, 2H), 3.63 (s, 2H), 2.87–2.80 (m, 4H), 2.50 (td, J = 11.9, 2.3 Hz, 2H), 2.08 (td, J = 13.2, 4.6 Hz, 2H), 1.93–1.87 (m, 2H), 1.84–1.77 (m, 1H), 1.00–0.95 (m, 2H), 0.82–0.77 (m, 2H).
Synthesis of 1′-((5-(Difluoromethyl)-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (13) (Scheme 1)
Ethyl 5-(Difluoromethyl)-1-phenyl-1H-pyrazole-3-carboxylate (65)
Ethyl 5,5-difluoro-2,4-dioxo-pentanoate (56) (1.00 g, 5.15 mmol) and phenylhydrazine (557 mg, 5.15 mmol) were dissolved in EtOH (40 mL) according to General Method 3a. Work-up and purification afforded an intermediate compound. This material was then dissolved in THF, 2 M HCl was added, and the mixture was heated to reflux for 18 h. The volatiles were removed in vacuo and the product was extracted with DCM, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound as a colorless oil (850 mg, 62%). LCMS (ESI) m/z: 267 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.55 (s, 6H), 6.63 (t, J = 53.4 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.2 Hz, 3H).
5-(Difluoromethyl)-1-phenyl-1H-pyrazole-3-carbaldehyde (73)
Ethyl 5-(difluoromethyl)-1-phenyl-1H-pyrazole-3-carboxylate (65) (187 mg, 0.70 mmol) and DIBAL (1 M in DCM) (1.76 mL, 1.76 mmol) were combined in DCM (10 mL) with MnO2 (1.22 g, 14.05 mmol) added in the second step according to General Method 6b. Work-up and purification afforded the title compound as a colorless oil (122 mg, 74%).
1′-((5-(Difluoromethyl)-1-phenyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (13)
Spiro[isochromane-1,4′-piperidine] (62 mg, 0.30 mmol), 5-(difluoromethyl)-1-phenyl-1H-pyrazole-3-carbaldehyde (73) (61 mg, 0.27 mmol), and sodium triacetoxyborohydride (116 mg, 0.55 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound as a colorless gum (10 mg, 8%). LCMS (ESI) m/z: 428 (M + H)+. HRMS (ESI) m/z: calcd for C24H26F2N3O [M + H+], 410.2053; found, 410.2054. 1H NMR (500 MHz, CDCl3): δ 7.56–7.44 (m, 5H), 7.26–7.19 (m, 2H), 7.17 (td, J = 7.0, 2.0 Hz, 1H), 7.11 (d, J = 7.5 Hz, 1H), 6.80 (s, 1H), 6.65 (t, J = 54 Hz, 3.93 (t, J = 5.5, 2H), 3.74 (s, 2H), 2.89–2.83 (m, 4H), 2.56 (t, J = 12.2 Hz, 2H), 2.11 (td, J = 13.2, 4.2 Hz, 2H), 1.94 (d, J = 13.2 Hz, 2H).
Synthesis of 1′-((1-Phenyl-5-(Trifluoromethyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (14) (Scheme 1)
Ethyl 1-Phenyl-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (66)
Phenylhydrazine (510 mg, 4.71 mmol) and ethyl 5,5,5-trifluoro-2,4-dioxo-pentanoate (57) (1.00 g, 4.71 mmol) were dissolved in EtOH (40 mL) according to General Method 3a. Work-up and purification afforded an intermediate compound. This material was then dissolved in THF, 2 M HCl was added, and the mixture was heated to reflux for 18 h. The volatiles were removed in vacuo, and the product was extracted with DCM, dried via a hydrophobic frit, and concentrated in vacuo. The crude material was purified by flash chromatography (10–50% EtOAc in heptane) to afford the title compound as a yellow oil (456 mg, 34%). LCMS (ESI) m/z: 285 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.54–7.47 (m, 5H), 7.33 (s, 1H), 4.45 (q, J = 7.2, 2H), 1.41 (t, J = 7.2, 3H).
1-Phenyl-5-(Trifluoromethyl)-1H-pyrazole-3-carbaldehyde (74)
Ethyl 1-phenyl-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (66) (417 mg, 1.47 mmol) and DIBAL (1 M in DCM) (3.67 mL, 3.67 mmol) were combined in DCM (10 mL) with MnO2 (273 mg, 3.14 mmol) added in the second step according to General Method 6b. Work-up afforded the crude product which was used without further purification.
1′-((1-Phenyl-5-(trifluoromethyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (14)
Spiro[isochromane-1,4′-piperidine] (49 mg, 0.24 mmol), 1-phenyl-5-(trifluoromethyl)-1H-pyrazole-3-carbaldehyde (74) (52 mg, 0.22 mmol), and sodium triacetoxyborohydride (92 mg, 0.43 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound as a colorless gum (20 mg, 21%). LCMS (ESI) m/z: 428 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.51–7.43 (m, 5H), 7.23–7.17 (m, 1H), 7.09 (d, J = 7.6 Hz, 1H), 6.84 (s, 1H), 3.90 (t, J = 5.5 Hz, 2H), 3.70 (s, 2H), 2.85–2.80 (m, 4H), 2.53 (td, 12.0, 2.5 Hz, 2H), 2.08 (td, 13.3, 4.4 Hz, 2H), 1.95–1.89 (m, 2H).
Synthesis of (4-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)phenyl)methanol (15) (Scheme 1)
1′-((5-Methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (78)
To 5-methyl-1H-pyrazole-3-carbaldehyde (1.35 g, 12.30 mmol) in NMP (125 mL) were added spiro[isochromane-1,4′-piperidine] (2.50 g, 12.30 mmol) and a few drops of acetic acid. After 1 h at rt, sodium triacetoxyborohydride (5.21 mg, 24.60 mmol) was added. The reaction mixture was stirred at rt for 18 h. The reaction was quenched with NaHCO3 (saturated aqueous solution), and the product was extracted with EtOAc, washed with brine, dried (Na2SO4), and concentrated in vacuo. The residue was loaded onto a 20 g SCX cartridge eluting with MeOH and then 20% 2 N NH3/MeOH. The volatiles were removed in vacuo to afford the title compound as a pale yellow solid (320 mg, 8%). The initial MeOH washings were concentrated in vacuo, and the resulting material was purified by flash chromatography (0–5% MeOH in DCM) to afford a second batch of title compound as a pale yellow solid (1.60 g, 42%). 1H NMR (400 MHz, CDCl3): δ 7.22–7.21 (m, 2H), 7.18–7.14 (m, 1H), 7.10 (d, J = 7.34 Hz), 6.02 (s, 1H), 3.91 (t, J = 5.5 Hz, 2H), 3.61 (s, 2H), 2.84 (t, J = 5.5 Hz, 2H), 2.78–2.75 (m, 2H), 2.49 (t, J = 11.5 Hz, 2H), 2.31 (s, 3H), 2.06 (td, J = 13.5, 3.8 Hz, 2H), 1.93–1.90 (m, 2H). LCMS (ESI) m/z: 298 (M + H)+.
(4-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)phenyl)methanol (15)
1′-[(5-methyl-1H-pyrazol-3-yl)methyl]spiro[isochromane-1,4′-piperidine] (78) (100 mg, 0.34 mmol), [4-(hydroxymethyl)phenyl]boronic acid (102 mg, 0.67 mmol), and copper acetate (92 mg, 0.50 mmol) were combined in DCM (3 mL) and pyridine (0.50 mL, 6.20 mmol) according to General Method 2. Work-up and purification afforded the title compound (30 mg, 21%). HRMS (ESI) m/z: calcd for C25H30N3O2 [M + H+], 404.2338; found, 404.2365. 1H NMR (400 MHz, CDCl3): δ 7.43 (s, 4H), 7.25–7.09 (m, 4H), 6.24 (s, 1H), 4.76 (s, 2H), 3.92 (dd, J = 5.5, 5.5 Hz, 2H), 3.68 (s, 2H), 2.89–2.82 (m, 4H), 2.59–2.51 (m, 3H), 2.34 (s, 3H), 2.16–2.06 (m, 2H), 1.92 (d, J = 12.2 Hz, 2H).
Synthesis of 1′-((5-Methyl-1-(p-tolyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (16) (Scheme 1)
1′-[(5-methyl-1H-pyrazol-3-yl)methyl]spiro[isochromane-1,4′-piperidine] (78) (50 mg, 0.17 mmol), p-tolylboronic acid (46 mg, 0.34 mmol), and copper acetate (46 mg, 0.25 mmol) were combined in DCM (1 mL) and pyridine (0.27 mL) according to General Method 2. Work-up and purification afforded the title compound as a yellow gum (15 mg, 22%). LCMS (ESI) m/z: 388 (M + H) +. 1H NMR (400 MHz, CDCl3): δ 7.36–7.32 (m, 2H), 7.28–7.22 (m, 3H), 7.20 (td, J = 6.83, 1.57 Hz, 1H), 7.15 (td, J = 7.32, 1.8 Hz, 1H), 7.11–7.08 (m, 1H), 6.22 (d, J = 0.5 Hz, 1H), 3.9 (t, J = 5.5 Hz, 2H), 6.37 (s, 2H), 2.90–2.83 (m, 4H), 2.53 (td, J = 12.90, 2.44 Hz, 2H), 2.41 (s, 3H), 2.33 (d, J = 0.7 Hz, 3H), 2.10 (td, J = 13.40, 4.47 Hz, 2H), 1.95–1.89 (m, 2H).
Synthesis of 1′-((5-Methyl-1-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (17) (Scheme 1)
Ethyl 5-Methyl-1-(4-(trifluoromethoxy)phenyl)-1H-pyrazole-3-carboxylate (67)
[4-(Trifluoromethoxy)phenyl]hydrazine hydrochloride (4.34 g, 18.97 mmol) and ethyl 2,4-dioxopentanoate (58) (3.00 g, 18.97 mmol) were dissolved in AcOH (25 mL) according to General Method 3b. Work-up and purification afforded the title compound (3.00 g, 48%). 1H NMR (400 MHz, CDCl3): δ 7.55–7.52 (m, 2H), 7.35 (d, J = 8.3 Hz, 2H), 6.77 (s, 1H), 4.44 (q, J = 7.1 Hz, 2H), 2.37 (s, 3H).
5-Methyl-1-(4-(trifluoromethoxy)phenyl)-1H-pyrazole-3-carbaldehyde (75)
Ethyl 5-methyl-1-[4-(trifluoromethoxy)phenyl]pyrazole-3-carboxylate (67) (2.60 g, 8.27 mmol) and DIBAL (1 M in DCM) (20.68 mL, 20.68 mmol) were combined in DCM (25 mL) according to General Method 6a. Work-up and purification afforded the title compound as a pale yellow oil (2.00 g, 85%). 1H NMR (400 MHz, CDCl3): δ 10.01 (s, 1H), 7.58–7.55 (m, 2H), 7.41 (d, J = 8.3 Hz, 2H), 6.76 (s, 1H), 2.41 (s, 3H).
1′-((5-Methyl-1-(4-(trifluoromethoxy)phenyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (17)
Spiro[isochromane-1,4′-piperidine] (250 mg, 1.23 mmol), 5-methyl-1-[4-(trifluoromethoxy)phenyl]-1H-pyrazole-3-carbaldehyde (75) (332 mg, 1.23 mmol), and sodium triacetoxyborohydride (521 mg, 2.46 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound (300 mg, 51%). HRMS (ESI) m/z: calcd for C25H27F3N3O2 [M + H+], 458.2055; found, 458.2073. 1H NMR (400 MHz, CDCl3): δ 7.52 (d, J = 8.8 Hz, 2H), 7.32 (d, J = 8.8 Hz, 2H), 7.23–7.08 (m, 4H), 6.26 (s, 1H), 3.92 (dd, J = 5.5, 5.5 Hz, 2H), 3.66 (s, 2H), 2.88–2.83 (m, 4H), 2.54 (dd, J = 11.3, 11.3 Hz, 2H), 2.37 (s, 3H), 2.15–2.05 (m, 2H), 1.94 (d, J = 12.9 Hz, 2H).
Synthesis of 1′-((1-(6-Methoxypyridin-3-yl)-5-methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (18) (Scheme 1)
Ethyl 1-(6-Methoxypyridin-3-yl)-5-methyl-1H-pyrazole-3-carboxylate (68)
Ethyl 5-methyl-1H-pyrazole-3-carboxylate (60) (500 mg, 3.24 mmol), (6-methoxypyridin-3-yl)boronic acid (992 mg, 6.49 mmol), and copper acetate (884 mg, 4.86 mmol) were combined in DCM (10 mL) according to General Method 2. Work-up and purification afforded the title compound (340 mg, 38%). 1H NMR (400 MHz, CDCl3): δ 8.23 (d, J = 2.7 Hz, 1H), 7.65 (dd, J = 2.7, 8.8 Hz, 1H), 6.85–6.81 (m, 2H), 4.26 (q, J = 7.1 Hz, 2H), 4.00 (s, 3H), 2.38 (s, 3H), 1.29 (dd, J = 7.2, 7.2 Hz, 3H).
1-(6-Methoxypyridin-3-yl)-5-methyl-1H-pyrazole-3-carbaldehyde (76)
Ethyl 1-(6-methoxy-3-pyridyl)-5-methyl-1H-pyrazole-3-carboxylate (68) (380 mg, 1.45 mmol) and DIBAL (1 M in DCM) (3.64 mL, 3.64 mmol) were combined in DCM (10 mL) according to General Method 6a. Work-up and purification afforded the title compound (270 mg, 81%). 1H NMR (400 MHz, CDCl3): δ 10.00 (s, 1H), 8.31–8.30 (m, 1H), 7.72 (dd, J = 2.8, 8.8 Hz, 1H), 6.92 (d, J = 8.8 Hz, 1H), 6.75 (s, 1H), 4.03 (s, 3H), 2.36 (s, 3H).
1′-((1-(6-Methoxypyridin-3-yl)-5-methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (18)
Spiro[isochromane-1,4′-piperidine] (120 mg, 0.59 mmol), 1-(6-methoxy-3-pyridyl)-5-methyl-1H-pyrazole-3-carbaldehyde (76) (128 mg, 0.59 mmol), and sodium triacetoxyborohydride (250 mg, 1.18 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound (150 mg, 60%). HRMS (ESI) m/z: calcd for C24H29N4O2 [M + H+], 405.2288; found, 405.2292. 1H NMR (400 MHz, CDCl3): δ 8.25 (d, J = 2.2 Hz, 1H), 7.72 (dd, J = 2.4, 8.8 Hz, 1H), 7.23–7.09 (m, 4H), 6.85 (d, J = 8.8 Hz, 1H), 6.24 (s, 1H), 4.00 (s, 3H), 3.92 (dd, J = 5.5, 5.5 Hz, 2H), 3.66 (s, 2H), 2.88–2.83 (m, 4H), 2.53 (dd, J = 11.4, 11.4 Hz, 2H), 2.32 (s, 3H), 2.15–2.06 (m, 2H), 1.93 (d, J = 13.1 Hz, 2H).
Synthesis of 1′-((1-(6-(Difluoromethoxy)pyridin-3-yl)-5-methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (19) (Scheme 1)
1′-[(5-methyl-1H-pyrazol-3-yl)methyl]spiro[isochromane-1,4′-piperidine] (78) (100 mg, 0.34 mmol), 2-(difluoromethoxy)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (182 mg, 0.67 mmol), and copper acetate (92 mg, 0.50 mmol) were combined in DCM (3 mL) and pyridine (0.50 mL, 6.20 mmol) according to General Method 2. Work-up and purification afforded the title compound (10 mg, 6%). HRMS (ESI) m/z: calcd for C24H27F2N4O2 [M + H+], 441.2102; found, 441.2108. 1H NMR (400 MHz, CDCl3): δ 8.32 (d, J = 2.5 Hz, 1H), 7.91 (dd, J = 2.7, 8.7 Hz, 1H), 7.23–7.02 (m, 5H), 6.27 (s, 1H), 3.92 (dd, J = 5.5, 5.5 Hz, 2H), 3.66 (s, 2H), 2.85 (dd, J = 5.5, 5.5 Hz, 4H), 2.54 (dd, J = 11.0, 11.0 Hz, 2H), 2.36 (s, 3H), 2.14–2.07 (m, 2H), 1.93 (d, J = 12.0 Hz, 2H).
Synthesis of 1′-((5-Methyl-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (20) (Scheme 1)
Ethyl 5-Methyl-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazole-3-carboxylate (69)
[6-(Trifluoromethyl)-3-pyridyl]hydrazine (2.46 g, 13.91 mmol) and ethyl 2,4-dioxopentanoate (59) (2.20 g, 13.91 mmol) were dissolved in AcOH (20 mL) according to General Method 3b. Work-up and purification afforded the title compound (2.70 g, 62%). 1H NMR (400 MHz, CDCl3): δ 8.83 (d, J = 2.4 Hz, 1H), 8.01 (dd, J = 2.1, 8.3 Hz, 1H), 7.80 (d, J = 8.3 Hz, 1H), 6.93 (s, 1H), 4.30 (q, J = 7.1 Hz, 2H), 2.40 (s, 3H), 1.32 (dd, J = 7.1, 7.1 Hz, 3H).
5-Methyl-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazole-3-carbaldehyde (77)
Ethyl 5-methyl-1-[6-(trifluoromethyl)-3-pyridyl]-1H-pyrazole-3-carboxylate (69) (1.45 mg, 4.85 mmol) and DIBAL (1 M in DCM) (12.11 mL, 12.11 mmol) were combined in DCM (10 mL) according to General Method 6a. Work-up and purification afforded the title compound (1.00 g, 77%). 1H NMR (400 MHz, CDCl3): δ 10.04 (s, 1H), 8.98 (d, J = 2.3 Hz, 1H), 8.13 (dd, J = 2.2, 8.3 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 6.82 (s, 1H), 2.51 (s, 3H).
1′-((5-Methyl-1-(6-(trifluoromethyl)pyridin-3-yl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (20)
Spiro[isochromane-1,4′-piperidine] (40 mg, 0.20 mmol), 5-methyl-1-[6-(trifluoromethyl)-3-pyridyl]-1H-pyrazole-3-carbaldehyde (77) (50 mg, 0.20 mmol), and sodium triacetoxyborohydride (83 mg, 0.39 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded the title compound as a colorless gum (40 mg, 43%). HRMS (ESI) m/z: calcd for C24H26F3N4O [M + H+], 443.2058; found, 443.2039. 1H NMR (400 MHz, CDCl3): δ 8.93 (d, J = 2.2 Hz, 1H), 8.10 (dd, J = 2.2, 8.5 Hz, 1H), 7.82 (d, J = 8.5 Hz, 1H), 7.22–7.09 (m, 4H), 6.33 (s, 1H), 3.92 (dd, J = 5.5, 5.5 Hz, 2H), 3.67 (s, 2H), 2.86 (dd, J = 5.5, 5.5 Hz, 4H), 2.59–2.51 (m, 2H), 2.48 (s, 3H), 2.15–2.03 (m, 2H), 1.93 (d, J = 12.4 Hz, 2H).
Synthesis of 2-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (21) (Scheme 1)
Spiro[isochromane-1,4′-piperidine] (71 mg, 0.35 mmol), tert-butyl 2-(3-formyl-5-methyl-1H-pyrazol-1-yl)benzoate (100 mg, 0.35 mmol), and sodium triacetoxyborohydride (148 mg, 0.70 mmol) were combined in DCM (2 mL) according to General Method 1. Work-up and purification afforded tert-butyl 2-(5-methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoate as a colorless gum (125 mg, 68%). 1H NMR (500 MHz, CDCl3): δ 7.91 (dd, J = 1.7, 7.6 Hz, 1H), 7.62–7.49 (m, 2H), 7.36 (d, J = 7.6 Hz, 1H), 7.25–7.09 (m, 3H), 5.32 (s, 1H), 4.72 (s, 1H), 3.90 (dd, J = 5.5, 5.5 Hz, 2H), 2.84 (dd, J = 5.4, 5.4 Hz, 2H), 2.57–2.56 (m, 1H), 2.18 (s, 4H), 1.93 (d, J = 13.4 Hz, 2H), 1.38 (d, J = 3.2 Hz, 12H), 1.30–1.25 (m, 1H). tert-Butyl 2-[5-methyl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)-1H-pyrazol-1-yl]benzoate (125 mg, 0.26 mmol) was then stirred in TFA (2 mL) at rt for 18 h and then concentrated in vacuo. The residue was loaded onto a 1 g SCX cartridge, eluting with 3 M NH3/MeOH. The crude product was purified by preparative HPLC (XBridge column, 0.1% NH4OH modifier) to afford the title compound (80 mg, 69%). HRMS (ESI) m/z: calcd for C25H28N3O3 [M + H+], 418.2125; found, 418.2120. 1H NMR (400 MHz, CDCl3): δ 7.97 (dd, J = 1.7, 7.4 Hz, 1H), 7.55–7.45 (m, 2H), 7.36 (dd, J = 1.4, 7.6 Hz, 1H), 7.25–7.05 (m, 4H), 6.37 (s, 1H), 4.01 (s, 2H), 3.81 (dd, J = 5.4, 5.4 Hz, 2H), 3.34 (dd, J = 2.1, 9.1 Hz, 2H), 3.07 (dd, J = 11.6, 11.6 Hz, 2H), 2.79 (dd, J = 5.3, 5.3 Hz, 2H), 2.58–2.49 (m, 2H), 2.31 (s, 3H), 1.92 (d, J = 14 Hz, 2H).
Synthesis of 3-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (22) (Scheme 2)
3-(3-(Ethoxycarbonyl)-5-methyl-1H-pyrazol-1-yl)benzoic Acid (87)
Ethyl 2,4-dioxopentanoate (79) (10.29 g, 65.07 mmol) was dissolved in EtOH (100 mL) and 3-hydrazinobenzoic acid (9.90 g, 65.07 mmol) was added. The reaction was heated to reflux for 1 h and then allowed to cool to rt. The solvent was removed in vacuo and H2O was added. The product was extracted with EtOAc, dried (hydrophobic frit), and concentrated in vacuo to afford a mixture of isomers (approximately 1:4 in favor of the desired isomer) (16.48 g, 60 mmol total). No attempt was made to separate the regioisomers at this stage, and the crude material was taken through to the next step without further purification. LCMS (ESI+) m/z: 275 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.20–8.12 (m, 2H), 7.74 (d, J = 8.0, 1H), 7.59 (t, J = 7.8, 1H), 6.76 (s, 1H), 4.41 (q, J = 7.1, 2H), 1.39 (t, J = 7.1, 3H).
Ethyl 1-(3-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazole-3-carboxylate (93)
A mixture of 3-(3-ethoxycarbonyl-5-methyl-1H-pyrazol-1-yl)benzoic acid and 3-(5-ethoxycarbonyl-3-methyl-1H-pyrazol-1-yl)benzoic acid (87) (5.00 g, 18.23 mmol in total as a mixture of isomers) was dissolved in thionyl chloride (3.99 mL, 54.69 mmol), and three drops of pyridine were added. The mixture was heated to 50 °C for 1 h and then allowed to cool to rt. The mixture was concentrated in vacuo and the resulting oil was dissolved in DCM (20 mL). Ethanolamine (2.19 mL, 36.46 mmol) was added dropwise and the mixture was stirred for 3 h at rt. Further, thionyl chloride (3.99 mL, 54.69 mmol) was added to convert the OH to Cl. This mixture was stirred at rt for 18 h. The reaction was quenched with the dropwise addition of H2O. The aqueous layer was basified with 2 M NaOH and extracted with DCM. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford a brown oil. The crude material was purified by flash chromatography (10–50% EtOAc in heptane). The second compound to elute was the desired isomer. This solid was dissolved in THF, cooled to 0 °C, and sodium hydride (60% in mineral oil) (729 mg, 18.23 mmol) was added. The reaction mixture was allowed to warm to rt and stirred for 2 h. The reaction was quenched carefully with water at 0 °C. The volatiles were removed in vacuo and the product was extracted with EtOAc, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (2.1 g, 38%). LCMS (ESI) m/z: 300 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.03–7.98 (m, 2H), 7.60 (d, J = 7.9 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 6.73 (s, 1H), 4.48–4.36 (m, 4H), 4.07 (t, J = 9.5 Hz, 2H), 2.34 (s, 3H), 1.39 (t, J = 7.0 Hz, 3H).
1-(3-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazole-3-carbaldehyde (99)
Ethyl 1-[3-(4,5-dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazole-3-carboxylate (93) (2.10 g, 7.02 mmol) and DIBAL (1 M in DCM) (17.54 mL, 17.54 mmol) were combined in DCM (20 mL) with MnO2 (6.10 g, 70.16 mmol) added in the second step according to General Method 5b. Work-up and purification afforded the title compound, which was used without further purification.
1′-((1-(3-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (105)
Spiro[isochromane-1,4′-piperidine] (80 mg, 0.39 mmol), 1-[3-(4,5-dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazole-3-carbaldehyde (99) (100 mg, 0.39 mmol), and sodium triacetoxyborohydride (166 mg, 0.78 mmol) were combined in DCM (20 mL) according to General Method 1. Work-up and purification afforded the title compound (30 mg, 16%). LCMS (ESI) m/z: 442 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 8.02 (s, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.24–7.10 (m, 3H), 7.08 (d, J = 7.5 Hz, 1H), 6.24 (s, 1H), 4.45 (t, J = 9.5 Hz, 2H), 4.08 (t, J = 9.5 Hz, 2H), 3.92–3.87 (m, 2H), 3.66 (s, 2H), 2.89–2.79 (m, 4H), 2.53 (t, J = 11.6 Hz, 2H), 2.36 (s, 3H), 2.09 (t, J = 13.3 Hz, 2H, 1.91 (d, J = 13.3 Hz, 2H).
3-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (22)
1′-[[1-[3-(4,5-Dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (105) (30 mg, 0.07 mmol) was dissolved in 3 M HCl according to General Method 3. Work-up and purification afforded the title compound (19 mg, 66%). LCMS (ESI) m/z: 418 (M + H)+. HRMS (ESI) m/z: calcd for C25H28N3O3 [M + H+], 418.2130; found, 418.2133. 1H NMR (400 MHz, CDCl3): δ 8.24–8.16 (m, 2H), 7.53 (d, J = 4.2 Hz, 2H), 7.21–7.02 (m, 4H), 6.44 (s, 1H), 4.17 (s, 2H), 3.88–3.83 (m, 2H), 3.44 (d, J = 10.9 Hz, 2H), 3.13–3.03 (m, 2H), 2.82–2.76 (m, 2H), 2.53 (t, J = 13.3 Hz, 2H), 2.32 (s, 3H), 1.98 (d, J = 14.3 Hz, 2H).
Synthesis of 4-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (23) (Scheme 2)
4-(3-(Ethoxycarbonyl)-5-methyl-1H-pyrazol-1-yl)benzoic Acid (88)
Ethyl 2,4-dioxopentanoate (10.39 g, 65.73 mmol) was dissolved in EtOH (100 mL) and 4-hydrazinobenzoic acid (80) (10.0 g, 65.73 mmol) was added. The reaction mixture was heated to reflux for 2 h and then allowed to cool to rt. The solvent was removed in vacuo, H2O was added, and the product was extracted with EtOAc. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford a mixture of isomers (approximately 1:4 in favor of the desired isomer) 4-(5-ethoxycarbonyl-3-methyl-1H-pyrazol-1-yl)benzoic acid and 4-(3-ethoxycarbonyl-5-methyl-1H-pyrazol-1-yl)benzoic acid. No attempt was made to separate the regioisomers at this stage and the crude material was taken onto the next step without further purification. LCMS (ESI) m/z: 275 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.24 (d, J = 8.3, 2H), 7.63 (d, J = 8.3, 2H), 6.78 (s, 1H), 4.43 (q, J = 7.1, 2H), 2.41 (s, 3H), 1.41 (t, J = 7.1, 3H) (major isomer reported).
Ethyl 1-(4-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazole-3-carboxylate (94)
A mixture of 4-(3-ethoxycarbonyl-5-methyl-1H-pyrazol-1-yl)benzoic acid and 4-(5-ethoxycarbonyl-3-methyl-1H-pyrazol-1-yl)benzoic acid (88) (18.82 g, 68.62 mmol in total as a mixture of isomers) was dissolved in thionyl chloride (20.02 mL, 274.48 mmol), and three drops of pyridine were added. The mixture was heated to 50 °C for 2 h and then allowed to cool to rt. The mixture was concentrated in vacuo, and the resulting oil was dissolved in DCM (20 mL). Ethanolamine (8.26 mL, 137.24 mmol) was added dropwise and the mixture was stirred for 3 h at rt. Further thionyl chloride (20.01 mL, 274.48 mmol) was added to convert the OH to Cl. This mixture was stirred at rt for 18 h. The reaction was quenched with the dropwise addition of H2O. The aqueous layer was basified with 2 M NaOH and extracted with EtOAc. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford a brown oil. The crude material was purified by flash chromatography (10–50% EtOAc in heptane). The second compound to elute was the desired isomer, ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-methyl-1H-pyrazole-3-carboxylate (10.62 g, 44%). LCMS (ESI) m/z: 336 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H), 6.75 (s, 1H), 4.42 (q, J = 7.1 Hz, 2H), 3.86–3.80 (m, 2H), 3.79–3.74 (m, 2H), 2.37 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H). Ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-methyl-1H-pyrazole-3-carboxylate (10.62 g, 31.63 mmol) was dissolved in THF (50 mL) and cooled to 0 °C. NaH (759 mg, 31.63 mmol) was added portionwise waiting for the effervescence to cease each time. The reaction was allowed to stir at 0 °C for 3 h and then carefully quenched by adding H2O dropwise. The mixture was allowed to warm to rt and TBME was added. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound, which was used without further purification. LCMS (ESI) m/z: 300 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.06 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 6.75 (s, 2H), 4.47 (t, J = 9.6 Hz, 2H), 4.42 (q, J = 7.1, 2H), 4.09 (t, J = 9.6 Hz, 2H), 2.37 (s, 3H), 1.40 (t, J = 7.1 Hz, 3H).
1-(4-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazole-3-carbaldehyde (100)
Ethyl 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazole-3-carboxylate (94) (9.75 g, 32.57 mmol) and DIBAL (1 M in DCM) (6.51 mL, 6.51 mmol) were combined in DCM (90 mL) with MnO2 (14.16 g, 162.87 mmol) added in second step according to General Method 5b. Work-up and purification afforded the title compound (5.14 g, 59%). LCMS (ESI) m/z: 256 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 10.00 (s, 1H), 8.11 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 6.73 (s, 1H), 4.48 (t, J = 9.5 Hz, 2H), 4.10 (t, J = 9.5 Hz, 2H), 2.40 (s, 3H).
1′-((1-(4-(4,5-Dihydrooxazol-2-yl)phenyl)-5-methyl-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (106)
Spiro[isochromane-1,4′-piperidine] (1.79 g, 8.81 mmol), 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazole-3-carbaldehyde (100) (2.5 g, 8.81 mmol), and sodium triacetoxyborohydride (3.74 g, 17.63 mmol) were combined in DCM (4 mL) according to General Method 1. The crude material was purified by flash chromatography on a KPNH column (eluting with 10–50% EtOAc in heptane) to afford the title compound (1.99 g, 48%). LCMS (ESI) m/z: 443 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.03 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.22–7.15 (m, 2H), 7.13 (t, J = 7.3 Hz, 1H), 7.08 (d, J = 7.3 Hz, 1H), 6.24 (s, 1H), 4.46 (t, J = 9.5 Hz, 2H), 4.09 (t, J = 9.5 Hz, 2H), 3.92–3.87 (m, 2H), 3.65 (s, 2H), 2.87–2.80 (m, 4H), 2.52 (t, J = 11.8 Hz, 2H), 1.90 (d, J = 13.4 Hz, 2H).
4-(5-Methyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (23)
1′-[[1-[4-(4,5-Dihydrooxazol-2-yl)phenyl]-5-methyl-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (106) (1.9 g, 4.29 mmol) was dissolved in 3 M HCl according to General Method 4. Work-up and purification afforded the title compound (1 g, 55%). LCMS (ESI) m/z: 418 (M + H)+. HRMS (ESI) m/z: calcd for C25H28N3O3 [M + H+], 418.2130; found, 418.2159. 1H NMR (400 MHz, CD3OD): δ 8.11 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 7.24–7.11 (m, 4H), 6.48 (s, 1H), 4.30 (s, 1H), 3.94 (t, J = 5.4 Hz, 2H), 3.45 (d, J = 11.4 Hz, 2H), 3.36 (d, J = 12.5 Hz, 2H), 2.87–2.80 (m, 2H), 2.40 (s, 3H), 2.31 (td, J = 13.7, 3.8 Hz, 2H), 2.11 (d, J = 14.6 Hz, 2H).
Synthesis of 4-(5-Ethyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (24) (Scheme 2)
Lithium 4-(3-(Ethoxycarbonyl)-5-ethyl-1H-pyrazol-1-yl)benzoate (87)
4-Aminobenzoic acid (2.00 g, 14.58 mmol) was suspended in H2O (30 mL) and cooled to 0 °C. Concentrated H2SO4 (15 mL, 14.58 mmol) was added dropwise to this suspension, followed by the dropwise addition of NaNO2 (5% aqueous solution) (20 mL, 14.58 mmol). The reaction mixture was stirred for 30 min at 0 °C, then NaBF4 (17% aqueous solution) (12 mL, 14.58 mmol) was added, and the reaction was stirred for further 30 min. A solution of ethyl 2-chloro-3-oxo-butanoate (2.40 g, 14.58 mmol) in MeOH (75 mL) was added to the reaction mixture, which was then allowed to warm to rt and stirred for 3 h. The resulting precipitate was filtered, washed with H2O, and air-dried to afford (E)-4-(2-(1-chloro-2-ethoxy-2-oxoethylidene)hydrazinyl)benzoic acid (3.00 g, 72%). 1H NMR (400 MHz, DMSO): δ 12.65–12.65 (m, 1H), 10.84 (s, 1H), 7.92 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.7 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 1.31 (dd, J = 7.1, 7.1 Hz, 3H). (E)-4-(2-(1-Chloro-2-ethoxy-2-oxoethylidene)hydrazinyl)benzoic acid (1.30 g, 4.80 mmol) and 1-diethoxyphosphorylbutan-2-one (1.00 g, 4.80 mmol) were mixed in diglyme (15 mL) and LiOH (504 mg, 12.01 mmol) was added. The reaction mixture was stirred at rt for 18 h. The reaction mixture was concentrated in vacuo to afford the title compound and was used directly in the next step without further purification.
Ethyl 1-(4-(4,5-Dihydrooxazol-2-yl)phenyl-5-ethyl-1H-pyrazole-3-carboxylate) (95)
Lithium 4-(3-ethoxycarbonyl-5-ethyl-1H-pyrazol-1-yl)benzoate (89) (1.40 g, 4.76 mmol) was mixed in thionyl chloride (3 mL, 18.66 mmol) and heated to reflux for 2 h. The reaction mixture was allowed to cool to rt and then concentrated in vacuo. The resulting orange solid was suspended in DCM (20 mL) and cooled to 0 °C. 2-Chloroethanamine hydrochloride (607 mg, 5.23 mmol) and DIPEA (3.32 mL, 19.03 mmol) were added, and the reaction mixture was allowed to warm up to rt for 18 h. The reaction was quenched with H2O, passed through a hydrophobic frit, and concentrated in vacuo. The crude material was purified by flash chromatography (eluting with 0–100% EtOAc in heptane) to afford ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-ethyl-1H-pyrazole-3-carboxylate (1.00 g, 57%). 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.6 Hz, 2H), 7.58 (d, J = 8.6 Hz, 2H), 6.82 (s, 1H), 6.67 (d, J = 5.0 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.88–3.78 (m, 4H), 2.71 (q, J = 7.5 Hz, 2H), 1.43 (dd, J = 7.1, 7.1 Hz, 3H), 1.30–1.25 (m, 3H). Ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-ethyl-1H-pyrazole-3-carboxylate (1.00 g, 2.86 mmol) was dissolved in THF (30 mL) and cooled to 0 °C. NaH (126 mg, 3.14 mmol) was added, and the reaction mixture was allowed to warm to rt for 18 h and then heated to 50 °C for 1 h. The reaction was allowed to cool to rt and then quenched with H2O. The volatiles were removed in vacuo, and the product was extracted with EtOAc. The combined organics were washed with brine, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (820 mg, 87%). 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 8.6 Hz, 2H), 7.54 (d, J = 8.7 Hz, 2H), 6.81 (s, 1H), 4.53–4.42 (m, 4H), 4.12 (dd, J = 9.5, 9.5 Hz, 2H), 2.71 (q, J = 7.4 Hz, 2H), 1.43 (dd, J = 7.1, 7.1 Hz, 3H), 1.27 (dd, J = 7.5, 7.5 Hz, 3H).
1-(4-(4,5-Dihydrooxazol-2-yl)phenyl)-5-ethyl-1H-pyrazole-3-carbaldehyde (101)
Ethyl 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-ethyl-1H-pyrazole-3-carboxylate (95) (820 mg, 2.62 mmol) and DIBAL (1 M in DCM) (6.54 mL, 6.54 mmol) were combined in DCM (25 mL) according to General Method 5a. Work-up and purification afforded the title compound (600 mg, 81%). 1H NMR (400 MHz, CDCl3): δ 10.03 (s, 1H), 8.13 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 6.80 (s, 1H), 4.51 (dd, J = 9.5, 9.5 Hz, 2H), 4.14 (dd, J = 9.5, 9.5 Hz, 2H), 2.74 (q, J = 7.5 Hz, 2H), 1.28 (dd, J = 7.5, 7.5 Hz, 3H).
4-(5-Ethyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (24)
Spiro[isochromane-1,4′-piperidine] (175 mg, 0.86 mmol), 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-ethyl-1H-pyrazole-3-carbaldehyde (101) (232 mg, 0.86 mmol), and sodium triacetoxyborohydride (365 mg, 1.72 mmol) were combined in DCM (5 mL) according to General Method 1. The crude oxazoline intermediate was dissolved in 3 M HCl (3 mL) according to General Method 3. Work-up and purification afforded 4-[5-ethyl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)-1H-pyrazol-1-yl]benzoic acid (200 mg, 51%). LCMS (ESI) m/z: 432.2 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.5 Hz, 2H), 7.37–7.30 (m, 2H), 7.22–7.10 (m, 4H), 6.37 (s, 1H), 4.09 (s, 2H), 3.94 (dd, J = 5.4, 5.4 Hz, 2H), 3.48–3.48 (m, 2H), 3.05 (dd, J = 11.4, 11.4 Hz, 2H), 2.87 (dd, J = 5.2, 5.2 Hz, 2H), 2.73–2.58 (m, 4H), 2.04 (d, J = 13.7 Hz, 2H), 1.19 (dd, J = 7.5, 7.5 Hz, 3H).
Synthesis of 4-(5-Isopropyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (25) (Scheme 2)
1,1-Diethoxy-5-methyl-hexane-2,4-dione (81)
Ethyl 2,2-diethoxyacetate (4.2 mL, 23 mmol) was added portionwise to a solution of 3-methylbutan-2-one (2 g, 23 mmol) in toluene (10 mL) at 0 °C. NaH (60% dispersion, 2.5 mL g, 46 mmol) was added portionwise, and the mixture was stirred at 0 °C for 2 h. H2O was added dropwise until the reaction was quenched, followed by 3 M HCl solution, dropwise, until the solution was pH 5. The organics were extracted with DCM, passed through a hydrophobic frit, and concentrated in vacuo to afford the title compound, which was used in the next step with no further purification.
4-(3-Formyl-5-isopropyl-1H-pyrazol-1-yl)benzoic Acid (84)
1,1-Diethoxy-5-methyl-hexane-2,4-dione (83) (386 mg, 1.78 mmol) and 4-hydrazinobenzoic acid (80) (272 mg, 1.78 mmol) were dissolved in EtOH (2 mL) and H2O (2 mL). The mixture was stirred at 35 °C for 18 h and then allowed to cool to rt and concentrated in vacuo to afford an orange solid. The crude material was triturated with Et2O and filtered to afford the title compound (349 mg, 72%). LCMS (ESI) m/z: 259 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 10.01 (s, 1H), 8.30 (d, J = 8.6 Hz, 2H), 7.62 (d, J = 8.6 Hz, 2H), 6.81 (s, 1H), 3.11 (sept, J = 6.8 Hz, 1H), 1.23 (d, J = 6.8 Hz, 6H).
4-(5-Isopropyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (25)
Spiro[isochromane-1,4′-piperidine] (39 mg, 0.19 mmol), 4-(3-formyl-5-isopropyl-1H-pyrazol-1-yl)benzoic acid (84) (50 mg, 0.19 mmol), and sodium triacetoxyborohydride (82 mg, 0.39 mmol) were combined in DCM (20 mL) according to General Method 1. Work-up and purification afforded the title compound (20 mg, 23%). LCMS (ESI) m/z: 446 (M + H)+. HRMS (ESI) m/z: calcd for C27H32N3O3 [M + H+], 446.2443; found, 446.2423. 1H NMR (500 MHz, CDCl3): δ 8.08 (d, J = 8.4 Hz, 2H), 7.30–7.26 (m, 3H), 7.18 (td, J = 7.4, 1.4 Hz, 1H), 7.14 (td, J = 7.4, 1.4 Hz, 1H), 7.09 (d, J = 7.4 Hz, 1H), 6.30 (s, 1H), 4.03 (s, 2H), 3.92 (t, J = 5.5 Hz, 2H), 3.48 (d, J = 10.5 Hz, 2H), 3.09–2.98 (m, 3H), 2.85 (t, J = 5.4 Hz, 2H), 2.66–2.57 (m, 2H), 2.03 (d, J = 13.8 Hz, 2H), 1.11 (d, J = 6.8 Hz, 6H).
Synthesis of 4-(5-(tert-Butyl)-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (26) (Scheme 2)
4-(5-(tert-Butyl)-3-(ethoxycarbonyl)-1H-pyrazol-1-yl)benzoic Acid (90)
Ethyl 5,5-dimethyl-2,4-dioxo-hexanoate (2.00 g, 9.99 mmol) was dissolved in EtOH (20 mL) and 4-hydrazinobenzoic acid (80) (1.52 g, 9.99 mmol) was added. The mixture was heated to reflux for 1 h and then allowed to cool to rt, and the solvent was removed in vacuo. H2O was added and the product was extracted with EtOAc. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (3.12 g, 99%). LCMS (ESI) m/z: 317 (M + H)+. 1H NMR (500 MHz, CD3OD): δ 8.21 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 2H), 6.79 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 2H), 1.21 (s, 9H).
Ethyl 5-(tert-Butyl)-1-(4-(4,5-dihydrooxazol-2-yl)phenyl)-1H-pyrazole-3-carboxylate (96)
4-(5-tert-Butyl-3-ethoxycarbonyl-1H-pyrazol-1-yl)benzoic acid (90) (3.12 g, 9.86 mmol) was dissolved in DCM (20 mL), and HOBt (1.60 g, 11.84 mmol), EDC.HCl (2.27 g, 11.84 mmol), DIPEA (6.87 mL, 39.45 mmol), and 2-chloroethanamine hydrochloride (1.37 g, 11.84 mmol) were added. The reaction was stirred at rt for 1 h. The reaction mixture was washed with H2O extracting with further DCM. The organics were dried (hydrophobic frit) and the solvent was removed in vacuo to afford an orange oil. The crude material was purified by flash chromatography (10–50% EtOAc in heptane) to afford ethyl 5-tert-butyl-1-[4-(2-chloroethylcarbamoyl)phenyl]-1H-pyrazole-3-carboxylate (1.76 g, 47%). LCMS (ESI) m/z: 378 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H), 6.90–6.38 (m, 1H), 6.76 (s, 1H), 4.41 (q, J = 7.1 Hz, 2H), 3.85–3.79 (m, 2H), 3.78–3.73 (m, 2H), 1.39 (t, J = 7.1 Hz, 3H), 1.19 (s, 9H). Ethyl 5-tert-butyl-1-[4-(2-chloroethylcarbamoyl)phenyl]-1H-pyrazole-3-carboxylate (1.76 g, 4.66 mmol) was dissolved in THF (20 mL) and cooled to 0 °C. NaH (186 mg, 4.66 mmol) was then added, and the reaction mixture was stirred at 0 °C for 1 h. H2O was added carefully to quench the reaction, and the volatiles were removed in vacuo. The product was extracted with TBME, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (1.00 g, 63%). LCMS (ESI) m/z: 342 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 6.76 (s, 1H), 4.48 (t, J = 9.5 Hz, 2H), 4.40 (q, J = 7.1 Hz, 2H), 4.10 (t, J = 9.5 Hz, 2H), 1.38 (t, J = 7.1 Hz, 3H), 1.19 (s, 9H).
5-(tert-Butyl)-1-(4-(4,5-dihydrooxazol-2-yl)phenyl)-1H-pyrazole-3-carbaldehyde (102)
Ethyl 5-tert-butyl-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-1H-pyrazole-3-carboxylate (96) (1.00 g, 2.93 mmol) and DIBAL (1 M in DCM) (6.51 mL, 6.51 mmol) were combined in DCM (5 mL) with MnO2 (2.55 g, 29.29 mmol) added in the second step according to General Method 5b. Work-up and purification afforded the title compound (624 mg, 68%). LCMS (ESI) m/z: 298 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 9.95 (s, 1H), 8.10 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 6.75 (s, 1H), 4.50 (t, J = 9.5 Hz, 2H), 4.12 (t, J = 9.6 Hz, 2H), 1.20 (s, 9H).
1′-((5-(tert-Butyl)-1-(4-(4,5-dihydrooxazol-2-yl)phenyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (107)
Spiro[isochromane-1,4′-piperidine] (41 mg, 0.20 mmol), 5-tert-butyl-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-1H-pyrazole-3-carbaldehyde (102) (60 mg, 0.20 mmol), and sodium triacetoxyborohydride (86 mg, 0.40 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound (41 mg, 40%). LCMS (ESI) m/z: 485 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.02 (d, J = 8.3 Hz, 2H), 7.44 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 7.7 Hz, 1H), 7.18 (t, J = 7.7 Hz, 1H), 7.13 (td, J = 7.3 Hz, 1.3, 1H), 7.08 (d, J = 7.3 Hz, 1H), 6.22 (s, 1H), 4.47 (t, J = 9.6 Hz, 2H), 4.10 (t, J = 9.6 Hz, 2H), 3.90 (t, J = 5.5 Hz, 2H), 3.61 (s, 2H), 2.88–2.80 (m, 4H), 2.50 (t, J = 12.1 Hz, 2H), 2.09 (td, J = 13.1, 4.3 Hz, 2H), 1.90 (d, J = 13.1 Hz, 2H), 1.18 (s, 9H).
4-(5-(tert-Butyl)-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (26)
1′-[[5-tert-butyl-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (107) (41 mg, 0.08 mmol) was dissolved in 3 M HCl according to General Method 4. Work-up and purification afforded the title compound (15 mg, 37%). LCMS (ESI) m/z: 459 (M + H)+. HRMS (ESI) m/z: calcd for C28H34N3O3 [M + H+], 460.2600; found, 460.2613. 1H NMR (500 MHz, d6-DMSO): δ 8.05 (d, J = 8.3 Hz, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 7.4 Hz, 1H), 7.17 (t, J = 7.4 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.09 (J = 7.4 Hz, 1H), 6.25 (s, 1H), 3.85–3.80 (m, 2H), 3.53 (br s, 2H), 2.83–2.71 (m, 4H), 2.47–2.39 (m, 2H), 2.01–1.91 (m, 2H), 1.81 (d, J = 13.1 Hz, 2H), 1.14 (s, 9H).
Synthesis of 4-(5-(Difluoromethyl)-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (27) (Scheme 2)
4-(5-(Difluoromethyl)-3-(ethoxycarbonyl)-1H-pyrazol-1-yl)benzoic Acid (91)
Ethyl 5,5-difluoro-2,4-dioxo-pentanoate (2.5 g, 12.88 mmol) was dissolved in EtOH (20 mL) and AcOH (5 mL) and 4-hydrazinobenzoic acid (80) (1.96 g, 12.88 mmol) was added. The reaction was heated to reflux for 18 h and then allowed to cool to rt, and the solvent was removed in vacuo. H2O was added, and the product was extracted with EtOAc, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (4.45 g, 100% in total as a mixture of isomers) and used in the next step without further purification.
5-(Difluoromethyl)-1-(4-(4,5-dihydrooxazol-2-yl)phenyl)-1H-pyrazole-3-carbaldehyde (97)
A mixture of 4-[5-(difluoromethyl)-3-ethoxycarbonyl-1H-pyrazol-1-yl]benzoic acid and 4-[3-(difluoromethyl)-5-ethoxycarbonyl-1H-pyrazol-1-yl]benzoic acid (91) (4.45 g, 14.34 mmol in total as a mixture of isomers) was dissolved in DCM (50 mL) and HOBt (2.87 g, 21.23 mmol), EDC.HCl (3.30 g, 17.21 mmol), DIPEA (7.49 mL, 43.03 mmol), and 2-chloroethanamine hydrochloride (2.00 g 17.21 mmol) were added. The reaction was stirred at rt for 18 h. H2O was added, extracting with further DCM. The combined organics were dried (hydrophobic frit), and the solvent was removed in vacuo to afford a dark orange oil. The crude material was purified by flash chromatography (10–50% EtOAc in heptane). The second compound to elute was the desired isomer, ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-(difluoromethyl)-1H-pyrazole-3-carboxylate (470 mg, 9%). 1H NMR (500 MHz, CDCl3): δ 7.94 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 7.27 (s, 1H), 6.65 (t, J = 53.5 Hz, 1H), 4.45 (q, J = 7.1 Hz, 2H), 3.87–3.81 (m, 2H), 3.79–3.75 (m, 2H), 1.41 (t, J = 7.1 Hz, 3H). Ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-(difluoromethyl)-1H-pyrazole-3-carboxylate (470 mg, 1.26 mmol) was dissolved in THF (25 mL), cooled to 0 °C, and NaH (202 mg, 5.06 mmol) was added. The reaction was stirred for 2 h at 0 °C and then quenched by the dropwise addition of H2O. The mixture was allowed to warm to rt, and the volatiles were removed in vacuo. The pH was carefully adjusted to pH 6 using 3 M HCl, and the product was extracted with EtOAc, dried (hydrophobic frit), and concentrated in vacuo to afford a yellow oil. The aqueous phase was also concentrated in vacuo and combined with the material obtained from the EtOAc extraction to afford the title compound, which was used in the next step without further purification.
5-(Difluoromethyl)-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]pyrazole-3-carbaldehyde (103).
Ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-(difluoromethyl)-1H-pyrazole-3-carboxylate (97) and DIBAL (1 M in DCM) (2.77 mL, 2.77 mmol) were combined in DCM (30 mL) with MnO2 (1.20 g, 13.83 mmol) added in the second step according to General Method 5b. Further, DIBAL (1 M in DCM) (2.77 mL, 2.77 mmol) was needed in this case for reaction completion and oxidation with MnO2 only required 2 h for completion before formation of side products. Work-up and purification afforded the title compound (148 mg, 35%). LCMS (ESI) m/z: 292 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 10.07 (s, 1H), 8.14 (d, J = 8.7 Hz, 2H), 7.62 (d, J = 8.7 Hz, 2H), 6.68 (t, J = 53.5 Hz, 1H), 4.50 (t, J = 9.6 Hz, 2H), 4.12 (t, J = 9.6 Hz, 2H).
1′-((5-(Difluoromethyl)-1-(4-(4,5-dihydrooxazol-2-yl)phenyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (108)
Spiro[isochromane-1,4′-piperidine] (52 mg, 0.25 mmol), 5-(difluoromethyl)-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-1H-pyrazole-3-carbaldehyde (103) (74 mg, 0.25 mmol), and sodium triacetoxyborohydride (108 mg, 0.51 mmol) were combined in DCM (20 mL) according to General Method 1. Work-up and purification afforded the title compound (45 mg, 37%). LCMS (ESI) m/z: 479 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 8.7 Hz, 2H), 7.22–7.16 (m, 2H), 7.16–7.12 (m, 1H), 7.08 (d, J = 7.5 Hz, 1H), 6.80 (s, 1H), 6.66 (t, J = 53.5 Hz, 1H), 4.47 (t, J = 9.6 Hz, 2H), 4.10 (t, J = 9.6 Hz, 2H), 3.90 (t, J = 5.5 Hz, 2H), 3.71 (s, 2H), 2.85–2.80 (m, 4H), 2.53 (t, J = 12.0 Hz, 2H), 2.08 (td, J = 13.4, 4.3 Hz, 2H), 1.91 (d, J = 13.4 Hz, 2H).
4-(5-(Difluoromethyl)-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (27)
1′-[[5-(Difluoromethyl)-1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (108) (45 mg, 0.09 mmol) was dissolved in 3 M HCl (3 mL) according to General Method 3. Work-up and purification afforded 4-[5-(difluoromethyl)-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)-1H-pyrazol-1-yl]benzoic acid (19 mg, 44%). LCMS (ESI) m/z: 454 (M + H)+. HRMS (ESI) m/z: calcd for C25H26F2N3O3 [M + H+], 454.1916; found, 454.1929. 1H NMR (500 MHz, CDCl3): δ 8.00 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 7.7 Hz, 1H), 7.20–7.13 (m, 2H), 7.09 (d, J = 7.3 Hz, 1H), 6.77 (s, 1H), 6.64 (t, J = 53.5 Hz, 1H), 4.02 (s, 2H), 3.96–3.91 (m, 2H), 3.43 (d, J = 10.0 Hz, 2H), 2.98 (t, J = 11.9 Hz, 2H), 2.88–2.82 (m, 2H), 2.52 (t, J = 13.5 Hz, 2H), 2.03 (d, J = 13.5 Hz, 2H).
Synthesis of 4-(3-(Spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-5-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic Acid (28) (Scheme 2)
4-(3-(Ethoxycarbonyl)-5-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic Acid (92)
4-Hydrazinobenzoic acid (80) (1.43 g, 9.43 mmol) and ethyl 5,5,5-trifluoro-2,4-dioxo-pentanoate (2.00 g, 9.43 mmol) were dissolved in EtOH (20 mL) and AcOH (5 mL) according to General Method 3a. Work-up and purification afforded the title compound, which was used in the next step with no further purification. LCMS (ESI) m/z: 329 (M + H)+. 1H NMR (500 MHz, CD3OD): δ 7.95 (d, J = 9.0 Hz, 2H), 7.65 (d, J = 9.0 Hz, 2H), 4.34 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H).
Ethyl 1-[4-(4,5-Dihydrooxazol-2-yl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (98)
4-[3-ethoxycarbonyl-5-(trifluoromethyl)-1H-pyrazol-1-yl]benzoic acid (92) (3.97 g, 12.10 mmol) was dissolved in DCM (20 mL), and EDC.HCl (2.78 g, 14.51 mmol), DIPEA (6.25 mg, 48.38 mmol), HOBt (1.96 g, 14.51 mmol), and 2-chloroethanamine hydrochloride (1.68 g, 14.51 mmol) were added. The reaction was stirred at rt overnight. H2O was then added and the mixture was passed through a hydrophobic frit. The solvent was removed in vacuo to afford a dark orange oil. The crude material was purified by flash chromatography (10–50% EtOAc in heptane). The second fraction to elute afforded the desired isomer, ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (467 mg, 10%). LCMS (ESI) m/z: 390 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.42 (d, J = 9.0 Hz, 2H), 7.35 (d, J = 9.0 Hz, 2H), 4.33 (q, J = 7.1 Hz, 2H), 3.67–3.58 (m, 4H), 1.36 (t, J = 7.1 Hz, 3H). Ethyl 1-[4-(2-chloroethylcarbamoyl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (467 mg, 1.20 mmol) was dissolved in THF (25 mL) and cooled to 0 °C. Sodium hydride (197 mg, 4.92 mmol) was added, and the reaction was stirred at 0 °C for 1 h, allowed to warm to rt for 2 h, and then heated to reflux for 1 h. The mixture was allowed to cool to rt and was quenched with H2O dropwise. The reaction mixture was concentrated in vacuo to afford the title compound, which was used without further purification.
1-[4-(4,5-Dihydrooxazol-2-yl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carbaldehyde (104)
Ethyl 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carboxylate (98) and DIBAL (1 M in DCM) (3.00 mL, 3.00 mmol) were combined in DCM (50 mL) according to General Method 6b. Further DIBAL (1 M in DCM) (1.20 mL, 1.20 mmol) was added in this case, but the reaction was not pushed to completion. LiAlH4 (1.20 mL, 1.20 mmol) was therefore added and the reaction was allowed to warm to rt for 18 h before adding further LiAlH4 (1.20 mL, 1.20 mmol) and stirring at rt for 2 h. MnO2 (1.04 g, 11.99 mmol) was added in the second step according to General Method 6b. Work-up and purification afforded the title compound as a colorless oil (45 mg, 12%). LCMS (ESI) m/z: 310 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 10.06 (s, 1H), 8.13 (d, J = 8.7 Hz, 2H), 7.59 (d, J = 8.7 Hz, 2H), 5.30 (s, 1H), 4.50 (t, J = 9.6 Hz, 2H), 4.12 (t, J = 9.6 Hz, 2H).
1′-((1-(4-(4,5-Dihydrooxazol-2-yl)phenyl)-5-(trifluoromethyl)-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (109)
Spiro[isochromane-1,4′-piperidine] (30 mg, 0.15 mmol), 1-[4-(4,5-dihydrooxazol-2-yl)phenyl]-5-(trifluoromethyl)-1H-pyrazole-3-carbaldehyde (104) (45 mg, 0.15 mmol), and sodium triacetoxyborohydride (62 mg, 0.29 mmol) were combined in DCM (20 mL) according to General Method 1. Work-up and purification afforded the title compound (32 mg, 44%). LCMS (ESI) m/z: 497 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 8.08–8.04 (m, 2H), 7.56 (d, J = 8.6 Hz, 2H), 7.22–7.17 (m, 2H), 7.16–7.13 (m, 1H), 7.09 (d, J = 7.5 Hz, 1H), 6.87 (s, 1H), 4.47 (t, J = 9.6 Hz, 2H), 4.10 (t, J = 9.6 Hz, 2H), 3.90 (t, J = 5.5 Hz, 2H), 3.70 (s, 2H), 2.85–2.78 (m, 2H), 2.54 (t, J = 11.7 Hz, 2H), 2.08 (td, J = 13.2, 3.7 Hz, 2H), 1.92 (d, J = 13.7 Hz, 2H).
4-(3-(Spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-5-(trifluoromethyl)-1H-pyrazol-1-yl)benzoic Acid (28)
1′-[[1-[4-(4,5-Dihydrooxazol-2-yl)phenyl]-5-(trifluoromethyl)-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (109) (32 mg, 0.06 mmol) was dissolved in 3 M HCl according to General Method 4. Work-up afforded no product. The aqueous phase was concentrated in vacuo and purified by preparative HPLC (XBridge column, 0.1% NH4OH modifier) to afford the title compound (5 mg, 66%). LCMS (ESI) m/z: 472 (M + H)+. HRMS (ESI) m/z: calcd for C25H25F3N3O3 [M + H+], 472.1848; found, 472.1851. 1H NMR (500 MHz, CDCl3): δ 8.46 (d, J = 8.5 Hz, 2H), 7.95 (d, J = 8.5 Hz, 2H), 7.74 (t, J = 53.5 Hz, 1H), 7.73 (t, J = 7.2 Hz, 1H), 7.43 (td, J = 8.7, 2.7 Hz, 1H), 7.38 (dd, J = 9.7, 2.7 Hz, 1H), 4.25 (t, J = 5.5 Hz, 2H), 4.03 (s, 2H), 3.19 (t, J = 5.5 Hz, 2H), 3.15 (d, J = 10.5 Hz, 2H), 2.84 (t, J = 11.9 Hz, 2H), 2.41–2.33 (m, 2H), 2.22 (d, J = 13.3 Hz, 2H).
Synthesis of 4-(5-Cyclopropyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (29) (Scheme 2)
1-Cyclopropyl-4,4-diethoxybutane-1,3-dione (82)
1-Cyclopropylethanone (11.8 mL, 119 mmol) was dissolved in toluene (100 mL) and cooled to 0 °C. NaH (60% dispersion, 9.5 g, 238 mmol) was added portionwise and stirred at 0 °C for 30 min. Ethyl 2,2-diethoxyacetate (21 mL, 119 mmol) was added dropwise, and the reaction was stirred at 0 °C for 2 h, after which the reaction was allowed to warm to rt and left to stir for 1 h. The reaction was cooled to 0 °C and H2O was added dropwise until the reaction was quenched, followed by 3 M HCl solution, dropwise, until the solution was pH 2. NH4Cl (saturated solution) was added to neutralize, and the aqueous phase was extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated in vacuo to afford the title compound, which was used in the next step with no further purification.
4-(5-Cyclopropyl-3-formyl-1H-pyrazol-1-yl)benzoic Acid (85)
1-Cyclopropyl-4,4-diethoxy-butane-1,3-dione (82) (995 mg, 4.64 mmol) was dissolved in EtOH (2 mL) and H2O (2 mL), and 4-hydrazinobenzoic acid (642 mg, 4.22 mmol) was added. The reaction mixture was stirred at 35 °C for 18 h and then allowed to cool to rt and concentrated in vacuo to afford an orange solid. The solid was triturated with Et2O, and the yellow solid filtered and dried to afford 4-(5-cyclopropyl-3-formyl-1H-pyrazol-1-yl)benzoic acid (443 mg, 39%). LCMS (ESI) m/z: 257 (M + H)+. 1H NMR (500 MHz, d6-DMSO): δ 13.17 (br s, 1H), 9.92 (s, 1H), 8.16–8.12 (m, 2H), 7.89–7.86 (m, 2H), 6.70 (s, 1H), 1.96–1.89 (m, 1H), 1.04–0.97 (m, 2H), 0.87–0.82 (m, 2H).
4-(5-Cyclopropyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (27)
Spiro[isochromane-1,4′-piperidine] (10.73 g, 52.80 mmol), 4-(5-cyclopropyl-3-formyl-1H-pyrazol-1-yl)benzoic acid (85) (12.30 g, 48.00 mmol), and sodium triacetoxyborohydride (20.35 g, 95.99 mmol) were combined in DCM (4 mL) according to General Method 1. After work-up, the crude material was passed through a silica plug (washing with 0–30% MeOH in EtOAc) to afford 4-[5-cyclopropyl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)-1H-pyrazol-1-yl]benzoic acid (15.00 g, 70%). LCMS (ESI) m/z: 444 (M + H)+. 1H NMR (400 MHz, DMSO-d6): δ 12.74 (br s, 1H), 8.09–8.05 (m, 2H), 7.83–7.79 (m, 2H), 7.27–7.06 (m, 4H), 6.17 (s, 1H), 3.82 (t, J = 5.4 Hz, 2H), 3.51 (s, 2H), 2.79–2.65 (m, 4H), 2.40 (t, J = 11.9 Hz, 2H), 1.98–1.89 (m, 4H), 1.78 (d, J = 13.2 Hz, 2H), 1.04–0.99 (m, 2H), 0.80–0.75 (m, 2H).
Synthesis of 4-(5-Cyclopropyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (29) Hydrochloride (Scheme 2)
4-[5-Cyclopropyl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)-1H-pyrazol-1-yl]benzoic acid (29) (35 g, 79 mmol) was suspended in MeOH (50 mL) and 4 M HCl in dioxane (49 mL, 197 mmol) was added. The suspended solids went into the solution and the reaction was stirred at rt for 2 h. The precipitated solid was filtered and washed with a small amount of cold MeOH and dried. The solid was then recrystallized from 10% H2O in EtOH (∼1.70 L) to afford the title compound as the HCl salt (17 g, 44%). LCMS (ESI) m/z: 444 (M + H)+. HRMS (ESI) m/z: calcd for C27H30N3O3 [M + H+], 444.2287; found, 444.2278. 1H NMR (400 MHz, DMSO-d6): δ 13.21 (br s, 1H), 10.58 (br s, 1H), 8.14–8.11 (m, 2H), 7.87–7.84 (m, 2H), 7.27–7.12 (m, 4H), 6.48 (s, 1H), 4.38 (s, 2H), 3.87 (t, J = 5.3 Hz, 2H), 3.43–3.37 (m, 2H), 3.30–3.20 (m, 2H), 2.78 (t, J = 5.3 Hz, 2H), 2.41–2.33 (m, 2H), 2.06–1.95 (m, 3H), 1.08–1.03 (m, 2H), 0.79–0.75 (m, 2H).
Synthesis of 4-(5-Cyclobutyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (30) (Scheme 2)
1-Cyclobutyl-4,4-diethoxybutane-1,3-dione (85)
To a solution of 1-cyclobutylethanone (5 g, 51 mmol) in toluene (43 mL) cooled to 0 °C was added NaH (60% dispersion, 4 g, 102 mmol) portionwise. The reaction was stirred at 0 °C for 10 min before the dropwise addition of ethyl 2,2-diethoxyacetate (9.1 mL, 51 mmol). The reaction was stirred at 0 °C for further 10 min before being allowed to warm to rt. After 1 h, the reaction started to exotherm, and the reaction was cooled to 0 °C for an additional 1 h. Water was added dropwise until the reaction was quenched, followed by 3 M HCl until the solution was pH 4. The organics were extracted with DCM, passed through a hydrophobic frit, and concentrated in vacuo to afford the title compound, which was used in the next step with no further purification.
4-(5-Cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoic Acid (86)
1-Cyclobutyl-4,4-diethoxy-butane-1,3-dione (83) (4.80 g, 21.03 mmol) was dissolved in EtOH (2 mL) and H2O (2 mL), and 4-hydrazinobenzoic acid (3.20 g, 21.03 mmol) were added. The mixture was stirred at 35 °C for 18 and then allowed to cool to rt and concentrated in vacuo to afford a mixture of 4-(3-cyclobutyl-5-formyl-pyrazol-1-yl)benzoic acid and 4-(5-cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoic acid (5.15 g, 86% total). No attempt was made to separate isomers at this stage, and the mixture was used without further purification. LCMS (ESI) m/z: 271 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 11.85 (br s, 1H), 10.04 (s, 1H), 8.29 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 6.91 (s, 1H), 3.57 (quint, J = 8.6 Hz, 1H), 2.49–1.90 (m, 6H) (desired isomer reported). 4-(5-Cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoic acid and 4-(3-cyclobutyl-5-formyl-pyrazol-1-yl)benzoic acid (5.15 g, 19.05 mmol total as a mixture of isomers) were dissolved in tert-butanol (20 mL). DMAP (6.99 g, 57.16 mmol) and EDC.HCl (7.31 g, 38.11 mmol) were added. The mixture was heated to reflux for 2 h and then allowed to cool to rt and poured into H2O. The product was extracted with EtOAc, dried (hydrophobic frit), and concentrated in vacuo. The crude material was purified by flash chromatography (0–20% EtOAc in heptane) to afford the transesterified methyl ester, methyl 4-(5-cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoate (641 mg, 12%). LCMS (ESI) m/z: 285 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 9.87 (s, 1H), 8.17 (d, J = 8.6 Hz, 2H), 7.58 (d, J = 8.6 Hz, 2H), 7.02 (s, 1H), 3.66 (quint, J = 8.6 Hz, 1H), 2.45–2.37 (m, 2H), 2.32–2.23 (m, 2H), 2.13–2.02 (m, 1H), 2.01–1.91 (m, 1H). Methyl 4-(5-cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoate (641 mg, 2.25 mmol) was dissolved in MeOH, and 2 M NaOH (11.27 mL, 22.55 mmol) was added. The mixture was stirred at rt for 2 h and then neutralized with 3 M HCl and extracted with DCM. The organics were separated, dried (hydrophobic frit), and concentrated in vacuo to afford the title compound (632 mg, 100%). LCMS (ESI) m/z: 271 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 10.02 (s, 1H), 8.27 (d, J = 8.6 Hz, 2H), 7.59 (d, J = 8.6 Hz, 2H), 6.89 (s, 1H), 3.56 (quint, J = 8.4 Hz, 1H), 2.39–2.29 (m, 2H), 2.19 (quint, J = 9.2 Hz, 2H), 2.00 (quint, 9.2 Hz, 1H), 1.98–1.90 (m, 1H).
4-(5-Cyclobutyl-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (30)
Spiro[isochromane-1,4′-piperidine] (38 mg, 0.19 mmol), 4-(5-cyclobutyl-3-formyl-1H-pyrazol-1-yl)benzoic acid (86) (50 mg, 0.19 mmol), and sodium triacetoxyborohydride (78 mg, 0.37 mmol) were combined in DCM (20 mL) according to General Method 1. Work-up and purification afforded the title compound (16 mg, 18%). LCMS (ESI) m/z: 458 (M + H)+. HRMS (ESI) m/z: calcd for C28H32N3O3 [M + H+], 458.2433; found, 458.2438. 1H NMR (500 MHz, CDCl3): δ 8.03 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 7.18 (td, J = 7.3, 1.3 Hz, 1H), 6.40 (s, 1H), 4.01 (s, 2H), 3.93 (t, J = 5.5 Hz, 2H), 3.52–3.44 (m, 3H), 3.00 (t, J = 12.2 Hz, 2H), 2.85 (t, J = 5.4 Hz, 2H), 2.60 (t, J = 13.7 Hz, 2H), 2.28–1.89 (m, 2H), 2.13–1.99 (m, 2H), 1.95–1.86 (m, 1H), 1.86–1.77 (m, 1H).
Synthesis of 4-(5-Methoxy-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (31) (Scheme 3)
Methyl 1-(4-Bromophenyl)-5-hydroxy-1H-pyrazole-3-carboxylate (110)
Dimethyl but-2-ynedioate (61) (3.29 mL, 26.73 mmol) was dissolved in Et2O (50 mL), and a suspension of 4-bromophenylhydrazine (5.00 g, 26.73 mmol) in Et2O (50 mL) was added slowly over 30 min. The reaction mixture was stirred at rt for 1 h and then concentrated in vacuo. The residue was redissolved in MeOH (100 mL), NaOMe (25% wt) (23 mL, 106.93 mmol) was added, and the reaction mixture was stirred at rt for 18 h. The mixture was concentrated in vacuo, and then 6 M HCl (10 mL) was added. The resulting precipitate was filtered and washed with DCM and Et2O. The solid was redissolved in MeOH, 10 drops of H2SO4 were added, and the mixture was heated to reflux for 4 h and then allowed to cool to rt. The resulting precipitate was filtered and washed with ice-cold MeOH and air-dried to afford the title compound (5.65 g, 64%). LCMS (ESI) m/z: 297/299 (M + H)+. 1H NMR (400 MHz, DMSO-d6): δ 12.46 (br s, 1H), 7.76–7.67 (m, 4H), 5.99 (s, 1H), 3.37 (br s, 1H).
Methyl 1-(4-Bromophenyl)-5-methoxy-1H-pyrazole-3-carboxylate (114)
Methyl 1-(4-bromophenyl)-5-hydroxy-1H-pyrazole-3-carboxylate (110) (250 mg, 0.84 mmol) was dissolved in DMF (5 mL) and K2CO3 (174 mg, 1.26 mmol) was added. The reaction mixture was cooled to 0 °C, and iodomethane (0.06 mL, 0.93 mmol) was added dropwise. The reaction was allowed to warm to rt and stirred at this temperature for 2 h. EtOAc and H2O were added, and the organics were separated and washed with H20, NaHCO3 (saturated solution), and brine before concentrating in vacuo. The crude material was purified by flash chromatography (10–20% EtOAc in heptane) to afford the title compound (162 mg, 62%). LCMS (ESI) m/z: 311/313 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.63 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.9 Hz, 2H), 6.23 (s, 1H), 3.99 (s, 3H), 3.94 (s, 3H).
1-(4-Bromophenyl)-5-methoxy-1H-pyrazole-3-carbaldehyde (118)
Methyl 1-(4-bromophenyl)-5-methoxy-1H-pyrazole-3-carboxylate (114) (162 mg, 0.52 mmol) and DIBAL (1 M in DCM) (0.52 mL, 0.52 mmol) were combined in THF (5 mL) according to General Method 5b. Further DIBAL (1 M in DCM) (0.52 mL, 0.52 mmol) was added in this case to push the reaction to completion. MnO2 (453 mg, 5.21 mmol) was added in the second step according to General Method 5b. Work-up and purification afforded the title compound (146 mg, 100%). LCMS (ESI) m/z: 281/283 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 9.88 (s, 1H), 7.66 (d, J = 9.0 Hz, 2H), 7.60 (d, J = 9.0 Hz, 2H), 6.18 (s, 1H), 4.00 (s, 3H).
1′-((1-(4-Bromophenyl)-5-methoxy-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (122)
Spiro[isochromane-1,4′-piperidine] (181 mg, 0.89 mmol), 1-(4-bromophenyl)-5-methoxy-1H-pyrazole-3-carbaldehyde (118) (250 mg, 0.89 mmol), and sodium triacetoxyborohydride (377 mg, 1.78 mmol) were combined in DCM (5 mL) according to General Method 1. Work-up and purification afforded the title compound (287 mg, 68%). LCMS (ESI) m/z: 468/470 (M + H)+. 1H NMR (500 MHz, CDCl3): δ 7.61 (d, J = 8.9 Hz, 2H), 7.52 (d, J = 8.9 Hz, 2H), 7.24–7.18 (m, 2H), 7.17–7.13 (m, 1H), 7.08 (s, J = 7.4 Hz, 1H), 5.85 (s, 1H), 3.96 (s, 3H), 3.88 (t, J = 5.5 Hz, 2H), 3.81–3.70 (m, 2H), 3.08–2.96 (m, 2H), 2.82 (t, J = 5.5 Hz, 2H), 2.77–2.65 (m, 2H), 2.31–2.14 (m, 2H), 1.94 (d, J = 13.8 Hz, 2H).
4-(5-Methoxy-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (31)
1′-[[1-(4-Bromophenyl)-5-methoxy-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (122) (58 mg, 0.12 mmol), N-formylsaccharin (31 mg, 0.15 mL), Pd(OAc)2 (0.8 mg, 0.004 mmol), Xantphos (3 mg, 0.006 mmol), and KF (18 mg, 0.31 mmol) were combined in DMF (1 mL) according to General Method 5. Work-up and purification afforded the title compound (11 mg, 21%). LCMS (ESI) m/z: 434 (M + H)+. HRMS (ESI) m/z: calcd for C25H28N3O4 [M + H+], 434.2080; found, 434.2077. 1H NMR (400 MHz, CDCl3): δ 8.05 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 7.9 Hz, 1H), 7.21–7.11 (m, 2H), 7.08 (d, J = 7.4 Hz, 1H), 5.85 (s, 1H), 3.94–3.88 (m, 7H), 3.35 (d, J = 10.8 Hz, 2H), 2.92 (t, J = 11.9 Hz, 2H), 2.84 (t, J = 5.3 Hz, 2H), 2.46 (t, J = 13.0 Hz, 2H), 2.00 (d, J = 14.0 Hz, 2H.
Synthesis of 4-(5-Ethoxy-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (32) (Scheme 3)
Methyl 1-(4-Bromophenyl)-5-ethoxy-1H-pyrazole-3-carboxylate (115)
Methyl 1-(4-bromophenyl)-5-hydroxy-1H-pyrazole-3-carboxylate (110) (500 mg, 1.68 mmol) was dissolved in DMF (5 mL) and K2CO3 (349 mg, 2.52 mmol) was added. The reaction mixture was cooled to 0 °C, and iodoethane (0.13 mL, 1.68 mmol) was added dropwise. The reaction was allowed to warm to rt and stirred for 2 h. EtOAc and H2O were added. The organics were separated and washed with H20, NaHCO3 (saturated aqueous solution), and brine and concentrated in vacuo. The crude material was purified by flash chromatography (10–20% EtOAc in heptane) to afford the title compound (369 mg, 67%). LCMS (ESI) m/z: 369/371 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.9 Hz, 2H), 6.20 (s, 1H), 4.22 (q, J = 7.1 Hz, 2H), 3.93 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H).
1-(4-Bromophenyl)-5-ethoxy-1H-pyrazole-3-carbaldehyde (119)
Methyl 1-(4-bromophenyl)-5-ethoxy-1H-pyrazole-3-carboxylate (115) (369 mg, 1.13 mmol) and DIBAL (1.36 mL, 1.36 mmol) were combined in THF (5 mL) according to General Method 6b. Further, DIBAL (2.72 mL, 2.72 mmol) was added in this case to push the reaction to completion. MnO2 (987 mg, 11.45 mmol) was added in the second step according to General Method 6b. Work-up and purification afforded the title compound (164 mg, 47%). LCMS (ESI) m/z: 295/297 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 9.88 (s, 1H), 7.68 (d, J = 9.0 Hz, 2H), 7.60 (d, J = 9.0 Hz, 2H), 6.15 (s, 1H), 4.23 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.1 Hz, 2H).
1′-((1-(4-Bromophenyl)-5-ethoxy-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (123)
Spiro[isochromane-1,4′-piperidine] (56 mg, 0.28 mmol), 1-(4-bromophenyl)-5-ethoxy-1H-pyrazole-3-carbaldehyde (119) (82 mg, 0.28 mmol), and sodium triacetoxyborohydride (118 mg, 0.56 mmol) were combined in DCM (10 mL) according to General Method 1. Work-up followed by flash chromatography (10–100% EtOAc in heptane) afforded the title compound (96 mg, 72%). LCMS (ESI) m/z: 482/484 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 8.9 Hz, 2H), 7.51 (d, J = 8.9 Hz, 2H), 7.23–7.12 (m, 3H), 7.08 (d, J = 7.5 Hz, 1H), 5.76 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.89 (t, J = 5.5 Hz, 2H), 3.65 (s, 2H), 2.96–2.87 (m, 2H), 2.82 (t, J = 5.5 Hz, 2H), 2.66–2.54 (m, 2H), 2.20–2.07 (m, 2H), 1.91 (d, J = 14.0 Hz, 2H), 1.46 (t, J = 7.1 Hz, 3H).
4-(5-Ethoxy-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (32)
1′-[[1-(4-Bromophenyl)-5-ethoxy-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (123) (96 mg, 0.20 mmol), Pd(OAc)2 (1 mg, 0.01 mmol), Xantphos (5 mg, 0.01 mmol), KF (29 mg, 0.50 mmol), and N-formylsaccharin (50 mg, 0.24 mmol) were combined in DMF (1 mL) according to General Method 5. Work-up and purification afforded the title compound (10 mg, 11%). LCMS (ESI) m/z: 448 (M + H)+. HRMS (ESI) m/z: calcd for C26H30N3O4 [M + H]+: 448.2236; found, 448.2249. 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.6 Hz, 2H), 7.25–7.21 (m, 1H), 7.20–7.11 (m, 2H), 7.08 (d, J = 7.5 Hz, 1H), 5.84 (s, 1H), 4.11 (q, J = 7.1 Hz, 2H), 3.94–3.88 (m, 2H), 3.33 (d, J = 10.4 Hz, 2H), 2.92 (t, J = 12.1 Hz, 2H), 2.84 (t, J = 5.5 Hz, 2H), 2.45 (t, J = 13.4 Hz, 2H), 2.00 (d, J = 14.0 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H).
Synthesis of 4-[5-Pyrrolidin-1-yl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)pyrazol-1-yl]benzoic Acid (33) (Scheme 3)
Methyl 1-(4-Bromophenyl)-5-chloro-4-formyl-1H-pyrazole-3-carboxylate (111)
POCl3 (2 mL, 1.68 mmol) was added cautiously to methyl 1-(4-bromophenyl)-5-hydroxy-pyrazole-3-carboxylate (120) (500 mg, 1.68 mmol), and then DMF (0.16 mL, 2.02 mmol) was added. The reaction mixture was heated to 100 °C for 2 h and then allowed to cool to rt, added to a H2O/ice mixture, and stirred for 1 h. The resulting precipitate was filtered, washed with H2O and heptane, and air-dried. The solid was dissolved in DCM and passed through a hydrophobic frit. The filtrate was concentrated in vacuo to afford the title compound as a beige solid (450 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 10.54 (s, 1H), 7.72–7.69 (m, 2H), 7.49–7.46 (m, 2H), 4.05 (s, 3H).
Methyl 1-(4-Bromophenyl)-4-formyl-5-(pyrrolidin-1-yl)-1H-pyrazole-3-carboxylate (112)
Methyl 1-(4-bromophenyl)-5-chloro-4-formyl-pyrazole-3-carboxylate (111) (600 mg, 1.75 mmol) and pyrrolidine (248 mg, 3.49 mmol) were taken up in DMF (3 mL) before the addition of K2CO3 (483 mg, 3.49 mmol). The reaction mixture was heated to 120 °C in the microwave for 1 h and then quenched with water. The product was extracted with DCM, dried (hydrophobic frit), and concentrated in vacuo. The crude material was purified by flash chromatography (0–80% EtOAc in heptane) to afford the title compound (270 mg, 39%). LCMS (ESI) m/z: 378, 380 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 10.37 (s, 1H), 7.62–7.58 (m, 2H), 7.38–7.45 (m, 2H), 3.97 (s, 3H), 3.26–3.22 (m, 4H), 1.89–1.86 (m, 4H).
Methyl 1-(4-Bromophenyl)-5-(pyrrolidin-1-yl)-1H-pyrazole-3-carboxylate (116)
To a solution of p-TsOH (24 mg, 0.13 mmol) in MeOH (3 mL) was added methyl 1-(4-bromophenyl)-4-formyl-5-pyrrolidin-1-yl-pyrazole-3-carboxylate (112) (220 mg, 0.58 mmol). The reaction mixture was heated to 120 °C in the microwave for 1 h. The reaction was concentrated in vacuo to afford the title compound, which was used in the next step with no further purification (200 mg, 77%). LCMS (ESI) m/z: 350, 352 (M + H)+. 1H NMR (400 MHz, CDCl3): δ 7.59–7.50 (m, 4H), 6.21 (s, 1H), 3.92 (s, 3H), 3.00 (m, 4H), 1.88–1.85 (m, 4H).
1-(4-Bromophenyl)-5-(pyrrolidin-1-yl)-1H-pyrazole-3-carbaldehyde (120)
Methyl 1-(4-bromophenyl)-5-pyrrolidin-1-yl-pyrazole-3-carboxylate (116) (200 mg, 0.57 mmol) was dissolved in DCM (10 mL) and cooled to −78 °C under N2. DIBAL (1 M in DCM, 1.43 mL, 1.43 mmol) was then added dropwise over 20 min and the reaction was stirred for a further 1 h at −78 °C. The reaction was quenched with 5 mL of 1:1 MeOH/H2O, allowed to warm up to rt, and then passed through a hydrophobic frit and concentrated in vacuo to the title compound, which was used in the next step with no further purification (166 mg, 81%). LCMS (ESI) m/z: 320, 322 (M + H)+.1H NMR (400 MHz, CDCl3): δ 9.87 (s, 1H), 7.63–7.59 (m, 2H), 7.56–7.53 (m, 2H), 6.15 (s, 1H), 3.00 (m, 4H), 1.89–1.86 (m, 4H).
1′-[[1-(4-Bromophenyl)-5-pyrrolidin-1-yl-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (124)
Spiro[isochromane-1,4′-piperidine] (50 mg, 0.25 mmol), 1-(4-bromophenyl)-5-pyrrolidin-1-yl-1H-pyrazole-3-carbaldehyde (120) (88 mg, 0.25 mmol), and sodium triacetoxyborohydride (104 mg, 0.49 mmol) were combined in DCM (3 mL) according to General Method 1. Work-up followed by purification afforded the title compound (62 mg, 47%). HRMS (ESI) m/z: calcd for C27H32BrN4O [M + H]+: 509.1670; found, 509.1756. 1H NMR (400 MHz, CHCl3): δ 7.54–7.53 (m, 4H), 7.24–7.08 (m, 4H), 5.75 (s, 1H), 3.91 (t, J = 5.5 Hz, 2H), 3.58 (s, 2H), 3.00 (t, J = 6.6 Hz, 4H), 2.89–2.81 (m, 4H), 2.56–2.48 (m, 2H), 2.14–2.01 (m, 2H), 1.93–1.84 (m, 6H).
4-[5-Pyrrolidin-1-yl-3-(spiro[isochromane-1,4′-piperidine]-1′-ylmethyl)pyrazol-1-yl]benzoic Acid (33)
1′-[[1-(4-Bromophenyl)-5-pyrrolidin-1-yl-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (124) (62 mg, 0.12 mmol), N-formylsaccharin (31 mg, 0.15 mmol), Pd(OAc)2 (0.8 mg, 0.004 mmol), Xantphos (3 mg, 0.006 mmol), and KF (18 mg, 0.31 mmol) were combined in DMF (2 mL) according to General Method 5. Work-up and purification afforded the title compound (4 mg, 6%). HRMS (ESI) m/z: calcd for C28H33N4O3 [M + H]+: 473.2547; found, 473.2545. 1H NMR (400 MHz, D6-DMSO): δ 8.02 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.7 Hz, 2H), 7.22–7.07 (m, 4H), 5.83 (s, 1H), 3.81 (dd, J = 5.5, 5.5 Hz, 2H), 3.44 (s, 2H), 3.33 (s, 1H), 2.96 (dd, J = 6.4, 6.4 Hz, 4H), 2.73 (dd, J = 5.4, 5.4 Hz, 4H), 2.42–2.33 (m, 2H), 1.97–1.89 (m, 2H), 1.84–1.76 (m, 6H).
Synthesis of 4-(5-Morpholino-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (34) (Scheme 3)
Methyl 1-(4-Bromophenyl)-4-formyl-5-morpholino-1H-pyrazole-3-carboxylate (113)
Methyl 1-(4-bromophenyl)-5-chloro-4-formyl-1H-pyrazole-3-carboxylate (111) (100 mg, 0.29 mmol) and K2CO3 (80 mg, 0.58 mmol) were mixed in DMF (1 mL) and morpholine (0.05 mL, 0.58 mmol) was added. The reaction mixture was heated to 120 °C in the microwave for 1 h and then quenched with H2O and extracted with DCM. The organics were concentrated in vacuo, and the crude material was purified by flash chromatography (0–80% EtOAc in heptane) to afford the title compound (37 mg, 31%). 1H NMR (400 MHz, CDCl3): δ 10.44 (s, 1H), 7.66 (d, J = 8.8 Hz, 2H), 7.42 (d, J = 8.7 Hz, 2H), 3.99 (s, 3H), 3.73–3.69 (m, 4H), 3.11 (dd, J = 4.6, 4.6 Hz, 4H).
Methyl 1-(4-Bromophenyl)-5-morpholino-1H-pyrazole-3-carboxylate (117)
Methyl 1-(4-bromophenyl)-4-formyl-5-morpholino-1H-pyrazole-3-carboxylate (113) (250 mg, 0.63 mmol) was mixed in MeOH (5 mL) and p-TsOH (24 mg, 0.13 mmol) was added. The reaction mixture was heated to 120 °C in the microwave for 1 h and then concentrated in vacuo to afford the title compound (200 mg, 78%). 1H NMR (400 MHz, CDCl3): δ 7.73 (2H, d, J = 8.8 Hz), 7.61 (2H, d, J = 8.9 Hz), 6.47 (1H, s), 3.95 (3H, s), 3.77–3.73 (4H, m), 2.92–2.89 (4H, m).
1-(4-Bromophenyl)-5-morpholino-1H-pyrazole-3-carbaldehyde (121)
Methyl 1-(4-bromophenyl)-5-morpholino-1H-pyrazole-3-carboxylate (117) (275 mg, 0.75 mmol) and DIBAL (1 M in DCM) (1.88 mL, 1.88 mmol) were combined in DCM (5 mL) according to General Method 6a. Work-up afforded the title compound, which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 9.93 (1H, s), 7.78–7.56 (4H, m), 6.42 (1H, s), 3.78–3.74 (4H, m), 2.93–2.88 (4H, m). Used crude—contaminated with the benzyl alcohol.
1′-((1-(4-Bromophenyl)-5-morpholino-1H-pyrazol-3-yl)methyl)spiro[isochromane-1,4′-piperidine] (125)
Spiro[isochromane-1,4′-piperidine] (78 mg, 0.38 mmol), 1-(4-bromophenyl)-5-morpholino-1H-pyrazole-3-carbaldehyde (121) (215 mg, 0.38 mmol), and sodium triacetoxyborohydride (163 mg, 0.77 mmol) were combined in DCM (5 mL) according to General Method 1. Work-up and purification afforded the title compound (200 mg, 95%). 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.24–7.10 (m, 4H), 5.97 (s, 1H), 3.92 (m, 2H), 3.78–3.75 (m, 4H), 3.61 (s, 2H), 2.93–2.83 (m, 8H), 2.57–2.50 (m, 2H), 2.15–2.03 (m, 2H), 1.93 (d, J = 12.5 Hz, 2H).
4-(5-Morpholino-3-(spiro[isochromane-1,4′-piperidin]-1′-ylmethyl)-1H-pyrazol-1-yl)benzoic Acid (34)
1′-[[1-(4-Bromophenyl)-5-morpholino-1H-pyrazol-3-yl]methyl]spiro[isochromane-1,4′-piperidine] (125) (150 mg, 0.29 mmol), Pd(OAc)2 (2 mg, 0.01 mmol), Xantphos (7 mg, 0.01 mmol), KF (42 mg, 0.72 mmol), and N-formylsaccharin (73 mg, 0.34 mmol) were combined in DMF (2 mL) according to General Method 5. Work-up and purification afforded the title compound (8 mg, 5%). HRMS (ESI) m/z: calcd for C28H33N4O4 [M + H+], 498.2502; found, 489.2498. 1H NMR (400 MHz, D6-DMSO): δ 8.05–7.95 (m, 4H), 7.23–7.08 (m, 4H), 6.05 (s, 1H), 3.82 (dd, J = 5.4, 5.4 Hz, 2H), 3.70 (dd, J = 4.5, 4.5 Hz, 4H), 3.46 (s, 2H), 2.84 (dd, J = 4.5, 4.5 Hz, 4H), 2.75–2.71 (m, 4H), 2.41–2.33 (m, 2H), 1.97–1.90 (m, 2H), 1.78 (d, J = 12.7 Hz, 2H).
Synthesis of 4-(5-Cyclopropyl-3-((6-fluorospiro[isochromane-1,4′-piperidin]-1′-yl)methyl)-1H-pyrazol-1-yl)benzoic Acid (35) (Scheme 2)
4-(5-Cyclopropyl-3-((6-fluorospiro[isochromane-1,4′-piperidin]-1′-yl)methyl)-1H-pyrazol-1-yl)benzoic Acid (35)
6-Fluorospiro[isochromane-1,4′-piperidine] (50) (216 mg, 0.98 mmol), 4-(5-cyclopropyl-3-formyl-1H-pyrazol-1-yl)benzoic acid (85) (250 mg, 0.98 mmol), and sodium triacetoxyborohydride (414 mg, 1.95 mmol) were combined in DCM (4 mL) according to General Method 1. Work-up and purification afforded the title compound (261 mg, 57%). LCMS (ESI) m/z: 462 (M + H)+. HRMS (ESI) m/z: calcd for C27H29FN3O3 [M + H+], 462.2193; found, 462.2220. 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.25 (dd, J = 8.7, 5.5 Hz, 1H), 6.87 (td, J = 8.7, 2.7 Hz, 1H), 6.78 (dd, J = 9.2, 2.7 Hz, 1H), 6.06 (s, 1H), 3.97 (s, 2H), 3.90 (t, J = 5.5 Hz, 2H), 3.41 (d, J = 10.7 Hz, 2H), 2.94 (t, J = 12.2 Hz, 2H), 2.85–2.79 (m, 2H), 2.54 (td, J = 13.9, 4.0 Hz, 2H), 1.99 (d, J = 13.9 Hz, 2H), 1.86–1.78 (m, 1H), 0.99–0.93 (m, 2H), 0.74–0.69 (m, 2H).
Acknowledgments
We thank Lucy Ellis, Fred Simeons, Yoko Shishikura, Liam Ferguson, Lorna Campbell, James Roberts, Susan Lawrie, Alex Cookson, Kirsty Cookson, Emma Gutcher, Desiree Zeller, James Ahn, Torey Alling, Lena Anoshchenko, Bryan Grogan, Megha Gupta, Junitta Guzman, Yulia Ovechkina, David Roberts, Catherine Shelton, Bjorn Sunde, Dean Thompson, and Anisa Tracy for technical assistance. This work was funded in part by an award to P.G.W. from the Bill and Melinda Gates Foundation (OPP1066891) and Wellcome Trust (100195/Z/12/Z); by awards from BMG&F to T.P. (OPP1024038) and A.J.L./G.T.R. (OPP1126594); by Eli Lilly and Company in support of the mission of the Lilly TB Drug Discovery Initiative; and an award to T.P. by NIAID of the National Institutes of Health (R01AI099188). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05589.
Author Present Address
∇ Center for Global Infectious Disease Research, Seattle Children’s Research Institute, 307 Westlake Avenue N, Seattle, WA 98109, USA
Author Contributions
P.C.R., M.H., and P.A.T. contributed equally to medicinal chemistry design and planning. M.H., P.A.T., M.T., L.A.T.C., A.L.M., S.H.D., D.M., M.M., and N.C. contributed to molecule synthesis. J.E., A.K., S.A.B., L.F., D.J., J.J., A.K., S.M., C.A.E., D.S., L.M., G.T.R., A.J.L., S.R.G., and T.P. planned/executed biology studies. O.E., J.R., P.S., L.S., G.F., D.J.K., and K.D.R. planned/executed DMPK/safety studies. P.C.R., M.H., P.A.T., J.E., A.K., O.E., F.Z., T.M., P.A.H., J.O., S.R.G., P.G.W., and T.P. contributed to monthly project planning meetings. P.C.R., P.A, T., M.T., L.A.T.C., and S.R.G. wrote draft manuscript. A.K., C.A.E., D.S., G.T.R., A.J.L., D.J.K., T.M., P.A.H., K.D.R., P.G.W., and T.P. edited the manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Gordon S. V.; Parish T. Microbe Profile: Mycobacterium tuberculosis: Humanity’s deadly microbial foe. Microbiology 2018, 164, 437–439. 10.1099/mic.0.000601. [DOI] [PubMed] [Google Scholar]
- WHO . Global Tuberculosis Report 2020; World Health Organization: Geneva (Switzerland), 2020; ISBN 978-92-4-001313-1.
- Zumla A.; George A.; Sharma V.; Herbert R. H. N.; Oxley A.; Oliver M.; Oliver M. The WHO 2014 Global tuberculosis report-further to go. Lancet Global Health 2015, 3, E10–E12. 10.1016/S2214-109x(14)70361-4. [DOI] [PubMed] [Google Scholar]
- Dheda K.; Barry C. E.; Maartens G. Tuberculosis. Lancet 2016, 387, 1211–1226. 10.1016/s0140-6736(15)00151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh C. R. Jr.; Barry C. E. 3rd; Lange C. Treatment of Tuberculosis. N. Engl. J. Med. 2015, 373, 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- Lechartier B.; Rybniker J.; Zumla A.; Cole S. T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014, 6, 158–168. 10.1002/emmm.201201772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kana B. D.; Karakousis P. C.; Parish T.; Dick T. Future target-based drug discovery for tuberculosis?. Tuberculosis 2014, 94, 551–556. 10.1016/j.tube.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Smith P. W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg. Med. Chem. 2015, 23, 5087–5097. 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Zuniga E. S.; Early J.; Parish T. The future for early-stage tuberculosis drug discovery. Future Microbiol. 2015, 10, 217–229. 10.2217/fmb.14.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman R. C. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis?. Tuberculosis 2013, 93, 569–588. 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- Mdluli K.; Kaneko T.; Upton A. The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harbor Perspect. Med. 2015, 5, a021154. 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Upadhyay A.; Fontes F. L.; North E. J.; Wang Y.; Crans D. C.; Grzegorzewicz A. E.; Jones V.; Franzblau S. G.; Lee R. E.; Crick D. C.; Jackson M. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G.; Consalvi S.; Biava M. MmpL3 Inhibitors: Diverse Chemical Scaffolds Inhibit the Same Target. Mini-Rev. Med. Chem. 2016, 16, 1274–1283. 10.2174/1389557516666160118105319. [DOI] [PubMed] [Google Scholar]
- Sacksteder K. A.; Protopopova M.; Barry C. E. 3rd; Andries K.; Nacy C. A. Discovery and development of SQ109: a new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa V.; Poce G.; Canseco J. O.; Buroni S.; Pasca M. R.; Biava M.; Raju R. M.; Porretta G. C.; Alfonso S.; Battilocchio C.; Javid B.; Sorrentino F.; Ioerger T. R.; Sacchettini J. C.; Manetti F.; Botta M.; De Logu A.; Rubin E. J.; De Rossi E. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. 10.1128/AAC.05270-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poce G.; Bates R. H.; Alfonso S.; Cocozza M.; Porretta G. C.; Ballell L.; Rullas J.; Ortega F.; De Logu A.; Agus E.; La Rosa V.; Pasca M. R.; De Rossi E.; Wae B.; Franzblau S. G.; Manetti F.; Botta M.; Biava M. Improved BM212 MmpL3 inhibitor analogue shows efficacy in acute murine model of tuberculosis infection. PLoS One 2013, 8, e56980. 10.1371/journal.pone.0056980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; Fitzgerald M.; Dai H.; Zhang L.; Hung D. T. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 2012, 7, 1377–1384. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remuiñán M. J.; Pérez-Herrán E.; Rullás J.; Alemparte C.; Martínez-Hoyos M.; Dow D. J.; Afari J.; Mehta N.; Esquivias J.; Jiménez E.; Ortega-Muro F.; Fraile-Gabaldón M. T.; Spivey V. L.; Loman N. J.; Pallen M. J.; Constantinidou C.; Minick D. J.; Cacho M.; Rebollo-López M. J.; González C.; Sousa V.; Angulo-Barturen I.; Mendoza-Losana A.; Barros D.; Besra G. S.; Ballell L.; Cammack N. Tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide and N-benzyl-6’,7’-dihydrospiro[piperidine-4,4’-thieno[3,2-c]pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLoS One 2013, 8, e60933. 10.1371/journal.pone.0060933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondreddi R. R.; Jiricek J.; Rao S. P. S.; Lakshminarayana S. B.; Camacho L. R.; Rao R.; Herve M.; Bifani P.; Ma N. L.; Kuhen K.; Goh A.; Chatterjee A. K.; Dick T.; Diagana T. T.; Manjunatha U. H.; Smith P. W. Design, synthesis, and biological evaluation of indole-2-carboxamides: a promising class of antituberculosis agents. J. Med. Chem. 2013, 56, 8849–8859. 10.1021/jm4012774. [DOI] [PubMed] [Google Scholar]
- Lun S.; Guo H.; Onajole O. K.; Pieroni M.; Gunosewoyo H.; Chen G.; Tipparaju S. K.; Ammerman N. C.; Kozikowski A. P.; Bishai W. R. Indoleamides are active against drug-resistant Mycobacterium tuberculosis. Nat. Commun. 2013, 4, 2907. 10.1038/ncomms3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzegorzewicz A. E.; Pham H.; Gundi V. A. K. B.; Scherman M. S.; North E. J.; Hess T.; Jones V.; Gruppo V.; Born S. E. M.; Korduláková J.; Chavadi S. S.; Morisseau C.; Lenaerts A. J.; Lee R. E.; McNeil M. R.; Jackson M. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H.; Williams J. T.; Coulson G. B.; Haiderer E. R.; Abramovitch R. B. HC2091 Kills Mycobacterium tuberculosis by Targeting the MmpL3 Mycolic Acid Transporter. Antimicrob. Agents Chemother. 2018, 62, e02459. 10.1128/AAC.02459-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud C.; Daher W.; Johansen M. D.; Roquet-Banères F.; Blaise M.; Onajole O. K.; Kozikowski A. P.; Herrmann J.-L.; Dziadek J.; Gobis K.; Kremer L. Active Benzimidazole Derivatives Targeting the MmpL3 Transporter in Mycobacterium abscessus. ACS Infect. Dis. 2020, 6, 324–337. 10.1021/acsinfecdis.9b00389. [DOI] [PubMed] [Google Scholar]
- Dal Molin M.; Selchow P.; Schäfle D.; Tschumi A.; Ryckmans T.; Laage-Witt S.; Sander P. Identification of novel scaffolds targeting Mycobacterium tuberculosis. J. Mol. Med. 2019, 97, 1601–1613. 10.1007/s00109-019-01840-7. [DOI] [PubMed] [Google Scholar]
- Poce G.; Consalvi S.; Venditti G.; Alfonso S.; Desideri N.; Fernandez-Menendez R.; Bates R. H.; Ballell L.; Barros Aguirre D.; Rullas J.; De Logu A.; Gardner M.; Ioerger T. R.; Rubin E. J.; Biava M. Novel Pyrazole-Containing Compounds Active against Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2019, 10, 1423–1429. 10.1021/acsmedchemlett.9b00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont C.; Chen Y.; Xu Z.; Roquet-Banères F.; Blaise M.; Witt A.-K.; Dubar F.; Biot C.; Guérardel Y.; Maurer F. P.; Chng S.-S.; Kremer L. A piperidinol-containing molecule is active against Mycobacterium tuberculosis by inhibiting the mycolic acid flippase activity of MmpL3. J. Biol. Chem. 2019, 294, 17512–17523. 10.1074/jbc.RA119.010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. T.; Haiderer E. R.; Coulson G. B.; Conner K. N.; Ellsworth E.; Chen C.; Alvarez-Cabrera N.; Li W.; Jackson M.; Dick T.; Abramovitch R. B. Identification of New MmpL3 Inhibitors by Untargeted and Targeted Mutant Screens Defines MmpL3 Domains with Differential Resistance. Antimicrob. Agents Chemother. 2019, 63, e00547. 10.1128/AAC.00547-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korycka-Machala M.; Viljoen A.; Pawelczyk J.; Borowka P.; Dziadek B.; Gobis K.; Brzostek A.; Kawka M.; Blaise M.; Strapagiel D.; Kremer L.; Dziadek J. 1H-Benzo[d]Imidazole Derivatives Affect MmpL3 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e00441. 10.1128/AAC.00441-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia A.; Baiget J.; Cacho M.; Pérez A.; Ortega-Guerra M.; Nxumalo W.; Khanye S. D.; Rullas J.; Ortega F.; Jiménez E.; Pérez-Herrán E.; Fraile-Gabaldón M. T.; Esquivias J.; Fernandez R.; Fernández E.; Encinas L.; Alonso M.; Giordano I.; Rivero C.; Miguel-Siles J.; Osende J. G.; Badiola K. A.; Rutledge P. J.; Todd M. H.; Remuiñán M.; Alemparte C. Easy-To-Synthesize Spirocyclic Compounds Possess Remarkable in Vivo Activity against Mycobacterium tuberculosis. J. Med. Chem. 2018, 61, 11327–11340. 10.1021/acs.jmedchem.8b01533. [DOI] [PubMed] [Google Scholar]
- Hill A. P.; Young R. J. Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discovery Today 2010, 15, 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Bayliss M. K.; Butler J.; Feldman P. L.; Green D. V. S.; Leeson P. D.; Palovich M. R.; Taylor A. J. Quality guidelines for oral drug candidates: dose, solubility and lipophilicity. Drug Discovery Today 2016, 21, 1719–1727. 10.1016/j.drudis.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Young R. J. Molecular Property Design: Does Everyone Get It?. ACS Med. Chem. Lett. 2015, 6, 722–725. 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. J.; Green D. V. S.; Luscombe C. N.; Hill A. P. Getting physical in drug discovery II: the impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discovery Today 2011, 16, 822–830. 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Miller M. M.; Liu Y.; Jiang J.; Johnson J. A.; Kamau M.; Nirschl D. S.; Wang Y.; Harikrishnan L.; Taylor D. S.; Chen A. Y. A.; Yin X.; Seethala R.; Peterson T. L.; Zvyaga T.; Zhang J.; Huang C. S.; Wexler R. R.; Poss M. A.; Michael Lawrence R.; Adam L. P.; Salvati M. E. Identification of a potent and metabolically stable series of fluorinated diphenylpyridylethanamine-based cholesteryl ester transfer protein inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6503–6508. 10.1016/j.bmcl.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Bonn B.; Masimirembwa C. M.; Castagnoli N. Jr. Exploration of catalytic properties of CYP2D6 and CYP3A4 through metabolic studies of levorphanol and levallorphan. Drug Metab. Dispos. 2010, 38, 187–199. 10.1124/dmd.109.028670. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. 10.1021/jm060379l. [DOI] [PubMed] [Google Scholar]
- Badiola K. A.; Quan D. H.; Triccas J. A.; Todd M. H. Efficient Synthesis and Anti-Tubercular Activity of a Series of Spirocycles: An Exercise in Open Science. PLoS One 2014, 9, e111782. 10.1371/journal.pone.0111782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T.; Konishi H.; Manabe K. Palladium-Catalyzed Fluorocarbonylation Using N-Formylsaccharin as CO Source: General Access to Carboxylic Acid Derivatives. Org. Lett. 2013, 15, 5370–5373. 10.1021/ol4026815. [DOI] [PubMed] [Google Scholar]
- Ollinger J.; Bailey M. A.; Moraski G. C.; Casey A.; Florio S.; Alling T.; Miller M. J.; Parish T. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS One 2013, 8, e60531. 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridal T. R.; Margulis M.; Wang X.; Donio M.; Sorota S. Comparison of human Ether-a-go-go related gene screening assays based on IonWorks Quattro and thallium flux. Assay Drug Dev. Technol. 2010, 8, 755–765. 10.1089/adt.2010.0267. [DOI] [PubMed] [Google Scholar]
- Bowes J.; Brown A. J.; Hamon J.; Jarolimek W.; Sridhar A.; Waldron G.; Whitebread S. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discovery 2012, 11, 909–922. 10.1038/nrd3845. [DOI] [PubMed] [Google Scholar]
- Lenaerts A. J.; Gruppo V.; Marietta K. S.; Johnson C. M.; Driscoll D. K.; Tompkins N. M.; Rose J. D.; Reynolds R. C.; Orme I. M. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob. Agents Chemother. 2005, 49, 2294–2301. 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.