

Abstract
Compared to the significant number of studies reporting altered abundance and function of drug transporters at the blood-brain barrier (BBB) in Alzheimer’s disease (AD), the impact of AD on the abundance of intestinal drug transporters and the subsequent effects on oral drug absorption have received little attention. We have reported altered abundance of some small intestinal drug transporters in a familial mouse model of AD, however, whether this leads to altered oral drug absorption is unknown. The current study examined plasma concentrations of caffeine and diazepam (markers for transcellular passive transport), digoxin (P-glycoprotein substrate) and valsartan (multidrug resistance-associated protein 2 substrate) following oral administration to 8-10 months old female wild-type (WT) and APPswe/PSEN1dE9 (APP/PS1) transgenic mice, a commonly used mouse model of familial AD. Plasma exposure of valsartan and digoxin was significantly (p < 0.05) lower in APP/PS1 animals compared to WT mice, while the plasma concentrations of the passive transcellular markers caffeine and diazepam did not differ significantly between the two genotypes. To assess whether the reduced oral absorption of valsartan and digoxin was due to decreased intestinal transport, the ex vivo transport of the above-mentioned drugs and mannitol (a marker of paracellular transport) across the jejunum of WT and APP/PS1 mice was assessed over 120 min. In line with the in vivo absorption studies, the permeability of caffeine and diazepam did not differ significantly between WT and APP/PS1 mice. The permeability of 3H-digoxin through APP/PS1 mouse jejunum was lower than that measured through WT jejunum; the average amount (relative to dose applied) permeating the tissue over 120 min was 0.22 ± 0.11% (mean ± SD) for APP/PS1 jejunum and 0.85 ± 0.3% for WT jejunum. A 1.9-fold reduction in the average amount of valsartan permeating the jejunum of APP/PS1 mice relative to WT mice also was detected. Although no apparent morphological alterations were observed in jejunal tissue of APP/PS1 mice, the permeability of 14C-mannitol across jejunum from APP/PS1 mice was lower than that across WT jejunum (Papp = 10.7 ± 3.7 ×10−6 cm/s and 6.0 ± 3.4 ×10−6 cm/s, respectively), suggesting tightened paracellular junctions in APP/PS1 mice. These studies are the first to demonstrate, in APP/PS1 mice, reduced intestinal permeability and absorption of drugs commonly prescribed to people with AD for their comorbidities. Modified dosing regimens may be necessary for selected drugs to ensure their plasma concentrations remain in the effective range, if these findings translate to people with AD.
Keywords: Alzheimer’s disease, intestinal transport, oral absorption, drug transporters
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia, which accounts for up to 75% of all dementia cases1. Given that age is the most common risk factor for dementia, it is not surprising that individuals with AD are prescribed multiple medicines for comorbidities, such as hypertension, hypercholesterolaemia and diabetes2, 3. People with dementia have up to five or more comorbidities and may be prescribed 10 more medicines than those without dementia4. These medicines are prescribed to individuals with AD using the same dosing regimens as those used in individuals without AD. This dosing is under the assumption that absorption, distribution, metabolism and excretion (ADME) of drugs are not altered in this disease, which is not surprising given that AD is generally considered a disease of the central nervous system (CNS). However, if the processes of drug absorption and/or disposition are indeed altered in AD, it is possible that people with AD may be at a greater risk of unexpected adverse effects or suboptimal efficacy of these non-AD medicines.
Given that AD mainly affects the CNS, there have been many studies assessing the abundance and function of drug transporters at the blood-brain barrier (BBB), with reports of modified abundance and function of these key determinants of CNS drug exposure5, 6, 7. For example, decreased BBB abundance of P-glycoprotein (P-gp) was detected in mouse models of AD and humans with AD6, 7, which was associated with increased brain exposure to the P-gp substrate verapamil relative to healthy age-matched subjects8. The abundance of breast cancer resistance protein (BCRP) at the BBB, another significant drug efflux transporter, was reported to be increased in a mouse model of AD and in people with AD9, which would likely impact the brain uptake of compounds that are BCRP substrates. While there is significant knowledge about the abundance and function of drug transport proteins at the AD BBB, less focus has been given to elucidating drug transporter abundance and function in peripheral organs or biological barriers, albeit a small number of reports suggest altered pharmacokinetics of drugs in preclinical models of AD. For example, relative to wild-type (WT) mice, we demonstrated that plasma concentrations of PBT2, a preclinical drug candidate for AD, were 60% lower in Tg2576 mice (a mouse model of familial AD) following oral administration. Given that plasma concentrations of PBT2 were only measured at one time point (120 min post-dose), no mechanistic insight into the reason for the reduced plasma exposure of this preclinical drug candidate was possible10. To our knowledge, there has only been one clinical study that reported altered oral drug exposure in individuals with AD; plasma concentrations of rivastigmine were significantly higher following oral administration to people with AD compared to healthy volunteers11. These differences in rivastigmine pharmacokinetics were attributed to changes in oral absorption because the ability to metabolize rivastigmine was suggested not to differ in individuals with AD.
As a result of these preclinical and clinical observations, and given that altered hepatic cytochrome Cyp2b-, Cyp2e1-, Cyp3a- and Cyp4a-associated activities have been reported in Tg2576 mice12, we recently compared the abundance of a number of drug transporters in the small intestine of WT and APP/PS1 mice, a mouse model of familial AD. Utilizing a quantitative transporter absolute proteomics (QTAP) approach, we demonstrated that the abundance of several transporters and enzymes was significantly altered in the small intestine of APP/PS1 mice relative to WT mice, including a 2.3-fold increase in multidrug resistance-associated protein 2 (Mrp2), a 1.9-fold decrease in monocarboxylate transporter 1 (Mct1), and a 3.6-fold increase in UDP-glucuronosyltransferase 2b513. The abundance of P-glycoprotein (P-gp) was not significantly different between WT and APP/PS1 mice. In addition to altered transporter abundance at the small intestine, we also recently reported that the renal abundance of Mrp2, organic anion transporter 3, and organic cation transporter 2 was significantly higher in APP/PS1 mice relative to WT mice14. Taken together, these results indicate that the processes governing intestinal absorption, metabolism and renal excretion are likely to be affected in AD.
While we have demonstrated altered abundance of small intestinal transporters, a systematic evaluation of the impact of this disease on the intestinal permeability of drugs, oral absorption and subsequently brain exposure of drugs in APP/PS1 mice has not been reported. The aim of this study, therefore, was to assess the intestinal permeability and oral absorption of a panel of drugs that permeate the small intestine via different mechanisms, including a substrate of Mrp2 (given the significant up-regulation of Mrp2), and markers of passive paracellular and transcellular diffusion. Valsartan was chosen as a substrate of Mrp215, 16, mannitol was selected as a marker of paracellular permeability, and caffeine and diazepam were selected as markers of transcellular diffusion (reflecting both a hydrophilic and lipophilic marker, respectively)17–19. In addition to assessing plasma concentrations following oral administration, the brain concentrations of each drug were measured to assess whether previously-reported alterations in BBB structure and function in AD influenced the extent of brain uptake20. For this reason, digoxin also was included in this study as a substrate of P-gp21, 22, given the previous reports of reduced BBB abundance of P-gp in mouse models of AD and in humans with AD23, 24. Comparing the in vivo and ex vivo oral absorption of these probe compounds provides insight into how people with AD potentially handle medicines differently relative to people without AD, and whether dose modifications are required to ensure medicine safety and efficacy. In this study, the animal model used to assess alterations in oral drug absorption is the same mouse model used to demonstrate altered transporter abundance via QTAP (i.e. APP/PS1 mice). As this model is routinely used to investigate AD-associated pathogenesis, it is considered a relevant preclinical model to assess pharmacokinetic changes that are associated with this disease25, 26.
Materials and methods
Materials.
14C-mannitol, 14C-caffeine, 3H-digoxin and 3H-diazepam were obtained from American Radiolabeled Chemicals, Inc (Saint Louis, MO). Ultima Gold™ liquid scintillation cocktail was purchased from Perkin Elmer (Boston, MA). Valsartan was purchased from Sapphire Bioscience (Redfern, New South Wales, Australia). Digoxin, diazepam and ammonium acetate were obtained from Sigma-Aldrich Pty Ltd (Castle Hill, New South Wales, Australia). HPLC or LC-MS grade acetonitrile and methanol were all purchased from Merck KGaA (Darmstadt, Germany).
Animals.
Animal experiments were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee and performed in accordance with the National Health and Medical Research Council guidelines for the care and use of animals for scientific purposes. Female APP/PS1 (B6C3-Tg (APPswe, PSEN1dE9)85Dbo/Mmjax) and age-matched littermate WT mice were obtained from Jackson Laboratories at 2-3 months of age and were housed with ad libitum access to standard rodent chow and water until they were 8-10 months old (22 - 40 g). This mouse model exhibits significant brain accumulation of amyloid-beta (Aβ), the protein implicated in the pathology associated with human AD, due to insertion of human transgenes responsible for overproduction of this protein (i.e., amyloid precursor protein (APP) and presenilin 1 (PS1))27. In addition to accumulation of brain Aβ, the mice exhibit cognitive dysfunction and, therefore, are considered a relevant model to study AD-associated pathogenesis25. Animals were used for experiments at 8-10 months given that APP/PS1 mice exhibit robust Aβ accumulation and cognitive deficit at this age25–27.
Oral administration of probe compounds to mice.
On the day of the experiment, WT and APP/PS1 mice were dosed by oral gavage with either caffeine (5 mg/kg in MilliQ water), diazepam (30 mg/kg in milli Q water containing 1% w/v carboxymethylcellulose sodium), digoxin (0.2 mg/kg in water containing 40% v/v propylene glycol and 10% v/v ethanol) or valsartan (15 mg/kg in water containing 40% v/v propylene glycol and 10% v/v ethanol). These doses have been administered to mice before and appeared to be well tolerated in animals. At each post-dose time point, ranging from 0.25 – 6 h, mice were anaesthetized with gaseous isoflurane, and blood samples (500 μL) were collected by cardiac puncture and immediately centrifuged to obtain plasma. Mice were sacrificed by cervical dislocation and brain samples were collected. Both plasma and brain samples were stored at −20°C until analysis for drug quantification by the developed and validated HPLC or LC MS/MS assays (Supporting Information). The concentration of each of the probe compounds in brain parenchyma were corrected for cerebral vascular contamination using a microvascular volume we determined previously in C57BL/6J mice using 14C-inulin (i.e. 0.017 mL/g)28. It should be noted that this vascular volume is similar to the vascular volume reported in 3×Tg AD mice (one mouse model of AD)5. A brain-to-plasma concentration ratio (B:P ratio) was then calculated by the formula:
Ex vivo permeability studies.
Mouse jejunal segments from WT and APP/PS1 mice were used to assess the intestinal permeability of the probe compounds. As previously described29, mice were anaesthetized with gaseous isoflurane, the jejunal segments were collected, and the mice were then sacrificed by cervical dislocation. The jejunal segments were washed with ice-cold Krebs Bicarbonate-buffered Ringers (KBR) solution at pH 6.0 and immediately mounted onto modified Ussing chambers (diffusional area 0.12 cm2) with the mucosal side facing the donor chamber. KBR solution (5 mL) was added into both the donor and receptor chambers and bubbled with 5% CO2/95% O2 for 15 min. Fresh KBR solution (5 mL) then replaced the solution in the donor and receptor chambers and 0.5 μCi of radiolabelled compound (i.e., 14C-mannitol, 14C-caffeine, 3H-digoxin and 3H-diazepam) or valsartan (20 μg/mL), in separate experiments, was added into the donor chambers. It was not possible to source valsartan in a radioactive form, and for this reason, an unlabelled form of valsartan was used in the ex vivo study. At the designated time points up to 120 min, 200 μL of receptor chamber solution was collected and replaced with an equal volume of fresh KBR. Samples (10 μL) also were collected from the donor chambers at the commencement of the experiment to quantify the initial compound concentration in the donor chamber. At the end of the study, the concentration of compounds in the collected samples were quantified using liquid scintillation counting (Tri-carb 2800TR, Perkin Elmer, USA) for radioactive compounds, or the validated HPLC assay for valsartan (Supporting Information). The apparent permeability coefficient (Papp) (cm/s) of each compound was calculated using the equation:
where dQ/dt is the rate of appearance of compound in the receptor chamber measured from the linear portion of a plot of the cumulative amount transported vs time, C is the concentration of the compound in the donor chamber, and A is the cross-sectional area of the tissue.
Histological analysis.
In order to evaluate whether general morphological alterations occur in the small intestine to explain some of the functional changes observed, WT and APP/PS1 mice were anesthetized with gaseous isoflurane and perfused transcardially with 10 mL of ice-cold phosphate-buffered saline (PBS) and 20 mL of ice-cold 4% v/v paraformaldehyde (PFA) at 10 mL/min. Jejunal tissue from both WT and APP/PS1 mice was then collected and fixed in 4% v/v PFA overnight at 4 ˚C. Sectioning and histological examination of small intestinal tissues were performed at Melbourne Histology Platform and Australian Phenomics Network (The University of Melbourne, Australia). Briefly, tissues were embedded in paraffin blocks, cut into 5 μm sections, and then stained with hematoxylin and eosin (H&E). The histological sections were then observed under a light microscope and photographed using the 3D Histech Pannoramic Scan II Slide scanner fitted with a x 20 Zeiss objective (Carl Zeiss Pty Ltd, New South Wales, Australia).
Statistical analysis.
The results were presented as mean ± SD unless stated otherwise. The area under the plasma concentration- and brain concentration-time curves of each compound from time zero to the last time point (AUCplasma and AUCbrain) and their associated errors were calculated using Bailer’s approach30, because of the destructive sampling technique required to obtain brain homogenate concentrations at each time point. A z-test was used to test for significant differences in AUCplasma and AUCbrain of each probe compound between WT and APP/PS1 mice. A z value > 1.96 was considered to be a significant difference between genotypes. Statistical comparisons of the B:P ratio, the Papp and the percentage of applied dose permeating jejunal tissue from WT and APP/PS1 mice were undertaken using Student’s t-tests (IBM SPSS Statistics for Windows, version 23.0; Armonk, NY). Statistical significance was set as p < 0.05.
Results
APP/PS1 mice exhibited altered oral absorption of probe compounds.
A total of eight HPLC and LC-MS/MS assays for the determination of the plasma and brain homogenate concentrations of the probe compounds were developed and validated, the details of which are provided in the Supporting Information. As shown in Fig. 1A and Table 1, the plasma concentrations of caffeine following oral administration did not differ between WT and APP/PS1 mice. Similarly, no significant difference between the oral absorption of diazepam was observed between WT and APP/PS1 mice (p > 0.05) as shown in Fig. 2A and Table 1. Following oral administration of valsartan, plasma concentrations reached a maximum at 1 h in both WT and APP/PS1 mice, however, a significantly lower AUCplasma was observed in APP/PS1 mice compared to WT mice (z > 1.96) (Fig. 3A, Table 1). The plasma concentrations of digoxin following oral administration peaked at 0.5 h in both WT and APP/PS1 mice. Despite a similar time required to reach maximum plasma concentrations (Fig. 4A), there was a substantially lower systemic exposure to digoxin in APP/PS1 mice relative to WT mice, as observed for valsartan, with a z value of 2.14 when comparing AUCplasma between WT and APP/PS1 mice (Table 1).
Figure 1.
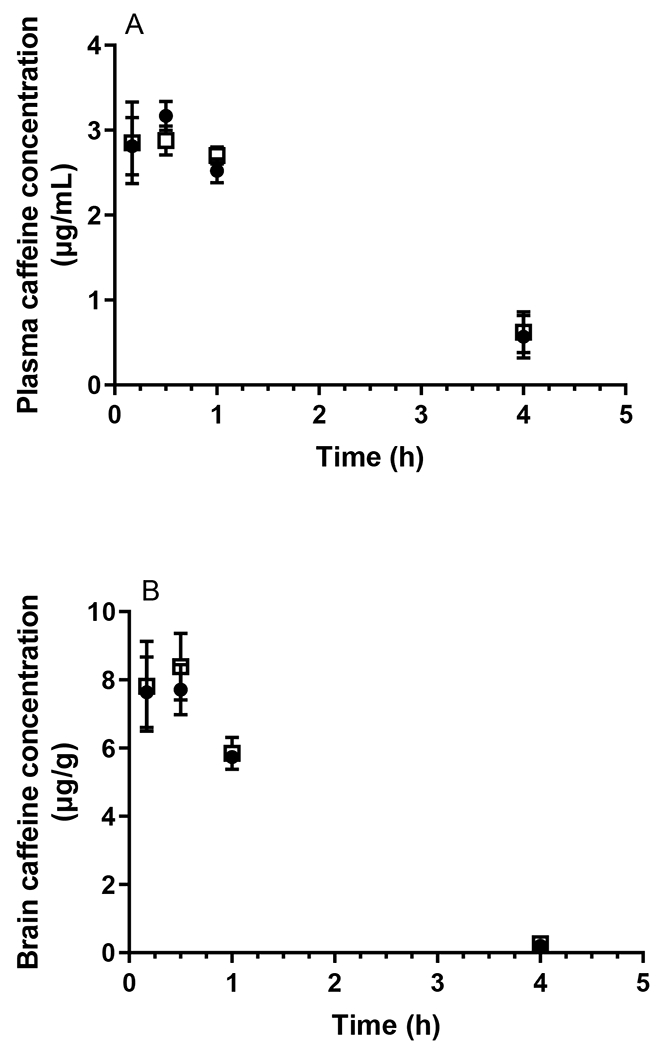
Plasma (A) and brain (B) concentrations of caffeine following oral administration (5 mg/kg) to WT (closed circles) and APP/PS1 mice (open squares). Data are presented as mean ± SD (n=3).
Table 1.
The AUCplasma, AUCbrain and z values of probe compounds following oral administration to WT and APP/PS1 mice. Data are presented as mean ± SD (n=3).
| Compound | AUCplasma (ng·h/mL) | AUCbrain (ng·h/g) | ||||
|---|---|---|---|---|---|---|
| WT | APP/PS1 | z value | WT | APP/PS1 | z value | |
| Caffeine | 10380 ± 738 | 11289 ± 681 | 0.90 | 21520 ± 224 | 22178 ± 356 | 1.56 |
| Diazepam | 356.6 ± 35.6 | 343.3 ± 29.2 | 0.29 | 119.5 ± 5.5 | 117.7 ± 18.2 | 0.10 |
| Valsartan | 1960 ± 231 | 1388 ± 105 | 2.26* | NA | NA | |
| Digoxin | 199.4 ± 12.7 | 154.1 ± 16.8 | 2.14* | NA | NA | |
NA: AUC value was not able to be calculated due to the detection limit of the brain assay.
z > 1.96 indicates a significant difference in AUCplasma or AUCbrain between WT and APP/PS1 mice.
Figure 2.
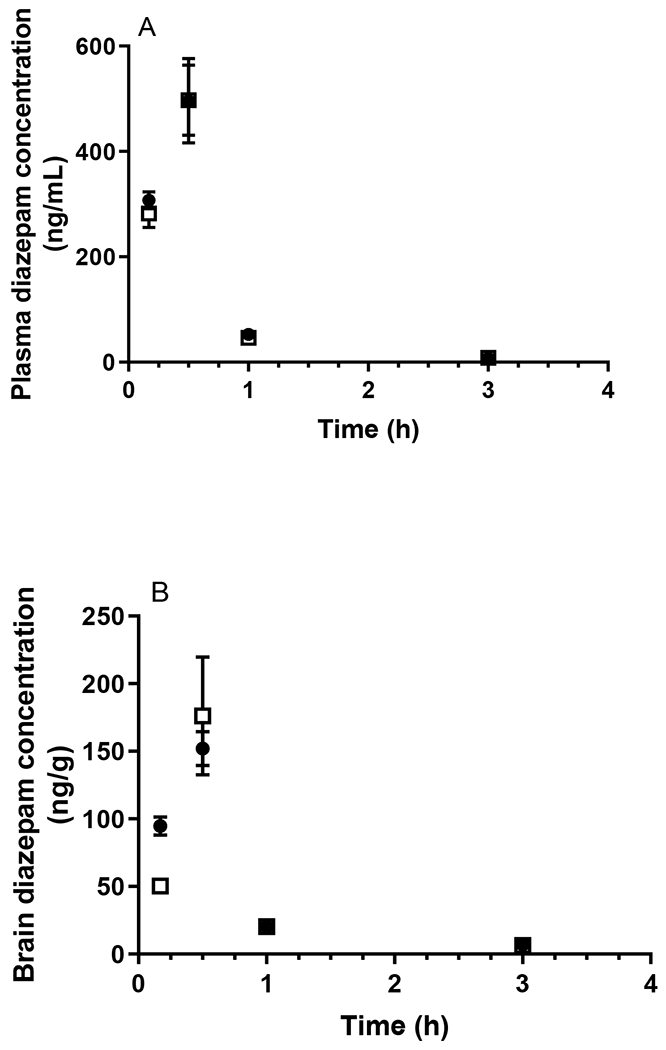
Plasma (A) and brain (B) concentrations of diazepam following oral administration (30 mg/kg) to WT (closed circles) and APP/PS1 mice (open squares). Data are presented as mean ± SD (n=3).
Figure 3.
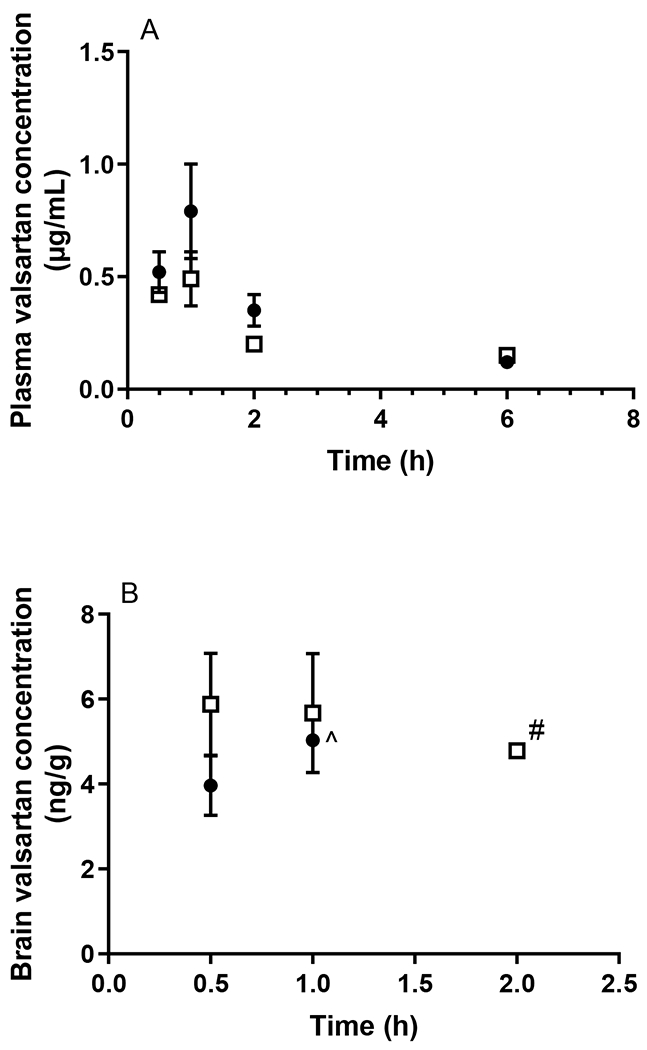
Plasma (A) and brain (B) concentrations of valsartan following oral administration (0.2 mg/kg) to WT (closed circles) and APP/PS1 mice (open squares). ^ Brain concentrations of valsartan were only detectable in one WT mouse at 1 h and were undetectable at later time points. # Brain concentrations of valsartan were only detectable in two APP/PS1 mice at 2 h and were undetectable at later time points. Data are presented as mean ± SD (n=3).
Figure 4.
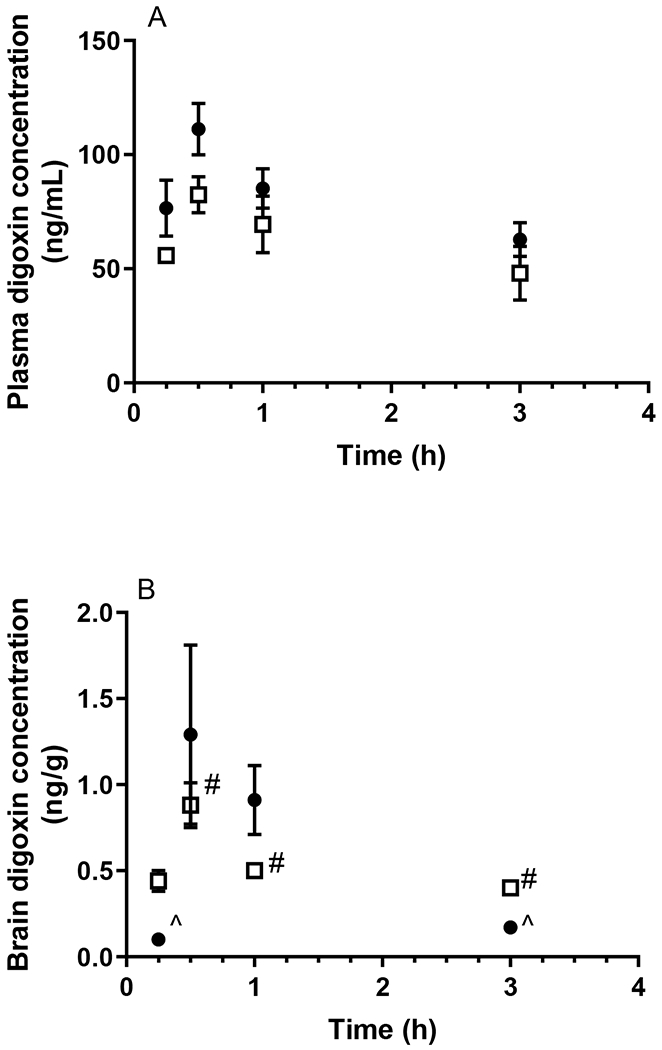
Plasma (A) and brain (B) concentrations of digoxin following oral administration (15 mg/kg) to WT (closed circles) and APP/PS1 mice (open squares). ^ Brain concentrations of digoxin were only detectable in one WT mouse at 0.25 and 3 h. # Brain concentrations of digoxin were only detectable in two APP/PS1 mice at 0.5, 1 and 3 h. Data are presented as mean ± SD (n=3).
In addition to measuring plasma concentrations following oral administration, the brain concentrations of each probe compound were assessed using the validated brain homogenate assays. The total brain exposure to caffeine did not differ between WT and APP/PS1 mice (Fig 1B) with respective average AUCbrain values of 21.52 and 22.18 μg·h/g, as shown in Table 1. The B:P ratio of caffeine in WT animals ranged between 1.07-2.78 over the sampling period; similar ratios were detected in APP/PS1 mice (ranging from 0.63-2.82). Overall, there was no significant difference in AUCbrain of diazepam between WT and APP/PS1 mice with a z value of 0.10. While the B:P ratio of diazepam was similar between WT and APP/PS1 mice at most post-dose time points (ranging from 0.31-1.07 in WT mice and 0.18-0.81 in APP/PS1 mice), the B:P ratio of diazepam in APP/PS1 mice (0.18 ± 0.05) was significantly (p < 0.05) lower relative to that in WT mice (0.31 ± 0.07) at the first post-dose time point (10 min), albeit no differences in diazepam plasma concentrations between the two genotypes were observed at this time point (Fig. 2B). Due to the brain assay detection limit of valsartan (i.e. 3 ng/g), brain concentrations of valsartan following oral administration were not detectable after 1 h in WT mice, and negligible after 2 h in both WT and APP/PS1 mice (Fig. 3B). However, the brain uptake of valsartan and B: P ratios at 0.5 h were not significantly different between WT and APP/PS1 mice (p > 0.05) with average values of 0.008 in WT mice and 0.012 in APP/PS1 mice. Similarly, the brain concentrations of digoxin in some samples at each post-dose time point were not detectable due to the assay limit (i.e., 0.6 ng/g) (Fig. 4B). Therefore, a statistical comparison of B:P ratio at each time point and overall digoxin AUCbrain comparison between WT and APP/PS1 animals could not be determined.
Permeability of probe compounds differs between jejunal tissue from WT and APP/PS1 mice.
To ascertain whether the altered oral absorption of digoxin and valsartan resulted from changes in intestinal permeability, the transport of these probe drugs as well as diazepam and caffeine was assessed in jejunal tissue obtained from WT and APP/PS1 mice. In addition, the permeability of 14C-mannitol was assessed to evaluate whether the paracellular route of transport was modified in this intestinal tissue. The permeability profiles of the five probe compounds through jejunal tissues obtained from WT and APP/PS1 mice are shown in Fig. 5, and the Papp values and the percentage of applied amount absorbed are reported in Table 2. In line with the in vivo absorption studies, the appearance of 14C-caffeine and 3H-diazepam in the receptor chamber did not differ significantly whether the jejunal tissue was from WT or APP/PS1 mice. The average amount of 14C-caffeine absorbed across the jejunal tissue from WT and APP/PS1 mice over a 2 h period was 0.40% and 0.46%, respectively. Similarly, the average amount of 3H-diazepam absorbed across the jejunal tissue from WT or APP/PS1 mice at the end of the 2 h period was 0.42% and 0.52%, respectively. A HPLC assay for the determination of valsartan in KBR buffer was developed and validated, the details of which are provided in the Supporting Information. A significant decrease in valsartan permeability was detected through the jejunum from APP/PS1 mice compared to WT mice (p < 0.05); the average amount of valsartan absorbed across jejunal tissue from APP/PS1 mice was only approximately one-half of that across jejunal tissue from WT mice. In line with that observed with in vivo absorption studies, a substantially reduced permeability of 3H-digoxin through the jejunum from APP/PS1 mice also was observed compared to WT mice (p < 0.05), with the average Papp values (×10−6 cm/s) being 14.5 and 64.2, respectively. The average amount of 14C-mannitol absorbed through jejunal tissue from APP/PS1 mice over a 2 h period (i.e. 0.20%) was significantly lower than the amount absorbed across jejunal tissue from WT mice (p < 0.05), suggesting a tightened paracellular space in the jejunum of APP/PS1 mice.
Figure 5.
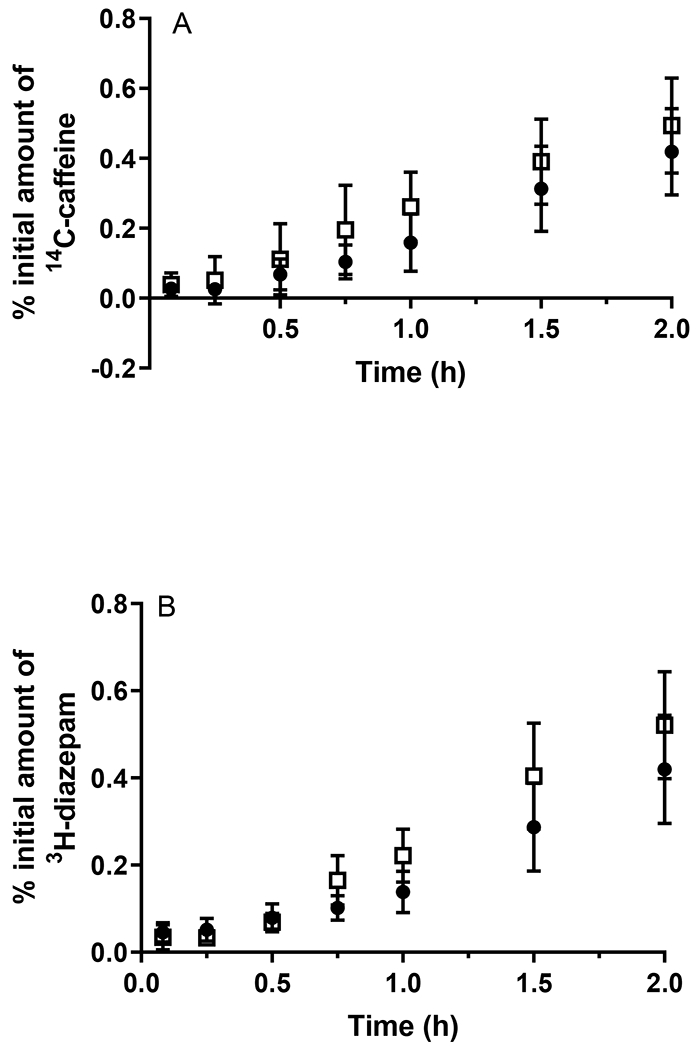
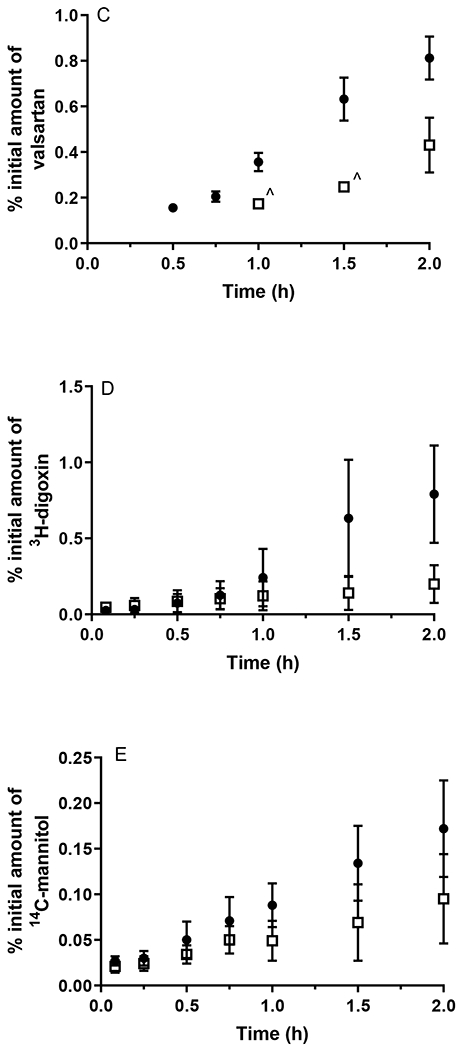
The appearance of (A) 14C-caffeine, (B) 3H-diazepam, (C) valsartan, (D) 3H-digoxin, and (E) 14C-mannitol in the receptor chamber following transport across the jejunum from WT (closed circles) and APP/PS1 mice (open squares) after application of a dose of 0.5 μCi (radioactive compounds) or 20 μg/mL (valsartan). ^ Concentrations of valsartan were only detectable in 2 replicates. Data are presented as mean ± SD (n = 6-9).
Table 2.
Apparent permeability coefficients (Papp) and percentage (%) of the initial amount of compounds absorbed after 120 min through jejunal tissue from WT and APP/PS1 mice. Data are presented as mean ± SD (n=4-9).
| Compound | Papp (×10−6 cm/s) | % of initial amount at 2 h | ||
|---|---|---|---|---|
| WT | APP/PS1 | WT | APP/PS1 | |
| 14C-Caffeine | 31.4 ± 10.1 | 27.2 ± 13.8 | 0.40 ± 0.09 | 0.46 ± 0.14 |
| 3H-Diazepam | 29.1 ± 9.7 | 34.1 ± 9.2 | 0.42 ± 0.12 | 0.52 ± 0.12 |
| Valsartan | 55.8 ± 16.7 | NA | 0.81 ± 0.19 | 0.43 ± 0.23* |
| 3H-Digoxin | 64.2 ± 39.4 | 14.5 ± 11.0* | 0.85 ± 0.30 | 0.22 ± 0.11* |
| 14C-Mannitol | 10.7 ± 3.7 | 6.0 ± 3.4* | 0.17 ± 0.05 | 0.12 ± 0.04* |
NA: Steady state was not reached and the Papp value was not able to be calculated.
p < 0.05 indicates a statistically significant difference between WT and APP/PS1 mice.
Small intestinal tissue from APP/PS1 mice exhibited no apparent morphological alterations.
We have previously reported a compensatory thickening in the cerebrovascular basement membrane in a triple transgenic mouse model of AD, which affected the rate of BBB transport of some therapeutics5. Therefore, it was considered important to evaluate whether there were morphological differences between small intestinal tissue from WT and APP/PS1 mice. As shown in Fig. 6, jejunal sections from WT mice demonstrated typical mucosal villi and submucosal layers, similar to that found in jejunal sections from APP/PS1 mice. The length of the villi did not appear to be changed in the APP/PS1 mice and the shape of the villi also was not affected. The thickness of the jejunal mucosal membrane appeared to be similar between WT and APP/PS1 animals.
Figure 6.
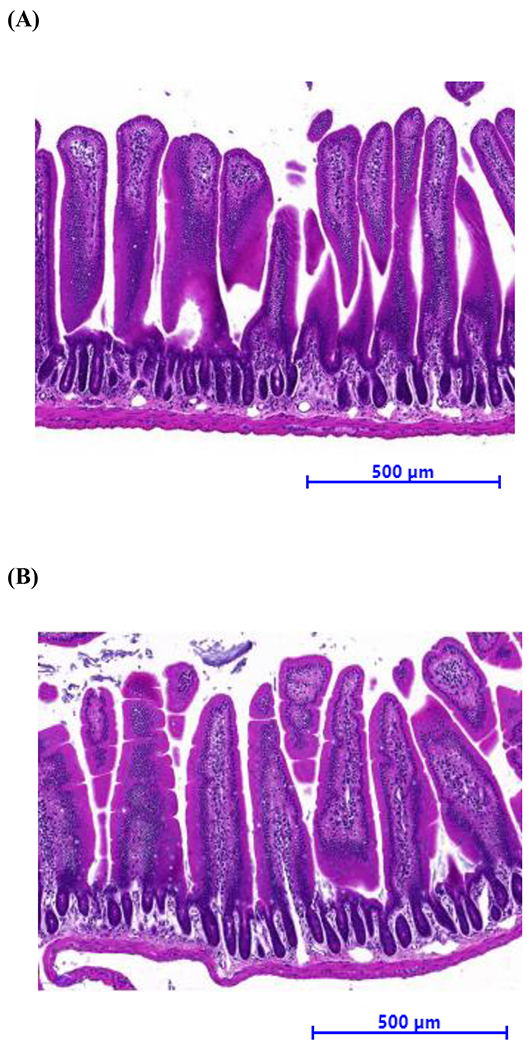
Representative histological appearance of jejunum slices (× 15 magnification) from 8-10-month-old (A) WT and (B) APP/PS1mice with H&E staining.
Discussion
People with AD are routinely prescribed medicines for their comorbidities at doses similar to those prescribed for people without AD, based on the assumption that AD does not alter drug absorption or disposition. We previously reported that the abundance of small intestinal and renal drug transporters was altered in APP/PS1 mice relative to WT mice, and growing evidence in both individuals with AD and animal models of AD suggests that oral absorption of medicines may differ in AD6, 11, 13. However, there has been no systematic evaluation of oral absorption processes in the same animal model using a panel of drugs that are absorbed via different mechanisms, which was the intent of this study. To our knowledge, this is the first study to systematically assess these processes in the same mouse model of familial AD. A better understanding of how oral drug absorption is altered in AD may lead to approaches to maximize the effectiveness and reduce the potential toxicity of the multiple medicines prescribed to individuals with AD. The five model compounds assessed in the current study were chosen based on their mechanisms of transport across the small intestine. Caffeine and diazepam were selected as markers of transcellular diffusion with hydrophilic and lipophilic characteristics, respectively; valsartan was chosen as a Mrp2 substrate; digoxin represented a model P-gp substrate; and mannitol was included (only in in vitro studies) to assess whether there were modifications in paracellular transport in APP/PS1 mice. It should be noted that the reported plasma concentrations of these probe compounds are the total drug concentrations and not unbound concentrations, which is the fraction responsible for pharmacological and toxicological activity. Concentrations of plasma proteins including albumin and alpha 1-acid glycoprotein are reduced in AD humans31–33. This aspect was not considered in the interpretation of the results presented in this study.
The oral absorption and small intestinal permeability of both caffeine and diazepam, two commonly-reported passive diffusion markers, were not different between WT and APP/PS1 mice, suggesting that jejunal passive diffusion was not affected in APP/PS1 mice. While these two drugs were used as markers of passive diffusion, they also undergo extensive hepatic metabolism, and therefore, potential changes to Cyp activity in APP/PS1 mice should be considered when interpreting the results. Our previous study13 has demonstrated that the abundance of intestinal and hepatic Cyp1a2 (the major enzyme involved in caffeine metabolism)34 was unmodified in APP/PS1 mice compared to WT mice, which agrees with the lack of change in caffeine absorption. Data are lacking in relation to the abundance of Cyp2c19 and Cyp3a4 (enzymes involved in diazepam metabolism) in APP/PS1 mice, however, given that the oral absorption of diazepam was unaffected, the levels of these enzymes are unexpected to be altered in this mouse model. Although the mice used in the studies were at an advanced age, it is important to note that the Papp of caffeine across the jejunum from WT mice was similar to values reported in other studies where jejunal tissue from 2-3-month-old Wistar rats was used35. The absorption of caffeine following an oral dose of 5 mg/kg in the present study was comparable to a previously-reported study where 8-14-week-old mice received the same oral dose exhibiting similar Cmax values appearing at 0.5 h post-dose in both studies36. Similarly, the Papp of diazepam across the aged WT and APP/PS1 jejunum was comparable to values reported using 2-3-month-old Sprague-Dawley rat jejunum37. These comparisons suggest that the age of the mice used in these studies did not appear to impact oral absorption processes given that plasma exposure observed is comparable to younger rats/mice.
Assessing the oral absorption of a Mrp2 substrate was crucial in this study given that our previous data revealed that the jejunum of APP/PS1 mice exhibited a significant 2.3-fold increase in Mrp2 abundance13. Although the permeability of valsartan is facilitated through the small intestine by other transporters such as organic anion transporter and organic anion transporting polypeptide, valsartan is a recognized substrate for Mrp2 in both humans and rodents and the intestinal absorption of valsartan has been shown to be inversely correlated with the abundance of Mrp2 in rodents15, 16. Consistent with an increase in the intestinal abundance of Mrp2, both the permeability of valsartan across the jejunum from APP/PS1 mice and the plasma exposure to valsartan in APP/PS1 mice following oral dosing were significantly lower than that observed in WT animals. These studies suggest that the increased abundance of Mrp2, which may be due to AD-associated intestinal inflammation, is associated with reduced absorption of a Mrp2 substrate. However, to confirm that the increase in small intestinal Mrp2 in APP/PS1 mice is responsible for the reduced absorption of valsartan, it would be necessary to undertake a number of additional studies including assessing the oral absorption and intestinal permeability of a larger number of Mrp2 substrates such as cisplatin, indinavir and ceftriaxone38. It should also be noted that valsartan has been reported to be a substrate of human OATP1B1, OATP1B3, OAT1 and OAT316. While there is no evidence of valsartan being a substrate of the murine orthologues, it is possible that any change in abundance and/or function of these transporters in APP/PS1 mice would impact valsartan absorption and contribute to the reduced plasma levels observed in this study. We have previously measured the levels of Oatp1b2 (murine orthologues for OATP1B1 and OATP1B3), Oat 1 and Oat 3 in the small intestine from both WT and APP/PS1 mice using QTAP, however, the concentrations of these transporters were below the detection limit (i.e. 0.06 fmol/μg)13 and therefore we cannot conclude that their abundance was indeed altered in APP/PS1 mice. In addition to being a substrate of transporters, valsartan has been reported to be a substrate of Cyp2c939. Any changes in Cyp2c9 abundance may, therefore, theoretically contribute to the reduced plasma concentrations of valsartan observed in this study. Indeed, the function of other Cyp enzymes has been reported to be modified in another mouse model of AD (i.e. Tg2576 mice)12. Even if Cyp2c9 abundance was significantly increased in APP/PS1 mice, this is unlikely to be a major contributor to the lower plasma concentrations of valsartan observed in these studies given that valsartan has been reported not to be extensively metabolised40.
Digoxin is a well-known substrate for P-gp and is often used to assess P-gp function at biological barriers21. Though the total intestinal abundance of P-gp did not differ between WT and APP/PS1 mice in our previous study13, it was considered important to assess whether the brain uptake of this P-gp substrate was altered given the previous reports of reduced BBB abundance of P-gp in AD. As part of assessing this, the oral exposure and intestinal permeability of digoxin also were evaluated. The Papp of digoxin across the WT jejunum was notably higher than the values we previously reported in 6-8-week-old Swiss outbred mouse jejunum26. This difference is most likely due to the greater age of the mice in these studies (i.e. 8-10-months) given that mouse intestinal P-gp abundance changes with age41 and reduced P-gp abundance and activity in multiple tissues including intestine and brain have been reported in both aged animals and humans42–44. Both the ex vivo permeability of 3H-digoxin through the jejunum of APP/PS1 mice and the plasma concentrations of digoxin following oral dosing to APP/PS1 mice were substantially decreased compared to that observed in WT mice. There are several possible explanations for these observations. Firstly, though the overall intestinal abundance of P-gp was not significantly different between WT and APP/PS1 mice13, the localization of P-gp may be altered in the AD small intestine, with more P-gp present at the luminal surface of the jejunum, therefore, resulting in increased efflux of digoxin into the intestinal lumen. Secondly, digoxin is a substrate of Oatp1a445 and Ostα/β46 transporters, which are expressed in the mouse small intestine. Though further investigation will be required, the abundance and/or function of Oatp1a4 and Ostα/β may be impacted in AD, which may then lead to altered permeability and absorption of digoxin across the small intestine.
While the reduced intestinal permeability of valsartan could be explained by increased Mrp2 function, the unexpected reduction in digoxin permeability (with no change in P-gp abundance) led us to investigate whether paracellular diffusion was reduced, contributing to the reduced absorption of digoxin, and in part, valsartan. To address this, the permeability of 14C-mannitol, a commonly used marker for the paracellular route, was assessed. The permeability of 14C-mannitol across the jejunum from WT mice was similar to what we reported previously in intestinal tissue obtained from 6-8-week-old Swiss outbred mice29. Unexpectedly, the amount of 14C-mannitol permeating the jejunum from APP/PS1 mice was significantly reduced. These data suggest that there appears to be a tightened paracellular space in the intestinal tissue from this mouse model, which may, in part, contribute to the reduced permeability of valsartan and digoxin across the jejunum from APP/PS1 mice. Unlike at the BBB, where there are multiple reports of modified abundance of tight junction proteins as a result of AD pathology47, 48, few studies have focused on assessing intestinal permeability in AD. It is reported that gut microbiota is altered in human AD, which is associated with inflammation49, 50, and indeed in inflammation, increased abundance of small intestinal tight junction proteins has been reported. For example, increased abundance of claudin-1, one of the major tight junction proteins responsible for the maintenance of intestinal integrity, has been observed in individuals with ulcerative colitis51, a disease associated with intestinal inflammation. In another study, the abundance of claudin-1 was significantly elevated in mucosal samples from patients with inflammatory bowel disease, and correlated well with inflammatory activity52. Therefore, it is possible that the reduced permeability of mannitol observed in the current study is a result of increased tight junction protein abundance in the small intestinal tissue, however, this requires further investigation. While speculative, another possible reason for the increased paracellular integrity in these tissues may result from the increased plasma levels of deoxycholic acid (DCA) in APP/PS1 mice53. DCA has been shown to increase the transepithelial electrical resistance in Caco-2 cells and reduce the permeability of 10 kDa dextran54. Therefore, the substantially increased concentrations of DCA in APP/PS1 mice also may be responsible, in part, for reduced permeability of the small intestine53.
It is well accepted that the integrity of the BBB and the abundance of some transporters such as glucose transporter 1 and P-gp at the BBB are affected in AD, which can cause altered BBB trafficking of drugs24, 55. Having shown that the oral absorption of some compounds was reduced in APP/PS1 mice, it was considered important to assess whether brain exposure of the model drugs was affected in APP/PS1 mice. As was the case with the plasma concentrations, the brain homogenate concentrations reflected total brain concentration and not unbound brain concentrations. Therefore, to more effectively predict whether pharmacological or toxicological effects of the model drugs would be different in disease state, it would be important to measure unbound brain concentrations using a technique such as intracerebral microdialysis. Unlike the passively-diffusing marker caffeine, where there was no difference in brain concentrations at any time when comparing WT and APP/PS1 mice, diazepam exhibited a significant reduction in brain uptake only at the first time point (i.e. 10 min). Given that the plasma concentrations of diazepam were not different between WT and APP/PS1 mice at this time point, this result suggests that only the rate of diazepam brain uptake, and not the extent of diazepam brain uptake may be reduced. This is indeed consistent with what we have previously demonstrated using an in situ transcardiac perfusion technique in 3xTG AD mice, another mouse model of familial AD5. In our previous study, the reduced rate of BBB transport of diazepam was attributed to a thickening of the cerebrovascular basement membrane. It is unknown whether the cerebrovascular basement membrane is thicker in APP/PS1 mice relative to WT mice as has been observed in 3xTG AD mice5 and humans56, however, this could contribute to the apparent reduction in the rate of brain uptake of diazepam observed in the present study. As has been observed by others using the AβPP transgenic mice model of AD57, the extent of brain uptake of diazepam was not reduced in APP/PS1 mice. Taken together, these findings support our observations that the brain uptake of diazepam was only reduced at the first time point (i.e. reduced rate) but not at all subsequent time points (i.e. no change in extent). The brain uptake of valsartan appeared to be higher in APP/PS1 mice as brain concentrations of valsartan were only detectable in APP/PS1 mice at the 2 h post-dose, although a statistical comparison could not be made. While a conclusion on whether the brain uptake of valsartan differs between genotypes is not possible based on the current data, further studies could assess whether the abundance of Mrp2 is altered in AD, particularly given that cerebral abundance of Mrp1 has been reported to be elevated in AD58. Due to the limitations associated with the sensitivity of the digoxin brain homogenate assay, it was not possible to detect digoxin brain concentrations at any post-dose time point, so no conclusion regarding differences in digoxin brain uptake between WT and APP/PS1 mice was drawn. However, given that the extent of digoxin transport across the BBB was not reported to be modified following subcutaneous administration in a mouse of model of AD (2-5-month-old AβPP-transgenic mice)57, the brain uptake of digoxin following oral administration was not expected to be different between the two genotypes.
While it is not known if these results correlate directly to humans, this study has identified that oral drug absorption of selected drugs is reduced in the APP/PS1 transgenic AD mouse model. It has been noted that the APP/PS1 transgenic mouse model displays a high pathophysiological resemblance to the human AD population59, and so it is possible that these phenomena will be observed clinically. The knowledge gained from this study provides the basis for future research focusing specifically on humans and the way in which drug doses may need to be altered in individuals with AD. Although clinical studies are required, the implication of this study is that additional therapeutic drug monitoring may be required in individuals with AD because it is possible that doses of medicines may result in reduced plasma concentrations and sub-optimal efficacy. Further investigations into this phenomenon will aid in optimizing medicine safety and use, thereby improving the quality of life for patients by avoiding sub-optimal treatment in the AD population.
Supplementary Material
Development and validation of HPLC and LC-MS/MS assays for the determination of probe compounds in KBR solution, mouse plasma and brain homogenate
Acknowledgements
The authors acknowledge PharmAlliance (an alliance between the Pharmacy Schools of the University of North Carolina at Chapel Hill, Monash University and University College London) and Bethlehem Griffiths Research Foundation for the financial support for this research. LJ holds an Alzheimer’s Association (US) Research Fellowship. KLRB is supported, in part, by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R35 GM122576. The authors thank Laura Leone for the valuable assistance in the histological examination.
References
- 1.Qiu C; Kivipelto M; von Strauss E Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci 2009, 11, (2), 111–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldstein FC; Ashley AV; Endeshaw YW; Hanfelt J; Lah JJ; Levey AI Effects of hypertension and hypercholesterolemia on cognitive functioning in patients with Alzheimer disease. Alzheimer Dis Assoc Disord 2008, 22, (4), 336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee HJ; Seo HI; Cha HY; Yang YJ; Kwon SH; Yang SJ Diabetes and Alzheimer’s disease: mechanisms and nutritional aspects. Clin Nutr Res 2018, 7, (4), 229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clague F; Mercer SW; McLean G; Reynish E; Guthrie B Comorbidity and polypharmacy in people with dementia: insights from a large, population-based cross-sectional analysis of primary care data. Age Ageing 2017, 46, (1), 33–9. [DOI] [PubMed] [Google Scholar]
- 5.Mehta DC; Short JL; Nicolazzo JA Altered brain uptake of therapeutics in a triple transgenic mouse model of Alzheimer’s disease. Pharm Res 2013, 30, (11), 2868–79. [DOI] [PubMed] [Google Scholar]
- 6.Hartz AM; Miller DS; Bauer B Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol 2010, 77, (5), 715–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeynes B; Provias J An investigation into the role of P-glycoprotein in Alzheimer’s disease lesion pathogenesis. Neurosci Lett 2011, 487, (3), 389–93. [DOI] [PubMed] [Google Scholar]
- 8.Deo AK; Borson S; Link JM; Domino K; Eary JF; Ke B; Richards TL; Mankoff DA; Minoshima S; O’Sullivan F; Eyal S; Hsiao P; Maravilla K; Unadkat JD Activity of P-glycoprotein, a beta-amyloid transporter at the blood-brain barrier, is compromised in patients with mild Alzheimer disease. J Nucl Med 2014, 55, (7), 1106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong HQ; Callaghan D; Jones A; Bai JY; Rasquinha I; Smith C; Pei K; Walker D; Lue LF; Stanimirovic D; Zhang WD ABCG2 is upregulated in Alzheimer’s brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for a beta(1–40) peptides. J Neurosci 2009, 29, (17), 5463–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adlard PA; Cherny RA; Finkelstein DI; Gautier E; Robb E; Cortes M; Volitakis I; Liu X; Smith JP; Perez K; Laughton K; Li QX; Charman SA; Nicolazzo JA; Wilkins S; Deleva K; Lynch T; Kok G; Ritchie CW; Tanzi RE; Cappai R; Masters CL; Barnham KJ; Bush AI Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial A beta. Neuron 2008, 59, (1), 43–55. [DOI] [PubMed] [Google Scholar]
- 11.Polinsky RJ Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther 1998, 20, (4), 634–47. [DOI] [PubMed] [Google Scholar]
- 12.Van Ess PJ; Pedersen WA; Culmsee C; Mattson MP; Blouin RA Elevated hepatic and depressed renal cytochrome P450 activity in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurochem 2002, 80, (4), 571–8. [DOI] [PubMed] [Google Scholar]
- 13.Pan Y; Omori K; Ali I; Tachikawa M; Terasaki T; Brouwer KLR; Nicolazzo JA Altered expression of small intestinal drug transporters and hepatic metabolic enzymes in a mouse model of familial Alzheimer’s disease. Mol Pharmaceut 2018, 15, (9), 4073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan Y; Omori K; Ali I; Tachikawa M; Terasaki T; Brouwer KLR; Nicolazzo JA Increased expression of renal drug transporters in a mouse model of familial Alzheimer’s disease. J Pharm Sci 2019, 108, (7), 2484–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Londero AS; Arana MR; Perdomo VG; Tocchetti GN; Zecchinati F; Ghanem CI; Ruiz ML; Rigalli JP; Mottino AD; Garcia F; Villanueva SSM Intestinal multidrug resistance-associated protein 2 is down-regulated in fructose-fed rats. J Nutr Biochem 2017, 40, 178–86. [DOI] [PubMed] [Google Scholar]
- 16.Hanna I; Alexander N; Crouthamel MH; Davis J; Natrillo A; Tran P; Vapurcuyan A; Zhu B Transport properties of valsartan, sacubitril and its active metabolite (LBQ657) as determinants of disposition. Xenobiotica 2018, 48, (3), 300–13. [DOI] [PubMed] [Google Scholar]
- 17.Menard S; Lebreton C; Schumann M; Matysiak-Budnik T; Dugave C; Bouhnik Y; Malamut G; Cellier C; Allez M; Crenn P; Schulzke JD; Cerf-Bensussan N; Heyman M Paracellular versus transcellular intestinal permeability to gliadin peptides in active celiac disease. Am J Pathol 2012, 180, (2), 608–15. [DOI] [PubMed] [Google Scholar]
- 18.Volpe DA Application of method suitability for drug permeability classification. AAPS J 2010, 12, (4), 670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kansara V; Mitra AK Evaluation of an ex vivo model implication for carrier-mediated retinal drug delivery. Curr Eye Res 2006, 31, (5), 415–26. [DOI] [PubMed] [Google Scholar]
- 20.Montagne A; Zhao Z; Zlokovic BV Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 2017, 214, (11), 3151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes J; Crowe A Inhibition of P-glycoprotein-mediated efflux of digoxin and its metabolites by macrolide antibiotics. J Pharmacol Sci 2010, 113, (4), 315–24. [DOI] [PubMed] [Google Scholar]
- 22.Greiner B; Eichelbaum M; Fritz P; Kreichgauer HP; Von Richter O; Zundler J; Kroemer HK The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 1999, 104, (2), 147–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Assema DM; Lubberink M; Bauer M; van der Flier WM; Schuit RC; Windhorst AD; Comans EF; Hoetjes NJ; Tolboom N; Langer O; Muller M; Scheltens P; Lammertsma AA; van Berckel BN Blood-brain barrier P-glycoprotein function in Alzheimer’s disease. Brain 2012, 135, (Pt 1), 181–9. [DOI] [PubMed] [Google Scholar]
- 24.Nicolazzo JA; Banks WA Decreased blood-brain barrier expression of P-glycoprotein in Alzheimer’s disease: impact on pathogenesis and brain access of therapeutic agents. Ther Deliv 2011, 2, (7), 841–4. [DOI] [PubMed] [Google Scholar]
- 25.Serneels L; Van Biervliet J; Craessaerts K; Dejaegere T; Horre K; Van Houtvin T; Esselmann H; Paul S; Schafer MK; Berezovska O; Hyman BT; Sprangers B; Sciot R; Moons L; Jucker M; Yang Z; May PC; Karran E; Wiltfang J; D’Hooge R; De Strooper B gamma-secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science 2009, 324, (5927), 639–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gengler S; Hamilton A; Holscher C Synaptic plasticity in the hippocampus of a APP/PS1 mouse model of Alzheimer’s disease is impaired in old but not young mice. PLoS One 2010, 5, (3), e9764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radde R; Bolmont T; Kaeser SA; Coomaraswamy J; Lindau D; Stoltze L; Calhoun ME; Jaggi F; Wolburg H; Gengler S; Haass C; Ghetti B; Czech C; Holscher C; Mathews PM; Jucker M Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 2006, 7, (9), 940–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicolazzo JA; Steuten JA; Charman SA; Taylor N; Davies PJ; Petrou S Brain uptake of diazepam and phenytoin in a genetic animal model of absence epilepsy. Clin Exp Pharmacol Physiol 2010, 37, (5–6), 647–9. [DOI] [PubMed] [Google Scholar]
- 29.Dube A; Nicolazzo JA; Larson I Chitosan nanoparticles enhance the intestinal absorption of the green tea catechins (+)-catechin and (−)-epigallocatechin gallate. Eur J Pharm Sci 2010, 41, (2), 219–25. [DOI] [PubMed] [Google Scholar]
- 30.Bailer AJ Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biop 1988, 16, (3), 303–9. [DOI] [PubMed] [Google Scholar]
- 31.Hye A; Riddoch-Contreras J; Baird AL; Ashton NJ; Bazenet C; Leung R; Westman E; Simmons A; Dobson R; Sattlecker M; Lupton M; Lunnon K; Keohane A; Ward M; Pike I; Zucht HD; Pepin D; Zheng W; Tunnicliffe A; Richardson J; Gauthier S; Soininen H; Kloszewska I; Mecocci P; Tsolaki M; Vellas B; Lovestone S Plasma proteins predict conversion to dementia from prodromal disease. Alzheimers Dement 2014, 10, (6), 799–807.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costa M; Horrillo R; Ortiz AM; Perez A; Mestre A; Ruiz A; Boada M; Grancha S Increased albumin oxidation in cerebrospinal fluid and plasma from Alzheimer’s disease patients. J Alzheimers Dis 2018, 63, (4), 1395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llewellyn DJ; Langa KM; Friedland RP; Lang IA Serum albumin concentration and cognitive impairment. Curr Alzheimer Res 2010, 7, (1), 91–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guessous I; Dobrinas M; Kutalik Z; Pruijm M; Ehret G; Maillard M; Bergmann S; Beckmann JS; Cusi D; Rizzi F; Cappuccio F; Cornuz J; Paccaud F; Mooser V; Gaspoz J-M; Waeber G; Burnier M; Vollenweider P; Eap CB; Bochud M Caffeine intake and CYP1A2 variants associated with high caffeine intake protect non-smokers from hypertension. Hum Mol Genet 2012, 21, (14), 3283–92. [DOI] [PubMed] [Google Scholar]
- 35.Da Silva LC; Da Silva TL; Antunes AH; Rezende KR A sensitive medium-throughput method to predict intestinal absorption in humans using rat intestinal tissue segments. J Pharm Sci 2015, 104, (9), 2807–12. [DOI] [PubMed] [Google Scholar]
- 36.Scheer N; McLaughlin LA; Rode A; Macleod AK; Henderson CJ; Wolf CR Deletion of 30 murine cytochrome p450 genes results in viable mice with compromised drug metabolism. Drug Metab Dispos 2014, 42, (6), 1022–30. [DOI] [PubMed] [Google Scholar]
- 37.Katneni K; Charman SA; Porter CJH Permeability assessment of poorly water-soluble compounds under solubilizing conditions: the reciprocal permeability approach. J Pharm Sci 2006, 95, (10), 2170–85. [DOI] [PubMed] [Google Scholar]
- 38.Jedlitschky G; Hoffmann U; Kroemer HK Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin Drug Metab Toxicol 2006, 2, (3), 351–66. [DOI] [PubMed] [Google Scholar]
- 39.Nakashima A; Kawashita H; Masuda N; Saxer C; Niina M; Nagae Y; Iwasaki K Identification of cytochrome P450 forms involved in the 4-hydroxylation of valsartan, a potent and specific angiotensin II receptor antagonist, in human liver microsomes. Xenobiotica 2005, 35, (6), 589–602. [DOI] [PubMed] [Google Scholar]
- 40.Waldmeier F; Flesch G; Mu Ller P; Winkler T; Kriemler HP; Buhlmayer P; De Gasparo M Pharmacokinetics, disposition and biotransformation of [14C]-radiolabelled valsartan in healthy male volunteers after a single oral dose. Xenobiotica 1997, 27, (1), 59–71. [DOI] [PubMed] [Google Scholar]
- 41.Mahmood B; Daood MJ; Hart C; Hansen TW; Watchko JF Ontogeny of P-glycoprotein in mouse intestine, liver, and kidney. J Investig Med 2001, 49, (3), 250–7. [DOI] [PubMed] [Google Scholar]
- 42.Guo M; Bughio S; Sun Y; Zhang Y; Dong L; Dai X; Wang L Age-related P-glycoprotein expression in the intestine and affecting the pharmacokinetics of orally administered enrofloxacin in broilers. PLoS One 2013, 8, (9), e74150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toornvliet R; van Berckel BNM; Luurtsema G; Lubberink M; Geldof AA; Bosch TM; Oerlemans R; Lammertsma AA; Franssen EJF Effect of age on functional P-glycoprotein in the blood-brain barrier measured by use of (R)-[11C]verapamil and positron emission tomography. Clin Pharmacol Ther 2006, 79, (6), 540–8. [DOI] [PubMed] [Google Scholar]
- 44.Rosati A; Maniori S; Decorti G; Candussio L; Giraldi T; Bartoli F Physiological regulation of P-glycoprotein, MRP1, MRP2 and cytochrome P450 3A2 during rat ontogeny. Dev Growth Differ 2003, 45, (4), 377–87. [DOI] [PubMed] [Google Scholar]
- 45.Takano J; Maeda K; Kusuhara H; Sugiyama Y Organic anion transporting polypeptide 1a4 is responsible for the hepatic uptake of cardiac glycosides in mice. Drug Metab Dispos 2018, 46, (5), 652–7. [DOI] [PubMed] [Google Scholar]
- 46.Seward DJ; Koh AS; Boyer JL; Ballatori N Functional complementation between a novel mammalian polygenic transport complex and an evolutionarily ancient organic solute transporter, OST alpha-OST beta. J Biol Chem 2003, 278, (30), 27473–82. [DOI] [PubMed] [Google Scholar]
- 47.Pan Y; Nicolazzo JA Impact of aging, Alzheimer’s disease and Parkinson’s disease on the blood-brain barrier transport of therapeutics. Adv Drug Deliv Rev 2018, 135, 62–74. [DOI] [PubMed] [Google Scholar]
- 48.Yamazaki Y; Shinohara M; Shinohara M; Yamazaki A; Murray ME; Liesinger AM; Heckman MG; Lesser ER; Parisi JE; Petersen RC; Dickson DW; Kanekiyo T; Bu G Selective loss of cortical endothelial tight junction proteins during Alzheimer’s disease progression. Brain 2019, 142, (4), 1077–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kowalski K; Mulak A Brain-gut-microbiota axis in Alzheimer’s disease. J Neurogastroenterol Motil 2019, 25, (1), 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai KSP; Liu CS; Rau A; Lanctot KL; Kohler CA; Pakosh M; Carvalho AF; Herrmann N Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry 2017, 88, (10), 876–82. [DOI] [PubMed] [Google Scholar]
- 51.Poritz LS; Harris LR 3rd; Kelly AA; Koltun WA Increase in the tight junction protein claudin-1 in intestinal inflammation. Dig Dis Sci 2011, 56, (10), 2802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weber CR; Nalle SC; Tretiakova M; Rubin DT; Turner JR Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab Invest 2008, 88, (10), 1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.MahmoudianDehkordi S; Arnold M; Nho K; Ahmad S; Jia W; Xie G; Louie G; Kueider-Paisley A; Moseley MA; Thompson JW; St John Williams L; Tenenbaum JD; Blach C; Baillie R; Han X; Bhattacharyya S; Toledo JB; Schafferer S; Klein S; Koal T; Risacher SL; Kling MA; Motsinger-Reif A; Rotroff DM; Jack J; Hankemeier T; Bennett DA; De Jager PL; Trojanowski JQ; Shaw LM; Weiner MW; Doraiswamy PM; van Duijn CM; Saykin AJ; Kastenmuller G; Kaddurah-Daouk R; Alzheimer’s Disease Neuroimaging I; the Alzheimer Disease Metabolomics, C. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement 2019, 15, (1), 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raimondi F; Santoro P; Barone MV; Pappacoda S; Barretta ML; Nanayakkara M; Apicella C; Capasso L; Paludetto R Bile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activation. Am J Physiol Gastrointest Liver Physiol 2008, 294, (4), G906–13. [DOI] [PubMed] [Google Scholar]
- 55.Szablewski L Glucose transporters in brain: in health and in Alzheimer’s Disease. J Alz Dis 2017, 55, (4), 1307–20. [DOI] [PubMed] [Google Scholar]
- 56.Claudio L Ultrastructural features of the blood-brain barrier in biopsy tissue from Alzheimer’s disease patients. Acta Neuropathol 1996, 91, (1), 6–14. [DOI] [PubMed] [Google Scholar]
- 57.Gustafsson S; Lindstrom V; Ingelsson M; Hammarlund-Udenaes M; Syvanen S Intact blood-brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha-synuclein pathology. Neuropharmacology 2018, 128, 482–91. [DOI] [PubMed] [Google Scholar]
- 58.Sultana R; Butterfield DA Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res 2004, 29, (12), 2215–20. [DOI] [PubMed] [Google Scholar]
- 59.Kitazawa M; Medeiros R; LaFerla FM Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Design 2012, 18, (8), 1131–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Development and validation of HPLC and LC-MS/MS assays for the determination of probe compounds in KBR solution, mouse plasma and brain homogenate