

Abstract
New approaches are needed to overcome intrinsic therapy resistance in glioblastoma (GBM). Because GBMs exhibit sexual dimorphism and are reported to express steroid hormone receptors, we reasoned that signaling through the androgen receptor (AR) could mediate therapy resistance in GBM, much as it does in AR-positive prostate and breast cancers. We found that nearly half of GBM cell lines, patient-derived xenografts and human tumors expressed AR at the transcript and protein level–with expression levels overlapping those of primary prostate cancer. Analysis of gene expression datasets also revealed that AR expression is higher in GBM patient samples than normal brain tissue. Multiple clinical-grade antiandrogens slowed the growth of and radiosensitized AR-positive GBM cell lines and patient-derived xenografts in vitro and in vivo. Antiandrogens blocked the ability of AR-positive GBM patient-derived xenografts to engage adaptive transcriptional programs following radiation and slowed the repair of radiation-induced DNA damage. These results suggest that combining blood-brain barrier permeable antiandrogens with radiation may have promise for patients with AR-positive GBMs.
Keywords: Glioblastoma, radiation, androgen receptor, enzalutamide, seviteronel, antiandrogens
INTRODUCTION
Glioblastoma (GBM) is the most common and aggressive primary brain tumor in adults (1). Despite multimodal treatment with surgery, radiation, and temozolomide (2), fewer than 10% of GBM patients survive more than 5 years after diagnosis. These poor outcomes are largely due to the radiation and chemotherapeutic resistance of GBM. Indeed, approximately 80% of GBMs recur within the high dose radiation field (3,4). Unfortunately, strategies to overcome radiation resistance in GBM have been largely unsuccessful to date (5,6).
To develop new strategies to radiosensitize GBM, we examined therapies that have successfully overcome radiation resistance in other cancers. The androgen receptor (AR) has long been implicated as a primary driver of prostate cancer growth and survival (7). Signaling through the AR mediates radiation resistance in prostate cancer by promoting the repair of radiation-induced DNA damage (8). Consistent with these findings, combining antiandrogens with radiotherapy significantly improves outcomes in men with aggressive prostate cancer (9). The AR also mediates radiation resistance and DNA repair in AR-positive breast cancers (10), which has prompted clinical trials of antiandrogens in non-prostate cancers (11).
Whether the AR represents a promising target in GBM is unknown. While the AR is not widely expressed in the central nervous system, there are case reports of GBMs expressing the AR and other steroid receptors (12,13). There are also sex-related biologic phenomena in GBM that could be related to active AR signaling in this tumor. Men, whose circulating testosterone levels are higher than women, have a higher risk of developing GBM (14). Further, women diagnosed with GBM have a small but distinct survival advantage compared to men (15,16). These findings could be due to lower circulating testosterone (and hence, lower AR signaling) in women with GBM.
We hypothesized that GBMs frequently express the AR, and AR-signaling may control growth and therapy resistance in GBM as it does in other cancers. Further, because most antiandrogens readily cross the blood brain barrier and are safely combined with radiation in other cancers, we reasoned that these drugs may have utility in AR-positive GBM. Herein, we report the frequent expression of AR at the RNA and protein level in GBM cell lines, patient-derived xenografts, and human tumors. We demonstrate that antiandrogens preferentially overcome radiation resistance in AR-positive GBM cell lines and patient-derived xenograft models. Mechanistic studies performed in vitro and in vivo point to altered DNA repair as a possible explanation for antiandrogen-mediated radiosensitization.
MATERIALS AND METHODS
Cell Culture and Drug Solutions
GBM cell lines were obtained from ATCC (LN18, T98G, U87, 8MGBA) or JCRB Cell Bank (AM38). All cell lines were tested for mycoplasma on a monthly basis (most recently: 2/2020) using the MycoAlert Mycoplasma Detection Kit (Cat# LT07–418, Lonza). GBM Patient-Derived Xenografts (PDXs) 14, 22, 26, 64, 76, and 110 were obtained from the Mayo Clinic Brain Tumor PDX National Resource. Cell lines were authenticated using short tandem repeat profiling at the University of Michigan Advanced Genomics Core (most recently: 2/2020) and cultured in DMEM (Cat# 11965–092, Gibco) supplemented with 10% FBS (Cat# S11550, ATLANTA biologicals), 2 mmol/L L-glutamine, penicillin, and streptomycin (Cat# 10378–016, Gibco). Cell lines were maintained in a 5% CO2 incubator. Seviteronel was obtained from Innocrin, and enzalutamide (HY-70002) was obtained from MedChem Express. Seviteronel and enzalutamide were prepared in DMSO (in vitro control) or 1% carboxymethylcellulose sodium, 0.1% Tween-80 (in vivo control).
Patient Cohorts and Gene Expression Data
AR expression data for the indicated cancers was obtained from The Cancer Genome Atlas (TCGA) Pan-Cancer Atlas dataset (17,18), available through cBioPortal for Cancer Genomics (https://www.cbioportal.org/). AR expression was binned to represent the percent of glioblastoma and prostate cancer patients with overlapping expression levels. In GBM, AR expression was compared by age, sex, and the indicated common molecular alterations. Further, the tumors with outlier “high AR” expression (mean + 2 SD) were compared to the rest of the tumors (“low AR” expressors) by common molecular alterations. AR expression was also compared across the classical, mesenchymal, and proneural subtypes, as defined by TCGA. AR expression data for Mayo Clinic Brain Tumor Patient-Derived Xenografts was obtained from cBioPortal. Normalized AR expression data for GBM cell lines was obtained from the Broad Institute Cancer Cell Line Encyclopedia (CCLE, https://portals.broadinstitute.org/ccle).
To compare AR expression levels between GBM and normal brain using a large number of samples, Affymetrix Human Genome U133 Plus 2.0 Array expression datasets were collected from Gene Expression Omnibus (GEO) and ArrayExpress (AE). Our list was derived from the combined glioblastoma and normal brain cohort assembled by Tang et al. (19). Pediatric, hypothalamus, and pituitary samples were excluded from analysis, creating a final group of 1343 samples (713 normal, 630 GBM). CEL files were processed using the single-channel array normalization (SCAN) of the SCAN.UPC package (version 2.28.0) in R software with default options. The SCAN function allows for comparison of expression profiles across datasets with output on a log2 scale centered at zero (20). BrainArray hgu133plus2hsentrezprobe (version 24.0.0) was used to covert probe sets to gene identifiers. Duplicate samples were identified by Euclidean distance and removed. Principle component analysis was performed using PCAtools (version 1.2.0).
Immunoblotting
Whole cell lysates or homogenized tumors were prepared in RIPA buffer (Cat# 89900, Thermo Scientific) supplemented with PhosSTOP phosphatase inhibitors and cOmplete protease inhibitor tablets (Cat# 04906845001, Roche) and processed as previously described (21). In cell lines, antibodies were used to detect AR (Cat# 5153 Cell Signaling Technology), GAPDH (Cat# 2118, Cell Signaling Technology) and β-actin (Cat# 47778, Santa Cruz Biotechnology), followed by HRP-conjugated secondary antibodies (Cat# 7074 and #7076, Cell Signaling Technology) and SuperSignal West Dura Extended Duration Substrate (Cat# 34076, Thermo Scientific). All human samples were handled according to Wake Forest IRB protocol #8427. In tumors, antibodies were used to detect AR (Cat# 5153, Cell Signaling Technology) and β-actin (Cat# A5441, Sigma), followed by HRP-conjugated secondary antibody (Cat# A8275, Sigma).
Immunohistochemistry
Tissues were fixed in 10% formalin and embedded in paraffin. Sections were cut at a thickness of 5 μm. Slides were heated at 65°C, de-paraffinized in xylene, and re-hydrated. Antigen retrieval was performed with 10 mM sodium citrate buffer (pH 6.0) by microwaving twice for 5 min. Endogenous peroxidase activity was quenched with Peroxide Blocking Kit (Cat# ACA500, ScyTek Laboratories). Slides were blocked and incubated with anti-AR antibody (Cat# 5153 Cell Signaling Technology) or matched isotype control overnight at 4°C. Slides were washed with PBS followed by incubation with polymer HRP anti-rabbit antibody (Vector Labs) for 20 min. After chromogenic detections by ImmPRESS NovaRED (Vector Labs), slides were counterstained with hematoxylin for 1 minute, dehydrated, cleared with xylene, and mounted with Permount (Cat# SP15–100, Fisher). Photomicrographs were taken with a 20x magnification lens on an Olympus IX70 microscope using a Retiga 2000R camera with ImagePro Plus v5.1 software.
Radiation
Radiation treatments were carried out using a Philips RT250 (Kimtron Medical) at a dose rate of approximately 2 Gy/min in the University of Michigan Experimental Irradiation Core. Dosimetry was carried out using an ionization chamber connected to an electrometer system that is directly traceable to a National Institute of Standards and Technology calibration. For tumor studies, animals were anesthetized with isoflurane and positioned such that the apex of each flank tumor was at the center of a 2.4 cm aperture in the secondary collimator, with the rest of the mouse shielded from radiation (22).
Colony formation, Clonogenic Survival & Viability Assays
For colony forming experiments, cells were plated at clonal density and treated with drug after 48 hours. After 10 to 14 days, cells were fixed with methanol-acetic acid and stained with brilliant blue. Colonies with 25 or more cells were scored. GI50 values were calculated as the drug concentration where colony formation reaches 50% when fit to the [Inhibitor] vs. normalized response (variable slope) curve. Clonogenic assays combining drug and radiation were performed as previously described (21,23). Proliferating cells were treated with drug 24 hours before irradiation (0 to 8 Gy) and replated at clonal density. After 10 to 14 days, cells were fixed with methanol-acetic acid and stained with brilliant blue. Colonies with 25 or more cells were scored, and survival curves were fitted using the linear quadratic equation. Radiation enhancement ratios were calculated as the ratio of the mean inactivation dose under control conditions divided by the mean inactivation dose under drug treatment conditions. All assays were performed in technical triplicate and repeated at least three times, unless otherwise indicated.
Immunofluorescence
Cells were cultured on coverslips and treated with seviteronel (10μM) or enzalutamide (20μM) for 24 hours before irradiation with 6 Gy. At 1, 6, and 24 hours after radiation, cells were fixed with 4% paraformaldehyde and processed for staining. γ-H2AX foci were detected with the Anti-phospho-Histone H2A.X antibody (mouse, Ser139, Cat# 05–636, Millipore) and Alexa Fluor 594 secondary (Goat Anti-Mouse IgG H&L, Cat# 150116, Abcam). Slides were imaged on an Olympus IX81 microscope with a 60× oil objective. Fields were chosen randomly based on DAPI staining. For quantitation, at least 50 cells from each of three independent experiments were visually scored for each condition. Cells with the indicated number of foci were scored as positive and compared for statistical analyses. Alkaline comet assays were also performed under a similar treatment schedule, and details can be found in the Supplementary Methods section.
Xenograft Studies
Animals were handled according to a protocol approved by the University of Michigan Committee for Use and Care of Animals. As indicated in the figures, T98G were resuspended in a 1:1 mixture of PBS:Matrigel (BD Biosciences) and injected subcutaneously into the bilateral dorsal flanks of C.B-17 SCID mice (4–6 weeks, female). Small pieces of GBM 26 tumor (obtained directly from the Mayo Clinic Brain Tumor PDX National Resource) were implanted into the flanks of C.B-17 SCID mice (4–6 weeks, female). Established tumors were serial passaged for experiments. All mice were maintained in specific pathogen-free conditions. Mice bearing T98G xenografts were implanted with 12.5 mg 60-day release 5-alpha-DHT pellets (Cat# SA-161, Innovative Research of America). Once tumor volumes reached ~70–100mm3, mice were randomized to receive vehicle control, drug treatment alone, radiation alone, or combined drug treatment and radiation. Drug treatments (150mg/kg seviteronel or 25mg/kg enzalutamide) were administered via oral gavage, starting 24 hours prior to the first dose of radiation. Radiation (2 Gy/fraction) was administered over daily fractions for the indicated number of days. Tumor size was measured three times per week using digital calipers and the formula (π/6)(Length × Width2). Body weight was monitored multiple times per week to assess weight loss during treatment.
RNA-seq
GBM26 tumors (4 per group, 16 total) were randomized to receive vehicle control, enzalutamide alone (25mg/kg), radiation alone (2Gy × 2 days), or combined drug treatment and radiation. Enzalutamide was administered via oral gavage, beginning 24 hours prior to the first dose of radiation. Four hours after the final drug treatment (two hours after RT), the tumors were snap frozen in liquid nitrogen and homogenized on dry ice with a mortar and pestle. RNA was extracted using the Monarch Total RNA Miniprep Kit (Cat# T2010S, New England BioLabs), according to the manufacturer protocol, with optional DNase I treatment. RNA integrity and quality were assessed by Agilent TapeStation (RIN > 9). Library preparation and sequencing was performed by the University of Michigan Advanced Genomics Core using standard poly-A enrichment. Library quality and final concentration was assessed by Agilent Bioanalyzer and run on an Illumina NovaSeq 6000 at a depth of 750 million × 150-bp paired-end reads.
Bioinformatics and Differential Expression Analysis
Bioinformatics analysis of RNAseq data was performed by the University of Michigan Bioinformatics Core using R software. FastQC (version v0.11.3) was used to assess quality of raw reads. Reads were then aligned to the UCSC hg19.fa reference genome using TopHat (version 2.0.13) and Bowtie2 (version 2.2.1.) (24,25). FastQC was used for a second round of quality control. The HTSeq (version 0.6.1) / DESeq2 (version 0.6.1) method of differential expression analysis was used (26). Counts were generated using HTSeq, and data were pre-filtered to remove genes with 0 counts in all samples. DESeq2 was used to perform normalization and determine differential expression. Genes were considered differentially expressed based on three criteria: test status = “OK”, FDR ≤ 0.05, and fold change ≥ ± 1.5. All fold changes are represented as the treatment condition compared to control. To analyze transcriptional changes in an initial list of 179 DNA repair genes of interest (27–30), a threshold was set at p < 0.05 in the radiation condition, and Log2 fold changes were compared between the groups.
Statistical Analysis
Data are presented as the mean ± SEM of at least three independent experiments, unless otherwise indicated. Statistical significance was determined by t-tests in GraphPad Prism Version 8 with use of the Holm-Sidak method to correct for multiple comparisons when appropriate. Comet assays were analyzed using the Kruskal-Wallis one-way ANOVA. The Chi-square test was used to determine statistical significance between AR-low and AR-high groups being compared by common molecular alterations. Time to tumor volume tripling was determined for each xenograft by identifying the earliest day on which it was at least three times as large as on the first day of radiation treatment. The Kaplan-Meier method was used to analyze time to tripling, and statistical differences were analyzed using the log-rank test. P value < 0.05 was considered statistically significant.
RESULTS
AR transcript is expressed in GBM tumors, patient-derived xenografts, and cell lines
In order to elucidate the potential role of the androgen receptor in GBM, we first assessed AR transcript levels in the TCGA dataset (Fig. 1A). As expected, AR transcript was highest in patients with prostate and breast cancers. About half of GBM patients express AR transcript at levels similar to levels seen in prostate cancer patients (Fig. 1B). To determine whether AR is differentially expressed in GBM and normal brain tissue, we examined 1343 samples from GEO/AE datasets with transcript data available for GBM tumors (n=713) or normal brain (n=630), which were normalized using single channel array normalization (SCAN) (20). After normalization, the expression profiles of the samples clustered into two distinct populations that corresponded to GBM and normal tissue (Supplementary Fig. S1A). AR transcript levels in GBM were significantly increased compared to normal brain (Fig. 1C), as others have previously shown using datasets with limited numbers of normal tissue samples (31). The magnitude of AR upregulation was similar to that of EGFR, which is well-known to be upregulated in GBM (Supplementary Fig. S1B). To determine if these findings were true in models of GBM appropriate for laboratory study, we examined AR expression in immortalized cell line and patient-derived xenograft (PDX) models of GBM. As in patient tumors, immortalized cell lines from the CCLE (32) and GBM PDXs from the Mayo PDX national resource (33) exhibited heterogeneous AR expression. While small numbers of cell lines and PDXs expressed undetectable AR transcript, most expressed AR at the transcript level (Fig. 1D–E).
Figure 1.
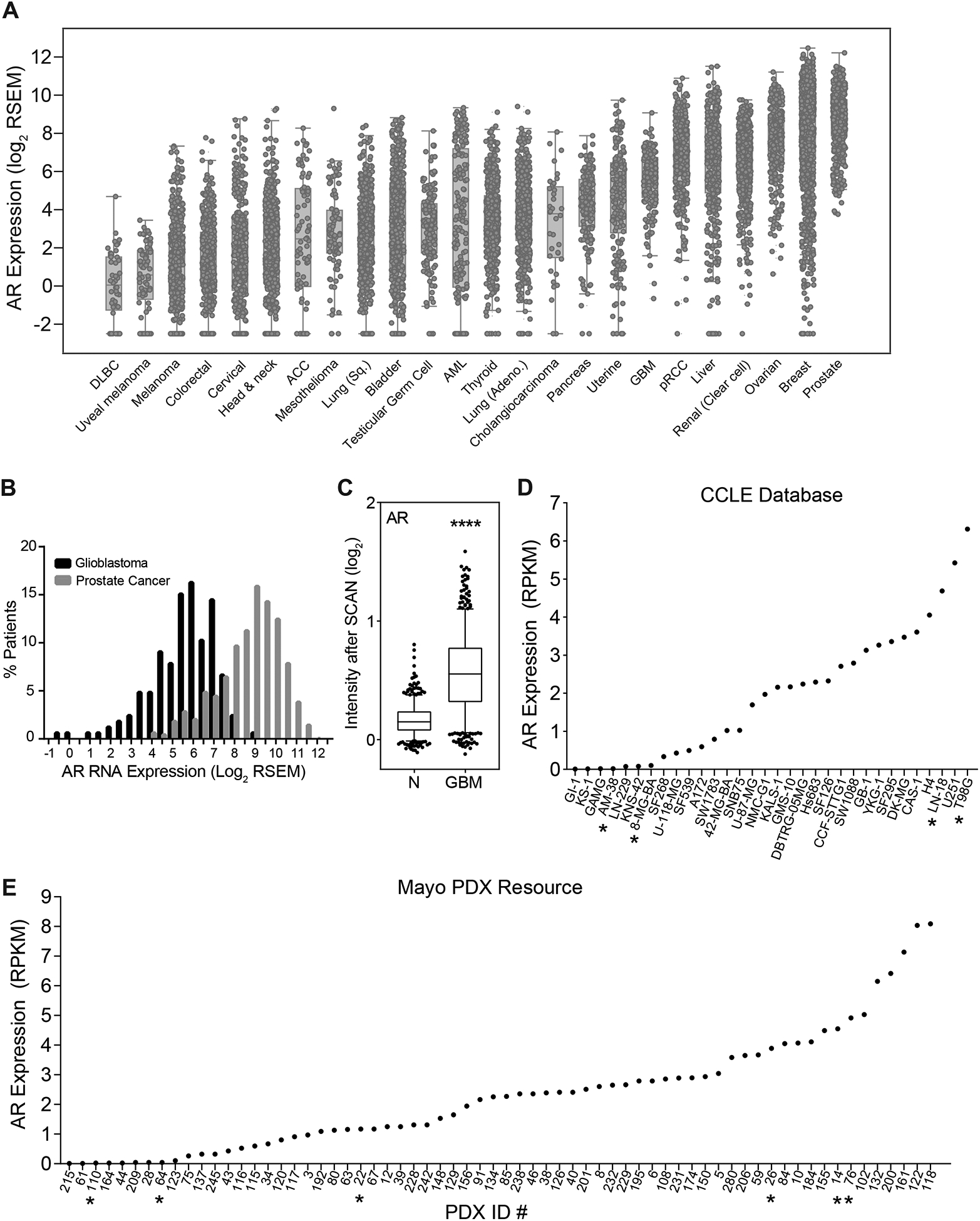
AR transcript is expressed in GBM tumors, cell lines and patient-derived xenografts. (A) AR expression data was downloaded from cBioPortal for 24 cancer types analyzed by TCGA (B). Histograms were generated from (A) to show the overlap of AR expression in GBM and prostate cancer. (C) AR expression in normal brain (N, n = 713) and glioblastoma (GBM, n = 630) samples from the combined GEO/AE dataset (****p < 0.0001, box plot whiskers mark the 5th and 95th percentiles). (D) AR transcript levels in GBM cell lines were downloaded from the Cancer Cell Line Encyclopedia (CCLE,* denotes the cell lines chosen for protein-level analysis: AM38, 8MGBA, LN18, T98G). (E) AR transcript levels as determined by RNA-seq analysis of tumors from the Mayo PDX resource (* denotes the PDXs that were chosen for protein-level analysis: 110, 64, 22, 26, 14, 76).
AR protein is expressed in GBM tumors, patient-derived xenografts, and cell lines
Based on the transcript analysis of cell lines from the CCLE (Figure 1D), we tested the AR protein expression of four GBM cell lines, two with undetectable AR transcript levels (8MGBA and AM38) and two with high AR transcript levels (LN18 and T98G). Consistent with transcript analysis, both LN18 and T98G expressed AR protein and are hereafter termed “AR-positive” while 8MGBA and AM38 did not and are referred to as “AR-negative.” The LNCaP prostate cancer cell line was used as a positive control for AR expression (Fig. 2A). U87MG had weak protein expression of AR and was not studied further. We performed a similar analysis using lysates of tumor tissue from GBM PDXs. GBMs 22, 64, and 110 were negative for AR protein expression (Fig. 2B), consistent with transcript levels. GBM 14 weakly expressed AR protein, in contrast to its high transcript levels. GBM 26 and 76 expressed high levels of AR protein, consistent with transcript levels, and we elected to study GBM 26 further because it was derived from a primary GBM. Finally, we analyzed AR protein in eight randomly selected patient tumors from the Wake Forest Brain Tumor Center of Excellence Tumor Bank (Fig 2C). Five samples were negative for AR protein (4607, 4441, 4673, 4525, 4474), one had low levels of AR (4536), and two had high levels of AR (4617, 4843). These results were recapitulated with immunohistochemistry staining. AR staining was strongly positive and nuclear in the prostate cancer positive control, negative in tumor 4673, which was AR-negative by immunoblotting and showed moderate cytoplasmic and nuclear staining in tumor 4617, which was positive by immunoblot (Fig. 2D). Together, these results suggest that nearly half of GBMs express AR at the transcript and protein level.
Figure 2.
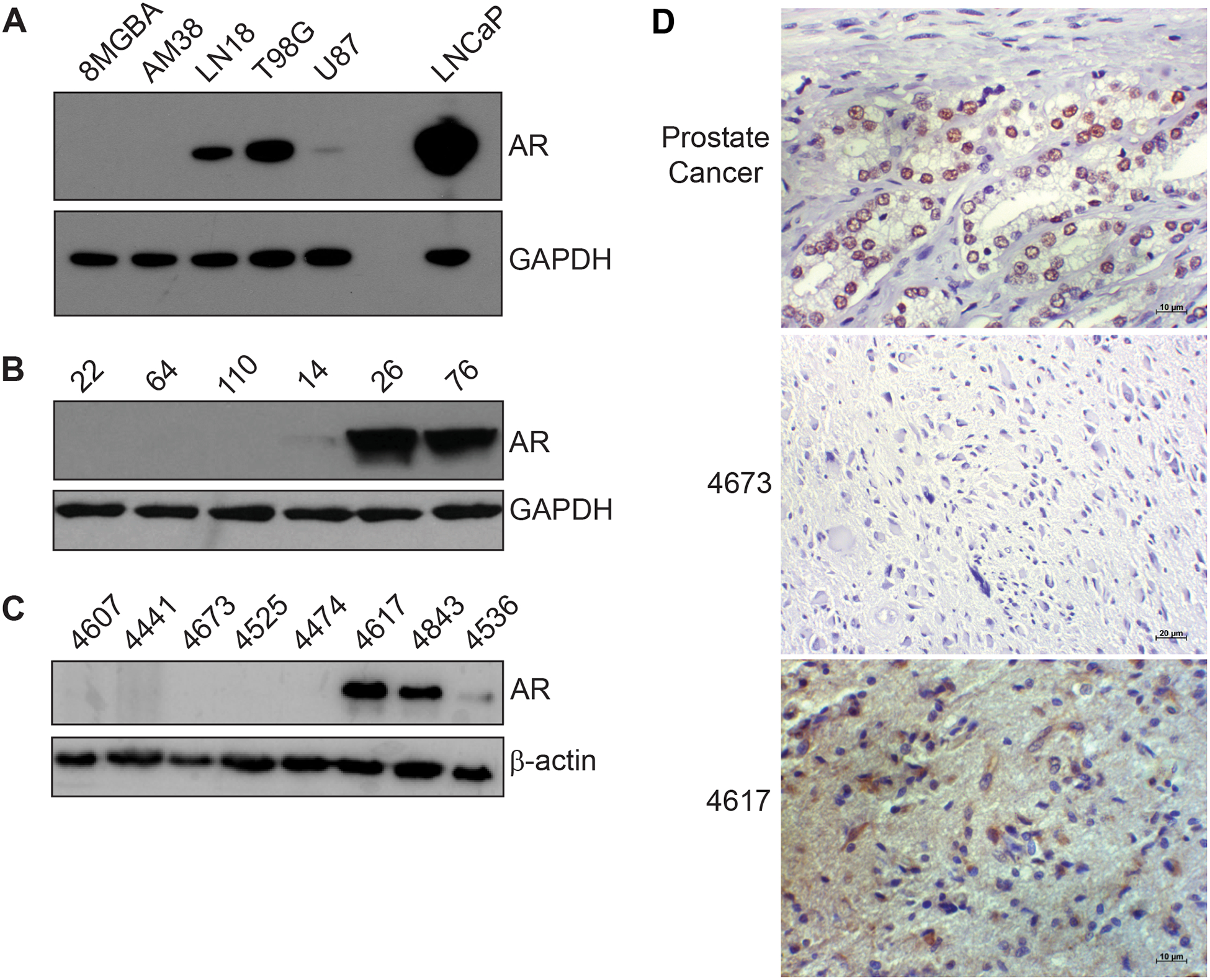
AR protein is expressed in GBM tumors, cell lines and patient-derived xenografts. (A), Cell lysates from the indicated cell lines were probed for AR protein expression. LNCaP is a prostate cancer cell line that was included as a positive control. (B) Cell lysates from the indicated PDXs from the Mayo PDX resource were probed for AR protein expression (C) Randomly selected lysates of tumors from the Wake Forest Brain Tumor Bank were probed for AR protein expression. (D) IHC staining of prostate cancer and patient tumors from the Wake Forest Brain Tumor Bank. Scale bars represent 10μm (prostate cancer, 4617) or 20μm (4673).
We next sought to determine whether AR expression was related to known clinical and molecular features of GBM (Supplementary Fig. S1C–H). AR transcript expression did not vary by sex or age (Fig. S1D–E). While AR expression was highest in the classical molecular subtype, it did not significantly vary by the presence or absence of known GBM oncogenic drivers (Fig. S1F). When we visually examined the AR expression of GBM tumors, we noted approximately 3% of TCGA tumors appeared to be AR high outliers (mean + 2 SD, Fig. S1G). While nearly all AR high outliers were MGMT methylated, they were not enriched for other known GBM oncogenic alterations (Fig. S1H). Thus, AR expression did not appear to be governed by any of the common molecular alterations in GBM, such as mutations in PTEN, TP53, NF1, PIK3R1, PIK3CA, and IDH1; homozygous deletions in CDKN2A, CDKN2B, and PTEN; and amplifications in EGFR and CDK4.
Treatment with antiandrogens selectively slows the growth of and radiosensitizes AR-positive GBM cell lines
To determine whether the observed AR expression in GBM could be exploited therapeutically, we selected two antiandrogens to test in AR-positive and negative GBM models – enzalutamide and seviteronel. Enzalutamide is a blood-brain barrier penetrant antiandrogen that is a potent AR antagonist and is FDA-approved to treat prostate cancer (34). Seviteronel is an investigational blood-brain penetrant drug that inhibits androgen synthesis by blocking CYP17 and also has direct AR antagonism (35,36). We first asked whether antiandrogens slowed the growth of AR-positive and negative GBM models. Treatment with seviteronel or enzalutamide (Fig. 3A–B) inhibited colony formation in the AR-positive GBM cell lines, as measured by GI50 values (Fig. 3C–D and G–H). These GI50 values (~3–4 μM for seviteronel, ~8–23 μM for enzalutamide) are higher in GBM than they are in prostate cancer models (~350nM for seviteronel and ~300nM for enzalutamide in LNCaP) (36), but they are clinically achievable (37). Colony formation in the AR-negative GBM cell lines was minimally inhibited or not inhibited by seviteronel or enzalutamide (Fig. 3E–F and I–J).
Figure 3.

Antiandrogens preferentially slow the growth of AR-positive cell line models of GBM. Exponentially growing GBM cell lines were treated with seviteronel (A, C-F) or enzalutamide (B, G-J) and analyzed for colony formation after 10–14 days. Individual experiments were performed in technical triplicate and representative experiments are shown in panels C-J. Error bars are the standard error of technical triplicate measurements. Dotted lines are curves fit to determine the GI50 concentration. Each experiment was independently conducted 3–4 times and indicated GI50 values are the averages from these 3–4 experiments ± the standard error. GI50 N.D. means GI50 could not be determined for at least one experiment.
We observed this same selectivity toward AR-expressing GBM models when we combined antiandrogens with radiation. When treated near GI50 concentrations of either seviteronel or enzalutamide, AR-positive cell lines were sensitized to radiation, with enhancement ratios ranging from 1.3 to 1.6 (Fig. 4A–B and E–F), while the AR-negative cell lines were not (Fig. 4C–D and G–H). Together, these results indicate that antiandrogens can slow the growth of and radiosensitize AR-positive but not AR-negative GBMs.
Figure 4.

Treatment with seviteronel or enzalutamide radiosensitizes AR-positive GBM cell lines. AR-positive (LN18, T98G) and AR-negative (8MGBA, AM38) cell lines were irradiated and treated with seviteronel (2.5 μM in LN18, 5μM in all other cell lines) or enzalutamide (10μM). Cells were replated at clonal density in technical triplicate and analyzed for colony formation 10–14 days later. Survival curves (A-H) are representative of n = 3 (LN18, sevi), n = 3 (T98G, sevi), n = 6 (8MGBA, sevi), n = 2 (AM38, sevi), n = 3 (LN18, enza), n = 3 (T98G, enza), n = 3 (8MGBA, enza), and n = 3 (AM38, enza) independent experiments. Error bars represent the SEM from technical triplicate measurements from the representative experiment. Enhancement ratios were calculated for each individual experiment based on the mean inactivating dose of radiation and we have reported the mean ± SEM from all experiments (*p < 0.05, **p < 0.01).
Antiandrogens radiosensitize AR-positive GBM xenografts in vivo
Having demonstrated that seviteronel and enzalutamide slow the growth of and radiosensitize AR-positive GBM cell lines in vitro, we then evaluated the effect of the drugs in vivo. Initially, AR-positive T98G tumors were grown in the bilateral flanks of female C.B-17 SCID mice with implanted dihydrotestosterone (DHT) pellets to mimic the high circulating testosterone levels of men. The mice were treated with seviteronel daily by oral gavage after tumors reached a volume of ~70–100mm3, with initiation of drug treatment 24 hours before the start of radiation. Drug treatment alone did not impact tumor tripling time, but combination with radiation significantly increased tumor tripling time compared to control and radiation alone (Fig. 5A–B), which indicates in vivo radiosensitization. During treatment, seviteronel was well tolerated and did not affect mouse weight (Supplementary Fig. S2C).
Figure 5.

Seviteronel and enzalutamide sensitize GBM xenografts to radiation and delay time to tumor tripling. AR-positive T98G (A-B) or AR-positive GBM26 (C-D) xenografts were serial passaged into C.B-17 SCID mice, and tumors were allowed to grow to ~70–100mm3. Treatment was then initiated as depicted in the four treatment groups (control, RT alone, drug alone, and RT + drug) with 9–14 tumors per condition. Schemas are included for the (A) T98G model with seviteronel treatment (**p < 0.01 vs. control; Φp < 0.05 vs. RT) and (B) GBM26 with enzalutamide treatment (***p < 0.001 and ****p < 0.00001 vs. control; Φp = 0.05 vs. RT). Tumor volume was tracked, and time to tumor tripling was plotted (as percent not tripled) using Kaplan Meier estimates (B, D).
We then asked if similar results could be obtained in patient-derived models of GBM, which better recapitulate the biology of human tumors and more faithfully reflect responses to therapy (33). AR-positive GBM26 PDX tumors were grown in the bilateral flanks of female C.B-17 SCID mice and treated with enzalutamide daily by oral gavage after reaching ~70–100mm3, with initiation of drug treatment 24 hours before the start of radiation. DHT pellets were omitted in this model, which mimics the situation in women with GBM. Single-agent enzalutamide significantly increased tumor tripling time compared to control, and combination treatment increased tumor tripling time compared to radiation alone (Fig. 5C–D). Like seviteronel, enzalutamide was well-tolerated and did not result in appreciable differences in mice weights (Supplementary Fig. S2D).
AR inhibition delays repair of radiation-induced DNA damage by blocking AR-positive GBMs from activating transcriptional programs
While the single agent effects of antiandrogens were modest in vivo, both seviteronel and enzalutamide radiosensitized AR-positive tumors in vitro and in vivo. To understand how antiandrogens might be exerting their radiosensitizing effects, we performed RNA-seq using the GBM26 model and the FDA-approved drug enzalutamide. We used a modified schedule (Fig. 6A) with enzalutamide treatment beginning 24hr before radiation, and two total fractions of radiation (2Gy/day) before analysis. Treatment with radiation caused the differential expression of 674 genes (FDR ≤ 0.05, and fold change ≥ ± 1.5). Adding enzalutamide to radiation decreased the number of differentially expressed genes to 106. Enzalutamide treatment by itself only caused the differential expression of 2 genes (Fig. 6B). Because we observed radiosensitization in vitro, we decided a priori to specifically focus on transcriptional changes genes related to DNA repair. Of these pre-specified genes, we identified 169 with Log2FC values for all conditions. RT significantly increased the expression of 13 genes related to DNA repair (threshold: p < 0.05 in RT vs control, Fig. 6C). Only 3 of these genes were still up-regulated in the RT + enzalutamide condition (DDB2, RRM2B, and XPC), and none changed significantly with enzalutamide treatment alone. Additionally, 64 genes related to DNA repair were decreased following RT (Supplementary Fig. S3). Only 4 genes were still decreased in the RT + enzalutamide condition, and none of the genes were significantly down-regulated by enzalutamide.
Figure 6.

Antiandrogens sensitize AR-positive GBMs to radiation by modulating DNA repair. GBM26 xenografts were serial passaged into C.B-17 SCID mice, and tumors were allowed to grow to ~70–100mm3. Treatment was then initiated as depicted (A) in the four treatment groups (control, RT alone, enzalutamide alone, and RT + enzalutamide combo) with 4 tumors per condition. (B) Common significant differentially expressed genes (FDR ≤ 0.05, and fold change ≥ ± 1.5) from treatment conditions (RT, enzalutamide, RT + enzalutamide combo) vs. control. (C) Heatmap of DNA repair genes (with threshold: p < 0.05 in RT vs control). Out of 169 prespecified genes with Log2FC values for all conditions, 13 were up-regulated in the RT condition (p < 0.05 in RT vs control), and 3 were up-regulated in the RT + enzalutamide combo condition (DDB2, RRM2B, and XPC; p < 0.05 in combo vs control). None of the genes were significantly changed with enzalutamide treatment alone. LN18 (D), T98G (E), 8MGBA (F), and AM38 (G) were treated with 10μM seviteronel, irradiated with 6 Gy 24 hours later, and fixed at the following time points post-RT: 1hr, 6hr, 24hr. Threshold for positivity was set as follows: 15 foci (8MGBA) and 20 foci (LN18, T98G, AM38). Minimum of 50 cells were scored per condition. Data plotted are the mean ± SEM of 3–6 experiments (*p < 0.05, **p < 0.01, or ****p < 0.0001 vs. RT). (H) Schematic representation of the role of the AR in GBMs (AR-positive) exposed to ionizing radiation. After radiation, AR promotes the DNA repair response leading to radiation resistance (green arrows). In the presence of antiandrogens, AR signaling is disrupted and radiation resistance is overcome (red bars).
These results suggested that antiandrogens were inhibiting the ability of AR-positive GBMs to engage the transcriptional programs needed to repair radiation-induced DNA damage. To examine the functional consequences of these findings, we assessed the ability of AR-positive and negative GBM cell lines to repair radiation-induced DNA damage by analyzing the formation and resolution of γ-H2AX foci, which is a marker of unresolved double-strand DNA breaks (38). Combined seviteronel and radiation treatment led to delayed repair of double strand DNA breaks in AR-positive but not AR-negative cell lines (Fig. 6D–G, Supplementary Fig. S4). While enzalutamide modestly slowed the repair of radiation-induced γ-H2AX foci, these changes were not significant (Supplementary Fig. S5).
To further characterize how seviteronel and enzalutamide affected the repair of other types of radiation-induced DNA damage, we employed the alkaline comet assay, which measures single-strand DNA breaks in addition to other types of DNA damage (39). Compared to treatment with RT alone, neither seviteronel nor enzalutamide combined with RT increased the amount of damage present at 1 hour post-RT, which is represented by the alkaline tail moment (Supplementary Fig. S6). There was an increased alkaline tail moment when either drug was combined with 6 Gy at 6 hours post-RT, though these changes were only significant in the LN18 cell line.
DISCUSSION
In this study, we aimed to overcome therapy resistance in GBM by applying a strategy that has been successfully used in other cancers, namely inhibiting AR activity. AR is expressed at the transcript and protein level in approximately half of GBM cell lines, patient-derived xenografts and primary patient tumors and the AR expression of GBM overlapped with levels seen in prostate cancer. Expression of AR did not vary by patient sex, age or underlying oncogenic alteration. Treatment with the antiandrogens enzalutamide and seviteronel slows the growth of and radiosensitizes AR-positive, but not AR-negative, GBM cell lines. This radiosensitization can be recapitulated in AR-positive xenografts by administration of seviteronel or enzalutamide with radiation. AR inhibition hindered AR-positive GBMs from engaging transcriptional programs needed to repair radiation-induced DNA damage, which resulted in prolonged unrepaired double-strand DNA breaks (Fig. 6H) and increased alkaline tail moments. Taken collectively, these results indicate that antiandrogens may have promise as a combination strategy with radiation for patients with AR-positive GBM.
The ability of antiandrogens to slow the repair of radiation-induced DNA damage in AR-positive GBM is consistent with mechanisms described in other AR-positive cancers. In prostate cancer, the AR is upregulated in response to ionizing radiation (40) and transcriptionally regulates multiple DNA repair genes. Thus, increased AR signaling after radiation can drive repair of DNA damage and contribute to radioresistance of prostate cancer and this process can be overcome by antiandrogens (8,41). In breast cancer, AR modulates DNA damage by activating kinases involved in DNA repair, such as DNA-PKcs, ATM, and Chk2 (10,42). Polkinghorn et al. previously identified 32 direct AR target genes involved in DNA repair (8). Two of these genes (RAD51C and HUS1) were upregulated in AR-positive GBMs following RT and this upregulation was reversed when radiation was combined with enzalutamide (Fig. 6C). These findings may explain the ability of antiandrogens to slow DNA repair and radiosensitize AR-positive GBMs.
Combined treatment with seviteronel and radiation delayed repair of double-strand DNA breaks in AR-positive cell lines to a greater extent than did enzalutamide. Yet, both compounds were able to radiosensitize AR-positive GBMs in vitro and in vivo. Seviteronel and enzalutamide impair AR function through different mechanisms, and they may have different effects on single-strand DNA (ssDNA) and dsDNA repair. Seviteronel is a selective CYP17 lyase inhibitor that can bind to AR and lock it in an inactive, unliganded conformation (35,36). Enzalutamide is a second-generation antiandrogen that inhibits binding of testosterone to AR. It also binds to the ligand-binding domain of AR with high affinity, impairing nuclear translocation, DNA binding, and recruitment of coactivators (34). Of the 10 DNA repair genes that enzalutamide blocks from radiation-induced upregulation, seven are directly involved in ssDNA repair. Thus, we reasoned that antiandrogens might radiosensitize AR-positive GBMs by inhibiting ssDNA break repair in addition to dsDNA breaks. The results of our alkaline comet studies support this hypothesis, but the greater effect in the LN18 cell line suggest that this regulation may vary between different models and/or patients.
Our results suggest that the best clinical use of antiandrogens for AR-positive GBM may be in combination with radiation. While both seviteronel and enzalutamide slowed the growth of AR-positive GBM in vitro, these antiandrogens only modestly slowed the growth of AR-positive GBMs in vivo when administered as monotherapy. The efficacy of both agents was more pronounced in vivo when combined with radiation. Consistent with these findings, enzalutamide caused minimal transcriptional changes when given as monotherapy but was able to stop the transcriptional reprogramming caused by radiation. One potential explanation for these results is that AR signaling is of minimal importance for the growth of AR-positive GBMs in vivo but is important for DNA repair in the setting of genotoxic agents such as radiation. A similar situation appears to exist in bladder cancer, where AR is expressed in a subset of tumors and associated with resistance to gemcitabine (43). This has informed a recent clinical trial for metastatic bladder cancer, where enzalutamide was combined with genotoxic agents, gemcitabine, and cisplatin. While the sample size in this study was small, combined enzalutamide, gemcitabine, and cisplatin were reasonably well tolerated and at least one patient with an AR-positive tumor achieved complete response to the therapy (44). Because antiandrogens affected transcripts involved in multiple DNA repair pathways, including base excision repair, there is a possibility that these agents will also potentiate the effects of other genotoxic agents in GBM such as temozolomide. Indeed, emerging evidence suggests that testosterone analogs may contribute to temozolomide resistance in GBM through AR-independent mechanisms (45,46). Thus, antiandrogens such as seviteronel and abiraterone that also inhibit androgen synthesis may be especially useful to overcome therapy resistance in GBM.
Our study has limitations. We studied multiple GBM models in vivo, but did so in the flank rather than intracranially. However, we believe that antiandrogens will have similar efficacy intracranially because of their brain penetrance. Enzalutamide and the structurally-related apalutamide achieve brain/plasma ratios 30%−60% in normal brain (47) and it is likely that concentrations in GBM tissue would be even higher due the disrupted blood-brain barrier. Whether combining antiandrogens and radiotherapy could be useful in other AR-positive brain tumors such as meningioma is not known (48). There is also a seizure-risk associated with enzalutamide due to off-target effects on the GABAA receptor (49). Because patients with GBM are at increased risk of seizure, other brain-penetrant anti-androgens such as seviteronel or apalutamide may be more appropriate agents for use in this population. Because we were focused on therapeutic applications, we intervened on the AR with anti-androgens rather than genetic approaches. While this strategy means that some of our observed phenotypes could be due to off-target drug effects, we believe this is unlikely due to the absence of effects on AR-negative GBM models.
In summary, we have found that many GBMs express AR at the transcript and protein level and that antiandrogens radiosensitize AR-positive GBMs in vitro and in vivo, in part by slowing DNA repair. These findings suggest that combining radiation and antiandrogens for patients with AR-positive GBM could warrant clinical investigation.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by grants from Innocrin Pharmaceuticals, Inc. and the Jones Family Foundation Fund within the Chad Carr Pediatric Brain Tumor Center to DRW. DRW is also funded by a University of Michigan Cancer Center Core grant (P30CA046592), as well as grants from the American Cancer Society, the Forbes Institute for Cancer Discovery, and the NCI (K08CA234416). WZ is supported by the Postdoctoral Translational Scholar Program (UL1TR002240) from the Michigan Institute for Clinical & Health Research of University of Michigan. We would like to thank the University of Michigan Advanced Genomics Core, the University of Michigan Bioinformatics Core, and Steven Kronenberg.
Footnotes
Disclosure of Potential Conflicts of Interest: Part of this work was funded by a research grant to DRW by Innocrin Pharmaceuticals, Inc. RES has received research grant funding from JAZZ Pharmaceuticals, the Southeastern Brain Tumor Foundation, and the American Society of Clinical Oncology Conquer Cancer Foundation for unrelated work and has received compensation for consulting from Novocure and Monteris Medical unrelated to this work. DRW also declares research grant funding from Agios for unrelated work. JRE and EBB were employees of Innocrin Pharmaceuticals, Inc., and shareholders in the company.
REFERENCES
- 1.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical Report: Primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro Oncol 2017;19:v1–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459–66. [DOI] [PubMed] [Google Scholar]
- 3.Brandes AA, Tosoni A, Franceschi E, Sotti G, Frezza G, Amista P, et al. Recurrence pattern after temozolomide concomitant with and adjuvant to radiotherapy in newly diagnosed patients with glioblastoma: correlation With MGMT promoter methylation status. J Clin Oncol 2009;27:1275–9. [DOI] [PubMed] [Google Scholar]
- 4.Gebhardt BJ, Dobelbower MC, Ennis WH, Bag AK, Markert JM, Fiveash JB. Patterns of failure for glioblastoma multiforme following limited-margin radiation and concurrent temozolomide. Radiat Oncol 2014;9:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peereboom DM, Shepard DR, Ahluwalia MS, Brewer CJ, Agarwal N, Stevens GHJ, et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J Neurooncol 2010;98:93–9. [DOI] [PubMed] [Google Scholar]
- 6.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 2014;370:709–22. [DOI] [PubMed] [Google Scholar]
- 7.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev 2004;25:276–308. [DOI] [PubMed] [Google Scholar]
- 8.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 2013;3:1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolla M, Van Tienhoven G, Warde P, Dubois JB, Mirimanoff R-O, Storme G, et al. External irradiation with or without long-term androgen suppression for prostate cancer with high metastatic risk: 10-year results of an EORTC randomised study. Lancet Oncol 2010;11:1066–73. [DOI] [PubMed] [Google Scholar]
- 10.Speers C, Zhao SG, Chandler B, Liu M, Wilder-Romans K, Olsen E, et al. Androgen receptor as a mediator and biomarker of radioresistance in triple-negative breast cancer. NPJ Breast Cancer 2017;3:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barton VN, Gordon MA, Richer JK, Elias A. Anti-androgen therapy in triple-negative breast cancer. Ther Adv Med Oncol 2016;8:305–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chung YG, Kim HK, Lee HK, Lee KC. Expression of androgen receptors in astrocytoma. J Korean Med Sci 1996;11:517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao D, Cheng C, Lan X, Xing R, Chen Z, Zhao H, et al. Regulation of p53wt glioma cell proliferation by androgen receptor-mediated inhibition of small VCP/p97-interacting protein expression. Oncotarget 2017;8:23142–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun T, Warrington NM, Rubin JB. Why does Jack, and not Jill, break his crown? Sex disparity in brain tumors. Biol Sex Differ 2012;3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96. [DOI] [PubMed] [Google Scholar]
- 16.Ostrom QT, Rubin JB, Lathia JD, Berens ME, Barnholtz-Sloan JS. Females have the survival advantage in glioblastoma. Neuro Oncol 2018;20:576–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455:1061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang J, He D, Yang P, He J, Zhang Y. Genome-wide expression profiling of glioblastoma using a large combined cohort. Sci Rep 2018;8:15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piccolo SR, Sun Y, Campbell JD, Lenburg ME, Bild AH, Johnson WE. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics 2012;100:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan MA, Parsels LA, Kollar LE, Normolle DP, Maybaum J, Lawrence TS. The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin Cancer Res 2008;14:5142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, et al. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015;17:757–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence TS. Ouabain sensitizes tumor cells but not normal cells to radiation. Int J Radiat Oncol Biol Phys 1988;15:953–8. [DOI] [PubMed] [Google Scholar]
- 24.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009;25:1105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang ZH, Jhaveri DJ, Marshall VM, Bauer DC, Edson J, Narayanan RK, et al. A comparative study of techniques for differential expression analysis on RNA-Seq data. PloS One 2014;9:e103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science 2001;291:1284–9. [DOI] [PubMed] [Google Scholar]
- 28.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes, 2005. Mutat Res 2005;577:275–83. [DOI] [PubMed] [Google Scholar]
- 29.Errol CF, Graham CW, Wolfram S, Richard DW, Roger AS, Tom E. DNA Repair and Mutagenesis, Second Edition American Society of Microbiology; 2006. [Google Scholar]
- 30.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer 2011;11:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zalcman N, Canello T, Ovadia H, Charbit H, Zelikovitch B, Mordechai A, et al. Androgen receptor: a potential therapeutic target for glioblastoma. Oncotarget 2018;9:19980–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao SG, Yu M, Spratt DE, Chang SL, Feng FY, Kim MM, et al. Xenograft-based platform-independent gene signatures to predict response to alkylating chemotherapy, radiation, and combination therapy for glioblastoma. Neuro Oncol 2019;21:1141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009;324:787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ. Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg Med Chem Lett 2014;24:2444–7. [DOI] [PubMed] [Google Scholar]
- 36.Norris JD, Ellison SJ, Baker JG, Stagg DB, Wardell SE, Park S, et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J Clin Invest 2017;127:2326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liston DR, Davis M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin Cancer Res 2017;23:3489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998;273:5858–68. [DOI] [PubMed] [Google Scholar]
- 39.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988;175:184–91. [DOI] [PubMed] [Google Scholar]
- 40.Spratt DE, Evans MJ, Davis BJ, Doran MG, Lee MX, Shah N, et al. Androgen Receptor Upregulation Mediates Radioresistance after Ionizing Radiation. Cancer Res 2015;75:4688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov 2013;3:1254–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Min A, Jang H, Kim S, Lee K-H, Kim DK, Suh KJ, et al. Androgen Receptor Inhibitor Enhances the Antitumor Effect of PARP Inhibitor in Breast Cancer Cells by Modulating DNA Damage Response. Mol Cancer Ther 2018;17:2507–18. [DOI] [PubMed] [Google Scholar]
- 43.Kashiwagi E, Ide H, Inoue S, Kawahara T, Zheng Y, Reis LO, et al. Androgen receptor activity modulates responses to cisplatin treatment in bladder cancer. Oncotarget 2016;7:49169–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gupta S, Dhillon J, Magliocco AM, Puskas J, Caceres G, Masawi F, et al. Results from a phase I/Ib trial of enzalutamide and gemcitabine and cisplatin in metastatic bladder cancer (mBC). J Clin Oncol 2019;37:471.30615550 [Google Scholar]
- 45.Chuang J-Y, Lo W-L, Ko C-Y, Chou S-Y, Chen R-M, Chang K-Y, et al. Upregulation of CYP17A1 by Sp1-mediated DNA demethylation confers temozolomide resistance through DHEA-mediated protection in glioma. Oncogenesis 2017;6:e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W-B, Chuang J-Y, Ko C-Y, Chang W-C, Hsu T-I. Dehydroepiandrosterone Induces Temozolomide Resistance Through Modulating Phosphorylation and Acetylation of Sp1 in Glioblastoma. Mol Neurobiol 2019;56:2301–13. [DOI] [PubMed] [Google Scholar]
- 47.Moilanen A-M, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep 2015;5:12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hess K, Spille DC, Wagner A, Stummer W, Paulus W, Brokinkel B. Letter: Brain Invasion in Meningiomas-Sex-Associated Differences are not Related to Estrogen- and Progesterone Receptor Expression. Neurosurgery 2017;81:E25–7. [DOI] [PubMed] [Google Scholar]
- 49.Foster WR, Car BD, Shi H, Levesque PC, Obermeier MT, Gan J, et al. Drug safety is a barrier to the discovery and development of new androgen receptor antagonists. Prostate 2011;71:480–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.