

Abstract
Introduction
In brain, extracellular vesicles (EVs) play an essential role in the neuron‐glia interface and ensure the crosstalk between the brain and the periphery. Some studies now link the pathway dysfunction of the EVs to apolipoprotein E gene variant (APOE ε4) and the risk of progression to Alzheimer's disease (AD). To better understand the role of APOE ε4 in pre‐clinical AD, we have determined levels of pathogenic, neurotrophic and inflammatory proteins in peripheral EVs (pEVs) and in plasma from cognitively impaired, no dementia (CIND) participants stratified upon the absence (APOE ε4‐) or the presence (APOE ε4+ ) of the ε4 allele of APOE.
Methods
Levels of 15 neurodegenerative, neurotrophic and neuroinflammatory proteins were quantified in pEVs and compared to their plasma levels from cognitively normal and CIND participants.
Results
Levels of neurotrophic and inflammatory markers were reduced in pEVs from APOE ε4+. The pentraxin‐2/α‐synuclein ratio measured in pEVs was able to predict AD 5 years before the onset among APOE ε4+‐CIND individuals.
Discussion
Our findings suggest an alteration of the endosomal pathway in APOE ε4+ and that pEVs pentraxin‐2/α‐synuclein ratio could serve as a useful early biomarker for AD susceptibility.
Keywords: Alzheimer's disease, biomarkers, DJ‐1, extracellular vesicles, lipocalin, pentraxin‐2, S100B, α‐synuclein
1. INTRODUCTION
Alzheimer's disease (AD) is the predominant form of dementia and the most common neurodegenerative disorder. 1 Advanced age and the presence of the ε4 allele of the apolipoprotein E gene (APOE) are the most relevant late‐onset sporadic AD‐promoting factors as they interact with the core mechanisms of this neurologic disorder. 2 , 3 ApoE is essential to modulate cerebral lipid homeostasis and neurogenesis with the ApoE4 isoform promoting less efficiently the transport of essential lipids to neurons. In addition, proteolytic degradation of the amyloid beta (Aβ) peptide is compromised in APOE ε4 carriers, thus increasing amyloidosis and the risk of progression to AD. 4 However, other mechanisms involving APOE ε4 in brain malfunction related to AD pathology remain to be explored.
With the lack of efficient pharmacotherapeutic options and early AD diagnosis, the challenge is now to identify preclinical AD biomarkers. In this context, subjective cognitive decline (SCD) was defined as a stage preceding mild cognitive impairment (MCI) and probably the earliest clinical sign of AD. 5 Before the establishment of a conceptual framework for research on SCD, various terms were used in the literature to refer to intermediate states between normal cognition and dementia such as “cognitive impairment, no dementia (CIND),” “subjective cognitive concerns,” “subjective cognitive impairment,” “subjective memory complaint,” and others. 6 , 7 In addition, common techniques for AD diagnosis such as neuroimaging (magnetic resonance imaging [MRI] and positron emission tomography [PET] scan) and cerebrospinal fluid (CSF) collection are expensive, invasive, and relatively accurate, which entails the development of more cost‐effectiveness and valid procedures. Therefore, identification of early and novel candidate blood‐based biomarkers emerges as one of the current major challenges.
Extracellular vesicles (EVs) are membrane‐shedding nanoparticles that are released from different cell types as mediators of cell‐to‐cell communication. EVs form a heterogeneous group with varying composition and metabolic fates and can transport and transfer various molecular components (eg, nucleic acids, proteins, and lipids) involved in the regulation of active signaling pathways. 8 In brain, EVs are involved in neuron‐glial interface, in neuroprotection, as well as in the dissemination of neuropathologic components (such as Aβ, tau) notably between the brain and periphery. 9 , 10 , 11 Recently, we demonstrated an early reduction of t‐tau and the amyloid precursor protein (APP) in plasma‐derived EVs (pEVs) isolated from patients with MCI and found that p‐tau181 and APP concentrations in pEVs were correlated to cognitive performances. 12
Cumulative evidence suggests that the dysfunctional endosomal‐lysosomal pathway is a prominent pathogenic mechanism in AD. 13 , 14 Neuronal endosomal changes were associated with the expression of the APOE ε4 allele in humanized mouse models as well as in humans. In transgenic APOE ε4 mice brain, the analysis of the endosomal‐lysosomal system revealed an age‐dependent increase in the number and size of early endosomes with an overexpression of genes involved in the normal processing of the endocytic pathway. 15 Another study reported that APOE ε4 impairs the insulin receptor trafficking by trapping it in the endosomes, leading to pathogenic cerebral insulin resistance associated with AD risk. 16 Moreover, it was described that APOE ε4 expression decreases EVs production in APOE ε4 mice and in post‐mortem tissue of neuropathologically healthy people. 17 These data strongly suggest that APOE ε4 may alter the endosomal trafficking and affect EVs composition and production from the endosomal pathway. In this context, targeting EVs‐derived biomarkers in pre‐clinical AD patients carrying APOE ε4 would be an innovative strategy to disclose the increased incidence of AD among APOE ε4 carriers and could eventually be used for early AD diagnosis.
The aim of this study was to determine whether changes in plasma and in pEVs of proteins involved in a variety of functions including neuroprotection, synaptic transmission, neuroinflammation, and neurovascular homeostasis, can predict the evolution of CIND patients to AD in APOE ε4 carriers.
RESEARCH IN CONTEXT
Systematic review: Previous studies suggest that the apolipoprotein E gene variant APOE ε4 might disturb lysosomal‐endosomal signaling pathway, which is a prominent pathogenic factor in Alzheimer's disease (AD). Our study provides the first attempt to disclose the interactions/crosstalk between the APOE ε4 variant and altered protein handling in peripheral extracellular vesicles (pEVs) and plasma from asymptomatic demented subjects that progress to AD.
Interpretation: Our multicenter longitudinal clinical cohort study shows differential regulation of neurotrophic and inflammatory factors according to the presence of the APOE ε4 variant with a better sensitivity for pEVs compared to plasma. Our findings demonstrate that dysfunctional synaptic transmission occurs earlier than amyloid beta (Aβ) deposition and tangle formation in APOE ε4+ preclinical patients at risk for developing AD and support the use of the neuronal pentraxin‐2/α‐synuclein ratio as a strong candidate to predict AD onset.
Future directions: Future investigations should consider larger sample size and include neuroimaging data to confirm the present finding.
2. METHODS
2.1. Study cohort
The present work was realized using data from the Canadian Study of Health and Aging (CSHA), a three‐phase, 10‐year multicenter longitudinal study. In CSHA‐1 (1991‐1992), participants were selected according to the Modified Mini‐Mental State (3MS) Examination followed by a self‐administered baseline risk factor questionnaire outlining the eligibility criteria (Table S1). A nurse, a physician, and a psychometrist clinically evaluated the participants. The physician and a neuropsychologist made independent preliminary diagnoses. A consensus diagnostic was thereafter reached between the two, according to the Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised (DSM)‐III‐R for dementia, 18 the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria (NINCDS‐ADRDA) for AD, 19 the World Health Organization International Classification of Diseases, 10th Revision criteria (ICD‐10) for vascular dementia (VaD), 20 and operational criteria for Lewy body dementia. 21 Similar recruitment and diagnostic processes were realized 5 and 10 years later in CSHA‐2 (1996‐97) and CSHA‐3 (2001‐02) respectively, according to new criteria such as the DSM, Fourth Edition, (DSM‐IV) for the diagnosis of dementia and AD, 22 and the National Institute of Neurological Disorders and Stroke‐Association Internationale pour la Recherche et l’Enseignement en Neurosciences (NINDS‐AIREN) criteria to define VaD. 23 Participants who did not meet the criteria for dementia or for cognitively healthy older people were classified as CIND.
Accordingly, our study includes a convenience subsample of 24 CIND participants from CSHA‐2 who were genotyped for the presence of the APOE ε4 allele and prospectively evaluated for AD at CSHA‐3. A subgroup of CIND‐ APOE ε4+ participants who developed AD (CIND ‐AD APOE ε4+) was further compared to age‐matched healthy controls who were selected with respect to inclusion and exclusion criteria (Table S1).
All participants gave consent for use of their blood sample for subsequent analyses. This study was approved by the ethical committee at INRS (CER19‐532).
2.2. Blood sampling
Blood samples were obtained from participants, only at CSHA‐2, on the same day of the clinical assessment in EDTA collection tubes and immediately centrifuged. Plasma samples were stored at −20°C at the National Microbiology Laboratory in Ottawa for 2 to 3 years, and then transferred in dry ice Styrofoam boxes to Saint‐Sacrement Hospital in Quebec City where they were placed at −80°C until analysis.
2.3. Isolation and characterization of extracellular vesicles
pEVs were extracted according to 12 , 24 , 25 and were characterized following the directive of the methodological guidelines to study EVs. 26
2.3.1. Nanoparticles tracking analysis
Various pEVs suspensions were diluted in ultra‐pure filtered water (1:1000) and injected into the Nanosight NS300 system (Malvern Panalytical instruments, Inc., UK) under the same operating conditions: syringe flow (100), camera level (14), detection threshold (5), capture duration (60s), number of capture (3), particles per frame (30 to 80), and temperature (23.5°C). Videos were stored and analyzed by an appropriate software (NTA 3.2) that tracks and relates the rate of Brownian motion to particle size and concentration.
2.3.2. Transmission electron microscopy
EVs suspensions were fixed with 2% paraformaldehyde prior to transfer on cast films of Formvar strengthened with a layer of evaporated carbon prepared on copper grids. Uranyl acetate (2%) was added as a negative staining reagent. Grids were washed and then analyzed using the Hitachi H‐7100 TEM instruments.
2.3.3. Western blot analysis
Blocked PVDF membranes containing equivalent amounts of pEVs proteins (10 μg) were incubated 16 hours at 4°C with the following primary antibodies: mouse anti‐tetraspanin CD63 and anti‐calnexin (1:500) (Santa Cruz Biotechnology, Inc.) and rabbit anti‐TSG101 (1:2000) (MyBiosource, USA). Membranes were then washed by 0.1% TBS‐Tween and incubated 1 hour at room temperature with the secondary antibodies HRP‐conjugated anti‐mouse (1:1000) and anti‐rabbit (1:2000) (Cell Signaling., Inc). Finally, membranes were stained with the enhanced chemiluminescence kit (Bio‐Rad) and visualized using the ChemiDoc imaging system.
2.3.4. Dual immune‐labeling of pEVs
Double‐labeling of pEVs with the lipophilic PKH67 dye (PKH67GL‐1KT, Millipore) and rabbit recombinant monoclonal TSG101 antibody conjugated to Alexa Fluor 594 was conducted as per manufacturer's instructions. Concisely, 100 μg of pEVs were pooled with 0.25 mL of PKH67 dissolved in dilution buffer and gently mixed at room temperature for 10 minutes. Staining was then blocked using exosome‐depleted FBS (1/4 V/V) (Life Technologies, USA) and the reaction mixture was placed into 100 kDa filter tube (Amicon UFC810024, Millipore) followed by centrifugation at 3000 g for 15 minutes. The obtained pellet was washed with PBS (1X) and supernatant was collected. The labeled pEVs fraction was further stained with fluorescent anti‐rabbit TSG101 coupled with Alexa Fluor 594 (1:500) (Cell Signaling., USA) for 1 hour at 25°C. Fluorescence excess was removed by filtration and by washing with PBS. Image analysis was performed using the Zeiss LSM780 laser scanning confocal microscope.
2.4. Luminex assay
The pEVs’ protein content of BDNF, APP, NSE, NPTX‐2, α‐Syn, DJ‐1, MMP‐9, S100B, PrGN, LCN‐2, and ANGPTL‐4 was determined by the multiplex Luminex assay (LXSAHM‐11, R&D Systems, Inc.,). Similarly, the accumulation of the protein fragments Aβ1 ‐40, Aβ1‐42 and different forms of the protein tau (t‐tau and p‐tau181) were evaluated by another Luminex assay (HNABTMAG‐68K, Millipore‐Sigma). Data were processed using an analytical software coupled to the Luminex 100/200 machine (Xponent 4.2, USA) and results were normalized according to the total pEVs’ protein amount. Some values were below the limit detection, which reduced the average number of participants per group but maintained adequate statistical analysis. Limit detection sensibility for the analytes is described (Table S2).
2.5. Statistical analysis
The Kolmogorov‐Smirnov and Shapiro‐Wilk tests were used to verify normal distribution. Statistical analysis was performed by Student's unpaired t test using the GraphPad Prism software version 6.0. Clinical and sociodemographic characteristics and correlation parameters analysis were carried out using the SPSS software (SPSS v18.0, Inc., IL, USA). The cognitive Mini‐Mental State Exam (MMSE) scores derived from the 3MS examination were used to compare cognitive performance of the participants. All results were given as means ± standard error of the mean (SEM) and the difference was considered statistically significant at P < .05.
3. RESULTS
The main clinical features of the study participants are summarized in the Table S3. As a critical risk factor for AD, age was matched between groups. There were no statistical differences in terms of sex distribution, education, and MMSE scores. To refine the heterogeneity of the disease, we have stratified patients according to the absence (APOE ε4‐) and the presence (APOE ε4+) of the APOE ε4 allele. Following the screening for dementia over a 5‐year period, a subgroup of APOE ε4+ CIND‐AD participants was compared to cognitively healthy older people. The main outcome clearly demonstrates a significant difference in cognitive scores. Meanwhile, similar records were obtained for education, age, and sex ratio (Table S4).
Different methods were employed to reflect the full spectrum of EVs properties (Figure 1A‐G). The morphology of pEVs was cup‐shaped (Figure 1A) and NTA analysis showed a distribution of particles sizes ranging from 30 to 260 nm (Figure 1B). Immunoblotting was used to detect specific EVs‐associated proteins (CD63 and TSG101), whereas calnexin was absent (Figure 1C). These results were further supported by confocal microscope imaging that clearly confirms the presence of the TSG101 marker as revealed by the merging with PKH67 fluorophore (green) and the specific TSG101 fluorescent antibody (red) (Figure 1D‐G).
FIGURE 1.
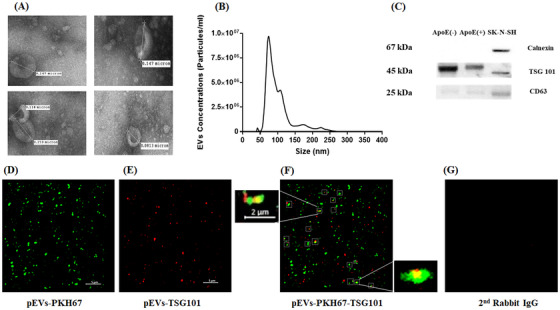
Characterization and visualization of pEVs. (A) TEM images of pEVs; magnification 40.000×. (B) Analysis of the size distribution and concentration. (C) Western blotting for exosomal proteins TSG101 and CD63. (D) Confocal imaging of EVs labeled with PKH67. (E) TSG101 antibodies using Alexa Fluor 594. (F) Co‐localization of PKH67 and TSG101. (G) Anti‐rabbit antibodies with pEVs
An equal number of pEVs was reported between CIND‐ APOE ε4‐ and CIND‐ APOE ε4+ participants (Figure 2A). Of interest, in pEVs subpopulations, the mean size was lower in APOE ε4+ carriers than noncarriers (Figure 2B). The total protein concentration in pEVs and in plasma was similar (Figure 2C‐D) and will be use to normalize the analyzed proteins levels.
FIGURE 2.
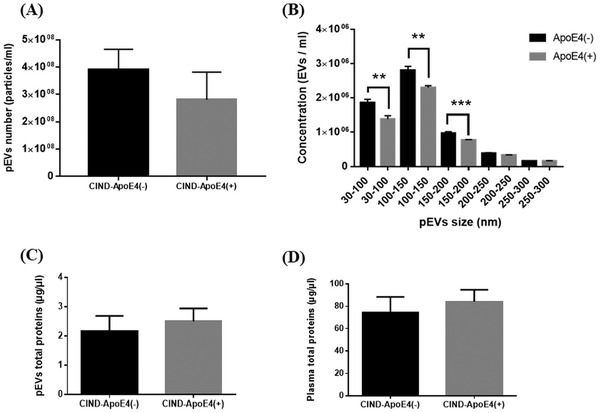
Effects of APOE ε4 variant on pEVs and plasma characteristics. (A) Effect on pEVs number. (B) Effect on pEVs size distribution. (C) Effect on pEVs protein content. (D) Effect on plasma protein content. ** P < .01, *** P < .001
The presence of APOE ε4 does not impact on the levels of Aβ1‐42, t‐tau, and p‐tau181, and the ratio Aβ1‐42/p‐tau181 in pEVs (Figure S1A‐D). In addition, pEVs Aβ1‐40 and plasma levels were not detectable in most samples (data not shown). The profile of inflammatory and trophic factors measured in CIND patients showed that APOE ε4 is associated with lower levels of neurotrophic markers such as DJ‐1, PrGN, and α‐Syn, with a higher discriminating capacity in pEVs compared to plasma except for α‐Syn (Figure 3A‐F). However, there was no difference between CIND‐APOE ε4‐ and APOE ε4+ in pEVs and plasma levels of NSE, APP, MMP‐9, and BDNF (Fig. S2A‐D). Of note, the concentrations of DJ‐1, PrGN, and NSE were lower, whereas α‐Syn and APP were higher in pEVs as compared to their plasma levels.
FIGURE 3.
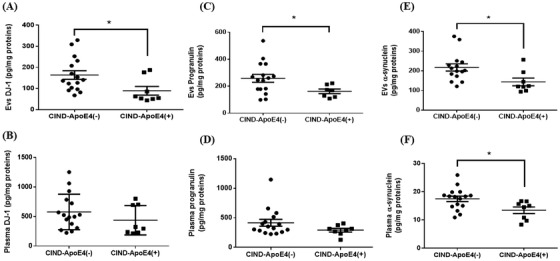
Levels of neurotrophic factors in pEVs and in plasma. Levels of DJ‐1, progranulin, and α‐synuclein measured in pEVs (A, C, E) and in plasma (B, D, F). Graph points indicate individual values for each participant. (−), ε4 non‐carriers; (+), ε4 carriers; apoE, apolipoprotein E; CIND, cognitively impaired, no dementia; EVs, extracellular vesicles. Statistical analysis was performed using the Student t test for DJ‐1 (EVs and plasma) and plasma α‐synuclein. For progranulin (EVs and plasma) and EV α‐synuclein, the Mann‐Whitney test was used. Values are mean ± SEM with * P < .05 versus CIND‐ApoE4(−)
For the first time, neuroinflammatory markers (LCN‐2, S100B, NPTX‐2, and ANGPTL‐4) were evidenced in pEVs, with the same range levels in pEVs and in plasma except for S100B, which was 100‐fold higher in pEVs (Figure 4A‐D). In CIND‐APOE ε4+, LCN‐2 levels were lower in plasma and in pEVs, whereas S100B, NPTX‐2, and ANGPTL‐4 were selectively lower in pEVs as compared to plasma (Figure 4A‐D).
FIGURE 4.

Levels of inflammatory factors in pEVs and in plasma. Levels of lipocalin, S100B, pentraxin‐2, and ANGPTL‐4 measured in pEVs (A, C, E, G) and in plasma (B, D, F, H). Graph points indicate individual values for every participant. (−), ε4 non‐carriers; (+), ε4 carriers; apoE, apolipoprotein E; CIND, cognitively impaired, no dementia; EVs, extracellular vesicles. Statistical analysis was performed using the Student t test for S100B and pentraxin‐2 (EVs and plasma) and plasma lipocalin and the Mann‐Whitney test for ANGPTL‐4 (EVs and plasma) and EVs lipocalin. Values are mean ± SEM with * P < .05 versus CIND‐ApoE4(−)
To further characterize the influence of the APOE ε4 variant on the changes of vesicular and plasma protein concentration, we have determined the linear regression causal relationship between the ε4 allele (as dependent variable), and the studied proteins (as independent variables). Accordingly, the presence of the APOE ε4 allele was negatively associated with the pEVs levels of NPTX‐2, ANGPTL‐4, DJ‐1, S100B, and PrGN as well as the plasma level of α‐Syn and LCN‐2 (Table 1).
TABLE 1.
Univariate correlation of plasma and EVs studied factors with APOE genotype among the study population
| NPTX‐2 EVs | ANGPTL‐4 EVs | DJ‐1 EVs | S100B EVs | PrGN EVs | α‐syn plasma | LCN‐2 plasma | ||
|---|---|---|---|---|---|---|---|---|
| ApoE | r | −.431 | −.415 | −.444 | −.518 | −.414 | −.475 | −.487 |
| p | .035* | .044* | .030* | .011* | .049* | .019* | .016* | |
| N | 24 | 24 | 24 | 23 | 23 | 24 | 24 |
ANGPTL‐4, angiopoietin‐like 4; EVs, extracellular vesicles; LCN‐2, lipocalin‐2; N, number of participants; NPTX‐2, pentraxin‐2; p, significance (2‐tailed); PrGN, progranulin; r, Pearson correlation coefficient; α‐syn, α‐synuclein.
, P < 0.05.
Our prospective follow‐up assessment revealed that among the eight CIND‐APOE ε4+ individuals, five developed AD, two developed VaD, and one remained CIND 5 years later. Concerning the CIND‐APOE ε4‐ group, six developed mixed type of dementia, five remained CIND, four developed VaD, and one developed AD 5 years later. The same protein profile was compared between the subgroup of five CIND ‐AD APOE ε4+ patients and cognitively healthy older people. α‐Syn level in pEVs was lower in the CIND ‐AD APOE ε4+ group (Figure 5A), whereas no differences were observed for other proteins (Fig. S3). Of interest, a strong correlation was observed between NPTX‐2 and α‐Syn levels (Figure 5B) with the NPTX‐2/α‐Syn ratio being higher in the CIND‐AD APOE ε4+ group and negatively correlated with the MMSE scores (Figure 5C‐D).
FIGURE 5.
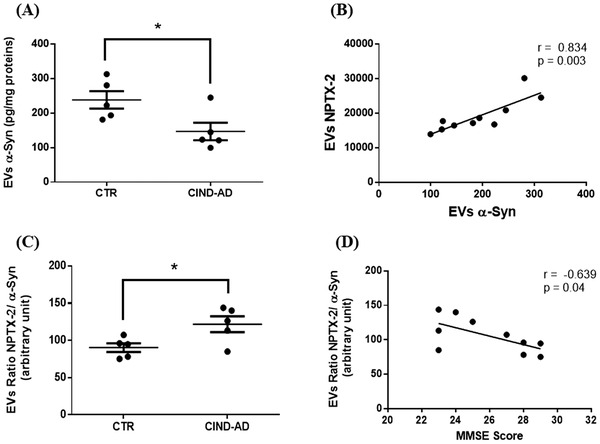
Levels and statistical correlation of pentraxin‐2 and α‐synuclein in pEVs with cognitive performance. (A) EVs levels of α‐Syn between controls and CIND‐AD patients. (B) Correlation between NPTX‐2 and α‐Syn levels in EVs. (C) EVs NPTX‐2/α‐Syn ratio between controls and CIND‐AD patients. (D) Correlation between EVs NPTX‐2/ α‐Syn ratio and MMSE score. Graph points indicate individual values for every participant. CIND‐AD, cognitively impaired, no dementia‐Alzheimer's disease; CTR, controls; EVs, extracellular vesicles; NPTX‐2, pentraxin‐2; p, significance (Student t test); r, Pearson correlation coefficient; α‐Syn, α‐synuclein. The correlation coefficient (Pearson r) and P values were determined using Pearson correlation. Statistical analysis was performed using the student t test for EVs α‐Syn levels and EVs NPTX‐2/α‐Syn ratio. Values are mean ± SEM
4. DISCUSSION
The inheritance of the APOE ε4 is the most important genetic risk factor associated with late‐onset AD and the major genetic predictor of sporadic AD progression, especially in patients with amnestic MCI. 27 To gain a thorough understanding of the role of APOE ε4 in AD susceptibility, we sought to determine its effects on the pEVs’ cargo proteins and in plasma from CIND participants.
We found that the presence of APOE ε4 did not affect the total density of pEVs. However, CIND‐ APOE ε4 participants released fewer pEVs in the range of 30 to 200 nm, which typically corresponds to EVs formed by an endosomal route. 28 The endocytic pathway plays an important role in Aß production, apoE function, and exosomes biogenesis. In brain, Cataldo et al. demonstrated that alteration of the endocytic pathway occurs very early intraneuronally, preceding Aβ deposition and AD neuropathological hallmarks. 29 In line with these intraneuronal changes, alteration of the endocytic pathway leads to decreased release of EVs in AD brain from APOE ε4 carriers. 17 Our results demonstrated that in periphery, pEVs release was lower up to 5 years before the clinical onset of AD, before changes in neuropathogenic proteins (Aβ and tau) in pEVs. Altogether these results demonstrated that in APOE ε4 carriers, EVs production was compromised very early in brain and in the periphery.
The impact of APOE ε4 on both pEVs number and protein cargo could be explained by pEVs enrichment in lipids. 30 EVs biogenesis depends largely on the interaction between the endosomal sorting complex required for the transport (ESCRT) and the membrane‐associated lipids (mainly cholesterol, sphingolipids, and ceramides). 8 Considering that the primary function of apoE is to transport cholesterol‐laden lipids that are essential to cell membrane structure and that APOE ε4 functions less efficiently than the APOE ε3 and APOE ε2 variants in this process, 31 suggest that APOE ε4 affect EVs formation and budding. Therefore, the pathophysiological crosstalk between APOE ε4 and the endosomal system could result in abnormal changes such as small particle size formation and selective protein processing in pre‐AD APOE ε4 carriers.
In CIND‐ APOE ε4+ participants, we found significant differences in trophic and inflammatory proteins in pEVs, which indicates early alteration of the pEVs cargo proteins and suggests an early reduction of cells to disseminate components by the endosomal‐lysosomal mechanism. Recently, we demonstrated that these proteins are released from neuronal‐derived EVs under stress conditions. 24 Because EVs were suggested to shuttle from the brain to the bloodstream by crossing the multiple layers of the blood‐brain barrier, 32 the pattern of these proteins in pEVs could be an indicator of the modifications of the inflammatory and neurotrophic status in the brain from CIND participants. In this context, a growing number of studies are focusing on isolating and analyzing the protein signature of plasma EVs enriched from brain origin. Of interest, some synaptic and neurotrophic proteins, as in our pEVs, were found to be altered in neuronal‐derived EVs. 32 , 33 However, the brain specificity of these EVs was based on the immunoreactivity of EVs surface marker proteins (specifically L1CAM), which are highly expressed in the CNS but also present in other peripheral cell types. Additional research efforts are needed to optimize methods that enable high‐yield capture of enriched brain EVs. Moreover, cumulative evidence now relates metabolic disorders to sporadic AD onset and can promote disease progression. 34 Hence, pEVs content might be relevant to disclose the role of systemic disorders in cerebral pathologies.
Our results revealed that some selected proteins (α‐Syn, NPTX‐2, S100B) were enriched in pEVs as compared to plasma and that pEVs displayed higher sensibility to detect protein variations, which suggest their use instead of plasma to reflect systemic changes. This is probably due to the presence of lipid bilayer membrane that surrounds and protects pEVs contents from the degradation by enzymes (ie, proteases), red blood cells, or liver and thus preserves them as a source of pathological and physiological information.
It was reported that NPTX‐1 was increased in plasma from MCI, which progressed to early AD as well as in brain and plasma from E4FAD mice (APOE ε4/ε4 / FAD+/−) following an infusion of Aß oligomers. 35 In pEVs, we did not observe any difference in NPTX‐2 level between healthy participants and CIND‐AD, which confirms that NPTX‐2 is increased only in the presence of an overexpression of Aβ, 36 which was not observed in CIND‐AD participants.
Misfolded proteins such as α‐Syn may spread through exosomes in the brain. 37 However, these pathological features are not limited to the brain; they can also be found in CSF. CSF α‐Syn levels were also associated with AD risk in preclinical and MCI individuals 38 , 39 and were increased in APOE ε4 MCI patients who progressed to AD. 40 pEVs α‐Syn could also be released by red blood cells, which highly expressed α‐Syn 41 and was found to be lower in APOE ε4 AD patients. 42 For the first time, we have evidence of the presence of α‐Syn in pEVs. In contrast to its CSF level, it is reduced in pEVs from CIND‐APOE ε4+ and from CIND‐AD APOE ε4+, indicating that its reduction in pEVs is observed up to 5 years before the clinical onset of AD. The observed decrease of the soluble form (monomers) could be related to an intracellular accumulation of α‐Syn in form of aggregates (polymeric forms). This finding is relevant, since experimental evidence has further linked α‐Syn accumulation to intracellular aggregation and hyperphosphorylation of tau 43 and also to the overproduction of Aβ via specific molecular interactions with presenilin 1. 44 Moreover, it has been suggested that apoE may be involved in the distribution of α‐Syn between the extracellular and intracellular matrix and that the APOE ε4 variant specifically reduces α‐Syn uptake. 45 These results underlie some biological processes that could explain the conversion of CIND patients to AD among APOE ε4 carriers.
α‐Syn is a presynaptic protein that controls exocytosis at various neurotransmitter systems in the brain by managing synaptic vesicles fusion, release, and recycling. 46 NPTX‐2 binds and induces clustering of postsynaptic ionotropic AMPA‐type glutamatergic receptors to regulate synaptic plasticity and to maintain long‐term potentiation. 47 , 48 Thus both synaptic proteins work simultaneously to ensure enhanced neuronal flux transmission. Our results suggest also that the NPTX‐2/α‐Syn ratio measured in pEVs allows an accurate classification of CIND participants that convert to AD. This ratio was related to the presence of the APOE ε4 allele and correlated with the MMSE scores. Taking into consideration that early memory decline in AD is attributed to synaptic loss, 49 , 50 this ratio may therefore be indicative of altered synaptic function and may reflect the extent of cognitive impairment between preclinical and clinical AD stages. Because it is broadly recommended in biomarker analysis to consider a combination of markers to obtain more informative data, the present ratio could provide a better sensibility assessment for early AD diagnosis.
5. CONCLUSIONS
This study provides comprehensive insight and enhances our knowledge of the emerging role of APOE ε4 in abnormal pEVs cargo proteins processing and the identification of blood‐based biomarkers. The measurement of ratio between NPTX‐2 and α‐Syn in pEVs might be an innovative strategy for monitoring the conversion of CIND patients to AD. However, it is important to note that the assessment of these markers only at the CIND stage might be insufficient to establish a solid pathological linkage. This implies the need to explore this ratio also in CIND patients after their conversion to AD in order to validate a cause‐and‐effect relationship. Another potential limitation of this study is the time span between plasma provision and marker assessment. In fact, plasma storage conditions can affect the level of some proteins, which tend to change when samples are frozen or exposed to different storage temperatures. However, the fluctuation of protein levels between different groups due to storage is limited because all of our plasma samples were kept in the same conditions. In addition, the sample size is adequate for statistical analysis but not large enough for epidemiological considerations. Therefore, these results require additional studies with larger samples of participants selected from the general population to establish this novel synapse‐derived ratio as a reliable biomarker to anticipate AD and an extensive analysis of pEV protein content with regard to the role of the APOE ε4 variant.
CONFLICT OF INTEREST
The authors report that they have no conflict of interest to disclose.
Supporting information
Figure S1. Levels of neurodegenerative factors in pEVs from CIND subjects. Levels of (A) Aβ1‐42, (B) p‐tau181, (C) t‐tau, (D) ratio of Aβ1‐42/p‐tau181.
Legends supplementary Figures
Figure S2. Levels of neurotrophic factors in pEVs and in plasma. Levels of NSE, APP, MMP9, and BDNF measured in pEVs (A, C, E, G) and in plasma (B, D, F, H).
Figure S3. Levels of neurotrophic and inflammatory factors in pEVs from healthy and CIND‐AD subjects.
Table S1 Inclusion and exclusion criteria of the study population
Table S2 Luminex limit detection sensitivity for all the studied proteins
Table S3 Clinical and sociodemographic characteristics of the studied subjects particiapnts
Table S4 Clinical and sociodemographic characteristics after stratification of the study population
ACKNOWLEDGMENTS
This study was supported by the Research Chair Louise & André Charron on Alzheimer's disease, the Armand‐Frappier Fondation, and MRIF (C.R.).
Ben Khedher MR, Haddad M, Laurin D, Ramassamy C. Apolipoprotein E4–driven effects on inflammatory and neurotrophic factors in peripheral extracellular vesicles from cognitively impaired, no demented participants converted to Alzheimer's disease. Alzheimer's Dement. 2021;7:e12124 10.1002/trc2.12124
REFERENCES
- 1. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;. 16(3):391‐460. [Google Scholar]
- 2. Edwards Iii GA, Gamez N, Escobedo G Jr, Calderon O, Moreno‐Gonzalez I. Modifiable risk factors for Alzheimer's disease. Front Aging Neurosci. 2019;11:146‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mattsson N, Groot C, Jansen WJ, et al. Prevalence of the apolipoprotein E ε4 allele in amyloid β positive subjects across the spectrum of Alzheimer's disease. Alzheimers Dement. 2018;14(7):913‐924. [DOI] [PubMed] [Google Scholar]
- 4. de Oliveira FF, Chen ES, Smith MC, Bertolucci PHF. Selected LDLR and APOE polymorphisms affect cognitive and functional response to lipophilic statins in Alzheimer's disease. J Mol Neurosci. 2020;70(10):1574‐1588. [DOI] [PubMed] [Google Scholar]
- 5. Jessen F, Amariglio RE, Buckley RF, et al. The characterisation of subjective cognitive decline. Lancet Neurol. 2020;19(3):271‐278.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jessen F, Amariglio RE, van Boxtel M, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer's disease. Alzheimers Dement. 2014;10(6):844‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lyketsos CG, Toone L, Tschanz JA, et al. Population‐based study of medical comorbidity in early dementia and “Cognitive Impairment, No Dementia (CIND)”: association with functional and cognitive impairment: the cache county study. Am J Geriat Psychiat. 2005;13(8):656‐664. [DOI] [PubMed] [Google Scholar]
- 8. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213‐228. [DOI] [PubMed] [Google Scholar]
- 9. Sardar Sinha M, Ansell‐Schultz A, Civitelli L, et al. Alzheimer's disease pathology propagation by exosomes containing toxic amyloid‐beta oligomers. Acta Neuropathol. 2018;136(1):41‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Skog J, Würdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. You Y, Ikezu T. Emerging roles of extracellular vesicles in neurodegenerative disorders. Neurobiology of Disease. 2019;130:104512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perrotte M, Haddad M, Le Page A, Frost EH, Fulöp T, Ramassamy C. Profile of pathogenic proteins in total circulating extracellular vesicles in mild cognitive impairment and during the progression of Alzheimer's disease. Neurobiol Aging. 2020;86:102‐111. [DOI] [PubMed] [Google Scholar]
- 13. de la Monte SM, Tong M. Brain metabolic dysfunction at the core of Alzheimer's disease. Biochem Pharmacol. 2014;88(4):548‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mathews PM, Levy E. Exosome production is key to neuronal endosomal pathway integrity in neurodegenerative diseases. Front Neurosci. 2019;13:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nuriel T, Peng KY, Ashok A, et al. The Endosomal‐lysosomal pathway is dysregulated by APOE4 expression in vivo. Front Neurosci. 2017;11:702‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao N, Liu C‐C, Van Ingelgom AJ, et al. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017;96(1):115‐129 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peng KY, Pérez‐González R, Alldred MJ, et al. Apolipoprotein E4 genotype compromises brain exosome production. Brain. 2018;142(1):163‐175.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM‐III‐R. 3rd ed. Washington DC: American Psychiatric Association; 1987. [Google Scholar]
- 19. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939‐944. [DOI] [PubMed] [Google Scholar]
- 20. World Health Organization . Tenth revision of the International Classification of Diseases, draft of chapter V, categories F00‐F99, mental, behavioural and developmental disorders In: Clinical Descriptions and Diagnostic Guidelines. Geneva: World Health Organization; 1987. [Google Scholar]
- 21. McKeith IG, Perry RH, Fairbairn AF, Jabeen S, Perry EK. Operational criteria for senile dementia of Lewy body type (SDLT). Psychol Med. 1992;22(4):911‐922. [DOI] [PubMed] [Google Scholar]
- 22. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders: DSM‐IV. 4th ed. Washington DC: American Psychiatric Association; 1994. [Google Scholar]
- 23. Román GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS‐AIREN International Workshop. Neurology. 1993;43(2):250‐260. [DOI] [PubMed] [Google Scholar]
- 24. Haddad M, Perrotte M, Khedher MRB, et al. Methylglyoxal and glyoxal as potential peripheral markers for MCI diagnosis and their effects on the expression of neurotrophic, inflammatory and neurodegenerative factors in neurons and in neuronal derived‐extracellular vesicles. Int J Mol Sci. 2019;20(19):4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haddad M, Perrotte M, Landri S, Lepage A, Fulop T, Ramassamy C. Circulating and extracellular vesicles levels of N‐(1‐Carboxymethyl)‐L‐lysine (CML) differentiate early to moderate Alzheimer's disease. J Alzheimers Dis. 2019;69(3):751‐762. [DOI] [PubMed] [Google Scholar]
- 26. Coumans FAW, Brisson AR, Buzas EI, et al. Methodological guidelines to study extracellular vesicles. Circ Res. 2017;120(10):1632‐1648. [DOI] [PubMed] [Google Scholar]
- 27. Scarabino D, Broggio E, Gambina G, Maida C, Gaudio MR, Corbo RM. Apolipoprotein E genotypes and plasma levels in mild cognitive impairment conversion to Alzheimer's disease: a follow‐up study. Am J Med Genet. 2016;171(8):1131‐1138. [DOI] [PubMed] [Google Scholar]
- 28. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cataldo AM, Peterhoff CM, Troncoso JC, Gomez‐Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157(1):277‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dinkins MB, Wang G, Bieberich E. Sphingolipid‐enriched extracellular vesicles and Alzheimer's disease: a decade of research. J Alzheimers Dis. 2017;60(3):757‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10(5):333‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mustapic M, Eitan E, Werner JK Jr, et al. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017;11:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goetzl EJ, Kapogiannis D, Schwartz JB, et al. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer's disease. FASEB J. 2016;30(12):4141‐4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease ‐ insights from amyloid‐β metabolism beyond the brain. Nat Rev Neurol. 2017;13(10):612‐623. [DOI] [PubMed] [Google Scholar]
- 35. Ma QL, Teng E, Zuo X, et al. Neuronal pentraxin 1: a synaptic‐derived plasma biomarker in Alzheimer's disease. Neurobiol Dis. 2018;114:120‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abad MA, Enguita M, DeGregorio‐Rocasolano N, Ferrer I, Trullas R. Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid‐β and is overexpressed in dystrophic neurites in Alzheimer's brain. J Neurosci. 2006;26(49):12735‐12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng T, Pu J, Chen Y, et al. Plasma exosomes spread and cluster around β‐Amyloid plaques in an animal model of Alzheimer's disease. Front Aging Neurosci. 2017;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berge G, Sando SB, Albrektsen G, et al. Alpha‐synuclein measured in cerebrospinal fluid from patients with Alzheimer's disease, mild cognitive impairment, or healthy controls: a two year follow‐up study. BMC Neurol. 2016;16(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vergallo A, Bun R‐S, Toschi N, et al. Association of cerebrospinal fluid alpha‐synuclein with total and phospho‐tau181 protein concentrations and brain amyloid load in cognitively normal subjective memory complainers stratified by Alzheimer's disease biomarkers. Alzheimers Dement. 2018;14(12):1623‐1631. [DOI] [PubMed] [Google Scholar]
- 40. Twohig D, Rodriguez‐Vieitez E, Sando SB, et al. The relevance of cerebrospinal fluid alpha‐synuclein levels to sporadic and familial Alzheimer's disease. Acta Neuropathol Commun. 2018;6(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Barbour R, Kling K, Anderson JP, et al. Red blood cells are the major source of alpha‐synuclein in blood. Neurodegener Dis. 2008;5(2):55‐59. [DOI] [PubMed] [Google Scholar]
- 42. Baldacci F, Daniele S, Piccarducci R, et al. Potential diagnostic value of red blood cells α‐synuclein heteroaggregates in Alzheimer's disease. Mol Neurobiol. 2019;56(9):6451‐6459. [DOI] [PubMed] [Google Scholar]
- 43. Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α‐synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31(21):7604‐7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winslow AR, Moussaud S, Zhu L, et al. Convergence of pathology in dementia with Lewy bodies and Alzheimer's disease: a role for the novel interaction of alpha‐synuclein and presenilin 1 in disease. Brain. 2014;137(Pt 7):1958‐1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ogaki K, Martens YA, Heckman MG, et al. Multiple system atrophy and apolipoprotein E. Mov Disord. 2018;33(4):647‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghiglieri V, Calabrese V, Calabresi P. Alpha‐synuclein: from early synaptic dysfunction to neurodegeneration. Front Neurol. 2018;9:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dong Z, Han H, Li H, et al. Long‐term potentiation decay and memory loss are mediated by AMPAR endocytosis. J Clin Invest. 2015;125(1):234‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elbaz I, Lerer‐Goldshtein T, Okamoto H, Appelbaum L. Reduced synaptic density and deficient locomotor response in neuronal activity‐regulated pentraxin 2a mutant zebrafish. FASEB J. 2015;29(4):1220‐1234. [DOI] [PubMed] [Google Scholar]
- 49. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789‐791. [DOI] [PubMed] [Google Scholar]
- 50. Xiao M‐F, Xu D, Craig MT, et al. NPTX2 and cognitive dysfunction in Alzheimer's Disease. eLife. 2017;6:e23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Levels of neurodegenerative factors in pEVs from CIND subjects. Levels of (A) Aβ1‐42, (B) p‐tau181, (C) t‐tau, (D) ratio of Aβ1‐42/p‐tau181.
Legends supplementary Figures
Figure S2. Levels of neurotrophic factors in pEVs and in plasma. Levels of NSE, APP, MMP9, and BDNF measured in pEVs (A, C, E, G) and in plasma (B, D, F, H).
Figure S3. Levels of neurotrophic and inflammatory factors in pEVs from healthy and CIND‐AD subjects.
Table S1 Inclusion and exclusion criteria of the study population
Table S2 Luminex limit detection sensitivity for all the studied proteins
Table S3 Clinical and sociodemographic characteristics of the studied subjects particiapnts
Table S4 Clinical and sociodemographic characteristics after stratification of the study population